

Abstract
There is growing interest in the epigenetic mechanisms that are dysregulated in cancer and other human pathologies. Under this broad umbrella, modulators of histone deacetylase (HDAC) activity have gained interest as both cancer chemopreventive and therapeutic agents. Of the first generation, FDA-approved HDAC inhibitors to have progressed to clinical trials, vorinostat represents a “direct acting” compound with structural features suitable for docking into the HDAC pocket, whereas romidepsin can be considered a prodrug that undergoes reductive metabolism to generate the active intermediate (a zinc-binding thiol). It is now evident that other agents, including those in the human diet, can be converted by metabolism to intermediates that affect HDAC activity. Examples are cited of short-chain fatty acids, seleno-α-keto acids, small molecule thiols, mercapturic acid metabolites, indoles, and polyphenols. The findings are discussed in the context of putative endogenous HDAC inhibitors generated by intermediary metabolism (e.g. pyruvate), the yin–yang of HDAC inhibition versus HDAC activation, and the screening assays that might be most appropriate for discovery of novel HDAC inhibitors in the future.
Keywords: Epigenetics, chromatin remodeling, protein acetylation, HDAC, chemoprevention, chemoprotection, cancer
Introduction
Epigenetics has arrived front-and-center on the popular landscape. A widely read current affairs magazine recently showed a cover image of double-stranded DNA being unzipped next to the words: “Why your DNA isn’t your destiny: the new science of epigenetics reveals how the choices you make can change your genes—and those of your kids.” Just how “new” this science really is can be debated, since it has its roots in the earliest discussions on nature versus nurture. Although Darwin argued that incremental changes underlie the process of natural selection and survival of the fittest, Lamarck postulated that some traits were acquired within a lifetime due to environmental pressures. Handel and Ramagopalan (2010) adopted the middle ground in stating that epigenetics allows for the “peaceful co-existence” of Darwinian and Lamarckian evolution, while emphasizing that the underlying mechanisms are now clearly implicated in disease susceptibility.
The US National Institutes of Health developed the Roadmap Epigenetics Program with the goal of studying human health and disease in the context of “changes in the regulation of gene activity and expression that are not dependent on gene sequence … both heritable changes in gene activity and expression, and long-term alterations in the transcriptional potential of a cell that are not necessarily heritable. Epigenetics refers to the study of single genes or sets of genes, whereas epigenomics refers to more global analyses of epigenetic changes across the entire genome” (http://nihroadmap.nih.gov/epigenomics/).
Articles related to this topic have appeared in the present journal, including a discussion on histone recognition by conserved structural folds (Yap and Zhou, 2010), decoding of the histone H4 lysine 20 methylation mark (Balakrishnan and Milavetz, 2010), and modifying chromatin architecture during the response to DNA breakage (Venkitaraman, 2010). The latter subject also falls under the umbrella of epigenetic changes implicated in cancer development, along with aberrant DNA methylation, altered profiles of microRNAs, and miswritten or misinterpreted histone modifications (Iorio and Croce, 2009; Chi et al., 2010; Poke et al., 2010; Sharma et al., 2010).
Histone modifications, histone deacetylases, and associated human pathologies
Posttranslational modifications to histones such as acetylation, methylation, phosphorylation, and ubiquitination play a pivotal role in the regulation of gene expression (Myzak and Dashwood, 2006a; Delage and Dashwood, 2008, 2009a,b; Lee et al., 2010a). These modifications alter chromatin structure and influence the binding of remodeling factors, transcription factors, co-activators, and co-repressors (Figure 1). For example, acetylation and deacetylation of histones is mediated by the opposing activities of two classes of enzymes: histone acetyl-transferases (HATs) and histone deacetylases (HDACs). HDACs include the zinc-dependent members of classes I, II, and IV, as well as the class III “sirtuins.” Among the class I HDACs, HDAC1, HDAC2, and HDAC8 are present mainly in the nucleus, whereas HDAC3 can be found in either the nucleus or the cytoplasm. HDAC3 may be associated in nuclear co-repressor complexes with protein partners such as N-Cor/SMRT (nuclear receptor co-repressor/silencing mediator for retinoid and thyroid hormone receptors) and HDAC4. The latter is designated as a class II HDAC, along with HDAC5, HDAC6, HDAC7, HDAC9, and HDAC 10, all of which can shuttle between the nucleus and cytoplasm, and tend to exhibit a more restricted tissue expression pattern than class I HDACs. HDAC11, currently the sole member of HDAC class IV, was identified by DNA sequence similarity; little is known about its major function(s) and possible redundancy with other HDACs (Yang and Seto, 2008). Classes I, II, and IV HDACs are inhibited to some degree by compounds such as trichostatin A (TSA).
Figure 1.

Interactions between histone-modifying enzymes and histone modifications associated with gene silencing and unsilencing. Gene activation (top) requires the recruitment of chromatin-remodeling complexes, histone acetyltransferases (HATs), and histone methyltransferases (HMTs, such as trithorax). During gene silencing, DNA methylation catalyzed by DNA methyltransferases (DNMTs) and methyl-binding domain proteins (e.g. methyl CpG binding protein 2, MeCP2) recruits histone deacetylases (HDACs) and HMTs to repress transcription. HDAC inhibitors trigger the release of HDACs and their co-repressor complexes, leading to an open chromatin state that is poised for gene activation. In cancer cells, epigenetic mechanisms affecting DNA methylation and histone marks silence tumor suppressors, cell cycle checkpoint regulators, and apoptosis inducers. For further details, see Delage and Dashwood (2008).
Class III HDACs are NAD+-dependent enzymes that lack the catalytic zinc atom and are generally TSA-insensitive. Their dependence on NAD+ links sirtuins to intermediary metabolism and to factors that affect NAD+/NADH ratios in cells. This topic connects basic aspects of intermediary metabolism to the modulation of HDAC activity. The reader is referred elsewhere for articles on sirtuins and metabolic signaling (Denu, 2007; Calabrese et al., 2008; Dittenhafer-Reed et al., 2010; Imai, 2010; Imai and Guarente, 2010; Kyrylenko and BaniAhmad, 2010; Silva and Wahlestedt, 2010; Yu and Auwerx, 2010).
In addition to metabolic signaling and metabolic disorders, HDACs have been implicated in diabetes (Lawless et al., 2009; Imai and Guarente, 2010), the cardiorenal axis and cardiovascular diseases (Bush and McKinsey, 2010; Colussi et al., 2010), psychiatric disorders (Stahl, 2010), neurodegenerative diseases (Dietz and Casaccia, 2010; Krainc, 2010; Ramadori and Coppari, 2010), chronic obstructive pulmonary disease (Barnes, 2010a), aging (Donmez and Guarente, 2010), and cancer (Marks and Xu, 2009; Biancotto et al., 2010; Mercurio et al., 2010). There is growing appreciation, therefore, for the importance of reversible protein acetylation in human health and disease.
Cellular targets of HDAC inhibitors include both histone and non-histone proteins. As a consequence, terms such as protein deacetylase, lysine (K) deacetylase (KDAC), and “KDAC inhibitor” have appeared in the literature (Gurard-Levin et al., 2010; Lundh et al., 2010; Singh et al., 2010a). HDAC6, for example, is a “KDAC” with a nuclear role in regulating the survivin gene promoter (Ma et al., 2005), but it also modulates the chaperone functions of heat shock protein 90 (Bali et al., 2005; Park et al., 2008; Kekatpure et al., 2009). HDAC6 acts as a tubulin deacetylase and master regulator of cellular responses to cytotoxic insults (Hubbert et al., 2002; Matthias et al., 2008). Effects on tubulin acetylation and protein trafficking link HDAC6 to various neurodegenerative disorders (Pandey et al., 2007; Ding et al., 2008; Rivieccio et al., 2009; Lee et al., 2010b). Thus, HDAC6 and other HDACs appear to influence protein misfolding/trafficking in the brain, as well as affecting neuronal cell differentiation and apoptosis via gene repression/de-repression.
Gene de-repression also provides a mechanistic basis for the use of HDAC inhibitors in cancer therapy. When HDACs remove the acetyl groups from histone tails (Figure 1), the resulting chromatin condensation leads to transcriptional repression (reviewed by Delage and Dashwood, 2008; Lee et al., 2010a). In cancer cells, this represents an important mechanism of gene silencing, shutting down the expression of critical players involved in cell survival, mitosis, nucleotide metabolism, and angiogenesis (Miyanaga et al., 2008; LaBonte et al., 2009). Since epigenetic modifications are potentially reversible, unlike the genetic changes that affect DNA sequence, they are desirable targets for therapeutic or chemopreventive strategies. Such an approach may be feasible in many different cancer types, and throughout the progression from early initiation to promotion and metastasis. By coaxing neoplastically transformed cells into re-expressing epigenetically silenced tumor suppressors, HDAC inhibitors trigger growth inhibition, cell cycle arrest, differentiation, and/or apoptosis. This can enhance the debulking of tumors by augmenting other cancer treatment modalities. Epigenetic modifications can also be early events in carcinogenesis; thus, prevention/reversal efforts might affect pre-neoplastic cells or early stages of tumorigenesis, before wholesale changes in histone posttranslational modifications and HDAC expression.
HDAC overexpression has been observed in a number of human primary cancers and cancer cell lines, including neuroblastoma (Oehme et al., 2009a,b), renal cancer (Fritzsche et al., 2008), prostate cancer (Patra et al., 2001; Abbas and Gupta, 2008), gastric cancer (Kim et al., 2003), and colorectal cancer (Mariadason, 2008; Ashktorab et al., 2009). In the latter case, for example, HDAC2 nuclear expression was detected at high levels in 82%, 62%, and 53% of human colorectal carcinomas, adenomas, and normal tissues, respectively (Ashktorab et al., 2009). Collectively, these and other studies provide evidence that perturbation of the balance between acetylation and deacetylation is an important factor in neoplastic transformation. Indirect evidence of the importance of acetylation status in tumorigenesis also comes from the observation that tumor cell growth can be halted or even reversed by HDAC inhibitors.
HDAC inhibitors and cancer therapeutics—role of metabolism
HDAC inhibitors were first identified and isolated from natural sources (reviewed by Yoshida et al., 2003). In the intervening two decades, the list of HDAC inhibitors has expanded to include hydroxamic acids, short-chain fatty acids, boronic acids, α-keto acids, cyclic tetrapeptides, benzamides, ketones, isothiocyanates, organosulfur compounds, selenium-based compounds and their metabolites, and other miscellaneous agents (Minucci and Pelicci, 2006; Delage and Dashwood, 2009a; Lane and Chabner, 2009; Nian et al., 2009a,b; Suzuki et al., 2009; Desai et al., 2010; Noureen et al., 2010). Based on the features of the active site pocket in the presence and absence of bound ligands (Finnin et al., 1999; Vannini et al., 2004, 2007; Somoza et al., 2004; Bottomley et al., 2008; Dowling et al., 2008; Schuetz et al., 2008; Ficner, 2009), and computational modeling in silico (Vannini et al., 2007; Nian et al., 2008, 2009b; Ortore et al., 2009; Suzuki et al., 2009; Wang, 2009; Oger et al., 2010), numerous HDAC inhibitor candidates have been identified. These compounds typically have a functional group that interacts with the zinc atom in the enzyme pocket, a spacer “arm” that fits into the channel near the active site, and in many (but not all cases) a cap group that associates with residues near the surface.
Before their mechanisms of action were elucidated, small molecule hydroxamic acids and cyclic tetrapeptides were observed to alter the differentiation status of cancer cells in culture (reviewed by Myzak and Dashwood, 2006b; Santini et al., 2007; Jones and Steinkühler, 2008). Yoshida et al. (1990) were the first to report on the potent HDAC inhibitory activity of TSA, a natural compound isolated from Streptomyces platensis. Subsequent studies showed that TSA reversed the morphological transformation of oncogenic ras-transformed NIH3T3 cells (Futamura et al., 1995). In addition, TSA increased global histone H3 and H4 acetylation, enhanced the expression of hepatocyte-specific genes, and induced hepatocyte differentiation in human hepatoma cells (Yamashita et al., 2003). In human embryonic kidney 293 (HEK293) cells, the glutathione S-transferase (GST) inhibitor ethacrynic acid potentiated the effects of TSA (Myzak et al., 2004), implicating the mercapturic acid pathway in the metabolism of this prototype HDAC inhibitor. The mercapturic acid pathway is a glutathione-dependent pathway that plays a critical role in the detoxification of a large number of foreign compounds (also known as xenobiotics). This pathway is modulated by many factors, including dietary constituents (Higdon et al., 2007; see also sulforaphane text below). In principle, therefore, nutrient interactions that induce the mercapturic acid pathway might lower the efficacy of TSA and structurally related HDAC inhibitors in vivo. This might account for the fact that TSA shows no effect in animal models due to its “metabolic instability” (Masuoka et al., 2008).
Due to these concerns, alternative hydroxamate-based HDAC inhibitors have been developed. Vorinostat (suberoylanilide hydroxamic acid, SAHA) has been described as hitting “the happy medium … potent enough to be useful and tolerated in patients” (Marks and Breslow, 2007). Early Phase I studies in humans suggested that vorinostat was well-tolerated (Kelly et al., 2003), had linear pharmacokinetics and good bioavailability (Kelly et al., 2005), and was effective in hematologic malignancies, including Hodgkin’s disease and subtypes of non-Hodgkin’s lymphoma (O’Connor et al., 2006). Phase 2 trials of vorinostat demonstrated activity in patients with cutaneous T-cell lymphoma (Duvic et al., 2007) and modest single-agent responses in patients with glioblastoma multiforme (Galanis et al., 2009). Other clinical trials have been conducted with vorinostat, alone and in combination with cancer therapeutic agents (Fouladi et al., 2010; Kadia et al., 2010; Ramalingam et al., 2010; Wilson et al., 2010). Marked interindividual pharmacokinetic variability has been observed with vorinostat, possibly related to pharmacogenetic influences on glucuronidation (Kang et al., 2010), or to dietary factors that modulate the mercapturic acid pathway (Higdon et al., 2007).
Like TSA, trapoxin was shown to induce morphological reversion in transformed NIH3T3 fibroblasts (Itazaki et al., 1990; Yoshida and Sugita, 1992). Subsequent work demonstrated that trapoxin was an irreversible HDAC inhibitor, and that chemical reduction of the epoxide group abolished the inhibitory activity (Kijima et al., 1993). The latter observation hinted at the possibility that reductive metabolism might play a role in lowering the efficacy of trapoxin and structurally related HDAC inhibitors in vivo. Trapoxin resembles TSA in lacking efficacy in animal models due to the “metabolic instability” of the parent compound (Masuoka et al., 2008).
On the other hand, cellular reduction of the disulfide bond in depsipeptide (FK228) generates a more active compound, most likely a mercaptobutenyl intermediate that fits into the HDAC pocket (Desai et al., 2010). This HDAC inhibitor was first isolated as a fermentation product from Chromobacterium violaceum (reviewed by Masuoka et al., 2008). FK228 has progressed to clinical trials under the name romidepsin, with evidence for “significant and sustainable single-agent activity and an acceptable safety profile” (Whittaker et al., 2010). Depsipeptide thus provided one of the earliest examples of metabolism generating an HDAC inhibitor, but other examples are now known, including the various compounds from dietary sources (see below).
HDAC inhibitors were discovered based on their ability to induce differentiation in cancer cells, and this continues to be an active area of research. For example, neuroblastoma cells differentiate in response to HDAC8-selective inhibitors or targeted knockdown of HDAC8 (Oehme et al., 2009b), and human leukemia differentiate after treatment with HDAC inhibitors FK228 and sodium phenylbutyrate (Savickiene et al., 2010). Sodium phenyl-butyrate has been used clinically in the treatment of disorders such as maple syrup urine disease (Brunetti-Pierri et al., 2010), and there is growing interest in the neuroprotective properties of this compound and its metabolites (Gardian et al., 2005; Ryu et al., 2005; Petri et al., 2006; Hogarth et al., 2007; Ebbel et al., 2010). A recently completed Phase 2 study in patients with amyotrophic lateral sclerosis (Lou Gehrig’s disease) concluded that blood levels of phenylbutyrate, and of its primary metabolite phenylacetate, increased with dosage, and that 9 g/day was effective for improving histone acetylation status (Cudkowicz et al., 2009). Phenylbutyrate shares structural features with the antiepileptic agent valproic acid (Göttlicher, 2004), and with the oldest known dietary HDAC inhibitor, butyrate.
Dietary HDAC inhibitors—role of metabolism
Short-chain fatty acids: HDAC inhibitors generated via gut fermentation of dietary fiber
Butyrate serves as the primary metabolic fuel for the colonocyte, where it can be present at up to millimolar concentrations in the gut (reviewed by Myzak and Dashwood, 2006b). This short-chain fatty acid is generated via the gut fermentation of dietary fiber, and it can be considered an early example of the role of metabolism in generating HDAC inhibitors. As in the case of TSA, butyrate was first reported to increase cell differentiation (Leder and Leder, 1975), and subsequently was shown to affect histone acetylation status (Riggs et al., 1977; Boffa et al., 1978). Like TSA, butyrate acts as a competitive HDAC inhibitor (Sekhavat et al., 2007). A Ki of 46 μM was reported for HDAC inhibition by butyrate in whole cell lysates of human MCF-7 breast cancer cells, compared with a Ki of 1 nM for TSA under the same conditions (Sekhavat et al., 2007). The difference in Ki values high-lights an important point, namely that dietary factors are much weaker HDAC ligands than the agents developed for cancer therapy. The possible relevance of this observation in the context of cancer prevention and treatment has been discussed elsewhere (Dashwood et al., 2006).
In erythroleukemia cells, 4-phenylbutyrate was a more effective HDAC inhibitor and a more potent inducer of histone acetylation than other structural analogs of butyrate, including 2- and 3-phenylbutyrate, 2-phenoxybutyrate, phenoxyacetate, cinnamate, and methoxycinnamate (Lea et al., 1999a). A prodrug form of butyrate, tributyrin, suppressed hepatocarcinogenesis in the rat and increased hepatic nuclear histone H3K9 acetylation levels (Kuroiwa-Trzmielina et al., 2009). Butyrate also was reported as the most relevant HDAC inhibitor formed in fermentations of human fecal slurry with apple pectin, and apple juice extracts produced butyrate and other unidentified HDAC inhibitors (Waldecker et al., 2008a,b).
Despite one case of remission in a child with acute myelogenous leukemia (Novogrodsky et al., 1983), therapeutic interventions with butyrate have been disappointing (Oki and Issa, 2006). Optimization of the route and length of administration of butyrate may increase its therapeutic effects. For example, in a randomized, double-blind cross-over study, daily rectal administration of butyrate was found to improve biomarkers of oxidative stress in the healthy human colon (Hamer et al., 2009). Combining butyrate with mesalazine produced a marked improvement in the symptoms and endoscopic appearance of the gut mucosa in ulcerative colitis patients (Assisi et al., 2008).
Chronic exposure to butyrate through the daily consumption of dietary fiber as “whole food” may also have significant chemopreventive effects over a lifetime (Pool-Zobel and Sauer, 2007). Consumption of whole grain foods made from high-amylose barley resulted in a 57% increase in fecal total short-chain fatty acids and a 91% higher excretion of butyrate (Bird et al., 2008). Soy oligosaccharide intake (3 g/day) increased the levels of short-chain fatty acids in women, such as propionate and butyrate, compared with women who had not consumed soy oligosaccharide (Bang et al., 2007). A cereal-based evening meal rich in indigestible carbohydrates was shown to increase plasma butyrate the next morning; the authors concluded that short-chain fatty acids, in particular butyrate, might account for the protection afforded by whole grains against cardiovascular disease and type 2 diabetes (Nilsson et al., 2010). An ongoing study in human volunteers seeks to examine the relationships between colonic cell turnover and early biomarkers of carcinogenesis, dietary fiber intake/fermentation, and global protein acetylation (Corfe et al., 2009). It will be of great interest if these findings can be related to altered HDAC activities and to the role of metabolism generating intermediates such as butyrate.
Organoselenium compounds: α-keto acid metabolites as HDAC inhibitors
Butyrate is the oldest known dietary HDAC inhibitor, but an interesting structural analog was recently discovered that pointed to a new class of selenium-based HDAC inhibitors. Thus, keto-methylselenobutyrate (KMSB) and its structural analog methylselenopyruvate (MSP) were identified as novel competitive HDAC inhibitors. Enzyme kinetic studies supported a competitive mechanism, with a Ki of 35 μM MSP with human HDAC8 (Lee et al., 2009; Nian et al., 2009b).
Seleno-α-keto acids are generated as metabolites of natural organoselenium compounds, including the major dietary forms methylselenocysteine (MSC) and selenom-ethionine (SM). The transamination of SM to KMSB, and of MSC to MSP, competes with a lyase-catalyzed pathway that produces methylselenol (Figure 2). The latter metabolite has been considered an important mediator of the anticancer effects of selenium compounds, acting on redox-sensitive signaling proteins and transcription factors to reduce the risk of cancer development and progression (Lü and Jiang, 2005; Jackson and Combs, 2008; Ohta and Suzuki, 2008; Tsuji et al., 2009; Pinto et al., 2010; Zeng et al., 2010).
Figure 2.

Working model for chemoprotection by organoselenium compounds. The production of methylselenol competes with a transamination reaction that generates seleno-α-keto acid metabolites as histone deacetylase (HDAC) inhibitors. For further details, see text and Lee et al. (2009) and Nian et al. (2009b).
Hepatic enzymes such as L-amino acid oxidase and glutamine transaminase-liver (GTL) produce the seleno-α-keto acid metabolites from the corresponding parent compounds in liver (Pinto et al., 2010), but in other tissues this reaction is catalyzed by glutamine transaminase-kidney (GTK). Interestingly, human colon and prostate cancer cells contain the enzyme GTK, which has high affinity for MSC but negligible activity toward SM as a substrate (Lee et al., 2009). As a consequence, in colon and prostate cancer cells SM is not readily converted to KMSB, whereas MSP is readily formed from MSC. Inhibitors of the pyridoxal phosphate group in GTK indicated that transamination is an important and necessary step for HDAC inhibition and histone hyper-acetylation by MSC. From molecular docking studies, the carbonyl group generated by the transamination reaction was predicted to interact in the HDAC pocket with a critical tyrosine residue and with the zinc atom (Figure 3, arrows). An amine group in the substrate interfered with docking and zinc-binding, thus explaining the lack of inhibition by MSC and SM parent compounds when added directly to HDAC activity assays in vitro.
Figure 3.

Molecular docking of seleno-α-keto acid metabolites in the histone deacetylase (HDAC) pocket. β-Keto-methyl-γ-selenobutyrate (KMSB) and β-methylselenopyruvate (MSP) contain a carbonyl group generated in the transamination reaction, see Figure 2. This carbonyl group is predicted to interact in the human HDAC8 pocket with a critical tyrosine 306 residue and the zinc atom. For details on the modeling procedure, see Nian et al. (2009b).
Further work is needed to corroborate whether these seleno-α-keto acid metabolites are generated in vivo, under conditions of normal dietary intake in foods such as Brazil nuts, garlic, seafood, and cruciferous vegetables, and below the threshold for selenium toxicity (http://lpi.oregonstate.edu/infocenter/minerals/selenium/). PubMed lists over 400 separate reviews on selenium and human health, including the conflicting evidence from various clinical trials (reviewed by Muecke et al., 2010). It is noteworthy that a large trial that was halted recently (Lippman et al., 2009) used SM, a form of selenium that is anticipated to generate methylselenol, but not KMSB, in tissues such as prostate and colon. MSC might have been a better candidate for the clinical trials, based on the new paradigm of HDAC inhibition.
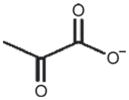
MSP has a pyruvate moiety, which raises an interesting question—does pyruvate itself act as an HDAC inhibitor, and do other α-keto acids generated as part of normal intermediary metabolism serve the role of endogenous HDAC inhibitors? There is evidence, in fact, to support such a possibility. Interestingly, the findings connect with the Warburg hypothesis (Warburg, 1956) and the divergent roles of nutrient transporters in normal cells and cancer cells. Thus, SLC5A8 and SLC5A12 are sodium-coupled monocarboxylate transporters (SCMTs) with important physiological functions in the gastrointestinal (GI) tract and other tissues (Ganapathy et al., 2009). These transporters exert a tumor suppressor function by regulating the intracellular concentrations of pyruvate, butyrate, and propionate. In cancer cells, silencing of SCMTs coupled with the conversion of pyruvate to lactate correlates with increased HDAC activity and reduced apoptosis (Ganapathy et al., 2009). When MCF-7 breast cancer cells were transfected with SLC5A8 cDNA, pyruvate-mediated apoptosis was triggered, a response also seen with butyrate and propionate, but not lactate. Lactate is produced in cancer cells as a result of the increased rate of glycolysis and the relatively low oxidation of pyruvate in mitochondria (Hockenbery, 2010; Israelsen and Vander Heiden, 2010; Sattler et al., 2010). Interestingly, pyruvate, butyrate, and propionate were identified as inhibitors of HDAC1 and HDAC3, whereas lactate had no effect on HDAC activity (Thangaraju et al., 2009a). Pyruvate and butyrate inhibited HDAC1 with IC50 values of 24 and 20 μM, and inhibited HDAC3 with IC50 values 80 and 75 μM, respectively. 3-Bromopyruvate, an alkylating agent with antitumor activity, also inhibited HDAC1 and HDAC3 in human breast cancer cells (Thangaraju et al., 2009b). This led to the intriguing hypothesis that cancer cells silence monocarboxylate transporters, and convert pyruvate to lactate, as a cooperative approach to circumventing pyruvate-mediated HDAC inhibition and apoptosis induction (Ganapathy et al., 2008).
Organosulfur compounds: small molecule thiols as HDAC inhibitors
Garlic, onions, shallots, and other members of the Allium family contain an interesting and complex range of water-soluble and fat-soluble organosulfur compounds, some of which have been implicated as cancer chemopreventive agents (Powolny and Singh, 2008; Iciek et al., 2009; Nian et al., 2009a; Gullett et al., 2010). Alliin (allylcysteine sulfoxide), allicin (allyl 2-propenethiosulfinate), S-allylcysteine (SAC), S-allylmercaptocysteine (SAMC), diallyl sulfide (DAS), diallyl disulfide (DADS), and diallyl trisulfide (DATS), as well as their metabolites allyl methyl sulfide (AMS), methyl mercaptan, and allyl mercaptan (AM) have been examined in the context of inhibition of carcinogen activation, induction of phase 2 detoxification pathways, and changes in cell differentiation and apoptosis pathways.
Over a decade ago, Lea and colleagues reported on the increased acetylation of histones in mouse erythroleukemia cells treated with DADS. Acetylation was also induced in rat hepatoma and human breast cancer cells by DADS and its metabolite, AM (Lea et al., 1999b). These observations were extended to other organosulfur compounds, including allyl isothiocyanate (Lea and Randolph, 2001; Lea et al., 2001) and SAMC (Lea et al., 2002). Increased histone acetylation in liver and Morris hepatoma 7777 was induced by treatment of rats with DADS, AM, and butanethiol (Lea and Randolph, 2001). In human colon cancer cells incubated with DADS, HDAC activity was inhibited and there was increased histone acetylation and p21WAF1 expression (Druesne et al., 2004a). Repetitive treatment of colon cancer cells with DADS induced prolonged hyperacetylation of histone H3 K14 (Druesne et al., 2004b). In rats given DADS by gavage or intracecal perfusion, increased histone acetylation was evident in normal colonocytes (Druesne-Pecollo et al., 2007, 2008).
Using HeLa nuclear extracts or purified human HDAC8 as source of enzyme, only AM inhibited HDAC activity in a concentration-dependent manner among several garlic-derived organosulfur compounds and their metabolites, including SAMC, SAC, DAS, DADS, DATS, AMS, and AM. Enzyme kinetics experiments coupled with computational modeling supported a competitive mechanism, with a Ki of 24 μM for AM with human HDAC8 (Nian et al., 2008). In the docked structure, the-SH group of AM was optimally positioned to interact with the zinc atom in the HDAC pocket (Figure 4). This paralleled the findings with other thiol-based HDAC inhibitors and their prodrug candidates (Suzuki et al., 2004, 2005; Sanda et al., 2007). Collectively, the studies with dietary organosulfur compounds support the hypothesis that a complex profile of water-soluble and lipid-soluble compounds is funneled by metabolism toward a small number of reactive thiols, with AM being the most effective HDAC inhibitor (Lea et al., 1999b; Nian et al., 2008). These findings do not preclude other mechanisms or molecular targets of dietary organosulfur compounds (Powolny and Singh, 2008; Iciek et al., 2009; Gullett et al., 2010).
Figure 4.

Molecular modeling of histone deacetylase (HDAC) 8-allyl mercaptan (AM) complex. AM is a small molecule thiol generated via the metabolism of organosulfur compounds in garlic, see Nian et al. (2008).
Isothiocyanates: mercapturic acid metabolites and HDAC inhibition
Brassica or cruciferous vegetables are a rich source of glucosinolates (Higdon et al., 2007). The hydrolysis of these glucosinolates by the plant enzyme myrosinase generates biologically active isothiocyanates and indoles. For example, broccoli and broccoli sprouts are a rich source of glucoraphanin, the precursor of sulforaphane (SFN). SFN is widely reported to exert anticancer effects in vitro and in vivo (Higdon et al., 2007; Juge et al., 2007; Clarke et al., 2008; Dinkova-Kostova and Talalay, 2008; Nian et al., 2009a; Valgimigli and Iori, 2009; Cheung and Kong, 2010; Gullett et al., 2010; Kwak and Kensler, 2010).
SFN was first discovered as a potent Phase 2 enzyme inducer (Zhang et al., 1992), acting via the Kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2 (Keap1/Nrf2) pathway and other anti-cancer mechanisms (reviewed in Clarke et al., 2008; Cheung and Kong, 2010; Gullett et al., 2010; Kwak and Kensler, 2010). A “one–two” chemoprotection paradigm has been proposed for SFN in which the electrophilic parent compound targets Keap1 to release Nrf2 into the nucleus, and the metabolites inhibit HDAC activity, leading to unsilencing of tumor suppressor genes that trigger cell cycle arrest and apoptosis (Dashwood et al., 2006; Dashwood and Ho, 2007).
Support for the latter hypothesis first came from experiments in HEK293 cells and human HCT116 colon cancer cells (Myzak et al., 2004), and subsequently in human prostate and breast cancer cells (Myzak et al., 2006; Pledgie-Tracy et al., 2007). Rather than the parent compound, SFN metabolites generated via the mercapturic acid pathway were implicated in the mechanism of HDAC inhibition (Figure 5). GST catalyzes formation of the SFN–glutathione (SFN-GSH) conjugate, which is then converted to other intermediates such as SFN–cysteine (SFN-Cys) and SFN–N-acetylcysteine (SFN-NAC). When cells were incubated with SFN and the cell-free media was added to the in vitro HDAC activity assay, concentration-dependent inhibition was evident (Myzak et al., 2004). This was attenuated when cells were pretreated with a GST inhibitor, ethacrynic acid, which blocks the first step in the mercapturic acid pathway. Notably, direct addition of SFN parent compound to the in vitro HDAC activity assay, with HeLa nuclear extracts as source of enzyme, had no inhibitory effect. Subsequent experiments provided evidence for the following order of HDAC inhibition in vitro: SFN-Cys > SFN-NAC > SFN-GSH ≫ SFN. By computational modeling, SFN-Cys fit the HDAC pocket and adopted a similar orientation as SAHA and TSA (Figure 5). The carboxylate group in SFN-Cys was predicted to form a bidentate ligand with the zinc atom, analogous to that seen for SAHA and TSA in the crystal structure (Finnin et al., 1999; Somoza et al., 2004).
Figure 5.

Metabolism of sulforaphane (SFN) via the mercapturic acid pathway generates intermediates such as SFN–glutathione (SFN-GSH), SFN–cysteine (SFN-Cys), and SFN–N-acetylcysteine (SFN-NAC). SFN-Cys was modeled to fit into the histone deacetylase (HDAC) pocket in a similar orientation as trichostatin A (TSA). A bidentate interaction is shown for SFN-Cys with the zinc atom, as occurs in the crystal structure containing TSA. See text and Myzak et al. (2004).
Since the–Cys group was predicted to enter into the HDAC pocket, other isothiocyanates that are metabolized via the mercapturic pathway were considered candidate HDAC inhibitors, including those found in pungent foods such as mustard, radish, horseradish, wasabi, and daikon (Higdon et al., 2007; Nian et al., 2009a; Verkerk et al., 2009; Yamasaki et al., 2009; Ernst et al., 2010). The mustard oil compound allyl isothiocya-nate was reported earlier to induce histone acetylation in mouse erythroleukemia cells, but with no apparent inhibition of HDAC activity (Lea et al., 2001). We confirmed the latter observation, while showing that longer-chain isothiocyanates inhibited HDAC activity in human colon cancer cells (Dashwood et al., 2006). The inhibition of HDAC activity increased with length of the spacer “arm” in the parent molecule, and was associated with H4K12 hyperacetylation, p21WAF induction, cell cycle arrest, and apoptosis (Rajendran et al., manuscript in preparation). As with SFN in vitro, none of the isothiocyanates were inhibitory when added directly to the HDAC assay in the presence of HeLa nuclear extracts, supporting the need for metabolites to be formed as the “ultimate” HDAC inhibitors.
The cap group in HDAC inhibitors lies close to the surface and can dictate specificity toward individual HDACs (Vannini et al., 2007; Nian et al., 2008, 2009b; Ortore et al., 2009; Suzuki et al., 2009; Wang, 2009; Oger et al., 2010). Interestingly, benzyl isothiocyanate (BITC) was reported to inhibit HDAC activity in human pancreatic carcinoma cells, and this was rescued by overexpression of HDAC1 or HDAC3 (Batra et al., 2010). Immunohistochemical staining of tumors from mice treated with BITC showed significantly reduced staining of HDAC1 and HDAC3 compared with controls (Batra et al., 2010).
In addition to HDAC expression and histone acetylation, other epigenetic marks may be involved. In human breast cancer cells, analyses of the human telomerase reverse transcriptase (hTERT) promoter revealed that SFN increased the levels of active chromatin marks, such as acetyl-H3, acetyl-H3K9, and acetyl-H4, while lowering repressive marks such as H3K9Me3 and H327Me3 (Meeran et al., 2010). Ma et al. also reported that phenylhexyl isothiocyanate (PHITC) inhibited HDAC activity in human leukemia cells, with evidence for increased histone acetylation, elevated H3K4 “active” methylation, and loss of H3K9 “repressive” methylation marks (Ma et al., 2006). PHITC reactivated aberrantly hypermethylated P15 gene expression in acute leukemia cells through changes in both DNA methylation and histone acetylation (Jiang et al., 2010). Moreover, in patients with acute leukemia, histone acetylation was virtually undetectable, but was reversed in the presence of PHITC (Xiao et al., 2010). Phenethyl isothiocyanate (PEITC), found in watercress, de-repressed the P21WAF1 promoter in prostate cancer cells via inhibition of HDAC activity, enhanced histone acetylation, and changes in histone methylation (Wang et al., 2008). Interestingly, PEITC was reported to overcome resistance to vorinostat in human leukemia cells (Hu et al., 2010), hinting at drug/diet interactions that augment HDAC inhibition and gene re-expression.
Indoles: acid condensation products that alter HDAC expression
As noted above, cruciferous vegetables contain glucosinolates such as glucoraphanin, the precursor of SFN, and glucobrassicin, the precursor of indole-3-carbinol (I3C). The latter compound and its acid condensation products, such as 3,3′-diindolylmethane (DIM), have been examined extensively for their cancer chemoprotective properties (Aggarwal and Ichikawa, 2005; Higdon et al., 2007; Weng et al., 2008; Ahmad et al., 2010). A recent report found that DIM selectively induced the proteasome-mediated degradation of class I HDACs in human colon cancer cells, without affecting class II HDACs (Li et al., 2010a). This distinguishes DIM, a dimer of I3C formed in vivo, from synthetic HDAC inhibitors centered around a 3-piperidin-3-ylindole moiety (Cho et al., 2010), and the 3-arylindeneindolin-2-one-based compounds that specifically target class III HDACs (Huber et al., 2010). Given that I3C generates a diverse array of oligomers in addition to DIM (Higdon et al., 2007), further studies appear to be warranted on dietary indoles and their effects on HDAC activity and turnover.
Polyphenols: pros and cons of HDAC modulation
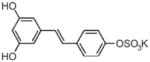
Dietary polyphenols such as resveratrol, quercetin, curcumin, and tea catechins have been examined as HDAC activators as well as HDAC inhibitors (Wood et al., 2004; Han, 2009; Imai, 2009, 2010; Chung et al., 2010; Imai and Guarente, 2010). A recent review summarized the debate surrounding “purported activators” of class III HDACs, such as SIRT1, in the context of therapeutic strategies related to aging, type II diabetes, and neurodegeneration (Dittenhafer-Reed et al., 2010).
In a rat liver cancer model, black tea polyphenols were reported to reduce significantly the expression levels of HDAC1 protein in liver and lung (Murugan et al., 2009). It was not clear whether other HDACs were affected, and whether alternative epigenetic mechanisms were involved, such as the inhibition of DNA methylation that has been reported for green tea catechins (Lee et al., 2005; Fang et al., 2007; Gilbert and Liu, 2010; Li and Tollefsbol, 2010). Polyphenols, including those in tea, undergo extensive metabolism in vivo to methylated and glucuronidated intermediates, as well as to novel breakdown products formed in the GI tract (Lee et al., 2002; Schantz et al., 2010; Sies, 2010; Stalmach et al., 2010a,b). Little if anything is known at present about how these intermediates affect HDAC activity.
Curcumin, and other curcuminoid polyphenols in Indian spices such as turmeric, have cancer chemoprotective properties (Aggarwal, 2010; Bar-Sela et al., 2010; Epstein et al., 2010; Padhye et al., 2010). There is growing interest in these compounds and their potential epigenetic mechanisms (Rahman, 2008; Chung et al., 2010; Fu and Kurzrock, 2010; Li et al., 2010b).
For example, selective loss of HDAC2 protein expression occurs in the pathogenesis of chronic obstructive pulmonary disease (COPD) (Barnes, 2009, 2010b; RajendrasozHan et al., 2009; Marwick et al., 2010), a situation exacerbated by cigarette smoke (Adenuga et al., 2009). In lung, HDAC2 deacetylates the glucocorticoid receptor (GR), an “off” mechanism that permits proinflammatory genes to be silenced (Figure 6). Curcumin treatment can help to maintain HDAC2 expression and activity, restoring corticosteroid function in monocytes exposed to oxidants (Meja et al., 2008). It is presently unclear whether this mechanism applies to curcumin metabolites such as di-, tetra-, and hexahydrocurcumin, the glucuronide and sulfate conjugates (Sharma et al., 2004; Hoehle et al., 2007; Dempe et al., 2008), and structural analogs such as dimethylcurcumin, 1,5-bis(3-pyridyl)-1,4-pentadien-3-one, and 3,5-bis-(2-fluorobenzylidene)-piperidinium-4-oneacetate (Steward and Gescher, 2008).
Figure 6.

Regulation of chromatin structure influences the expression of proinflammatory genes. In response to oxidative stress and proinflammatory conditions, signaling molecules such as NF-κB–p65 become activated and enter the nucleus, thereby recruiting HATs and their coactivator complexes to enhance gene activity. Corticosteroids and natural modulators of HAT and HDAC activities regulate the acetylation status of the glucocorticoid receptor (GR) and its ability to bind to the promoters of proinflammatory genes. For further details, see Delage and Dashwood (2009b).
As noted above, curcumin maintains rather than attenuates HDAC2 activity in lung and is beneficial in cases such as COPD, but this runs counter to the general paradigm of dietary HDAC inhibitors triggering gene de-repression as a beneficial outcome in cancer cells. There also exist dietary compounds that purportedly activate HDACs or inhibit HATs, such as resveratrol and theophylline (Delage and Dashwood, 2009b), which could theoretically up-regulate proinflammatory genes under conditions of oxidative stress. In the case of inflammatory bowel disease, curcumin has been described as having “bright prospects” due to it beneficial effects on cyclooxygenase, lipoxygenase, tumor necrosis factor, interferon, and nuclear factor kappa B (NF-κB) pathways (Hanai and Sugimoto, 2009). In GI tissues, mechanisms might exist to maintain HDAC2 (and other HDACs), which are not active under conditions of oxidative stress and chronic inflammation in the lung. The yin–yang of HDAC inhibition versus HDAC activation under conditions of oxidative stress, as well as normal conditions, warrants further investigation.
Miscellaneous agents: whole foods and HDAC inhibition
In addition to studying the HDAC inhibitory effects of isolated dietary constituents, such as SFN, the corresponding whole foods also have been examined. Consumption of a single cup of broccoli sprouts in human volunteers was shown to inhibit HDAC activity in circulating peripheral blood mononuclear cells (Myzak et al., 2007). Bitter melon (Momordica charantia), a plant that is both eaten and used medicinally, contains a protein MCP30 that inhibited HDAC1 activity and promoted histone acetylation in prostate cancer cells (Xiong et al., 2009). MCP30 was identified as a Type I ribosome-inactivating protein, which suppressed the growth of PC3 cells in vivo in a fashion similar to SFN (Myzak et al., 2007), with no effect on normal prostate cells. There is a major gap in the literature related to whole foods and their effects on HDAC activity, histone acetylation, and other epigenetic endpoints.
Concluding remarks
There is accumulating evidence to support the role of metabolism in generating modulators of HDAC activity (Table 1). HDAC inhibitor drugs developed to date are typically potent agents, some being “direct acting” and others requiring metabolism to be active. For compounds such as vorinostat, metabolism tends to lower the overall efficacy in vivo. However, there is a growing list of compounds, many from the human diet, that are converted by metabolism to the presumed “ultimate” HDAC inhibitor. The pharmacokinetic/pharmacodynamic distribution of HDAC inhibitors is also likely to be influenced by diet and nutritional status. A better understanding of this issue might clarify the interindividual variability observed with HDAC inhibitors in human subjects, and the potential for drug–diet interactions. For example, in patients treated with agents such as vorinostat, phenylbutyrate, or valproic acid, might additional benefit derive from HDAC inhibitor intake in the form of broccoli sprouts or other foods? Might the toxicity and drug resistance associated with some clinically used HDAC inhibitors be circumvented by lowering the dose, while supplementing with dietary HDAC inhibitors that must be metabolized to their active forms? The latter typically provides for a more sustained level of HDAC inhibition than the “fast-on/fast-off” agents currently used in the clinic (Chou et al., 2008). There is still much to learn about the epigenetic mechanisms that influence human health and disease susceptibility, and how these mechanisms are affected by diet and other lifestyle factors. Mainstream smoke, for example, in known to alter microRNA expression patterns in mouse lung, whereas PEITC and N-acetylcysteine given during pregnancy or weaning can normalize these microRNA profiles (Izzotti et al., 2010). Thus, microRNAs, DNA methylation, and histone status collectively comprise a cadre of epigenetic elements that can be modulated by dietary factors and their metabolites (Davis and Ross, 2007, 2008; Ross and Milner, 2007). In the future, an improved understanding of epigenetic mechanisms and their impact on human health and disease will depend on several avenues of research, including metabolism as a key to HDAC inhibition.
Table 1.
Summary of the role of metabolism in generating histone deacetylase (HDAC) inhibitors.
| Parent compound (non-dietary) | Metabolite(s) | HDAC-related mechanism(s) | Structure(s) of key intermediates/metabolites | References |
|---|---|---|---|---|
| Trichostatin A (TSA) | N-Demethylated trichostatin, trichostatic acid | N-Demethylated metabolite retained HDAC inhibitory activity while the acid did not. Mercapturic acid pathway lowers activity/efficacy in vivo? |
|
Elaut et al. (2002), Sanderson et al. (2004), Myzak et al. (2004) |
| Romidepsin | Reduced dithiol (4-reduced) | Class I HDAC inhibitor, FDA approved for CTCL. A prodrug converted to active metabolite HDAC inhibitor |
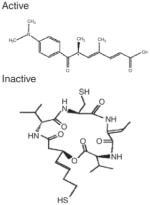
|
Furumai et al. (2002) |
| Vorinostat (SAHA) | SAHA-glucuronide | FDA-approved HDAC inhibitor, for CTCL. Phase II conjugation leads to PK variability; mercapturic acid pathway lowers activity in vivo? |
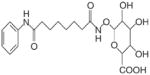
|
Kang et al. (2010) |
| Parent compound (dietary) | Metabolite(s) | HDAC-related mechanism(s) | Structures of key intermediates/metabolites | References |
| Dietary fiber, fat, alcohol | Short-chain fatty acids (butyrate, propionate) from gut fermentation | Competitive HDAC inhibition; butyrate Ki = 46 μM in MCF-7 whole cell lysate |
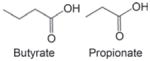
|
Boffa et al., (1978), Choudhury and Shukla (2008), Singh et al. (2010b), Sekhavat et al. (2007) |
| Sulforaphane (SFN), other diet-derived isothiocyanates | SFN–cysteine, mercapturic acid pathway intermediates | Reduces HDAC activity via direct and/or indirect mechanisms |
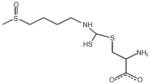
|
Myzak et al. (2004) |
| Indole-3-carbinol | Oligomers (3,3′-diindolylmethane and others) | Inhibits the expression of class I HDACs |
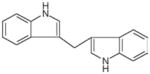
|
Li et al. (2010a), Higdon et al. (2007) |
| Organoselenium compounds (methyl selenocysteine) | Methyl selenopyruvate | Inhibits HDAC activity, Ki 35 μM hHDAC8 |
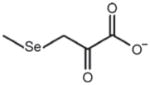
|
Nian et al. (2009b), Lee et al. (2009) |
| Glucose, other intermediates of intermediary metabolism | Pyruvate | HDAC3 (IC50 24 μM) and HDAC1 (IC50 80 μM) inhibition |
|
Thangaraju et al. (2009a) |
| Organosulfur compounds (diallyl disulfide, other garlic compounds) | Allyl mercaptan, endogenous small molecule thiols? | Competitive inhibitor, Ki 25 μM for hHDAC8; Sp3 increased on P21WAF1 promoter |
|
Nian et al. (2008), Lea et al. (1999b) |
| Resveratrol (and other dietary polyphenols) | 4′-O-Sulfate-resveratrol | Purported inducer of SIRT1, class III HDACs |
|
Calamini et al. (2010) |
Both dietary and non-dietary sources provide constituents which, through metabolism, can lead to intermediates with the ability to affect HDAC activity, chromatin silencing/unsilencing, and gene expression. See text and the references listed for further details.
CTCL, cutaneous T-cell lymphoma; hHDAC8, human HDAC8.
Abbreviations
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- N-Cor
nuclear repressor co-repressor
- SMRT
silencing mediator for retinoid and thyroid hormone receptors
- TSA
trichostatin A
- KDAC
lysine deacetylase
- FK228
depsipeptide
- KMSB
keto-methylselenobutyrate
- MSP
methylselenopyruvate
- MSC
methylselenocysteine
- SM
selenomethionine
- GTL
glutamine transaminase-liver
- GTK
glutamine transaminase-kidney
- SCMTs
sodium-coupled monocarboxylate transporters
- SAC
S-allylcysteine
- SAMC
S-allylmercaptocysteine
- DAS
diallyl sulfide
- DADS
diallyl disulfide
- DATS
diallyl trisulfide
- AMS
allyl methyl sulfide
- AM
allyl mercaptan
- SFN
sulforaphane
- Nrf2
nuclear factor erythroid 2-related factor 2
- Keap1
Kelch-like ECH-associated protein 1
- GST
glutathione S-transferase
- SFN-GSH
SFN–glutathione
- SFN-Cys
SFN–cysteine
- SFN-NAC
SFN–N-acetylcysteine
- SAHA
suberoylanilide hydroxamic acid
- BITC
benzyl isothiocyanate
- hTERT
human telomerase reverse transcriptase
- PHITC
phenylhexyl isothiocyanate
- PEITC
phenethyl isothiocyanate
- I3C
indole-3-carbinol
- DIM
3,3′-diindolylmethane
- SIRT1
sirtuin 1
- COPD
chronic obstructive pulmonary disease
- GR
glucocorticoid receptor
- NF-κB
nuclear factor kappa B
- GI
gastrointestinal
- HMTs
histone methyltransferases
- DNMTs
DNA methyltransferases
- MeCP2
methyl CpG-binding protein 2
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article. Research conducted in the authors’ laboratory is supported by NIH grants CA90890, CA65525, CA122906, CA122959, CA80176, and by NIEHS Center grant P30 ES00210.
References
- Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics. 2008;3:300–309. doi: 10.4161/epi.3.6.7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol. 2009;40:464–473. doi: 10.1165/rcmb.2008-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB. Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu Rev Nutr. 2010;30:173–199. doi: 10.1146/annurev.nutr.012809.104755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Sakr WA, Rahman KM. Anticancer properties of indole compounds: mechanism of apoptosis induction and role in chemotherapy. Curr Drug Targets. 2010;11:652–666. doi: 10.2174/138945010791170923. [DOI] [PubMed] [Google Scholar]
- Ashktorab H, Belgrave K, Hosseinkhah F, Brim H, Nouraie M, Takkikto M, Hewitt S, Lee EL, Dashwood RH, Smoot D. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig Dis Sci. 2009;54:2109–2117. doi: 10.1007/s10620-008-0601-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assisi RF GISDI Study Group. Combined butyric acid/mesalazine treatment in ulcerative colitis with mild–moderate activity. Results of a multicentre pilot study. Minerva Gastroenterol Dietol. 2008;54:231–238. [PubMed] [Google Scholar]
- Balakrishnan L, Milavetz B. Decoding the histone H4 lysine 20 methylation mark. Crit Rev Biochem Mol Biol. 2010;45:440–452. doi: 10.3109/10409238.2010.504700. [DOI] [PubMed] [Google Scholar]
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Bang MH, Chio OS, Kim WK. Soy oligosaccharide increases fecal bifidobacteria counts, short-chain fatty acids, and fecal lipid concentrations in young Korean women. J Med Food. 2007;10:366–370. doi: 10.1089/jmf.2005.096. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. New therapies for chronic obstructive pulmonary disease. Med Princ Pract. 2010a;19:330–338. doi: 10.1159/000316368. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. 2010b;120:76–85. doi: 10.1016/j.jsbmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. doi: 10.1146/annurev.physiol.010908.163257. [DOI] [PubMed] [Google Scholar]
- Bar-Sela G, Epelbaum R, Schaffer M. Curcumin as an anti-cancer agent: review of the gap between basic and clinical applications. Curr Med Chem. 2010;17:190–197. doi: 10.2174/092986710790149738. [DOI] [PubMed] [Google Scholar]
- Batra S, Sahu RP, Kandala PK, Srivastava SK. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol Cancer Ther. 2010;9:1596–1608. doi: 10.1158/1535-7163.MCT-09-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancotto C, Frigè G, Minucci S. Histone modification therapy of cancer. Adv Genet. 2010;70:341–386. doi: 10.1016/B978-0-12-380866-0.60013-7. [DOI] [PubMed] [Google Scholar]
- Bird AR, Vuaran MS, King RA, Noakes M, Keogh J, Morell MK, Topping DL. Wholegrain foods made from a novel high-amylose barley variety (Himalaya 292) improve indices of bowel health in human subjects. Br J Nutr. 2008;99:1032–1040. doi: 10.1017/S000711450783902X. [DOI] [PubMed] [Google Scholar]
- Boffa LC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem. 1978;253:3364–3366. [PubMed] [Google Scholar]
- Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkühler C, Gallinari P, Carfí A. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Lanpher B, Erez A, Ananieva EA, Islam M, Marini JC, Sun Q, Yu C, Hegde M, Li J, Wynn RM, Chuang DT, Hutson S, Lee B. Phenylbutyrate therapy for maple syrup urine disease. Hum Mol Genet. 2010;20:631–640. doi: 10.1093/hmg/ddq507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush EW, McKinsey TA. Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res. 2010;106:272–284. doi: 10.1161/CIRCRESAHA.109.209338. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Mancuso C, Pennisi G, Calafato S, Bellia F, Bates TE, Giuffrida Stella AM, Schapira T, Dinkova Kostova AT, Rizzarelli E. Cellular stress response: a novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem Res. 2008;33:2444–2471. doi: 10.1007/s11064-008-9775-9. [DOI] [PubMed] [Google Scholar]
- Calamini B, Ratia K, Malkowski MG, Cuendet M, Pezzuto JM, Santarsiero BD, Mesecar AD. Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Biochem J. 2010;429:273–282. doi: 10.1042/BJ20091857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Whitehead L, Li J, Chen CH, Jiang L, Vögtle M, Francotte E, Richert P, Wagner T, Traebert M, Lu Q, Cao X, Dumotier B, Fejzo J, Rajan S, Wang P, Yan-Neale Y, Shao W, Atadja P, Shultz M. Conformational refinement of hydroxamate-based histone deacetylase inhibitors and exploration of 3-piperidin-3-ylindole analogues of dacinostat (LAQ824) J Med Chem. 2010;53:2952–2963. doi: 10.1021/jm100007m. [DOI] [PubMed] [Google Scholar]
- Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury M, Shukla SD. Surrogate alcohols and their metabolites modify histone H3 acetylation: involvement of histone acetyl transferase and histone deacetylase. Alcohol Clin Exp Res. 2008;32:829–839. doi: 10.1111/j.1530-0277.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010;501:79–90. doi: 10.1016/j.abb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008;269:291–304. doi: 10.1016/j.canlet.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi C, Illi B, Rosati J, Spallotta F, Farsetti A, Grasselli A, Mai A, Capogrossi MC, Gaetano C. Histone deacetylase inhibitors: keeping momentum for neuromuscular and cardiovascular diseases treatment. Pharmacol Res. 2010;62:3–10. doi: 10.1016/j.phrs.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Corfe BM, Williams EA, Bury JP, Riley SA, Croucher LJ, Lai DY, Evans CA. A study protocol to investigate the relationship between dietary fibre intake and fermentation, colon cell turnover, global protein acetylation and early carcinogenesis: the FACT study. BMC Cancer. 2009;9:332. doi: 10.1186/1471-2407-9-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH, Jr, Zhang H, Schoenfeld DA, Shefner J, Matson S, Matson WR, Ferrante RJ. Northeast ALS and National VA ALS Research Consortiums. 2009. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler. 10:99–106. doi: 10.1080/17482960802320487. [DOI] [PubMed] [Google Scholar]
- Dashwood RH, Ho E. Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol. 2007;17:363–369. doi: 10.1016/j.semcancer.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–349. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CD, Ross SA. Dietary components impact histone modifications and cancer risk. Nutr Rev. 2007;65:88–94. doi: 10.1111/j.1753-4887.2007.tb00285.x. [DOI] [PubMed] [Google Scholar]
- Davis CD, Ross SA. Evidence for dietary regulation of microRNA expression in cancer cells. Nutr Rev. 2008;66:477–482. doi: 10.1111/j.1753-4887.2008.00080.x. [DOI] [PubMed] [Google Scholar]
- Delage B, Dashwood RH. Dietary manipulation of histone structure and function. Annu Rev Nutr. 2008;28:347–366. doi: 10.1146/annurev.nutr.28.061807.155354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delage B, Dashwood RH. Targeting the epigenome with dietary agents. In: Surh Y-J, editor. Dietary Modulation of Cell Signaling Pathways. Vol. 13. Boca Raton, FL: CRC Press; 2009a. pp. 337–369. [Google Scholar]
- Delage B, Dashwood RH. Nutrients, histone modifications, and chromatin remodeling in chronic inflammation. In: Choi S-W, Frisco S, editors. Nutrients and Epigenetics. Vol. 6. Boca Raton, FL: CRC Press; 2009b. pp. 127–154. [Google Scholar]
- Dempe JS, Pfeiffer E, Grimm AS, Metzler M. Metabolism of curcumin and induction of mitotic catastrophe in human cancer cells. Mol Nutr Food Res. 2008;52:1074–1081. doi: 10.1002/mnfr.200800029. [DOI] [PubMed] [Google Scholar]
- Denu JM. Vitamins and aging: pathways to NAD+ synthesis. Cell. 2007;129:453–454. doi: 10.1016/j.cell.2007.04.023. [DOI] [PubMed] [Google Scholar]
- Desai D, Salli U, Vrana KE, Amin S. SelSA, selenium analogs of SAHA as potent histone deacetylase inhibitors. Bioorg Med Chem Lett. 2010;20:2044–2047. doi: 10.1016/j.bmcl.2009.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz KC, Casaccia P. HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol Res. 2010;62:11–17. doi: 10.1016/j.phrs.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Dolan PJ, Johnson GV. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J Neurochem. 2008;106:2119–2130. doi: 10.1111/j.1471-4159.2008.05564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52 (Suppl 1):S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Feldman JL, Denu JM. Catalysis and mechanistic insights into sirtuin activation. Chembiochem. 2010;12:281–289. doi: 10.1002/cbic.201000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010;9:285–290. doi: 10.1111/j.1474-9726.2010.00548.x. [DOI] [PubMed] [Google Scholar]
- Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duée PH, Martel P, Chaumontet C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004a;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duée PH, Martel P, Chaumontet C. Repetitive treatments of colon HT-29 cells with diallyl disulfide induce a prolonged hyperacetylation of histone H3 K14. Ann N Y Acad Sci. 2004b;1030:612–621. doi: 10.1196/annals.1329.071. [DOI] [PubMed] [Google Scholar]
- Druesne-Pecollo N, Chaumontet C, Latino-Martel P. Diallyl disulfide increases histone acetylation in colon cells in vitro and in vivo. Nutr Rev. 2008;66 (Suppl 1):S39–S41. doi: 10.1111/j.1753-4887.2008.00066.x. [DOI] [PubMed] [Google Scholar]
- Druesne-Pecollo N, Chaumontet C, Pagniez A, Vaugelade P, Bruneau A, Thomas M, Cherbuy C, Duée PH, Martel P. In vivo treatment by diallyl disulfide increases histone acetylation in rat colonocytes. Biochem Biophys Res Commun. 2007;354:140–147. doi: 10.1016/j.bbrc.2006.12.158. [DOI] [PubMed] [Google Scholar]
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbel EN, Leymarie N, Schiavo S, Sharma S, Gevorkian S, Hersch S, Matson WR, Costello CE. Identification of phenylbutyrate-generated metabolites in Huntington disease patients using parallel liquid chromatography/electrochemical array/mass spectrometry and off-line tandem mass spectrometry. Anal Biochem. 2010;399:152–161. doi: 10.1016/j.ab.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elaut G, Török G, Vinken M, Laus G, Papeleu P, Tourwe D, Rogiers V. Major phase I biotransformation pathways of Trichostatin a in rat hepatocytes and in rat and human liver microsomes. Drug Metab Dispos. 2002;30:1320–1328. doi: 10.1124/dmd.30.12.1320. [DOI] [PubMed] [Google Scholar]
- Epstein J, Sanderson IR, Macdonald TT. Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr. 2010;103:1545–1557. doi: 10.1017/S0007114509993667. [DOI] [PubMed] [Google Scholar]
- Ernst IM, Wagner AE, Schuemann C, Storm N, Höppner W, Döring F, Stocker A, Rimbach G. Allyl-, butyl- and phenylethyl-isothiocyanate activate Nrf2 in cultured fibroblasts. Pharmacol Res. 2010;63:233–240. doi: 10.1016/j.phrs.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Fang M, Chen D, Yang CS. Dietary polyphenols may affect DNA methylation. J Nutr. 2007;123 (Suppl 1):223S–228S. doi: 10.1093/jn/137.1.223S. [DOI] [PubMed] [Google Scholar]
- Ficner R. Novel structural insights into class I and II histone deacetylases. Curr Top Med Chem. 2009;9:235–240. doi: 10.2174/156802609788085304. [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, Reid JM, Ames MM, Speights R, Ingle AM, Zwiebel J, Blaney SM, Adamson PC. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J Clin Oncol. 2010;28:3623–3629. doi: 10.1200/JCO.2009.25.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, Kristiansen G. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8:381. doi: 10.1186/1471-2407-8-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116:4670–4676. doi: 10.1002/cncr.25414. [DOI] [PubMed] [Google Scholar]
- Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- Futamura M, Monden Y, Okabe T, Fujita-Yoshigaki J, Yokoyama S, Nishimura S. Trichostatin A inhibits both ras-induced neurite outgrowth of PC12 cells and morphological transformation of NIH3T3 cells. Oncogene. 1995;10:1119–1123. [PubMed] [Google Scholar]
- Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, Richon VM, Scheithauer B, Giannini C, Flynn PJ, Moore DF, Jr, Zwiebel J, Buckner JC. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy V, Thangaraju M, Gopal E, Martin PM, Itagaki S, Miyauchi S, Prasad PD. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008;10:193–199. doi: 10.1208/s12248-008-9022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009;121:29–40. doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- Gilbert ER, Liu D. Flavonoids influence epigenetic-modifying enzyme activity: structure–function relationships and the therapeutic potential for cancer. Curr Med Chem. 2010;17:1756–1768. doi: 10.2174/092986710791111161. [DOI] [PubMed] [Google Scholar]
- Göttlicher M. Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol. 2004;83 (Suppl 1):S91–S92. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- Gullett NP, Ruhul Amin AR, Bayraktar S, Pezzuto JM, Shin DM, Khuri FR, Aggarwal BB, Surh YJ, Kucuk O. Cancer prevention with natural compounds. Semin Oncol. 2010;37:258–281. doi: 10.1053/j.seminoncol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Gurard-Levin ZA, Kilian KA, Kim J, Bähr K, Mrksich M. Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem Biol. 2010;5:863–873. doi: 10.1021/cb100088g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer HM, Jonkers DM, Bast A, Vanhoutvin SA, Fischer MA, Kodde A, Troost FJ, Venema K, Brummer RJ. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin Nutr. 2009;28:88–93. doi: 10.1016/j.clnu.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Han SH. Potential role of sirtuin as a therapeutic target for neurodegenerative diseases. J Clin Neurol. 2009;5:120–125. doi: 10.3988/jcn.2009.5.3.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai H, Sugimoto K. Curcumin has bright prospects for the treatment of inflammatory bowel disease. Curr Pharm Des. 2009;15:2087–2094. doi: 10.2174/138161209788489177. [DOI] [PubMed] [Google Scholar]
- Handel AE, Ramagopalan SV. Is Lamarckian evolution relevant to medicine? BMC Med Genet. 2010;11:73. doi: 10.1186/1471-2350-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery DM. Targeting mitochondria for cancer therapy. Environ Mol Mutagen. 2010;51:476–489. doi: 10.1002/em.20552. [DOI] [PubMed] [Google Scholar]
- Hoehle SI, Pfeiffer E, Metzler M. Glucuronidation of curcuminoids by human microsomal and recombinant UDP-glucuronosyltransferases. Mol Nutr Food Res. 2007;51:932–938. doi: 10.1002/mnfr.200600283. [DOI] [PubMed] [Google Scholar]
- Hogarth P, Lovrecic L, Krainc D. Sodium phenylbutyrate in Huntington’s disease: a dose-finding study. Mov Disord. 2007;22:1962–1964. doi: 10.1002/mds.21632. [DOI] [PubMed] [Google Scholar]
- Hu Y, Lu W, Chen G, Zhang H, Jia Y, Wei Y, Yang H, Zhang W, Fiskus W, Bhalla K, Keating M, Huang P, Garcia-Manero G. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood. 2010;116:2732–2741. doi: 10.1182/blood-2009-11-256354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Huber K, Schemies J, Uciechowska U, Wagner JM, Rumpf T, Lewrick F, Süss R, Sippl W, Jung M, Bracher F. Novel 3-arylideneindolin-2-ones as inhibitors of NAD+-dependent histone deacetylases (sirtuins) J Med Chem. 2010;53:1383–1386. doi: 10.1021/jm901055u. [DOI] [PubMed] [Google Scholar]
- Iciek M, Kwiecien I, Wlodek L. Biological properties of garlic and garlic-derived organosulfur compounds. Environ Mol Mutagen. 2009;50:247–265. doi: 10.1002/em.20474. [DOI] [PubMed] [Google Scholar]
- Imai S. A possibility of nutriceuticals as an anti-aging intervention: activation of sirtuins by promoting mammalian NAD biosynthesis. Pharmacol Res. 2010;62:42–47. doi: 10.1016/j.phrs.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S. SIRT1 and caloric restriction: an insight into possible trade-offs between robustness and frailty. Curr Opin Clin Nutr Metab Care. 2009;12:350–356. doi: 10.1097/MCO.0b013e32832c932d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 2010;31:212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27:5848–5856. doi: 10.1200/JCO.2009.24.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelsen WJ, Vander Heiden MG. ATP consumption promotes cancer metabolism. Cell. 2010;143:669–671. doi: 10.1016/j.cell.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Itazaki H, Nagashima K, Sugita K, Yoshida H, Kawamura Y, Yasuda Y, Matsumoto K, Ishii K, Uotani N, Nakai H. Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J Antibiot. 1990;43:1524–1532. doi: 10.7164/antibiotics.43.1524. [DOI] [PubMed] [Google Scholar]
- Izzotti A, Larghero P, Balansky R, Pfeffer U, Steele VE, De Flora S. Interplay between histopathological alterations, cigarette smoke and chemopreventive agents in defining microRNA profiles in mouse lung. Mutat Res. 2010 Oct 23; doi: 10.1016/j.mrfmmm.2010.10.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Jackson MI, Combs GF., Jr Selenium and anticarcinogenesis: underlying mechanisms. Curr Opin Clin Nutr Metab Care. 2008;11:718–726. doi: 10.1097/MCO.0b013e3283139674. [DOI] [PubMed] [Google Scholar]
- Jiang S, Ma X, Huang Y, Xu Y, Zheng R, Chiao JW. Reactivating aberrantly hypermethylated p15 gene in leukemic T cells by a phenylhexyl isothiocyanate mediated inter-active mechanism on DNA and chromatin. J Hematol Oncol. 2010;3:48. doi: 10.1186/1756-8722-3-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Steinkühler C. From natural products to small molecule ketone histone deacetylase inhibitors: development of new class specific agents. Curr Pharm Des. 2008;14:545–561. doi: 10.2174/138161208783885317. [DOI] [PubMed] [Google Scholar]
- Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. 2007;64:1105–1127. doi: 10.1007/s00018-007-6484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadia TM, Yang H, Ferrajoli A, Maddipotti S, Schroeder C, Madden TL, Holleran JL, Egorin MJ, Ravandi F, Thomas DA, Newsome W, Sanchez-Gonzalez B, Zwiebel JA, Espinoza-Delgado I, Kantarjian HM, Garcia-Manero G. A phase I study of vorinostat in combination with idarubicin in relapsed or refractory leukaemia. Br J Haematol. 2010;150:72–82. doi: 10.1111/j.1365-2141.2010.08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SP, Ramirez J, House L, Zhang W, Mirkov S, Liu W, Haverfield E, Ratain MJ. A pharmacogenetic study of vorinostat glucuronidation. Pharmacogenet Genomics. 2010;20:638–641. doi: 10.1097/FPC.0b013e32833e1b37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284:7436–7445. doi: 10.1074/jbc.M808999200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–3588. [PubMed] [Google Scholar]
- Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- Kim DH, Kim M, Kwon HJ. Histone deacetylase in carcinogenesis and its inhibitors as anti-cancer agents. J Biochem Mol Biol. 2003;36:110–119. doi: 10.5483/bmbrep.2003.36.1.110. [DOI] [PubMed] [Google Scholar]
- Krainc D. Clearance of mutant proteins as a therapeutic target in neurodegenerative diseases. Arch Neurol. 2010;67:388–392. doi: 10.1001/archneurol.2010.40. [DOI] [PubMed] [Google Scholar]
- Kuroiwa-Trzmielina J, de Conti A, Scolastici C, Pereira D, Horst MA, Purgatto E, Ong TP, Moreno FS. Chemoprevention of rat hepatocarcinogenesis with histone deacetylase inhibitors: efficacy of tributyrin, a butyric acid prodrug. Int J Cancer. 2009;124:2520–2527. doi: 10.1002/ijc.24212. [DOI] [PubMed] [Google Scholar]
- Kwak MK, Kensler TW. Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol. 2010;244:66–76. doi: 10.1016/j.taap.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrylenko S, Baniahmad A. Sirtuin family: a link to metabolic signaling and senescence. Curr Med Chem. 2010;17:2921–2932. doi: 10.2174/092986710792065009. [DOI] [PubMed] [Google Scholar]
- LaBonte MJ, Wilson PM, Fazzone W, Groshen S, Lenz HJ, Ladner RD. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med Genomics. 2009;2:67. doi: 10.1186/1755-8794-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- Lawless MW, O’Byrne KJ, Gray SG. Histone deacetylase inhibitors target diabetes via chromatin remodeling or as chemical chaperones? Curr Diabetes Rev. 2009;5:201–209. doi: 10.2174/157339909788920956. [DOI] [PubMed] [Google Scholar]
- Lea MA, Randolph VM. Induction of histone acetylation in rat liver and hepatoma by organosulfur compounds including diallyl disulfide. Anticancer Res. 2001;21:2841–2845. [PubMed] [Google Scholar]
- Lea MA, Randolph VM, Hodge SK. Induction of histone acetylation and growth regulation in eryrthroleukemia cells by 4-phenylbutyrate and structural analogs. Anticancer Res. 1999a;19:1971–1976. [PubMed] [Google Scholar]
- Lea MA, Randolph VM, Patel M. Increased acetylation of histones induced by diallyl disulfide and structurally related molecules. Int J Oncol. 1999b;15:347–352. doi: 10.3892/ijo.15.2.347. [DOI] [PubMed] [Google Scholar]
- Lea MA, Randolph VM, Lee JE, desBordes C. Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int J Cancer. 2001;92:784–789. doi: 10.1002/ijc.1277. [DOI] [PubMed] [Google Scholar]
- Lea MA, Rasheed M, Randolph VM, Khan F, Shareef A, desBordes C. Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutr Cancer. 2002;43:90–102. doi: 10.1207/S15327914NC431_11. [DOI] [PubMed] [Google Scholar]
- Leder A, Leder P. Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell. 1975;5:319–322. doi: 10.1016/0092-8674(75)90107-5. [DOI] [PubMed] [Google Scholar]
- Lee JI, Nian H, Cooper AJ, Sinha R, Dai J, Bisson WH, Dashwood RH, Pinto JT. Alpha-keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev Res (Phila) 2009;2:683–693. doi: 10.1158/1940-6207.CAPR-09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010a;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010b;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Maliakal P, Chen L, Meng X, Bondoc FY, Prabhu S, Lambert G, Mohr S, Yang CS. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiol Biomarkers Prev. 2002;11:1025–1032. [PubMed] [Google Scholar]
- Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68:1018–1030. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010a;70:646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kong D, Wang Z, Sarkar FH. Regulation of microRNAs by natural agents: an emerging field in chemoprevention and chemotherapy research. Pharm Res. 2010b;27:1027–1041. doi: 10.1007/s11095-010-0105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tollefsbol TO. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr Med Chem. 2010;17:2141–2151. doi: 10.2174/092986710791299966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, Parsons JK, Bearden JD, 3rd, Crawford ED, Goodman GE, Claudio J, Winquist E, Cook ED, Karp DD, Walther P, Lieber MM, Kristal AR, Darke AK, Arnold KB, Ganz PA, Santella RM, Albanes D, Taylor PR, Probstfield JL, Jagpal TJ, Crowley JJ, Meyskens FL, Jr, Baker LH, Coltman CA., Jr Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü J, Jiang C. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid Redox Signal. 2005;7:1715–1727. doi: 10.1089/ars.2005.7.1715. [DOI] [PubMed] [Google Scholar]
- Lundh M, Christensen DP, Rasmussen DN, Mascagni P, Dinarello CA, Billestrup N, Grunnet LG, Mandrup-Poulsen T. Lysine deacetylases are produced in pancreatic beta cells and are differentially regulated by proinflammatory cytokines. Diabetologia. 2010;53:2569–2578. doi: 10.1007/s00125-010-1892-8. [DOI] [PubMed] [Google Scholar]
- Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- Ma X, Fang Y, Beklemisheva A, Dai W, Feng J, Ahmed T, Liu D, Chiao JW. Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int J Oncol. 2006;28:1287–1293. [PubMed] [Google Scholar]
- Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3:28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwick JA, Adcock IM, Chung KF. Overcoming reduced glucocorticoid sensitivity in airway disease: molecular mechanisms and therapeutic approaches. Drugs. 2010;70:929–948. doi: 10.2165/10898520-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Masuoka Y, Shindoh N, Inamura N. Histone deacetylase inhibitors from microorganisms: the Astellas experience. Prog Drug Res. 2008;66:335, 337–335, 359. doi: 10.1007/978-3-7643-8595-8_7. [DOI] [PubMed] [Google Scholar]
- Matthias P, Yoshida M, Khochbin S. HDAC6 a new cellular stress surveillance factor. Cell Cycle. 2008;7:7–10. doi: 10.4161/cc.7.1.5186. [DOI] [PubMed] [Google Scholar]
- Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE. 2010;5:e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, Marwick JA, Chakravarty P, Fletcher D, Whittaker P, Megson IL, Kirkham PA, Rahman I. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol. 2008;39:312–323. doi: 10.1165/rcmb.2008-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62:18–34. doi: 10.1016/j.phrs.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Miyanaga A, Gemma A, Noro R, Kataoka K, Matsuda K, Nara M, Okano T, Seike M, Yoshimura A, Kawakami A, Uesaka H, Nakae H, Kudoh S. Antitumor activity of histone deacetylase inhibitors in non-small cell lung cancer cells: development of a molecular predictive model. Mol Cancer Ther. 2008;7:1923–1930. doi: 10.1158/1535-7163.MCT-07-2140. [DOI] [PubMed] [Google Scholar]
- Muecke R, Schomburg L, Buentzel J, Kisters K, Micke O German Working Group Trace Elements and Electrolytes in Oncology. Selenium or no selenium—that is the question in tumor patients: a new controversy. Integr Cancer Ther. 2010;9:136–141. doi: 10.1177/1534735410367648. [DOI] [PubMed] [Google Scholar]
- Murugan RS, Vinothini G, Hara Y, Nagini S. Black tea polyphenols target matrix metalloproteinases, RECK, proangiogenic molecules and histone deacetylase in a rat hepatocarcinogenesis model. Anticancer Res. 2009;29:2301–2305. [PubMed] [Google Scholar]
- Myzak MC, Dashwood RH. Chemoprotection by sulforaphane: keep one eye beyond Keap1. Cancer Lett. 2006a;233:208–218. doi: 10.1016/j.canlet.2005.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myzak MC, Dashwood RH. Histone deacetylases as targets for dietary cancer preventive agents: lessons learned with butyrate, diallyl disulfide, and sulforaphane. Curr Drug Targets. 2006b;7:443–452. doi: 10.2174/138945006776359467. [DOI] [PubMed] [Google Scholar]
- Myzak MC, Ho E, Dashwood RH. Dietary agents as histone deacetylase inhibitors. Mol Carcinog. 2006;45:443–446. doi: 10.1002/mc.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232:227–234. [PMC free article] [PubMed] [Google Scholar]
- Nian H, Delage B, Ho E, Dashwood RH. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ Mol Mutagen. 2009a;50:213–221. doi: 10.1002/em.20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian H, Bisson WH, Dashwood WM, Pinto JT, Dashwood RH. Alpha-keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis. 2009b;30:1416–1423. doi: 10.1093/carcin/bgp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian H, Delage B, Pinto JT, Dashwood RH. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis. 2008;29:1816–1824. doi: 10.1093/carcin/bgn165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson AC, Östman EM, Knudsen KE, Holst JJ, Björck IM. A cereal-based evening meal rich in indigestible carbohydrates increases plasma butyrate the next morning. J Nutr. 2010;140:1932–1936. doi: 10.3945/jn.110.123604. [DOI] [PubMed] [Google Scholar]
- Noureen N, Rashid H, Kalsoom S. Identification of type-specific anticancer histone deacetylase inhibitors: road to success. Cancer Chemother Pharmacol. 2010;66:625–633. doi: 10.1007/s00280-010-1324-y. [DOI] [PubMed] [Google Scholar]
- Novogrodsky A, Dvir A, Ravid A, Shkolnik T, Stenzel KH, Rubin AL, Zaizov R. Effect of polar organic compounds on leukemic cells. Butyrate-induced partial remission of acute myelogenous leukemia in a child. Cancer. 1983;51:9–14. doi: 10.1002/1097-0142(19830101)51:1<9::aid-cncr2820510104>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, Zelenetz AD, Frankel S, Richon V, Marks P, Kelly WK. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24:166–173. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- Oehme I, Deubzer HE, Lodrini M, Milde T, Witt O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Investig Drugs. 2009a;18:1605–1617. doi: 10.1517/14728220903241658. [DOI] [PubMed] [Google Scholar]
- Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M, Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009b;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- Oger F, Lecorgne A, Sala E, Nardese V, Demay F, Chevance S, Desravines DC, Aleksandrova N, Le Guével R, Lorenzi S, Beccari AR, Barath P, Hart DJ, Bondon A, Carettoni D, Simonneaux G, Salbert G. Biological and biophysical properties of the histone deacetylase inhibitor suberoylanilide hydroxamic acid are affected by the presence of short alkyl groups on the phenyl ring. J Med Chem. 2010;53:1937–1950. doi: 10.1021/jm901561u. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Suzuki KT. Methylation and demethylation of intermediates selenide and methylselenol in the metabolism of selenium. Toxicol Appl Pharmacol. 2008;226:169–177. doi: 10.1016/j.taap.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Oki Y, Issa JP. Review: recent clinical trials in epigenetic therapy. Rev Recent Clin Trials. 2006;1:169–182. doi: 10.2174/157488706776876490. [DOI] [PubMed] [Google Scholar]
- Ortore G, Di Colo F, Martinelli A. Docking of hydroxamic acids into HDAC1 and HDAC8: a rationalization of activity trends and selectivities. J Chem Inf Model. 2009;49:2774–2785. doi: 10.1021/ci900288e. [DOI] [PubMed] [Google Scholar]
- Padhye S, Chavan D, Pandey S, Deshpande J, Swamy KV, Sarkar FH. Perspectives on chemopreventive and therapeutic potential of curcumin analogs in medicinal chemistry. Mini Rev Med Chem. 2010;10:372–387. doi: 10.2174/138955710791330891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Park JH, Kim SH, Choi MC, Lee J, Oh DY, Im SA, Bang YJ, Kim TY. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun. 2008;368:318–322. doi: 10.1016/j.bbrc.2008.01.056. [DOI] [PubMed] [Google Scholar]
- Patra SK, Patra A, Dahiya R. Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun. 2001;287:705–713. doi: 10.1006/bbrc.2001.5639. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Pinto JT, Lee JI, Sinha R, Macewan ME, Cooper AJ. Chemopreventive mechanisms of alpha-keto acid metabolites of naturally occurring organoselenium compounds. Amino Acids. 2010 Apr 10; doi: 10.1007/s00726-010-0578-3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6:1013–1021. doi: 10.1158/1535-7163.MCT-06-0494. [DOI] [PubMed] [Google Scholar]
- Poke FS, Qadi A, Holloway AF. Reversing aberrant methylation patterns in cancer. Curr Med Chem. 2010;17:1246–1254. doi: 10.2174/092986710790936329. [DOI] [PubMed] [Google Scholar]
- Pool-Zobel BL, Sauer J. Overview of experimental data on reduction of colorectal cancer risk by inulin-type fructans. J Nutr. 2007;137:2580S–2584S. doi: 10.1093/jn/137.11.2580S. [DOI] [PubMed] [Google Scholar]
- Powolny AA, Singh SV. Multitargeted prevention and therapy of cancer by diallyl trisulfide and related Allium vegetable-derived organosulfur compounds. Cancer Lett. 2008;269:305–314. doi: 10.1016/j.canlet.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I. Dietary polyphenols mediated regulation of oxidative stress and chromatin remodeling in inflammation. Nutr Rev. 2008;66 (Suppl 1):S42–S45. doi: 10.1111/j.1753-4887.2008.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendrasozhan S, Yao H, Rahman I. Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD. 2009;6:291–297. doi: 10.1080/15412550903049132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadori G, Coppari R. Pharmacological manipulations of CNS sirtuins: potential effects on metabolic homeostasis. Pharmacol Res. 2010;62:48–54. doi: 10.1016/j.phrs.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam SS, Kummar S, Sarantopoulos J, Shibata S, LoRusso P, Yerk M, Holleran J, Lin Y, Beumer JH, Harvey RD, Ivy SP, Belani CP, Egorin MJ. Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol. 2010;28:4507–4512. doi: 10.1200/JCO.2010.30.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci USA. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SA, Milner JA. Epigenetic modulation and cancer: effect of metabolic syndrome? Am J Clin Nutr. 2007;86:s872–s877. doi: 10.1093/ajcn/86.3.872S. [DOI] [PubMed] [Google Scholar]
- Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- Sanda T, Okamoto T, Uchida Y, Nakagawa H, Iida S, Kayukawa S, Suzuki T, Oshizawa T, Suzuki T, Miyata N, Ueda R. Proteome analyses of the growth inhibitory effects of NCH-51, a novel histone deacetylase inhibitor, on lymphoid malignant cells. Leukemia. 2007;21:2344–2353. doi: 10.1038/sj.leu.2404902. [DOI] [PubMed] [Google Scholar]
- Sanderson L, Taylor GW, Aboagye EO, Alao JP, Latigo JR, Coombes RC, Vigushin DM. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab Dispos. 2004;32:1132–1138. doi: 10.1124/dmd.104.000638. [DOI] [PubMed] [Google Scholar]
- Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Curr Drug Metab. 2007;8:383–393. doi: 10.2174/138920007780655397. [DOI] [PubMed] [Google Scholar]
- Sattler UG, Hirschhaeuser F, Mueller-Klieser WF. Manipulation of glycolysis in malignant tumors: fantasy or therapy? Curr Med Chem. 2010;17:96–108. doi: 10.2174/092986710790112657. [DOI] [PubMed] [Google Scholar]
- Savickiene J, Treigyte G, Vistartaite G, Tunaitis V, Magnusson KE, Navakauskiene R. C/EBPa and PU.1 are involved in distinct differentiation responses of acute promyelocytic leukemia HL-60 and NB4 cells via chromatin remodeling. Differentiation. 2010;81:57–67. doi: 10.1016/j.diff.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Schantz M, Erk T, Richling E. Metabolism of green tea catechins by the human small intestine. Biotechnol J. 2010;5:1050–1059. doi: 10.1002/biot.201000214. [DOI] [PubMed] [Google Scholar]
- Schuetz A, Min J, Allali-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhavat A, Sun JM, Davie JR. Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem Cell Biol. 2007;85:751–758. doi: 10.1139/o07-145. [DOI] [PubMed] [Google Scholar]
- Sharma A, Heuck CJ, Fazzari MJ, Mehta J, Singhal S, Greally JM, Verma A. DNA methylation alterations in multiple myeloma as a model for epigenetic changes in cancer. Wiley Interdiscip Rev Syst Biol Med. 2010;2:654–669. doi: 10.1002/wsbm.89. [DOI] [PubMed] [Google Scholar]
- Sharma RA, Euden SA, Platton SL, Cooke DN, Shafayat A, Hewitt HR, Marczylo TH, Morgan B, Hemingway D, Plummer SM, Pirmohamed M, Gescher AJ, Steward WP. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10:6847–6854. doi: 10.1158/1078-0432.CCR-04-0744. [DOI] [PubMed] [Google Scholar]
- Sies H. Polyphenols and health: update and perspectives. Arch Biochem Biophys. 2010;501:2–5. doi: 10.1016/j.abb.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Silva JP, Wahlestedt C. Role of sirtuin 1 in metabolic regulation. Drug Discov Today. 2010;15:781–791. doi: 10.1016/j.drudis.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther. 2010a;10:935–954. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Thangaraju M, Prasad PD, Martin PM, Lambert NA, Boettger T, Offermanns S, Ganapathy V. Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. J Biol Chem. 2010b;285:27601–27608. doi: 10.1074/jbc.M110.102947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Stahl SM. Fooling mother nature: epigenetics and novel treatments for psychiatric disorders. CNS Spectr. 2010;15:358–365. doi: 10.1017/s1092852900029229. [DOI] [PubMed] [Google Scholar]
- Stalmach A, Mullen W, Steiling H, Williamson G, Lean ME, Crozier A. Absorption, metabolism, and excretion of green tea flavan-3-ols in humans with an ileostomy. Mol Nutr Food Res. 2010a;54:323–334. doi: 10.1002/mnfr.200900194. [DOI] [PubMed] [Google Scholar]
- Stalmach A, Steiling H, Williamson G, Crozier A. Bioavailability of chlorogenic acids following acute ingestion of coffee by humans with an ileostomy. Arch Biochem Biophys. 2010b;501:98–105. doi: 10.1016/j.abb.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Steward WP, Gescher AJ. Curcumin in cancer management: recent results of analogue design and clinical studies and desirable future research. Mol Nutr Food Res. 2008;52:1005–1009. doi: 10.1002/mnfr.200700148. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Suzuki T, Ota Y, Nakano T, Kurihara M, Okuda H, Yamori T, Tsumoto H, Nakagawa H, Miyata N. Design, synthesis, and biological activity of boronic acid-based histone deacetylase inhibitors. J Med Chem. 2009;52:2909–2922. doi: 10.1021/jm900125m. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kouketsu A, Matsuura A, Kohara A, Ninomiya S, Kohda K, Miyata N. Thiol-based SAHA analogues as potent histone deacetylase inhibitors. Bioorg Med Chem Lett. 2004;14:3313–3317. doi: 10.1016/j.bmcl.2004.03.063. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, Nakagawa H, Miyata N. Novel inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates. J Med Chem. 2005;48:1019–1032. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009a;417:379–389. doi: 10.1042/BJ20081132. [DOI] [PubMed] [Google Scholar]
- Thangaraju M, Karunakaran SK, Itagaki S, Gopal E, Elangovan S, Prasad PD, Ganapathy V. Transport by SLC5A8 with subsequent inhibition of histone deacetylase 1 (HDAC1) and HDAC3 underlies the antitumor activity of 3-bromopyruvate. Cancer. 2009b;115:4655–4666. doi: 10.1002/cncr.24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Suzuki N, Suzuki KT, Ogra Y. Selenium metabolism in rats with long-term ingestion of Se-methylselenocysteine using enriched stable isotopes. J Toxicol Sci. 2009;34:191–200. doi: 10.2131/jts.34.191. [DOI] [PubMed] [Google Scholar]
- Valgimigli L, Iori R. Antioxidant and pro-oxidant capacities of ITCs. Environ Mol Mutagen. 2009;50:222–237. doi: 10.1002/em.20468. [DOI] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR. Modifying chromatin architecture during the response to DNA breakage. Crit Rev Biochem Mol Biol. 2010;45:2–13. doi: 10.3109/10409230903325446. [DOI] [PubMed] [Google Scholar]
- Verkerk R, Schreiner M, Krumbein A, Ciska E, Holst B, Rowland I, De Schrijver R, Hansen M, Gerhäuser C, Mithen R, Dekker M. Glucosinolates in Brassica vegetables: the influence of the food supply chain on intake, bioavailability and human health. Mol Nutr Food Res. 2009;53 (Suppl 2):S219. doi: 10.1002/mnfr.200800065. [DOI] [PubMed] [Google Scholar]
- Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008a;19:587–593. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Waldecker M, Kautenburger T, Daumann H, Veeriah S, Will F, Dietrich H, Pool-Zobel BL, Schrenk D. Histone-deacetylase inhibition and butyrate formation: fecal slurry incubations with apple pectin and apple juice extracts. Nutrition. 2008b;24:366–374. doi: 10.1016/j.nut.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Wang D. Computational studies on the histone deacetylases and the design of selective histone deacetylase inhibitors. Curr Top Med Chem. 2009;9:241–256. doi: 10.2174/156802609788085287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, Feng J, Liu D, Chiao JW. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008;33:375–380. [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weng JR, Tsai CH, Kulp SK, Chen CS. Indole-3-carbinol as a chemopreventive and anti-cancer agent. Cancer Lett. 2008;262:153–163. doi: 10.1016/j.canlet.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- Wilson PM, El-Khoueiry A, Iqbal S, Fazzone W, LaBonte MJ, Groshen S, Yang D, Danenberg KD, Cole S, Kornacki M, Ladner RD, Lenz HJ. A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-FU-based chemotherapy. Cancer Chemother Pharmacol. 2010;65:979–988. doi: 10.1007/s00280-009-1236-x. [DOI] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- Xiao L, Huang Y, Zhen R, Chiao JW, Liu D, Ma X. Deficient histone acetylation in acute leukemia and the correction by an isothiocyanate. Acta Haematol. 2010;123:71–76. doi: 10.1159/000264628. [DOI] [PubMed] [Google Scholar]
- Xiong SD, Yu K, Liu XH, Yin LH, Kirschenbaum A, Yao S, Narla G, DiFeo A, Wu JB, Yuan Y, Ho SM, Lam YW, Levine AC. Ribosome-inactivating proteins isolated from dietary bitter melon induce apoptosis and inhibit histone deacetylase-1 selectively in premalignant and malignant prostate cancer cells. Int J Cancer. 2009;125:774–782. doi: 10.1002/ijc.24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Omi Y, Fujii N, Ozaki A, Nakama A, Sakakibara Y, Suiko M, Nishiyama K. Mustard oil in “Shibori Daikon” a variety of Japanese radish, selectively inhibits the proliferation of H-ras-transformed 3Y1 cells. Biosci Biotechnol Biochem. 2009;73:2217–2221. doi: 10.1271/bbb.90322. [DOI] [PubMed] [Google Scholar]
- Yamashita Y, Shimada M, Harimoto N, Rikimaru T, Shirabe K, Tanaka S, Sugimachi K. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer. 2003;103:572–576. doi: 10.1002/ijc.10699. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap KL, Zhou MM. Keeping it in the family: diverse histone recognition by conserved structural folds. Crit Rev Biochem Mol Biol. 2010;45:488–505. doi: 10.3109/10409238.2010.512001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Sugita K. A novel tetracyclic peptide, trapoxin, induces phenotypic change from transformed to normal in sis-oncogene-transformed NIH3T3 cells. Jpn J Cancer Res. 1992;83:324–328. doi: 10.1111/j.1349-7006.1992.tb00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- Yoshida M, Matsuyama A, Komatsu Y, Nishino N. From discovery to the coming generation of histone deacetylase inhibitors. Curr Med Chem. 2003;10:2351–2358. doi: 10.2174/0929867033456602. [DOI] [PubMed] [Google Scholar]
- Yu J, Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2010;62:35–41. doi: 10.1016/j.phrs.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Botnen JH, Briske-Anderson M. Deoxycholic acid and selenium metabolite methylselenol exert common and distinct effects on cell cycle, apoptosis, and MAP kinase pathway in HCT116 human colon cancer cells. Nutr Cancer. 2010;62:85–92. doi: 10.1080/01635580903191551. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]