

Abstract
The Yersinia adhesin A (YadA) is a collagen-binding trimeric autotransporter of Yersinia enterocolitica, an enteropathogen that causes a range of gastroenteric and systemic diseases, and YadA is essential for Y. enterocolitica virulence. Although previous studies suggest a specific binding site in collagen for YadA, we found that recombinant YadA binds to both major cyanogen bromide fragments of collagen type II and the collagen-like model peptide (Pro-Hyp-Gly)10 [(POG)10]. To further characterise the YadA–collagen interaction, we investigated the binding of YadA to (POG)10 and three other model peptides, (Pro-Pro-Gly)10 which lacks the hydroxyl groups of (POG)10, T3-785 which contains a stretch of the collagen type III sequence and Gly− which is similar to (POG)10 but lacks the central glycine. All the peptides except Gly− adopt a collagen-like triple-helical conformation at room temperature. All three triple-helical peptides bound to YadA, with (POG)10 being the tightest, whereas binding of Gly− was hardly detectable. The affinity of (POG)10 for YadA was 0.28 µM by isothermal titration calorimetry and 0.17 µM by surface plasmon resonance (SPR), similar to that of collagen type I. Our results show that a collagen-like triple-helical conformation, strengthened by the presence of hydroxyproline residues, is both necessary and sufficient for YadA binding.
Keywords: adhesion, collagen, triple-helical peptide, YadA, Yersinia enterocolitica
Introduction
Collagens are the major component of the extracellular matrix (ECM); they provide strength and rigidity to tissues and are also involved in cell attachment and bacterial adhesion (for a review, see Gelse et al., 2003). Diverse collagen types have a common, collagenous triple-helical conformation. A requirement for the triple helical structure is the motif X–Y-glycine (G), where 20% of the residues X and Y are occupied by the imino acids proline (P) and four-hydroxyproline (O). The amino acids in these positions contribute to the stability of triple helix. The sequence POG is the most stable, constituting 10% of native collagen (Ramshaw et al., 1998), and the most stable triple-helical model peptides consist only of POG repeats (Persikov et al., 2000).
Adhering to collagen is important for virulence in several human pathogenic bacteria, including Yersinia enterocolitica (Emody et al., 1989), which causes a range of gastroenteric symptoms. In some individuals, the primary infection is followed by sequelae such as reactive arthritis, iritis and erythema nodosum (Cover and Aber, 1989). The major collagen-binding protein of Y. enterocolitica is the Yersinia adhesin YadA. In addition, YadA mediates adherence to epithelial cells and binding to other ECM proteins, such as laminin and fibronectin, and is also implicated in other virulence-related functions, such as autoagglutination, serum resistance and phagocytosis resistance (reviewed by El Tahir and Skurnik, 2001). Yersinia enterocolitica requires YadA for full virulence and the collagen-binding ability of YadA has been specifically shown to be an essential virulence factor because loss of it leads to avirulence in mice (Roggenkamp et al., 1995; Tamm et al., 1993).
YadA is a homotrimeric protein composed of a C-terminal β-barrel membrane anchor and associated linker followed by a coiled-coil stalk, a neck region and a globular head domain in the N-terminal portion of the protein (Roggenkamp et al., 2003). Deletion mutation studies have assigned the collagen-binding ability of Y. enterocolitica YadA to the neck and globular head domain (Roggenkamp et al., 2003). We have shown that the neck and head region together form a single binding domain, the core of which is formed of three left-handed parallel β-rolls (Nummelin et al., 2004). The YadA–collagen interaction is known to be extremely stable and tolerate high temperatures and pH changes (Emody et al., 1989; Flugel et al., 1994), but the apparent affinity for collagen type I is only 0.28 µM (Nummelin et al., 2004). Collagen-binding experiments on site-directed mutants of YadA have identified several critical residues on the protein surface, but the actual mechanism of collagen binding is still unclear (Nummelin et al., 2004). YadA-binding determinants in collagen type I, a heterotrimeric molecule formed from two α1(I) chains and one α2(I) chain, and collagen type II, a homotrimer of α1(II) chains, have been suggested to reside in a conserved region found in the α1(I) and α1(II) chains, and not in the triple-helical shape of collagen trimers (Schulze-Koops et al., 1992; Schulze-Koops et al., 1995).
Synthetic collagen-like peptides can adopt a triple-helical structure and have proven to be excellent tools in biochemical and biophysical experiments (reviewed in Koide, 2005). The (POG)10 triple helix is stable at room temperature (RT) and its structure has been solved (Berisio et al., 2000; Sakakibara et al., 1973). While the (POG)10 peptide serves as a model for triple-helical conformation and shape, it does not contain sequence information. It is more compact than collagen, being more tightly supercoiled with a 75 (or left-handed 72) symmetry (Berisio et al., 2000; Okuyama et al., 2004), rather than the 107 (or left-handed 103) screw symmetry suggested for native collagen based on fibre diffraction experiments (Fraser et al., 1979). The same is true for the non-modified (PPG)10 peptide (Kramer et al., 1998; Okuyama et al., 1981). A naturally occurring sequence can be inserted into the model peptides and still retain the triple helical structure, if the natural sequence is surrounded with POG-triplets and the G at every third position is preserved. The T3-785 peptide has an imino acid-poor sequence from next to a collagenase site in human type III collagen in the middle of the peptide with three POG-triplets at each end: (POG)3-ITG-ARG-LAG-POG-(POG)3 (Kramer et al., 1999). The structure of T3-785 has been solved (Kramer et al., 1999) and it shows that the triple-helix in the central part of the peptide is not as tight as at the ends with POG-triplets, with the symmetry in the middle region close to 107. Finally, because the G in the collagen triplet is a requirement for stable triple helical structure, a modified (POG)10 peptide, (POG)4-PO-(POG)5 (Gly−), lacking one of the central glycines, is mostly monomeric even at +2°C and forms negligible amounts of triple helices at +25°C (Long et al., 1993).
In this study, we have investigated the binding of the collagen-like peptides (POG)10, (PPG)10, T3-785 and Gly− to recombinant collagen-binding domain of Y. enterocolitica YadA. (POG)10 is a standard triple-helical collagen-like model peptide, as is (PPG)10 though it lacks the hydroxyl groups of (POG)10. T3-785 was chosen for this study because the proposed YadA-binding regions in collagens type I and type II lie near collagenase sites (Schulze-Koops et al., 1995), and the specific sequence in T3-785 is also adjacent to a collagenase site in collagen type III (Kramer et al., 1999). T3-785 is thus a reasonable candidate to test the hypothesis presented in the previous study that a less stable triple helix close to collagenase sites might promote YadA binding. The effects of a triple-helical conformation can be controlled for with Gly−, which does not form appreciable amounts triple helices but has a near-identical sequence to (POG)10. Our results show that (POG)10 binds YadA as tightly as type I collagen does, with a Kd of 0.28 µM by ITC and 0.17 µM by SPR. The other peptides bind more weakly, and we could only determine a Kd for (PPG)10 by ITC, which was 10 µM. In particular, binding was hardly detectable for the non-triple-helical Gly−. Furthermore, we examined the binding of YadA-expressing Y. enterocolitica O:3 to these four peptides by enzyme-linked immunosorbent assay (ELISA). Finally, we used ELISA to compare the binding of recombinant YadA to collagen type II and (POG)10. As we expected and in contradiction to earlier studies (Schulze-Koops et al., 1992; Schulze-Koops et al., 1995), there is no evidence for a specific YadA-binding site. A triple-helical conformation is both necessary and sufficient for YadA binding.
Materials and methods
Bacteria
Yersinia enterocolitica serotype O:3 strain 6471/76 (YeO3) (Skurnik, 1984), which expresses wild-type (wt) YadA on the surface of the bacteria, was used in the ELISA assay. The YeO3-c strain, which lacks the virulence plasmid pYV and consequently does not express YadA, was used as a negative control (Skurnik, 1984). Recombinant proteins were produced in Escherichia coli M15(pREP4) (QIAGEN).
Expression vectors
YadA-binding domain (YadA26–241) was expressed using the pHN-1 plasmid (Nummelin et al., 2002). Mutant proteins were expressed using pHN-1-derived plasmids carrying single or double mutations (Nummelin et al., 2004). YadA24–378 was expressed from the plasmid pOP-1 (Nummelin et al., 2002).
Circular dichroism spectra of YadA mutants
YadA26–241 and mutant proteins were dialysed overnight against 5 mM phosphate buffer at pH 7.4 containing 5 mM NaCl, after which the proteins were diluted to approximately 0.5 mg/ml. Far-UV circular dichroism (CD) spectra between 250 and 195 nm were recorded with a Jasco J-715 instrument and a 1 mm cuvette at +20°C, with spectra based on the average of five measurements. The CD signal was converted into molar ellipticity values using the formula θ = CD/ n×c×l×10, where the CD signal is in millidegrees, n the number of residues, c the molar concentration of protein and l the length of the light path.
Collagen and collagen-like peptides
Collagens type I and type II were from Sigma. (POG)10 and (PPG)10 peptides were from Peptides International Inc., Louisville, KY, USA. The peptides Gly− and T3-785 used in the SPR measurements were synthesised in the Protein Microchemistry Laboratory at the Centre for Advanced Biotechnology and Medicine, Piscataway, NJ, USA (Kramer et al., 1999; Long et al., 1993). The same peptides used in ITC, whole cell ELISA and gel shift assays were purchased from Innovagen, Lund, Sweden.
Cyanogen bromide cleavage of collagen and purification of fragments
Five milligram of bovine collagen type II was dissolved in 500 µl of 70% formic acid and heated to 70°C for 10 min. Ten milligram of cyanogen bromide (CNBr) was added to the solution and incubated overnight at RT. We purified the resulting fragments by preparative SDS–PAGE, as previously reported (Retamal et al., 1999). Our protocol was as follows: we removed the well dividers of a 12% Tris–HCl gel (Bio-Rad) to create a large single well. After concentrating the sample and mixing with sample buffer, we applied the CNBr-cleaved collagen to the gel and ran it. After running, the gel was stained with BioSafe Coomassie (Bio-Rad) and the two major bands cut out. These gel strips were crushed with a spatula and the collagen fragments were extracted overnight into TBS solution (20 mM Tris pH 7.4, 150 mM NaCl) at RT. The following day, we sonicated the solution briefly and continued the extraction for another 3 h at +37°C. After this, the gel fragments were pelleted and the supernatant concentrated. Due to the insensitivity of collagen to standard concentration assays, we determined the concentrations of the fragments by SDS–PAGE and compared the staining intensities of the fragments with the intensities obtained by a dilution series of BSA. The identities of the purified fragments were verified by mass spectrometry.
Recombinant protein production
The collagen-binding fragment of YadA, YadA26–241 and YadA24–378 and mutant proteins were produced and purified as described earlier (Nummelin et al., 2002). Furthermore, the protein was produced in the same way as for crystallisation. Briefly, His-tagged protein was expressed in E.coli and purified from the cell lysate supernatant using Ni-NTA (QIAGEN) affinity chromatography as the only purification step. Additionally, for the protein used in the gel shift assays, we performed a size exclusion chromatography step on a Superdex200 26/60 column (GE Healthcare). The produced wt protein was trimeric, as evidenced by the elution profile in size exclusion chromatography, the wt CD spectrum and the fact the trimeric form of the protein was visible in SDS–PAGE analysis of purified YadA26–241 and YadA24–378.
Enzyme-linked immunosorbent assay (ELISA) with YadA-expressing bacteria
Binding of YadA-expressing bacteria to immobilised collagen-like peptides and type I collagen was measured using ELISA. We grew YadA-expressing Y. enterocolitica YeO3 and YadA-negative YeO3-c overnight in MedECa medium to ensure strong expression of YadA (Skurnik, 1985) and then diluted in PBS to approximately 2 × 108 cfu/ml, which corresponds to an optical density at 600 nm of 0.2 (El Tahir et al., 1997). We dissolved the peptides in phosphate-buffered saline (PBS; 10 mM sodium phosphate, 150 mM NaCl, pH 7.4) at a concentration of 200 µg/ml, applied 100 µl of these solutions to microtitre plate wells and incubated for 30 min at RT. Collagen type I and bovine serum albumin (BSA; both from Sigma), at the same concentration, were used as controls.
After incubation with the peptide solutions, we washed the wells three times with 150 µl of PBS and then blocked with 150 µl of PBS+3% BSA for half an hour at RT. Hundred microlitre of bacterial suspension was added to the wells and incubated for 30 min. The wells were washed as above, then 100 µl of primary antibody dilution (1:10) in PBS+3% BSA was added and incubated for 30 min at RT. The primary antibody was the Y. enterocolitica O:3 -specific monoclonal antibody A6 (Pekkola-Heino et al., 1987). After washes as above, we added 100 µl of secondary antibody (anti-mouse-HRP; DAKO P260) at a dilution of 1:2000 in PBS+3% BSA and incubated for 30 min at RT. The wells were washed three times with PBS, after which 100 µl of prepared HRP substrate solution (OPD; DAKO S-2045) was added. The reaction was allowed to progress for about 5 min and then stopped with the addition of 100 µl 0.5 M sulphuric acid, after which absorbances were read at 492 nm.
Elisa of recombinant YadA binding to collagens and CNBr fragments
Wells of a microtitre plate (Wallac HB Isoplate) were coated with 100 µl of solutions containing human collagens type I and type IV, bovine collagen type II, (POG)10, Gly− and BSA, each at 50 µg/ml and incubated for 45 min at RT. The wells were then blocked with 150 µl of 3% BSA in PBS for 30 min at RT. We added 100 µl of YadA24–378 at 50 µg/ml, diluted into PBS+3% BSA to the wells and incubated for 45 min. We included control wells where YadA was omitted. After washing twice with 150 µl PBS-T (PBS+0.1% Tween20) for 10 min, we added 100 µl of primary antibody 3G12 (Skurnik et al., 1994) at a dilution of 1:50 in PBS+3% BSA and incubated 30 min. We then washed the wells again as above before adding secondary antibody (anti-mouse IgG-HRP from sheep, GE Healthcare) at a dilution of 1:2000 in PBS+3% BSA. The wells were washed twice for 10 min with PBS-T, after which 100 µl of HRP substrate solution (SigmaFast OPD) was added. The reaction was stopped after 10 min by adding 50 µl of 2.5 M sulphuric acid and absorbances were read at 490 nm.
To investigate the binding of YadA to CNBr fragments of collagen type II, wells of a microtitre plate were coated as above with collagen type II, CNBr fragments of collagen type II, (POG)10 and BSA, each at 20 µg/ml. To assess the background levels, we included control wells with collagen type II and (POG)10 where YadA was omitted. Washes, antibody incubations and detection were performed as described above.
Gel shift assays
For the single concentration assay, 5 µg of YadA26–241 was mixed with 5 µg of each collagen-like peptide in a total volume of 10 µl of PBS. For the concentration series, we used 5 µg of YadA26–241, equal to a concentration of 3.5 µM in a total volume of 20 µl. We mixed peptide with YadA26–241 in a total of 10 µl of PBS. For the (POG)10 series, the concentrations were between 0.01 and 100 µM per trimer of peptide, calculated for a total volume of 20 µl. For T3-785 and (PPG)10, the concentration was between 0.1 and 150 µM. For all assays, 5 µg of YadA26–241 alone was run as a control. We added 10 µl of 2x native electrophoresis loading buffer (0.5 M Tris pH 8.5; 25% glycerol; 0.01% bromophenol blue) to each sample and applied the whole sample to a well in a 12% polyacrylamide gel buffered with Tris–HCl (Bio-Rad Ready Gel). The samples were run in native electrophoresis buffer (0.025 M Tris; 0.2 M glycine; pH 8.3) till the buffer front reached the lower edge of the gel at RT (approximately +21°C). For the single concentration assay, we also ran a gel at +5°C. After running, the gels were rinsed with water and stained with BioSafe Coomassie solution (Bio-Rad). After destaining with water, the gels were imaged with Gel Doc 2000 (Bio-Rad).
Surface plasmon resonance
SPR measurements were performed using a BIAcore 2000™ instrument (Biacore AB). A CM5 sensor chip and Amine Coupling Kit for the immobilization of YadA were also from Biacore AB. We coupled YadA26–241 to the chip according to the manufacturer’s instructions. After activation, 0.35 mg ml−1 (16 µM) YadA26–241 in 10 mM Na-acetate buffer, pH 5.1 was passed over the surface, resulting in 7000 resonance units (RU) coupled YadA.
The binding of collagen mimicking peptides to immobilised YadA26–241 was assayed qualitatively by running 4 µM of peptide in 10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.005% P-20 (running buffer) at a flow rate of 20 µl min−1 at +25°C over the surfaces. The binding of the peptide was monitored versus time. The background caused by peptide run over an uncoupled reference surface was subtracted from the data.
To measure the apparent binding constant of YadA-binding domain to (POG)10, the peptide was immobilised on the sensor chip surface. The coupling was performed using 0.1 mg ml−1 (POG)10 dissolved in 10 mM acetate buffer, pH 5.5. and a surface of 60 RUs of coupled peptide was used to determine the apparent binding constants to YadA and the mutants. The experiments were done at a flow rate of 20 µl min−1 in running buffer and analysis of SPR data was done as described in detail in Nummelin et al. (2004). Briefly, the binding was assumed to have reached equilibrium shortly before the end of the injection. The response at this point was corrected for by subtracting the response from an uncoupled reference surface. The corrected responses obtained at different protein concentrations were non-linearly fitted to a one-binding site model.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were done using VP-ITC from MicroCal LLC. To assay the interaction of (POG)10 with YadA26–241, degassed YadA in PBS buffer was put into the cell at a concentration of 4.5 µM per trimer. The (POG)10 was placed into the syringe at a concentration of 125 µM of trimer in PBS buffer. A series of 35 injections of 3 µl were performed with 240 s intervals at +25°C. For (PPG)10 and T3-785, the concentration of YadA26–241 was 50 µM and the peptide concentration was 350 µM, and the reaction was run at +21°C. The concentration of (monomeric) Gly− was 600 µM, with YadA26–241 at 50 µM. Data were analysed with Origin7 and integrated enthalpy data were fitted using a one-sites-binding model in the same programme.
Results
Surface plasmon resonance studies
To test the ability of YadA to bind specifically collagen-like triple helix, the binding of four different peptides, (POG)10, (PPG)10, Gly− and T3-785, to immobilised YadA26–241, was assayed with SPR, as shown by the sensorgrams of the binding of the peptides to 7000 RU YadA coupled surface (Fig. 1). The results show that only the triple-helical peptides (POG)10, (PPG)10 and T3-785 bound to YadA. The binding of YadA to Gly− was close to background levels. The levels of binding to YadA showed interesting differences: (POG)10 peptide bound with higher affinity than (PPG)10 and its dissociation from the surface was slower, as shown by the shape of the curve in the sensorgram (Fig. 1). The T3-785 peptide showed significantly decreased binding.
Fig. 1.

Binding of different collagen-like peptides to immobilised YadA26–241 by SPR. The curves represent resonance units (RUs) as a function of time. The injections start at 0 sec and end at 300 sec. The curve for (POG)10 binding is in blue, the curve for (PPG)10 is coloured green, the curve for T3-785 is magenta and the curve for Gly− is in light brown.
To try to identify a potential binding site for (POG)10, we investigated the binding of wt YadA and a set of surface mutants of YadA to (POG)10. These double mutations were targeted to charged and polar residues on the surface of YadA, and we had previously used the same set to study in the binding of collagen type I (Nummelin et al., 2004). The determination of binding constants to peptides using immobilised YadA proved difficult, because the SPR curves could not be fitted using the available models due to the slow dissociation of the peptides, particularly of (POG)10. Therefore, (POG)10 peptide was immobilised on a sensor chip, and the binding of YadA and the mutants to the peptide were measured (Table I). The peptide coupling gave a low response. As a result, we saw no signal for mutants V98D-N99A, H159A-H162A and E80A-K83A and the binding constants for those could not be determined. We believe that these mutants bind very weakly to (POG)10, i.e. with a dissociation constant (Kd) larger than 27 µM. Binding of wt YadA and other mutants, even with the low response, followed a one-to-one binding model. The apparent dissociation constant of wt YadA-binding domain to (POG)10 was 0.17 ± 0.02 µM.
Table I.
Apparent dissociation constants for the YadA–(POG)10 interaction for YadA surface mutants
| Mutant | Kapp per μM (SPR) | %wt | %wt to Col I (SPR) |
|---|---|---|---|
| wt | 0.17 ± 0.02 | 100 | 100 (0.28 ± 0.06 µM) |
| V98D, N99A | Nd | – | 4 |
| K108A, L110A | 4.6 ± 0.7 | 4 | 11 |
| Q124A, K125A | 0.67 ± 0.16 | 25 | 20 |
| H159A, H162A | Nd | – | 12 |
| K148A, D150A | 0.89 ± 0.025 | 19 | 15 |
| D180A, E182A | 27 ± 13.3 | 1 | 1 |
| E190A, S191A | 0.21 ± 0.041 | 81 | 50 |
| E80A, K83A | Nd | – | 20 |
| N166A, Y169A | 0.098 ± 0.038 | 174 | 17 |
| R133 | 1.28 ± 0.04 | 13 | 10 |
The first column on the left shows the mutations concerned (wt denotes wild type). The second column indicates the dissociation constant in micromoles per litre as measured by surface plasmon resonance (SPR). The third column shows the percentage of binding to (POG)10 compared to wt YadA. The fourth column gives the percentage of binding to type I collagen by SPR (from Nummelin et al., 2004). Nd, not determined.
The binding studies with the set of surface mutants to the (POG)10 peptide showed that mutations N166A, Y169A and K148A, and D150A did not have as large an effect on peptide binding as on collagen type I binding (Table I). The mutant D180A disrupts an intermonomeric contact, thus unfolding YadA, as seen in the CD spectrum of the mutant D180A-E182A (Supplementary data are available at PEDS online). The other mutants yielded CD spectra similar to that of wt YadA. However, mutants E80A-K83A, K108A-L110A and E190A-S191A display slightly altered spectra compared to the wt, but these do not reflect large alterations in the structure of YadA (Supplementary data are available at PEDS online for an explanation). As the mutants whose binding constants could not be determined (V98D-N99A, H159A-H162A and E80A-K83A) have very weak affinity to (POG)10, it seems that, unlike for collagen I, that the binding determinants for the peptide reside mostly in the distal (from the outer membrane) part of the YadA β-rolls of the collagen-binding domain (Fig. 2). This result may be due to the low amount of bound peptide on the sensor chip in the experiments, or an actual preference of the outermost part of YadA on the cell surface to bind a collagenous triple-helical conformation.
Fig. 2.
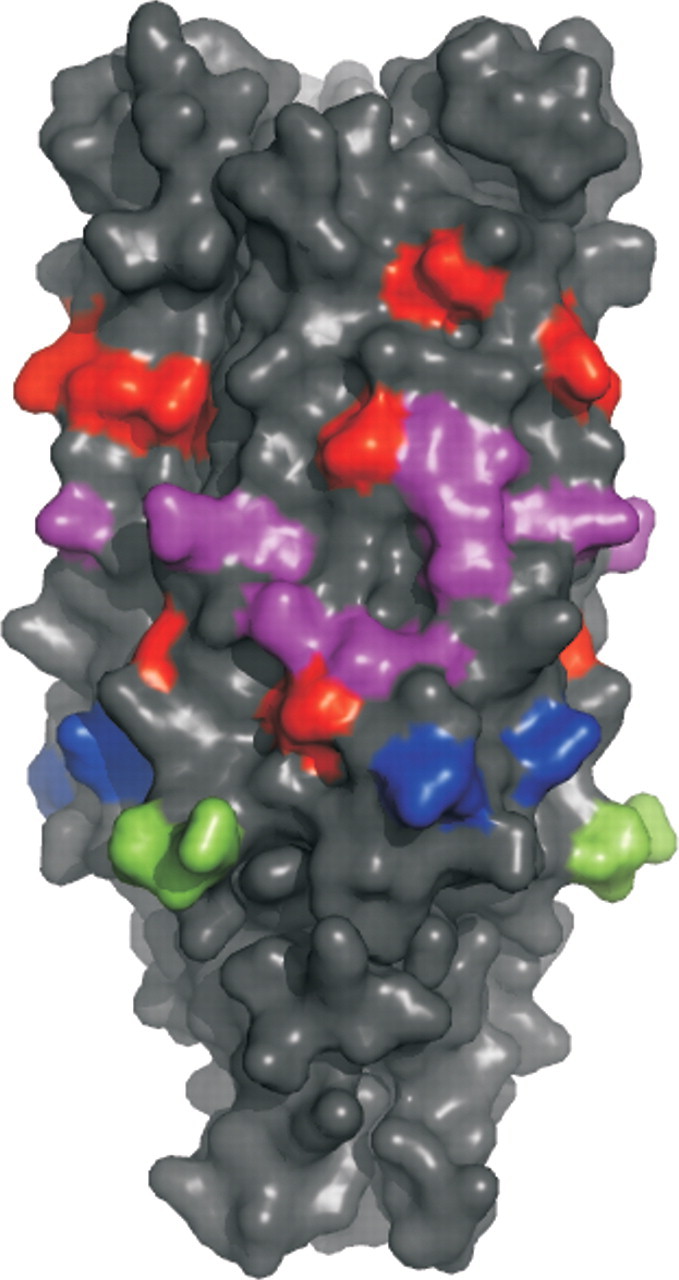
Effect of YadA surface mutations on (POG)10 binding. The surface mutants are coloured according to their effect on binding to immobilised (POG)10 peptide as measured with SPR. In the order of increasing effect on binding, the mutants are coloured in blue (Kd 0.098 µM), green (Kd 0.21 µM), purple (Kd 0.67–1.28 µM) and red (Kd 4.6–27 µM). The mutants which gave no signal (V98D-N99A; H159A-H162A; E80A-K83A) are assumed to have a large effect on peptide binding, and are therefore coloured red. The mutant D180A-E182A is not highlighted in the figure as it causes a major conformational change.
Isothermal titration calorimetry
To confirm the binding of YadA to (POG)10, the interaction was also tested with ITC (Fig. 3A). We obtained a dissociation constant (Kd) of 0.28 ± 0.05 µM (Table II) with this technique, which is in excellent agreement with the apparent binding constant determined using SPR. The other peptides did not yield as good binding isotherms, but we obtained clear enough data for (PPG)10 (Fig. 3B) to calculate an apparent dissociation constant of 10 µM (Table II). We were unable to determine a binding constant for T3-785 and Gly− even at high concentrations of trimeric YadA (0.05 mM) and peptide (350 µM of trimeric T3-785, 600 µM of monomeric Gly−; data not shown). Using higher concentrations of YadA and peptides would not have been practicable. For (POG)10, (PPG)10, and T3-785, the binding is endothermic, indicating that the interaction is driven by the displacement of structured water from the binding interface. For Gly−, only a slight exothermic reaction was seen that was at the level of the buffer control, i.e. close to the heat of dilution. This suggests that Gly− bound hardly at all.
Fig. 3.

Binding of (POG)10 and (PPG)10 to YadA26–241 by ITC. (A) Results for (POG)10 binding. (B) Results for (PPG)10 binding. The upper panel shows raw ITC data, the lower panel integrated enthalpy values with fitted curve.
Table II.
Calculated thermodynamic parameters for the YadA–(POG)10 and YadA–(PPG)10 interactions from the ITC experiments
| YadA26–241+(POG)10 | YadA26–241+(PPG)10 | |
|---|---|---|
| Model | OneSites | OneSites |
| N | 1.03 ± 0.021 | 0.126 ± 0.014 |
| K (M−1) | 3.58 × 106 ± 6.2 × 105 | 9.92 × 104 ± 8.1 × 103 |
| Kd (M) | 2.79 × 10−7 ± 0.499 × 10−7 | 1.01 × 10−5 ± 0.083 × 10−5 |
| ΔH (kJ/mol) | 34.45 ± 0.981 | 14.28 ± 1.80 |
| ΔS (J/mol) | 149 | 71.4 |
The model used was OneSites (one or more equivalent binding sites). N is the calculated stoichiometry, Kd the dissociation constant, ΔH the enthalpy and ΔS the entropy of the interaction. Kd was derived from the association constant (K) reported by the programme using the following formula: Kd=1/K.
Gel shift assays
To see the effects of complex formation between YadA and the peptides, we analysed the interactions by gel shift assay. In the assay, YadA26–241 was mixed with a molar excess of each peptide. YadA26–241 alone was run as a control. Gels were run at two different temperatures, RT (Fig. 4A) and +5°C to ensure the triple-helical conformation of the peptides. The +5°C run yielded similar results to the RT run (data not shown). In the gel, YadA mixed with Gly− ran similarly to YadA alone, suggesting no binding. There is a clear shift in the sample containing (POG)10, indicative of complex formation. T3-785 also shows a clear shift in this assay, and in fact a large proportion of protein appears not even to enter the gel, which suggests aggregation within the sample. Some aggregation also occurs within the (POG)10 sample. (PPG)10 exhibits a slight shift compared to unbound YadA.
Fig. 4.

Complex formation of YadA26–241 and collagen-like peptides assayed by gel shifts. The results are from gel shift assays run at room temperature. (A) Binding of different collagen-like peptides to YadA. YadA26–241 was mixed with a molar excess of peptide. The arrow marks the position of YadA in the absence of peptide ligand. (B) Concentration series for (POG)10 mixed with YadA at 3.5 µM. (C) Concentration series for T3-785. (D) Concentration series for (PPG)10.
To further investigate the aggregation seen in the T3-785 and (POG)10 runs, a concentration series of peptides was set up and mixed with a constant amount of YadA26–241 and run in native gels (Fig. 4B–D). The aggregation behaviour of (POG)10 disappeared at an approximately 30-fold excess of the peptide (Fig. 4B), which suggests that the aggregation was brought about by more than one YadA molecule binding to the same peptide. At saturating concentrations of (POG)10 this phenomenon is no longer detected. Similarly, the aggregation seen with T3-785 lessened at high concentrations (Fig. 4C), though it did not completely disappear at even a 50-fold excess of peptide, which suggests that saturation had not yet been reached. Estimating a binding constant for these two peptides based on this concentration series proved difficult due to the diffuse nature of the bands in the gel. However, some binding of (POG)10 to YadA is seen already at 0.1 µM, which is consistent with our other measurements for a Kd of approximately 0.3 µM. Based on the gel shift results, it appears that the Kd for T3-785 is at least an order of magnitude larger. We also ran a native gel with a concentration series of (PPG)10 (Fig. 4D). No aggregation was seen at any concentration, and the small shift of YadA on binding the peptide made interpretation of the gel difficult, so no affinity could be estimated. However, the staining of the YadA band intensified with increasing (PPG)10 concentration, indicative of peptide binding.
Binding of YadA-expressing bacteria to collagen-like peptides
To confirm that full-length, native YadA also binds triple-helical peptides, we tested the binding of wt YadA on the surface Y. enterocolitica to immobilized collagen peptides using an ELISA assay. As controls, we included collagen type I (positive control), a YadA-negative strain of Y. enterocolitica (negative control) and bovine serum albumin (background control). Additionally, we included a collagen control with no added bacteria to test background levels. The results (Fig. 5) indicate that with this technique too, (POG)10 binds most strongly of the four tested peptides, giving a signal of approximately two thirds of native collagen. The other peptides bound YadA-expressing bacteria more weakly, with (PPG)10 exhibiting the highest binding of the three at about one quarter of the level of collagen. As expected, Gly− bound bacteria barely above background levels, and T3-785 showed only marginally stronger binding. In all cases, the control strain YeO3-c bound weakly to all peptides, showing that any binding is YadA-mediated.
Fig. 5.

The binding of YadA-expressing cells to immobilized peptides measured by ELISA. Collagen and bovine serum albumin (BSA) are included as control. The columns show the mean absorbances measured at 492 nm from four replicate wells. The error bars denote standard deviations. YeO3 (data in black) is a YadA-positive strain and YeO3-c (data in dark gray) is a YadA-negative strain. The column in light grey shows the background given by collagen in the absence of cells.
Binding of YadA to CNBr fragments of collagen II
We investigated the binding of YadA to collagens type I, II and IV by ELISA. In these assays, we used a longer version of YadA, YadA24–378, which is recognised by the monoclonal antibody 3G12. This antibody does not bind to YadA26–241, and as the collagen-binding region resides in this fragment, the use of 3G12 with the longer version of YadA does not interfere with the YadA–collagen interaction. Our results (Fig. 6) show that YadA binds to the three types of collagen and (POG)10 at approximately the same level, whereas Gly− binds hardly at all, as expected. There are slight differences between the binding of the different collagen types, with type I giving the highest response, followed by type IV, and type II binding the weakest. (POG)10 binds at roughly the same level as collagen type II.
Fig. 6.

Binding of recombinant YadA to immobilised different types of collagens, (POG)10 and Gly- by ELISA. Collagens type I and type IV are human and collagen type II is from cattle. The columns show the mean absorbances at 490 nm from four replicate wells. The error bars denote standard deviations. Columns in black are the results of wells incubated in the presence of YadA, columns in light grey are controls without YadA.
We also performed ELISA using CNBr fragments generated from the α1(II) chain of bovine collagen type II. This protocol requires denaturation of collagen, as was done in previous publications reporting a specific binding site for YadA in collagen (Schulze-Koops et al., 1992; Schulze-Koops et al., 1995). We investigated the binding of YadA to the two largest CNBr fragments, CB10 (∼30 kDa) and CB11 (∼26 kDa) (Miller and Lunde, 1973), as was done in Schulze-Koops et al. (1995). The former contains the region suggested to contain the binding site for YadA, whereas the latter was not expected to bind YadA (Schulze-Koops et al., 1995).
In the assay, YadA24–378 clearly binds to collagen type II, both CNBr fragments and (POG)10 (Fig. 7). The level of binding is the same for collagen type II and (POG)10, and somewhat lower for CB10 and CB11. There is also a difference between the two fragments, with CB10 binding more strongly than CB11.
Fig. 7.

Binding of recombinant YadA to immobilized collagen type II, collagen CNBr fragments and (POG)10 by ELISA. CB10 and CB11 are the two largest fragments obtained by cyanogen bromide cleavage of bovine collagen type II. The columns show the mean absorbances at 490 nm from four replicate wells. The error bars denote standard deviations. Columns in black are the results of wells incubated in the presence of YadA and columns in light grey are controls without YadA.
Discussion
In this study, we investigated the binding of the collagen-binding domain of YadA to four different collagen-like peptides. Our results show that a collagen-like triple-helical conformation in conjunction with the presence of hydroxylated proline residues is both necessary and sufficient for YadA binding. This is in contrast to previous studies that have implicated specific collagen sequences to be binding determinants for YadA (Schulze-Koops et al., 1992; Schulze-Koops et al., 1995).
The binding studies with collagen-mimicking peptides show conclusively that YadA binds selectively to a triple-helical conformation. The apparent binding constant of the YadA–(POG)10 interaction, measured by SPR and ITC, was the same, within experimental error, as for collagen type I obtained by SPR. In addition, (POG)10 bound YadA strongly in the gel shift assay. These results all support the conclusion that a triple-helical structure is sufficient for YadA binding.
Whereas (POG)10 binds strongly to YadA, the other two triple-helical peptides, (PPG)10 and T3-785, bind more weakly. Although the trimeric forms of these two peptides have much lower melting temperatures, 24°C and 25°C, respectively, versus 58°C in (POG)10 (Kobayashi et al., 1970; Sakakibara et al., 1973; Li et al., 1993), the weaker binding does not appear to be the result of unstable trimers. This conclusion is supported by the gel shift assays, where the gels run at different temperatures gave the same results for the peptides. In particular, (PPG)10 showed only a small shift at both temperatures, indicating that triple helix stability is not an issue. For (POG)10 and T3-785, the observed shifts in Fig. 4A are due to aggregation effects, whereas with (PPG)10 only a small shift is seen. At saturating concentrations of (POG)10, the aggregation disappears and the shift becomes much smaller, similar to that of (PPG)10. As the only difference between (POG)10 and (PPG)10 is hydroxyproline content, our conclusion is that hydroxyproline residues promote aggregation, especially since T3-785 also contains POG repeats. However, in all cases the non-triple helix-forming Gly−, though containing an equal amount of hydroxyprolines to (POG)10, failed to bind YadA, indicating that a triple-helical conformation is absolutely required for complex formation and that the triple-helical structure is the major binding determinant.
YadA is a homotrimer, and so presents three equivalent binding sites on its surface. Our gel shift results support the notion that YadA is capable of binding more than one peptide (Fig. 4). Similarly, it appears that a single trimeric peptide can bind at least two molecules of YadA, either on opposite faces of the triple helix or at either end of the peptide. The aggregation seen in the gel shift assays disappears at saturation, which is only attained at a large molar excess of peptide. Also the slow dissociation of (POG)10 from immobilised YadA seen in SPR is supportive of multivalent binding. However, our ITC experiments suggest a single binding site for (POG)10 (Table II). If the microscopic binding constants for the three binding sites were the same, having three equivalent binding sites would result in three different macroscopic binding constants, with a difference of a factor of three between the first and second and second and third binding constants (Fletcher et al., 1970). We explain this discrepancy through the aggregation-inducing effect of (POG)10: the measured K is the first macroscopic binding constant. Aggregation can already be seen at a one-to-one molar ratio in the gel shift experiments, and so this probably happens during the ITC run as well. Aggregation has the effect of limiting accessibility of the peptide to YadA, so the apparent second and third binding constants would in this case be smaller than the expected one third and one ninth, respectively, of the first macroscopic binding constant. In addition, the criterion that the sites are independent of each other might not be fulfilled, and binding of (POG)10 at one site may well influence binding to adjacent sites. In this case, we suggest that the smaller binding constants are not observed, as the molar ratio range of the ITC experiment is not large enough to detect them. However, it is clear from the gel shift and SPR experiments that YadA does bind multiple peptides.
The peptide binding results from the biophysical methods was confirmed with the experiment with Y. enterocolitica cells expressing native YadA. The cells bound most strongly to (POG)10 and with lesser affinity to (PPG)10. The binding to T7-385 was weak, and that of Gly− was barely above background levels, which is consistent with our SPR data. In this assay, native collagen type I bound more strongly to the cells than (POG)10, despite the biophysical methods showing an affinity of the same magnitude as (POG)10. This is probably due to cooperative binding: collagen provides a much larger binding area than the peptide and can probably recruit numerous YadA molecules, and the high density of YadA on the surface of the cell makes it possible for several neighbouring molecules to bind the same strand of collagen. Such cooperative effects would explain why collagen appears to bind more strongly than (POG)10 to YadA expressed on the cell surface. The small size of the peptide allows at most only a couple of YadA molecules to bind, thus reducing cooperativity. This finding is supported by the ELISA experiment using recombinant YadA, where the protein bound equally well to collagen type II and (POG)10, as expected based on the affinities.
In the ELISA experiments with recombinant YadA, we also reproduced the results showing that the largest CNBr fragment of bovine collagen, CB10, binds more strongly to YadA than the other large fragment, CB11. CB10 contains the conserved region postulated to contain the binding site for YadA (Schulze-Koops et al., 1995). Indeed, YadA does bind to this fragment with about twice the apparent affinity as CB11. However, YadA also clearly binds to CB11 well above background levels. The fact that the CNBr fragments both bind YadA at a lower level than intact collagen and (POG)10 might be explained if the preparation of the fragments disrupts the triple-helical collagenous structure. Presumably renaturation of the triple helix is not global, and as a triple-helical conformation is required for YadA binding, this would account for the lower signals seen for the CNBr fragments. However, as the previous studies postulating a specific binding site for YadA in collagen also contained denaturing steps in the preparation of CNBr fragments, our results are comparable to those obtained in these studies (Schulze-Koops et al., 1992; Schulze-Koops et al., 1995).
Based on our results, it is evident that YadA does not require a specific sequence to bind collagen, and that a triple-helical conformation is sufficient for binding. Although YadA does bind more strongly to the CB10 fragment, it also binds to the CB11 fragment with appreciable affinity. This demonstrates that YadA can adhere to collagenous sequences in an unspecific manner, recognising the triple-helical structure and possibly hydroxyproline-rich segments.
Of course, there may be sequences in collagen that bind to YadA slightly tighter than (POG)10, just as there are sequences—e.g. (PPG)10 and T3-785—that bind more weakly. However, given that the concentration of POG repeats is much higher in collagen than that of any specific sequence, our results clearly demonstrate that YadA does not bind a specific sequence. On the contrary, our results suggest that other factors, such as compactness of the triple helix, imino acid content and prevalence of hydroxyproline residues, are more important binding determinants than the actual amino acid sequence. We, therefore, conclude that YadA is able to bind different regions of collagen in a non-specific manner and no binding determinants, other than a triple-helical structure rich in hydroxyprolines, are required. This type of non-specific binding may also be true of other collagen-binding trimeric autotransporters, such as EmaA from Aggregatibacter actinomycetemcomitans and BadA from Bartonella henselae, which both contain YadA-like globular domains (Mintz, 2004; Riess et al., 2004).
This view that YadA can bind rather unspecifically to a collagen-like triple helix is supported by work on other bacterial collagen-binding proteins, especially MSCRAMMs such as the staphylococcal protein CNA and ACE from Enterococcus faecalis, the structures of which have been solved (Symersky et al., 1997; Liu et al., 2007). In particular, CNA has been solved as a complex with a collagen like peptide (Zong et al., 2005). The Collagen Hug model proposed for CNA collagen binding suggests that the collagen triple helix is first recognised by a shallow trench in the N2 domain, after which the N1 domain closes the triple helix wrapping it between these two domains. The preferred ligand of CNA consist of POG repeats, and although the actual high-affinity peptide used in the study contained a central GPRGRT sequence, these do not directly participate in binding, and the authors postulate that a triple-helical shape is the major binding determinant for CNA (Zong et al., 2005). The residues involved in POG repeat binding in CNA are also largely conserved in ACE, and have indeed been shown to be important in collagen binding for that protein as well (Liu et al., 2007). Also the collagen-binding domain of type I collagenase from Clostridium histolyticum binds to both (POG)n and (PPG)n peptides, where n is large enough to allow triple helix formation at RT (Matsushita et al., 2001). It is therefore reasonable to suppose that the same could be true of YadA. Not having a single, high-specificity binding site makes sense in view of pathogenicity: bacteria are unlikely to come into contact with extended collagenous surfaces, and thus the likelihood of encountering a specific binding site is rather small. If bacterial proteins can bind collagen in a more general fashion, bacteria expressing these proteins can then adhere opportunistically to any exposed collagen surfaces. A major difference between CNA and YadA is of course the mechanism of collagen binding. The CNA Hug mechanism requires triple helical collagen monomers brought about by tissue injury, whereas the open binding surface of YadA makes it possible for Y. enterocolitica to adhere to fibrillar collagen. This mechanistic difference may reflect the nature of the two bacteria; Staphylococcus aureus causes infection through damaged tissues, whereas Y. enterocolitica invades through the enteric mucosa and is thus confronted with intact collagen fibrils.
Funding
This work was supported by the Academy of Finland (Nos 1105157, 1114752, 16815 and 6301440 to AG and 45820, 50441 and 42105 to MS) and the Sigrid Juselius Foundation (to AG).
Supplementary Material
Acknowledgements
Adrian Goldman is a member of the Biocentrum Helsinki organisation. We would like to thank Dr Hilkka Lankinen (Department of Virology, Haartman Institute, Helsinki University) for assistance with the SPR measurements. We would also like to thank Professor Peter C. Kahn (Department of Biochemistry and Microbiology, Rutgers University, NJ, USA) for help with interpreting the CD data. Mass spectrometry was performed as a service at the Protein Chemistry Core Facility of the Institute of Biotechnology, University of Helsinki.
Footnotes
Edited by David L. Ollis
References
- Berisio R., Vitagliano L., Mazzarella L., Zagari A. Biopolymers. 2000;56:8–13. doi: 10.1002/1097-0282(2000)56:1<8::AID-BIP1037>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Cover T.L., Aber R.C. N. Engl. J. Med. 1989;321:16–24. doi: 10.1056/NEJM198907063210104. [DOI] [PubMed] [Google Scholar]
- El Tahir Y., Toivanen P., Skurnik M. J. Immunoassay. 1997;18:165–183. doi: 10.1080/01971529708005811. [DOI] [PubMed] [Google Scholar]
- El Tahir Y., Skurnik M. Int. J. Med. Microbiol. 2001;291:209–218. doi: 10.1078/1438-4221-00119. [DOI] [PubMed] [Google Scholar]
- Emody L., Heesemann J., Wolf-Watz H., Skurnik M., Kapperud G., O’Toole P., Wadstrom T. J. Bacteriol. 1989;171:6674–6679. doi: 10.1128/jb.171.12.6674-6679.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J.E., Spector A.A., Ashbrook J.D. Biochemistry. 1970;9:4580–4587. doi: 10.1021/bi00825a018. [DOI] [PubMed] [Google Scholar]
- Flugel A., Schulze-Koops H., Heesemann J., Kuhn K., Sorokin L., Burkhardt H., von der Mark K., Emmrich F. J. Biol. Chem. 1994;269:29732–29738. [PubMed] [Google Scholar]
- Fraser R.D., MacRae T.P., Suzuki E. J. Mol. Biol. 1979;129:463–481. doi: 10.1016/0022-2836(79)90507-2. [DOI] [PubMed] [Google Scholar]
- Gelse K., Poschl E., Aigner T. Adv. Drug Deliv. Rev. 2003;55:1531–1546. doi: 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y., Sakai R., Kakiuchi K., Isemura T. Biopolymers. 1970;9:415–425. doi: 10.1002/bip.1970.360090404. [DOI] [PubMed] [Google Scholar]
- Koide T. Connect. Tissue Res. 2005;46:131–141. doi: 10.1080/03008200591008518. [DOI] [PubMed] [Google Scholar]
- Kramer R.Z., Vitagliano L., Bella J., Berisio R., Mazzarella L., Brodsky B., Zagari A., Berman H.M. J. Mol. Biol. 1998;280:623–638. doi: 10.1006/jmbi.1998.1881. [DOI] [PubMed] [Google Scholar]
- Kramer R.Z., Bella J., Mayville P., Brodsky B., Berman H.M. Nat. Struct. Biol. 1999;6:454–457. doi: 10.1038/8259. [DOI] [PubMed] [Google Scholar]
- Li M.H., Fan P., Brodsky B., Baum J. Biochemistry. 1993;32:7377–7387. doi: 10.1021/bi00080a007. [DOI] [PubMed] [Google Scholar]
- Liu Q., Ponnuraj K., Xu Y., Ganesh V.K., Sillanpaa J., Murray B.E., Narayana S.V., Hook M. J. Biol. Chem. 2007;282:19629–19637. doi: 10.1074/jbc.M611137200. [DOI] [PubMed] [Google Scholar]
- Long C.G., Braswell E., Zhu D., Apigo J., Baum J., Brodsky B. Biochemistry. 1993;32:11688–11695. doi: 10.1021/bi00094a027. [DOI] [PubMed] [Google Scholar]
- Matsushita O., Koide T., Kobayashi R., Nagata K., Okabe A. J. Biol. Chem. 2001;276:8761–8770. doi: 10.1074/jbc.M003450200. [DOI] [PubMed] [Google Scholar]
- Miller E.J., Lunde L.G. Biochemistry. 1973;12:3153–3159. doi: 10.1021/bi00741a003. [DOI] [PubMed] [Google Scholar]
- Mintz K.P. Microbiology. 2004;150:2677–2688. doi: 10.1099/mic.0.27110-0. [DOI] [PubMed] [Google Scholar]
- Nummelin H., El Tahir Y., Ollikka P., Skurnik M., Goldman A. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1042–1044. doi: 10.1107/s0907444902005231. [DOI] [PubMed] [Google Scholar]
- Nummelin H., Merckel M.C., Leo J.C., Lankinen H., Skurnik M., Goldman A. EMBO J. 2004;23:701–711. doi: 10.1038/sj.emboj.7600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama K., Okuyama K., Arnott S., Takayanagi M., Kakudo M. J. Mol. Biol. 1981;152:427–443. doi: 10.1016/0022-2836(81)90252-7. [DOI] [PubMed] [Google Scholar]
- Okuyama K., Hongo C., Fukushima R., Wu G., Narita H., Noguchi K., Tanaka Y., Nishino N. Biopolymers. 2004;76:367–377. doi: 10.1002/bip.20107. [DOI] [PubMed] [Google Scholar]
- Pekkola-Heino K., Viljanen M.K., Stahlberg T.H., Granfors K., Toivanen A. Acta Pathol. Microbiol. Immunol. Scand. C. 1987;95:27–34. doi: 10.1111/j.1699-0463.1987.tb00005.x. [DOI] [PubMed] [Google Scholar]
- Persikov A.V., Ramshaw J.A., Kirkpatrick A., Brodsky B. Biochemistry. 2000;39:14960–14967. doi: 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- Ramshaw J.A., Shah N.K., Brodsky B. J. Struct. Biol. 1998;122:86–91. doi: 10.1006/jsbi.1998.3977. [DOI] [PubMed] [Google Scholar]
- Retamal C.A., Thiebaut P., Alves E.W. Anal. Biochem. 1999;268:15–20. doi: 10.1006/abio.1998.2977. [DOI] [PubMed] [Google Scholar]
- Riess T., et al. J. Exp. Med. 2004;200:1267–1278. doi: 10.1084/jem.20040500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggenkamp A., Neuberger H.R., Flugel A., Schmoll T., Heesemann J. Mol. Microbiol. 1995;16:1207–1219. doi: 10.1111/j.1365-2958.1995.tb02343.x. [DOI] [PubMed] [Google Scholar]
- Roggenkamp A., Ackermann N., Jacobi C.A., Truelzsch K., Hoffmann H., Heesemann J. J. Bacteriol. 2003;185:3735–3744. doi: 10.1128/JB.185.13.3735-3744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara S., Inouye K., Shudo K., Kishida Y., Kobayashi Y., Prockop D.J. Biochim. Biophys. Acta. 1973;303:198–202. doi: 10.1016/0005-2795(73)90164-5. [DOI] [PubMed] [Google Scholar]
- Schulze-Koops H., Burkhardt H., Heesemann J., von der Mark K., Emmrich F. Infect. Immun. 1992;60:2153–2159. doi: 10.1128/iai.60.6.2153-2159.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Koops H., Burkhardt H., Heesemann J., von der Mark K., Emmrich F. Arthritis Rheum. 1995;38:1283–1289. doi: 10.1002/art.1780380917. [DOI] [PubMed] [Google Scholar]
- Skurnik M. J. Appl. Bacteriol. 1984;56:355–363. doi: 10.1111/j.1365-2672.1984.tb01362.x. [DOI] [PubMed] [Google Scholar]
- Skurnik M. Infect. Immun. 1985;47:183–190. doi: 10.1128/iai.47.1.183-190.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurnik M., El Tahir Y., Saarinen M., Jalkanen S., Toivanen P. Infect. Immun. 1994;62:1252–1261. doi: 10.1128/iai.62.4.1252-1261.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symersky J., et al. Nat. Struct. Biol. 1997;4:833–838. doi: 10.1038/nsb1097-833. [DOI] [PubMed] [Google Scholar]
- Tamm A., Tarkkanen A.M., Korhonen T.K., Kuusela P., Toivanen P., Skurnik M. Mol. Microbiol. 1993;10:995–1011. doi: 10.1111/j.1365-2958.1993.tb00971.x. [DOI] [PubMed] [Google Scholar]
- Zong Y., Xu Y., Liang X., Keene D.R., Hook A., Gurusiddappa S., Hook M., Narayana S.V. EMBO J. 2005;24:4224–4236. doi: 10.1038/sj.emboj.7600888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.