

Abstract
Background. Endothelial cells (ECs) embedded in 3D matrices [matrix-embedded endothelial cells (MEECs)] of denatured collagen implanted around vascular access anastomoses preserve luminal patency. MEEC implant efficacy depends on embedded EC health. As the uremic milieu inhibits proliferation and induces apoptosis of ECs, we examined whether uremia might impact MEECs.
Methods. ECs grown on 2D gelatin-coated polystyrene tissue culture plates (gTCPS) or in MEEC were treated with sera pooled from 20 healthy control or uremic patients with end-stage renal disease. EC viability was examined using 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide assay, cell counting and Trypan blue exclusion. Media conditioned (CM) with 2 and 3D-supported ECs were examined for its potential to inhibit vascular smooth muscle cell (vSMC) proliferation using 3[H] thymidine incorporation and cyclin D1 expression. ECs grown on gTCPS were treated with uremic serum filtered through matrices to examine if matrices retain uremic toxins or whether EC effects were cell mediated.
Results. Uremic serum significantly reduced viability and number of live, and increased dead ECs when grown on gTCPS, but not in MEECs. EC survival correlated with vSMC inhibition. While CM from ECs grown in gTCPS with uremic serum inhibited vSMC proliferation no better than uremic serum alone (22 versus 27%), MEEC CM inhibited vSMC proliferation by 47% (P = 0.0004). Cyclin D1 expression tracked with indices of vSMC proliferation. There was no significant difference in EC viability between EC treated with matrix-filtered or unfiltered uremic serum.
Conclusion. The viability, number and efficacy of MEECs were preserved in uremic serum compared to those of ECs on gTCPS. MEECs are protected from uremic toxicity, not from retention of uremic toxins by matrices, but likely from intrinsic changes in EC sensitivity to uremia. MEECs implanted at vascular access sites should inhibit neointimal hyperplasia in uremia. This study underscores the robustness of matrix embedding as a cell protectant, especially in hostile environments like uremia.
Keywords: endothelial dysfunction, matrix-embedded endothelial cells, uremia
Introduction
Vascular access is crucial for hemodialysis (HD) patients and the access dysfunction is the major cause of morbidity and mortality in these patients [1]. It is a leading reason for hospitalization among patients with end-stage renal disease (ESRD) and costs the United States $1billion annually [2]. Access dysfunction is a complex multifactorial process and is predominantly due to thrombosis and neointimal hyperplasia at the site of anastomosis [3].
Endothelial cell (EC) dysfunction is universal in patients with chronic renal failure (CRF) [4, 5]. Plasma from patients with ESRD, or various candidate uremic toxins, inhibit fundamental EC biological processes such as viability, proliferation, migration and wound healing [6, 7]. Abnormalities in these EC functions play a particularly crucial role in vascular remodeling, especially at the site of dialysis vascular access. Arteriovenous fistula and graft anastomoses require rapid proliferation of ECs to restore the barrier, permeability, biochemical regulatory functions of ECs in controlling vascular repair, local thrombosis, inflammation and neointimal hyperplasia [8]. Since uremia inhibits EC proliferation and migration, and eventually results in abnormal vascular remodeling, neointimal hyperplasia at the site of anastomosis of vascular access is not uncommon. This results in primary access failure and ineffective dialysis [9].
Embedding ECs within 3D gelatin matrices creates an engineered construct of matrix-embedded endothelial cells (MEECs) that allows preservation of EC viability for months, transport of cells with ease and facile placement in the perivascular space where they effectively mediate vascular repair [10]. MEECs inhibit neointimal hyperplasia and stenosis when implanted around sites of controlled vascular injury in animals [11] and thrombosis in clinical settings of vascular access [12]. ECs within these matrices are thought to exert their beneficial effects by secreting antiproliferative, antithrombotic and anti-inflammatory agents that promote vascular repair [13]. Thus, MEEC implants (Vascugel®; Pervasis Therapeutics) are being explored as therapeutic modalities to maintain vascular access patency.
Despite the wealth of preclinical data in a number of animal models and the recent demonstration of safety of MEECs in humans with ESRD [12], relatively little is known about the effects of uremia on MEECs. All previous preclinical studies were performed in a nonuremic environment [11, 13]. Since the uremic milieu is fundamentally different than normal in many respects—including biochemical, immunological and oxidation states—it is important to evaluate the efficacy of MEECs in the uremic milieu. As uremia reduces the viability and numbers of ECs grown on tissue culture polystyrene plates, we set out to evaluate whether uremia exerts a similar effect on the MEECs. This is especially important since the viability and functional potential of the MEECs are crucial for their optimal efficacy to preserve the patency of vascular access. This study compares EC versus MEEC viability, number and functional potential to inhibit vascular smooth muscle cell (vSMC) proliferation in the uremic milieu.
Materials and methods
Recruitment of study participants
Patients with ESRD on HD were recruited randomly from a pool of 150 patients undergoing maintenance HD at the DeVita Hemodialysis Center (Boston, MA). The protocol was approved both by Institutional Review Boards of Boston University Medical Center and Massachusetts Institute of Technology. Patients who were 20–75 years of age were recruited over a 6-month period and excluded if their hemoglobin concentration was <8 g/dL. Informed consent was obtained and 10 mL of blood collected prior to the next subsequent HD session. Control sera was from Research Blood Components Inc. (Boston, MA) for gender- and ethnicity-matched patients. Blood was collected in vacutainer tubes and serum separated by centrifuging clotted blood at 1100 g for 10 min at room temperature. Serum was filtered using 0.45 μM filters (Becton Dickinson), pooled and frozen in aliquots at −80°C. Blood urea nitrogen, creatinine and glucose were assayed in all patients, and control sera excluded with creatinine >1.0 mg/dL.
Cell culture and antibodies
Humanaortic endothelial cells (HAECs) and umbilical vein endothelial cells (HUVECs) (Promocell, Heidelberg, Germany) pooled from three donors were grown in endothelial growth medium-2 (EGM-2) (Promocell). EGM-2 was prepared by supplementing endothelial basal medium (EBM-2) with fetal bovine serum (FBS, 2%), hydrocortisone (1 μg/mL), fibroblast growth factor-1 (10 ng/mL), epidermal growth factor (5 ng/mL), insulin-like growth factor (20 ng/mL), ascorbic acid (1 μg/mL) and heparin (90 μg/mL). Tissue culture polystyrene plates coated with gelatin (0.1% gelatin A; Sigma, St. Louis, MO) served as 2D cell support (gTCPS). Human vSMCs were grown in MCDB131 medium (Gibco, Carlsbad, CA) with 10% FBS and 1% penicillin and streptomycin (Gibco). CD31 and VCAM-1 antibodies were acquired from Santacruz Biotechnology. Cyclin D1 and actin antibodies were from Biovision (Mountain View, California) and Invitrogen (Carlsbad, CA), respectively.
Generation of MEECs
MEECs were generated by culturing ECs within sterile denatured collagen matrices without cross linkers (Pfizer, New York, NY). The matrices were cut into 0.6 × 0.6 × 0.3 cm blocks and hydrated in EGM-2 at 37°C for 2–48 h (Figure 1a). An average pore size of 212 + 132 μm (Figure 1b) optimized growth of cells (Figure 1c). ECs were suspended in EGM-2 medium and 40 000 cells seeded onto hydrated matrix blocks and allowed to attach for 1.5 h. Matrices were transferred to free-standing 30-mL polypropylene tubes containing 10 mL of EGM-2, and cultured for up to 3 weeks at 5% CO2 at 37°C, with media changed every 48–72 h. Samples from each lot were digested with collagenase (Type I, Worthington Biochemicals) and cell seeding efficiency determined with a Z1 Coulter particle counter (Beckman Coulter; Fullerton, CA). At the end of 3 weeks, each matrix had ∼0.3 × 106 cells. MEECs are positive for CD31, Platelet endothelial cell adhesion molecule (PECAM) and vascular cell adhesion molecule (VCAM-1) both of which are EC markers confirming that MEECs maintain EC phenotype (Figure 1d and e). Matrices were separated randomly and grown in the EBM-2 without any growth factor supplements with 10% pooled control or uremic serum for 48 h.
Fig. 1.
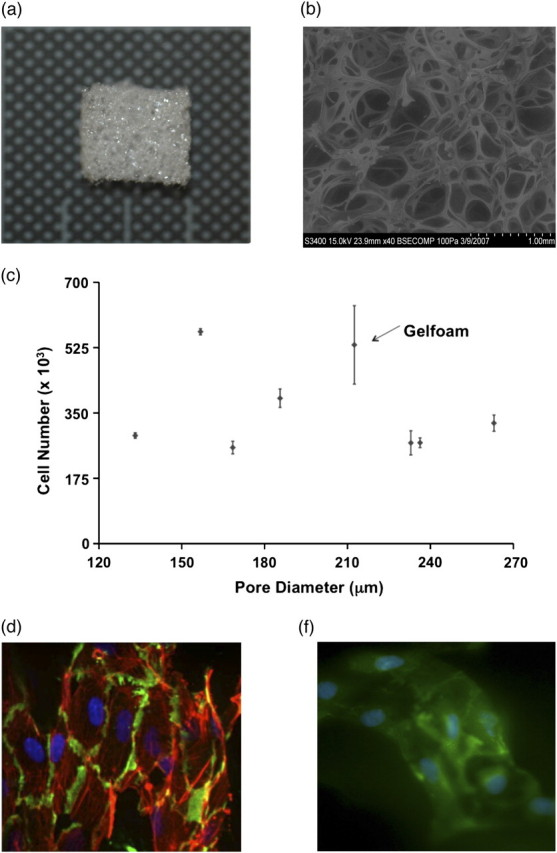
(a) The 3D gelatin matrix (Gelfoam) used in this study. Matrices were cut in 0.6 × 0.6 × 0.3 cm blocks seeded with HAECs at a density of 106 cells/mL. (b) The average pore size of the 3D Gelfoam matrix is 212 ± 132 μm. (c). 80 000 HAECs seeded on matrices with different pore size were allowed to grow for 3 weeks. There was no correlation between the pore size and the number of ECs, The pore size of 212 μm provided the highest density of ECs. (d) MEECs were fixed, cryosectioned, and stained for DAPI (nuclei), PECAM (green) and rhodamine phalloidin (red). (e) MEECs fixed and stained for DAPI (nuclei) VCAM-1 (green).
Preparation of scaffolds for staining
Gelfoam scaffolds were cultured for the desired time, rinsed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde overnight at 4°C. All subsequent steps were carried out on ice or at 4°C. Following fixation, scaffolds were washed thrice for 5 min with PBS followed by a 10 min incubation with 200 mM glycine in PBS to quench remaining free aldehyde groups. Scaffolds were again washed thrice for 5 min with PBS and then transferred to ice cold 18% sucrose in PBS for 3 h, followed by ice cold 30% sucrose for an additional 3 h. Scaffolds were then washed thoroughly with PBS and flash frozen with liquid nitrogen. Sections 20–60 μ thick were cryotome cut and captured on positively charged slides. (SuperFrost Plus; VWR) Sections were stored at −80°C for up to 3 weeks before staining.
Immunofluorescence
Slides were allowed to reach room temperature and a PAP pen (Electron Microscopy Sciences, Hatfield, PA) used to create a hydrophobic barrier around each section. Sections were washed twice with PBS and then permeabilized with 0.2% Triton X-100 in PBS for 10 min. Sections were blocked for 1 h in 1% bovine serum albumin (BSA) + 20% goat serum in PBS. Cells were immediately incubated in primary antibody to PECAM (1:50, mouse anti-porcine CD31; Serotec) or VCAM-1 (1:50, mouse monoclonal IgG1) in 1% BSA/PBS overnight at 4°C in a humidified chamber. Sections were then washed thrice for 5 min with 1% BSA/PBS and incubated for 1 h in the dark at room temperature with a fluorescent secondary antibody (Alexa 488 conjugated goat anti-mouse IgG, 1:75; Invitrogen) in 1% BSA/PBS. In some cases, a 1:200 dilution of rhodamine phalloidin was added to the secondary antibody solution. Finally, sections were washed thrice for 5 min with PBS and coverslipped using a fade resistant mounting medium containing DAPI, a blue fluorescent nuclear stain (Vectashield with DAPI, Vector Labs, Burlingame, CA). Stained samples were stored in the dark at 40C until imaged.
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide assay
EC viability and proliferation in matrices or on gTCPS was determined with 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT). About 0.15 × 106 ECs seeded into 35 mm gTCPS were grown in the incubator at 5% CO2 at 37°C. After 48 h, both the groups containing EC and MEECs had 0.2–0.3 × 106 ECs. The medium was then switched to EBM-2 without growth supplements and with 10% control or uremic serum for 48 h. The supernatants collected were labeled as conditioned media (CM). MTT (Roche) reduction by EC was determined using the manufacturer’s protocol. The blue formazan product was extracted with a sodium dodecyl sulfate (SDS)/dimethylformamide mixture and 100 μL of the solutions were transferred to 96-well plates and absorbance measured at a wavelength of 570 nm with background subtraction at 660 nm on a spectrophotometric plate reader (Molecular Devices Versamax tunable microplate reader).
Coulter counting and Trypan blue dye exclusion assay
EC number was determined by direct cell counting and Trypan blue (Sigma) exclusion. ECs seeded on gTCPS or within matrices were treated with control or uremic serum for 48 h and washed thrice in ice-cold PBS to complete removal of residual serum. ECs on gTCPS were trypsinized using 0.5% Trypsin ethylenediaminetetraacetic acid (EDTA) (Gibco) and resuspended in 1 mL of PBS. The total number of cells was determined using a Z1 Coulter particle counter (Beckman Coulter). The 3D matrices were digested with collagenase (Type I, Worthington Biochemicals) and cells harvested and examined [14]. For Trypan blue dye exclusion, 20 μL of cell suspension was added to 80 μL of 0.4% Trypan blue (Invitrogen), mixed and allowed to stand for 5 min at room temperature. About 20 μL of the trypan blue cell suspension was transferred to a Neubauer chamber (hemocytometer Hausser Scientific) and visualized under light microscopy (Nikon). Cells that took up the dye were considered dead and live cell number was calculated by subtracting the number of dead cells from the total number of counted cells.
vSMC proliferation assay
1 × 104 human vSMCs seeded in 48-well plates were synchronized overnight by serum starvation with 0.1% FBS. Medium was then switched for 16–18 h to the 5% human serum-treated CM collected from ECs on gTCPS or in MEECs. vSMCs grown with 5% control and uremic serum served as controls. One μCi of 3[H] thymidine (Perkin-Elmer, Boston, MA) was added to each well overnight and the cells were harvested after 16–24 h of treatment. Cells were washed with ice-cold PBS thrice followed by the precipitation of protein in 10% trichloroacetic acid in PBS for 30 min at 4°C, washed with 90% ethanol and solubilized in 1 mL of 0.25 M NaOH with 0.1% SDS for 1 h. Samples were added to scintillation cocktail and radioactivity measured by liquid scintillation counting (Beckman Coulter)
Immunoblotting
Cells were lysed in 50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 30 mM EDTA, 0.5% Triton X-100 with complete protease inhibitor (Roche). About 20 μg of protein samples were resolved on SDS–polyacrylamide gel electrophoresis and immunoblotted. The signal was developed using enhanced chemiluminescence substrate (Pierce Laboratories) according to the manufacturer’s instructions. The bands were normalized to β-actin and quantified using ImageJ (National Institutes of Health). Backgrounds were subtracted to assign densitometry values.
Statistical analysis
Parameteric data are represented as mean ± standard deviation and compared using two-tailed t-test with equal variances. The results were considered significant for P < 0.05.
Results
Patient characteristics
All but one of the 22 patients on HD approached consented and none required to be excluded for anemia. One blood sample was excluded, as it failed to clot. Two serum samples from 22 ‘healthy’ volunteers were excluded as their serum creatinine levels were 1.5 and 6.8 mg/dL, respectively. HD patients were 48 ± 13 (27–73 range) years old and were predominantly African-American males (Table 1). There was a significant difference between the ages and blood pressures between the control and uremic groups. Blood urea nitrogen and creatinine levels were significantly higher and hemoglobin levels were significantly lower in the uremic patients (Table 1).
Table 1.
Patient characteristics
| Control (N = 20) | Uremic (N = 20) | P-value | |
| Age | 25–59 (40)a | 27–73 (48) | 0.01 |
| Gender | |||
| Male | 15 | 15 | |
| Female | 5 | 5 | |
| Ethnic background | |||
| African-American | 16 | 16 | |
| Hispanic | 3 | 3 | |
| Asian Indian | 1 | 1 | |
| Caucasian | 0 | 0 | |
| Cause of renal failure | |||
| DM | 0 | 11 | |
| HTN | 0 | 5 | |
| Lupus nephritis | 0 | 1 | |
| Unknown | 0 | 3 | |
| Blood pressure (mm Hg) | |||
| Systolic blood pressure | 122.3 ± 10.5 | 169.65 ± 15.64 | 0.007 |
| Diastolic blood pressure | 77.45 ± 4.9 | 89.95 ± 6.9 | 0.003 |
| Hemoglobin (g/dL) | 13.8 ± 1.04 | 10.50 ± 1.39 | 0.0001 |
| Blood urea nitrogen (mg/dL) | 16.7 ± 2.73 | 76.85 ± 12.32 | 0.0001 |
| Serum creatinine (mg/dL) | 0.77 ± 0.11 | 6.27 ± 1.20 | 0.0001 |
| Serum glucose (mg/dL) | 73.65 ± 8.99 | 102.05 ± 27.28 | 0.0001 |
Data presented as range (average).
Uremic serum reduced the viability of ECs grown in gTCPS
ECs 0.25–0.3 × 106 grown on gTCPS were exposed to 10% control or uremic serum. Uremic serum reduced viability identically in HUVEC (32%, P = 0.0016 compared to control serum) and HAEC (30%, P = 0.006). Increasing serum concentration to 15% significantly increased EC viability by 20% (P = 0.006) in control serum but induced even greater EC damage with uremia. At this serum concentration, EC survival was 46% lower in uremic serum (P = 0.03) (Figure 2c).
Fig. 2.

Uremic serum reduces the viability of ECs grown on gTCPS. HUVECs (a) and HAECs (b) seeded in six-well plates were treated with 10% control or uremic serum in EBM2 for 48 h. The cells were washed and subjected to MTT assay. Mean results of six samples are shown. Error bars = SD. The Student’s t-test was applied to determine statistical significance. (c) HAECs seeded in the six-well plates were treated with 10 or 15% control or uremic serum for 48 h. The cells were subjected to MTT assay. Mean results of six samples are shown. Error bars = SD.
Uremic serum reduces the total number of ECs grown on gTCPS but not within 3D matrices
To determine if uremia halted cell proliferation or exerted toxic effects, we examined the numbers of live and dead cells under different serum conditions (Figure 3a). About 0.25–0.3 × 106 HAECs on gTCPS were exposed to 10% control or uremic serum. Consistent with EC viability (Figure 2a), uremic serum reduced the number of live cells after 48 h by 26% (P = 0.019) and increased cell death by 33% (P = 0.008). MEECs were resistant to uremic serum. For seeding densities equal to that when ECs were cultured on gTCPS, we observed no differences in the number of live or dead MEECs between control and uremic sera in MEEC (Figure 3b).
Fig. 3.

Uremic serum reduces number of ECs grown in gTCPS but not of MEECs. (a) HAECs seeded in six-well plate were treated with 10% control and uremic serum for 48 h. After trypsinization, the cells were counted with a Coulter counter and after Trypan blue staining. Mean results of six samples are shown. Error bars = SEM. (b) HAECs seeded in matrices were treated with 10% control and uremic serum for 48 h. The collagen was digested using collagenase and the cells were counted in Coulter counter and after Trypan blue staining. Mean results of five samples are shown. Error bars = SEM.
CM from MEECs grown in uremic serum inhibits vSMC proliferation more than that from ECs cultured on gTCPS
ECs inhibit vSMC proliferation [6, 7, 15, 16]. Uremic serum alone inhibited vSMC proliferation by 27% compared to control serum (P = 0.002). CM from ECs grown on gTCPS with uremic serum inhibited vSMC proliferation by 23% (P = 0.0009) compared to control serum (Figure 4a), thus not better than serum alone. MEECs retained their regulatory function even in uremic serum inhibiting vSMC proliferation by 46% (P = 0.0004) compared to the control serum (Figure 4a)—equivalent to expected inhibition by ECs in healthy environments [6, 7, 15, 16]. Levels of the critical cell cycle regulator cyclin D1 correlated with vSMC number. Cyclin D1 expression was reduced when vSMCs were treated with CM from MEECs compared to that from ECs on gTCPS and lowest with CM from MEECs in the presence of uremic serum (Figure 4b and c). This result indicates that uremic serum suppresses vSMC proliferation. However, CM from MEECs exerted maximal suppression of vSMC proliferation.
Fig. 4.
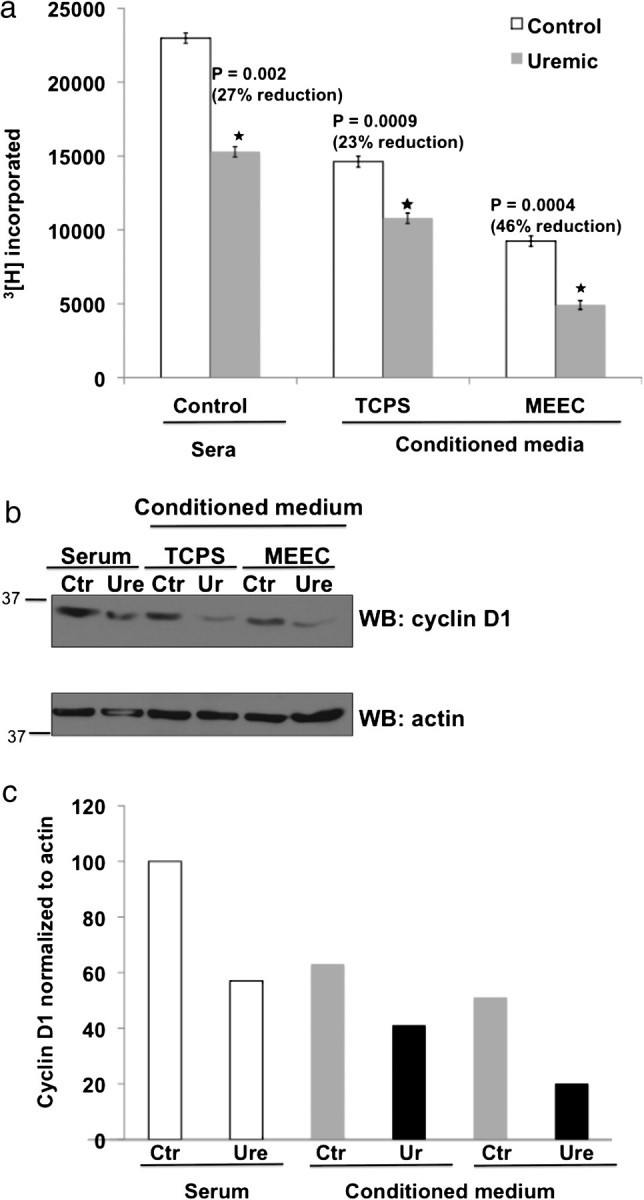
Conditioned medium obtained from MEECs has a greater inhibitory effect on vSMC proliferation compared to that from ECs grown on TCPS. (a) The supernatant of HACEs grown in 10% control or uremic sera for 48 h were considered to be CM. VSMCs seeded and serum starved in 0.1% FBS for 16 h were treated with CM diluted 1:2 (5% human serum) for 16 h followed by addition of 1 μCi 3[H] thymidine per well. The inclusion of 3[H] thymidine was assayed after 24 h of addition of radioactivity. Mean results of six samples are shown. Error bars = SD (b) Cell lysates of vSMCs treated for 48 h with 5% sera (control = Ctr, uremic = Ure) or CM from gTCPS and MEEC were probed for cyclin D1. Actin served as loading control. Representative immunoblot from two experiments. (c) Quantification of immunoblot using densitometry with cyclin D1 normalized to actin signal.
EC deplete matrices do not retain or prevent diffusion of uremic toxins
We observed enhanced survival of MEECs compared to ECs grown on gTCPS in uremic serum (Figures 2 and 3). To investigate the possibility that the matrices serve as a physical barrier or retain uremic toxins, HAECs grown on gTCPS were treated for 48 h with 10% uremic serum filtered through acellular matrices. Unfiltered serum served as control. There was no significant difference in EC survival between matrix-filtered or unfiltered serum (Figure 5). These data suggest the Gelfoam matrices do not serve as a physical barrier to the uremic solutes. It is likely that the ECs grown on Gelfoam matrices acquire morphological and functional changes that make them inherently resilient to uremic toxicity.
Fig. 5.

Uremic serum filtered through acellular matrices equally inhibits EC survival compared to unfiltered serum. HAECs grown on gTCPS were treated for 48 h with uremic serum 10% filtered through EC-deplete Gelfoam matrices (without ECs). Unfiltered serum served as control. EC survival was measured by MTT assay. Mean result of two experiments is shown. Error bars = SEM.
Discussion
Uremia-induced endothelial dysfunction affects several fundamental EC biological functions. Uremic serum adversely affects the viability, proliferation and regulatory potency of ECs grown in traditional 2D tissue culture but not when these same cells where embedded within 3D matrices (as MEECs). The interaction of ECs with a physiological substrata allows them to retain the full spectrum of their biological control properties even in settings of pathological stress and may even add to their efficacy in the setting such as uremia. These data offer specific insight as to how intact ECs withstand the initial insult of uremia, the impact of uremia in disrupting EC–basement membrane interactions and how MEECs can inhibit intimal hyperplasia in dialysis vascular access of uremic patients.
Endothelial dysfunction manifests in protean ways including reduced vasodilatory and permeability control capacity, increased secretion of inflammatory cytokines, upregulation of leukocyte adhesion molecules and a shift toward a prothrombotic profile [4, 5]. The uremic milieu adversely affects fundamental EC functions [17], e.g. via induction of apoptosis and inhibition of proliferation [6]. It has been traditionally thought that the clinical effects of uremia arise from accumulation of uremic toxins normally eliminated by the kidneys. There are numerous candidate uremic toxins including low molecular weight proteins, β2-microglobulin, leptin, advanced glycosylation end products (AGEs), parathyroid hormone, asymmetric dimethylarginine, p-cresol, indoxyl sulfate and homocysteine [7, 18]. A number of these compounds induce endothelial dysfunction. Individual uremic toxins such as oxalic acid, p-cresol and indoxyl sulfate reduce EC proliferation and migration significantly [19, 20]. AGEs and oxidized low-density lipoprotein are both increased in CRF patients and both reduce EC viability and induce EC apoptosis [21, 22]. Thus, the retention of uremic toxins within circulating serum and subsequent contact with vascular endothelium is considered to be a major mechanism for uremia-induced endothelial dysfunction. We validated these effects of uremic serum when ECs were grown on gelatin-coated tissue culture polystyrene and demonstrated concomitant loss of EC inhibition of VSMC proliferation (Figure 4a). It is likely that uremia has a general inhibitory effect on a range of cell types, and the untoward effects of uremia on EC biology can be ascribed to the direct toxicity of uremic toxins.
We generated MEECs by culturing ECs within commercially available Gelfoam matrices that contain compressed denatured collagen without exogenous cross linkers [13, 14]. The 212-μm pore size optimizes exchange of nutrients, discharge of metabolites and cell growth (Figure 1c). The number of ECs seeded on the matrices was chosen to optimize cell seeding and final outcome of the MEEC construct (S. Murikipudi, unpublished observations). We previously showed the growth curve of ECs within gelatin matrices over time [23]. ECs within the 3D structure of these porous matrices adopt an EC phenotype (Figure 1d and e) can be transported with ease and can be readily implanted within a range of animal models to regulate tissue repair including arteriovenous fistula models [13, 14, 23]. Allogeneic and even xenogeneic MEEC implants are effective cellular regulators in animals and humans without engendering an immune response because of the nature of the matrix substratum that supports the embedded ECs. Embedding of ECs within 3D matrices containing pores and microarchitecture of physiological dimensions likely mitigates excessive stress associated with injury, mechanical strain, denudation or exposure to pharmacological doses of growth factors and allow ECs to attain a state resistant to exposure to EC toxins [10, 24, 25].
Boston Medical Center is located in the inner city area of Boston consisting predominantly of an African-American population. We attempted to select age, gender and ethnic background matched controls. Therefore, African-American subjects dominate our study. The dialysis population typically consists of patients of advanced age and despite our best attempt it was difficult to get a matching control such an age range without other confounding ailments such as hypertension and diabetes. This explains the age discrepancy. The age of the patients from which sera were obtained can influence ECs by a number of conceivable mechanisms including increase in age-related factors such AGEs. Regardless, while ECs in 2D culture were sensitive to the older uremic serum, ECs in Gelfoam were protected from these effects. Thus, the age difference between the control and uremic groups did not influence the results in this study.
We confirmed that the protective effect of 3D substrata for a range of ECs. The total number of cells within a given plate or matrix is the net balance of cell proliferation, adhesion and death. Uremia inhibits EC proliferation, and increases apoptosis, and may well inhibit cell retention on culture surfaces. Thus, uremic serum should reduce the number of live cells in culture. Our observations of loss of EC number and function in 2D culture with exposure to uremic serum are consistent with that of other investigators [6, 19, 20] and at odds with others [26]. Consistent with almost 30% loss in EC viability with 10% uremic serum (Figure 3a), Cardinal et al. [6] reported that 20% uremic plasma reduced proliferation of human coronary artery ECs by 10% and increased apoptosis by 36%. Dou et al. [20] observed an inhibition of HUVEC proliferation ranging from 21 to 54% with p-cresol and indoxyl acetate. In contrast, Aznar-Salatti et al. [26] observed that the viability of HUVECs remains unaffected in the uremic serum. In a fascinating manner, they grew their HUVECs in M199 medium without supplemental growth factors and the absence of this growth stress may explain their findings. In the absence of growth stimuli like vascular endothelial growth factor or Fibroblast growth factor-2 ECs attain a different phenotype [27].
The MTT assay depends on the conversion of MTT dye to blue formazan by metabolically active cells. Cholesterol interferes with the formazan product and leads to spuriously low cell viability [28]. Since serum cholesterol levels are high in patients with CRF and the cholesterol content is likely to be increased in ECs in response to uremic serum [6], we further confirmed the reduced EC viability by trypan blue dye exclusion assay. A significant reduction in EC viability in uremic serum was corroborated with the reduction in the number of live ECs (Figures 2 and 3). This may explain [12] the observation why blood vessels from the uremic patients show wide areas of EC denudation [29].
Uremia is vasotoxic and uremic serum is toxic to ECs—how then does matrix embedding protect ECs? It is possible gelatin scaffolds retard the diffusion of the uremic toxins. Yet, neither the dimensions of the matrices nor our experimental findings support such a mechanical or physical effect (Figure 5d). Uremic serum passed through Gelfoam matrices is as toxic as unfiltered serum. Indeed, the matrix pore size of 132–212 μm (Figure 1b) should allow free diffusion of all the uremic toxins. Taken together, these data and calculations suggest that the matrices provide a protective environmental niche for ECs and enable ECs to retain their regulatory phenotype. Although ECs grown on gTCPS and MEECs exhibit similarities in many respects [11], there are significant differences. MEECs show different gene expression profiles compared to those of ECs grown on gTCPS [25]. For example, ECs grown on gTCPS have higher expression of integrins α1, α2b, αv and β3, while MEECs express more αx and β4 [25]—effects which will likely alter outside-to-in signaling. In addition, ECs on gTCPS and in three dimensional matrices have different intracellular signaling, such as in the JAK–STAT pathway [25]. Therefore, it is conceivable that MEECs exhibit altered inside-to-out and out-to-inside signaling to endow survival benefits, thus making MEECs more resilient to the uremic milieu. It is also plausible that ECs grown on Gelfoam matrices have different basement membrane compositions which may impart favorable survival benefit to MEECs. Overall, matrix-embedding bestows resilience to ECs by multiple potential mechanisms, which are under active investigation in our laboratory.
The present study is not without limitations. This study examines the effects of matrix embedding on EC phenotype in response to uremic stress. Therefore, all the other variables including the gelatin substratum and the numbers of ECs at the beginning of exposure to uremic serum were kept constant. However, the HAECs were grown in Gelfoam matrices for 3 weeks, while the ECs grown on gelatin-coated gTCPS for 48 h prior to the exposure to uremic or control serum. This inherent limitation arises from the impossibility of maintaining ECs for 3 weeks on gTCPS. It is likely that maintaining cells in such a 3D environment for 3 weeks alters the cell characteristics and that of the subendothelial matrix, making them more resilient to stress. Second, the in vivo uremic milieu is multifaceted and is difficult to model completely in vitro. The uremic serum used in these studies cannot therefore be considered to be identical to in vivo conditions as they likely lose the acid–base derangements and redox stress.
Uremia is a devastatingly toxic environment to ECs. Uremia-induced EC dysfunction adversely affects vascular remodeling that culminates in access failure. Maintaining vascular access patency is of paramount importance to the survival of ESRD patients. The technology of three dimensional matrix-embedding of ECs provides a means to generate constructs that can be implanted perivascularly to regain the multifaceted and potent endothelial control at the site of vascular injury to inhibit thrombosis and neointimal hyperplasia [12, 23]. Matrix Embedding protects ECs from uremic toxicity and allows them to function optimally even in the uremic toxins thus, these implantable construts are likely to enhance vascular access patency in CRF patients. Use of these MEEC constructs may help not only in resolving neointimal hyperplasia at the site of vascular access anastomoses but also understanding dynamics of tissue repair in uremia.
Acknowledgments
The authors thank Dr Laura Dember for help with IRB application at Boston Medical Center, Drs Dember, Adam Segal, Jasvinder Bhatia and Aylit Schultz (Boston Medical Center and Boston University School of Medicine) for patient samples and Dr Ramon Bongeio (Boston University School of Medicine) for in-depth review of the manuscript and insightful comments. This work was funded in part by R01 GM-49039 (ERE), NIDDK K08 DK080946 (VCC) from the National Institutes of Health and Young Investigator Award, National Kidney Foundation, USA (VCC). Part of this work will be presented at American Society of Nephrology annual meeting in November 2011 in Philadelphia, Pennsylvania.
References
- 1.Feldman HI, Kobrin S, Wasserstein A. Hemodialysis vascular access morbidity. J Am Soc Nephrol. 1996;7:523–535. doi: 10.1681/ASN.V74523. [DOI] [PubMed] [Google Scholar]
- 2.United States Renal Data System. Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United State, National Institute of Diabetes and Digestive and Kidney Disease. Bethesda, MD: 2007. [Google Scholar]
- 3.Roy-Chaudhury P, Spergel LM, Besarab A, et al. Biology of arteriovenous fistula failure. J Nephrol. 2007;20:150–163. [PubMed] [Google Scholar]
- 4.Yu Y, Lyons TJ. A lethal tetrad in diabetes: hyperglycemia, dyslipidemia, oxidative stress, and endothelial dysfunction. Am J Med Sci. 2005;330:227–232. doi: 10.1097/00000441-200511000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Dandona P, Chaudhuri A, Aljada A. Endothelial dysfunction and hypertension in diabetes mellitus. Med Clin North Am. 2004;88:911–931. doi: 10.1016/j.mcna.2004.04.006. x-xi. [DOI] [PubMed] [Google Scholar]
- 6.Cardinal H, Raymond MA, Hebert MJ, et al. Uraemic plasma decreases the expression of ABCA1, ABCG1 and cell-cycle genes in human coronary arterial endothelial cells. Nephrol Dial Transplant. 2007;22:409–416. doi: 10.1093/ndt/gfl619. [DOI] [PubMed] [Google Scholar]
- 7.Vanholder R, Argiles A, Baurmeister U, et al. Uremic toxicity: present state of the art. Int J Artif Organs. 2001;24:695–725. [PubMed] [Google Scholar]
- 8.Cowan DB, Langille BL. Cellular and molecular biology of vascular remodeling. Curr Opin Lipidol. 1996;7:94–100. doi: 10.1097/00041433-199604000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Roy-Chaudhury P, Sukhatme VP, Cheung AK. Hemodialysis vascular access dysfunction: a cellular and molecular viewpoint. J Am Soc Nephrol. 2006;17:1112–1127. doi: 10.1681/ASN.2005050615. [DOI] [PubMed] [Google Scholar]
- 10.Nathan A, Nugent MA, Edelman ER. Tissue engineered perivascular endothelial cell implants regulate vascular injury. Proc Natl Acad Sci U S A. 1995;92:8130–8134. doi: 10.1073/pnas.92.18.8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nugent HM, Edelman ER. Endothelial implants provide long-term control of vascular repair in a porcine model of arterial injury. J Surg Res. 2001;99:228–234. doi: 10.1006/jsre.2001.6198. [DOI] [PubMed] [Google Scholar]
- 12.Conte MS, Nugent HM, Gaccione P, et al. Multicenter phase I/II trial of the safety of allogeneic endothelial cell implants after the creation of arteriovenous access for hemodialysis use: the V-HEALTH study. J Vasc Surg. 2009;50:1359–1368. doi: 10.1016/j.jvs.2009.07.108. e1351. [DOI] [PubMed] [Google Scholar]
- 13.Nugent HM, Rogers C, Edelman ER. Endothelial implants inhibit intimal hyperplasia after porcine angioplasty. Circ Res. 1999;84:384–391. doi: 10.1161/01.res.84.4.384. [DOI] [PubMed] [Google Scholar]
- 14.Zani BG, Kojima K, Vacanti CA, et al. Tissue-engineered endothelial and epithelial implants differentially and synergistically regulate airway repair. Proc Natl Acad Sci U S A. 2008;105:7046–7051. doi: 10.1073/pnas.0802463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerini C, Dou L, Anfosso F, et al. P-cresol, a uremic retention solute, alters the endothelial barrier function in vitro. Thromb Haemost. 2004;92:140–150. doi: 10.1160/TH03-07-0491. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann MA, Lalla E, Lu Y, et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vanholder R, De Smet R, Glorieux G, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 18.Vanholder R, Glorieux G, Lameire N. New insights in uremic toxicity. Contrib Nephrol. 2005;149:315–324. doi: 10.1159/000085693. [DOI] [PubMed] [Google Scholar]
- 19.Recht PA, Tepedino GJ, Siecke NW, et al. Oxalic acid alters intracellular calcium in endothelial cells. Atherosclerosis. 2004;173:321–328. doi: 10.1016/j.atherosclerosis.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 20.Dou L, Bertrand E, Cerini C, et al. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004;65:442–451. doi: 10.1111/j.1523-1755.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- 21.Bolton CH, Downs LG, Victory JG, et al. Endothelial dysfunction in chronic renal failure: roles of lipoprotein oxidation and pro-inflammatory cytokines. Nephrol Dial Transplant. 2001;16:1189–1197. doi: 10.1093/ndt/16.6.1189. [DOI] [PubMed] [Google Scholar]
- 22.Wratten ML, Sereni L, Tetta C. Oxidation of albumin is enhanced in the presence of uremic toxins. Ren Fail. 2001;23:563–571. doi: 10.1081/jdi-100104738. [DOI] [PubMed] [Google Scholar]
- 23.Nugent HM, Groothuis A, Seifert P, et al. Perivascular endothelial implants inhibit intimal hyperplasia in a model of arteriovenous fistulae: a safety and efficacy study in the pig. J Vasc Res. 2002;39:524–533. doi: 10.1159/000067207. [DOI] [PubMed] [Google Scholar]
- 24.Methe H, Hess S, Edelman ER. The effect of three-dimensional matrix-embedding of endothelial cells on the humoral and cellular immune response. Semin Immunol. 2008 doi: 10.1016/j.smim.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Methe H, Edelman ER. Tissue engineering of endothelial cells and the immune response. Transplant Proc. 2006;38:3293–3299. doi: 10.1016/j.transproceed.2006.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aznar-Salatti J, Escolar G, Cases A, et al. Uraemic medium causes endothelial cell dysfunction characterized by an alteration of the properties of its subendothelial matrix. Nephrol Dial Transplant. 1995;10:2199–2204. doi: 10.1093/ndt/10.12.2199. [DOI] [PubMed] [Google Scholar]
- 27.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. 2002;90:1189–1196. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 28.Ahmad S, Ahmad A, Schneider KB, et al. Cholesterol interferes with the MTT assay in human epithelial-like (A549) and endothelial (HLMVE and HCAE) cells. Int J Toxicol. 2006;25:17–23. doi: 10.1080/10915810500488361. [DOI] [PubMed] [Google Scholar]
- 29.Amann K, Tyralla K, Gross ML, et al. Special characteristics of atherosclerosis in chronic renal failure. Clin Nephrol. 2003;60(Suppl 1):S13–S21. [PubMed] [Google Scholar]