

Abstract
The inward rectifier family of potassium (Kir) channels is comprised of at least 16 family members exhibiting broad and often overlapping cellular, tissue, or organ distributions. The discovery of disease-causing mutations in humans and experiments on knockout mice has underscored the importance of Kir channels in physiology and in some cases raised questions about their potential as drug targets. However, the paucity of potent and selective small-molecule modulators targeting specific family members has with few exceptions mired efforts to understand their physiology and assess their therapeutic potential. A growing body of evidence suggests that G protein-coupled inward rectifier K (GIRK) channels of the Kir3.X subfamily may represent novel targets for the treatment of atrial fibrillation. In an effort to expand the molecular pharmacology of GIRK, we performed a thallium (Tl+) flux-based high-throughput screen of a Kir1.1 inhibitor library for modulators of GIRK. One compound, termed VU573, exhibited 10-fold selectivity for GIRK over Kir1.1 (IC50 = 1.9 and 19 μM, respectively) and was therefore selected for further study. In electrophysiological experiments performed on Xenopus laevis oocytes and mammalian cells, VU573 inhibited Kir3.1/3.2 (neuronal GIRK) and Kir3.1/3.4 (cardiac GIRK) channels with equal potency and preferentially inhibited GIRK, Kir2.3, and Kir7.1 over Kir1.1 and Kir2.1.Tl+ flux assays were established for Kir2.3 and the M125R pore mutant of Kir7.1 to support medicinal chemistry efforts to develop more potent and selective analogs for these channels. The structure–activity relationships of VU573 revealed few analogs with improved potency, however two compounds retained most of their activity toward GIRK and Kir2.3 and lost activity toward Kir7.1. We anticipate that the VU573 series will be useful for exploring the physiology and structure–function relationships of these Kir channels.
Keywords: GIRK, pharmacology, screening, thallium flux, fluorescence, electrophysiology, high throughput
Introduction
The inward rectifier family of potassium (Kir) channels is comprised of at least 16 family members exhibiting unique functional and regulatory properties that enable them to carry out important functions in most organ systems (Hebert et al., 2005; Hibino et al., 2010). Kir channels preferentially pass current in the inward direction at voltages more negative than the Nernst electrochemical equilibrium potential for potassium (EK). Unlike their voltage-gated potassium channel counterparts, Kir channels lack discrete voltage-sensing domains that regulate pore opening. Rather, the rectification of the channel conductance-voltage relationship is due to voltage-dependent pore block by magnesium and polyamines at potentials more positive than EK. The degree of rectification varies widely between family members and is used to categorize them functionally into groups of strong versus weak rectifiers. In general, strong rectifiers are expressed in excitable cells such as neurons and muscle cells, whereas weak rectifiers are expressed in epithelial and other non-excitable cell types (Hibino et al., 2010). Some Kir channels are postulated therapeutic targets for common diseases (Bhave et al., 2010). Consequently, we have been working to develop small-molecule probes of clinically relevant Kir channels to provide sharper pharmacological tools with which to study their physiological roles and therapeutic potential.
The founding Kir channel family member Kir1.1, is a weak rectifier encoded by the gene KCNJ1 (Ho et al., 1993; Zhou et al., 1994). Kir1.1 is expressed almost exclusively in epithelial cells of the renal tubule where it critically regulates salt and water balance and hence blood volume and pressure (Welling and Ho, 2009). Autosomal recessive mutations in KCNJ1 give rise to antenatal Bartter syndrome, a severe salt and water wasting disorder characterized by hypokalemic metabolic alkalosis and low to normal blood pressure (Simon et al., 1996). In contrast, heterozygous carriers of KCNJ1 mutations have lower blood pressure but no overt evidence of disease (Ji et al., 2008). These genetic data raise the intriguing possibility that Kir1.1 represents a drug target for a novel class of diuretic. Consequently, our group recently employed high-throughput screening and medicinal chemistry to develop the first publicly disclosed small-molecule inhibitors of Kir1.1 (Lewis et al., 2009; Bhave et al., 2011). These, as well as inhibitors recently disclosed by investigators at Merck (Pasternak et al., 2010), should be instrumental in assessing the therapeutic potential of Kir1.1 for the management of hypertension.
Another emerging drug target in the Kir channel family is the G protein-coupled inward rectifier potassium (GIRK) channel, which is expressed in the heart and throughout the nervous system (Hibino et al., 2010). The major cardiac form of GIRK is a heterotetrameric channel comprised of Kir3.1 and Kir3.4 subunits, which is expressed primarily in atrial but not ventricular myocytes. In the nervous system, heteromeric GIRK channels are primarily formed by Kir3.1 and Kir3.2. In the basal state, GIRK channels exhibit a low open-state probability and therefore contribute little to the resting membrane potassium conductance and potential. In the heart, sympathetic release of acetylcholine (ACh) onto M2 muscarinic receptors leads to G protein-dependent opening of GIRK channels, potassium efflux, and consequent membrane hyperpolarization. The ACh-induced increase in potassium conductance slows the rate of membrane depolarization, action potential generation, and heart rate. In patients with chronic atrial fibrillation, GIRK channels become constitutively active in atrial cardiomyocytes through mechanisms that are incompletely understood (Dobrev et al., 2005; Voigt et al., 2007; Makary et al., 2011). This background current hyperpolarizes the membrane potential, abbreviates the action potential, and increases the availability sodium channels for activation. A growing body of experimental and clinical data support the notion that electrical remodeling sets up high-frequency re-entrant current sources that perpetuate atrial fibrillation, suggesting that a specific blocker of GIRK could have anti-arrhythmic actions without the ventricular side effects commonly associated with current therapies (reviewed in Wakili et al., 2011). Similarly, the other Kir channel family member Kir2.3 is enriched in atrial cardiomyocytes and may also be a pharmaceutical target for atrial fibrillation (Ehrlich, 2008).
The molecular pharmacology of GIRK and most other inward rectifiers is limited. A high-throughput screen using a voltage-sensitive dye identified analogs of amiloride (a K+ sparing diuretic) and propafenone (a class 1c anti-arrhythmic) that inhibit GIRK with sub-micromolar potencies (Walsh, 2010). These and several cardiac and neurological drugs exhibiting weak off-target activity toward GIRK (reviewed in Bhave et al., 2010) may be useful lead compounds for developing more specific inhibitors of GIRK. The bee venom toxin tertiapin is a nanomolar affinity GIRK antagonist that also inhibits Kir1.1 (Jin and Lu, 1998; Kitamura et al., 2000). Hashimoto et al. (2006) demonstrated that administration of tertiapin to cannulated dogs terminated induced atrial fibrillation, providing some of the first experimental evidence that GIRK is a target for anti-arrhythmic therapeutics (Hashimoto et al., 2006). Nissan Chemical Industries developed a benzopyrene derivative, termed NIP-142, which inhibits Kir3.1/3.4 GIRK channels with sub-micromolar affinity and the cardiac Kv1.5 delayed rectifier current with equal potency (Matsuda et al., 2006; Tanaka and Hashimoto, 2007). NIP-142 has shown efficacy in terminating induced atrial fibrillation in dogs, but it is unknown whether the improvement in sinus rhythm is due to inhibition of GIRK, Kv1.5, or both. However, the low nanomolar inhibitor NTC-801, which is highly selective for GIRK over other cardiac channels, was shown recently to be effective in several models of atrial fibrillation (Machida et al., 2011).
In an effort to further expand the molecular pharmacology of GIRK, we employed a Tl+ flux-based fluorescence assay to screen a Kir1.1 inhibitor focus library for antagonists of GIRK. One compound termed VU573 was found to preferentially inhibit GIRK (cardiac and neuronal forms), Kir2.3 and Kir7.1 over Kir1.1 and Kir2.1. We anticipate that VU573 and its analogs will be useful for investigating the physiology and structure–function relationships of inward rectifier potassium channels.
Materials and Methods
Expression vectors
Plasmids used in this study are from the following sources: rat Kir1.1 (NM_017023; Chun Jiang, Georgia State University, Atlanta, GA, USA), human Kir2.1 (NM_000891.2; Al George, Vanderbilt University School of Medicine, Nashville, TN, USA), human Kir7.1 (NM_002242.2; David Clapham, Harvard Medical School, Cambridge, MA, USA). Human Kir1.1 (NM_000220), Kir3.1 (NM_002239.2), Kir3.2 (NM_002240.2), Kir3.4 (NM_000890.3), and Kir2.3 (NM_152868) were purchased from OriGene Technologies (Rockville, MD, USA). The M125R mutant of Kir7.1 was created using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies). The open reading frame of Kir7.1 was fully sequenced to ensure that no spurious mutations were introduced during mutation of the intended codon.
Cell lines
Monoclonal mGluR8/GIRK/HEK cells stably expressing Kir3.1/3.2, the M4 muscarinic receptor and rat mGlu8a were cultured as described previously (Niswender et al., 2008). Polyclonal stable T-REx-HEK-293 cell lines expressing Kir2.3 and Kir7.1-M125R under the control of a tetracycline-inducible promoter were established essentially as described in detail elsewhere (Fallen et al., 2009; Lewis et al., 2009). Monoclonal Kir2.3 cell lines were isolated through limiting dilution in 384-well plates and testing for tetracycline-inducible Tl+ flux, as described below. Polyclonal Kir7.1-M125R cells were used in this study. The development of monoclonal C1 T-REx-HEK-293 cells expressing Kir1.1 (S44D) was described previously (Lewis et al., 2009). T-REx-HEK-293 lines were cultured in DMEM growth medium containing 10% FBS, 50 U/mL Penicillin, 50 μg/mL Streptomycin, 5 μg/mL Blasticidin S, and 250 μg/mL Hygromycin.
Two-electrode voltage-clamp analysis
Stage V–VI oocytes were isolated from gravid Xenopus laevis frogs using sterile surgical techniques under tricaine anesthesia. All methods were in accordance with the guidelines for the use of laboratory animals of Vanderbilt University School of Medicine. The oocyte follicle layer was removed by manual dissection following enzymatic treatment with 1 mg/mL collagenase (Type 1A Sigma) dissolved in calcium-free OR-2 of the following composition (in mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, with pH 7.5 adjusted with NaOH. Oocytes were allowed to recover overnight at 16°C in modified L-15 media containing gentamicin sulfate (25 mg/mL).
pcDNA3.1(+) vectors carrying Kir1.1, human Kir2.1, 2.3, and 7.1 cDNA were used to synthesize channel cRNA from a T7 RNA polymerase and nucleotides provided in the mMESSAGE mMACHINE kit (Ambion, Austin, TX, USA) after linearization of the expression constructs. cRNA was purified by LiCl precipitation, diluted in RNAase-free water and used for injections. The oocytes were injected with 0.5–10 ng of cRNA of Kir1.1, Kir2.1, Kir2.3, and Kir7.1 using a Drummond digital microdispenser. Oocytes were incubated at 16°C in modified L-15 for 24–72 h prior to Kir channel recordings.
Whole-cell currents were recorded from Xenopus oocytes injected with Kir channel cRNA using the two-electrode voltage-clamp (TEVC) technique. Current and voltage commands were generated with a GeneClamp 600 amplifier, a Digidata 1200 A/D converter, and pCLAMP 8.0 software (Molecular Devices, Sunnyvale, CA, USA). The bath was actively clamped to 0 mV using a VG-2A bath clamp (Molecular Devices). Electrodes pulled from borosilicate glass (Sutter Instruments, Novato, CA, USA) using a PP-830 vertical puller (Narishige International, Narishige, Japan) had resistances of 0.5–5.0 MΩ when filled with 3 M KCl. The standard bath solution contained (in mM): 85 NaCl, 5 KCl, 10 HEPES, 2 MgCl2, pH 7.4 with NaOH. After achieving stable current amplitude, VU573 was applied continuously. This was followed by application of the non-specific potassium channel blocker barium. For Kir current recordings, cells were voltage-clamped and stepped every 5 s from a holding potential of −70 to −100 mV for 200 ms, then ramped at a rate of 1.6 mV/ms to 60 mV before returning to −80 mV. All recordings were made at room temperature (20–23°C).
Whole-cell patch clamp electrophysiology
T-REx-HEK-293-Kir2.3 cells were patch clamped after overnight induction with tetracycline (1 μg/mL). GIRK (Kir3.1/3.2) and wild type Kir7.1 were studied in HEK-293 cells transiently co-transfected with the respective channel expression vectors and pcDNA3.1-EGFP (transfection marker) using Lipofectamine LTX/Plus reagent according to manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). The standard intracellular solution contained 135 mM KCl, 2 mM MgCl2, 1 mM EGTA, 10 mM HEPES-free acid, and 2 mM Na2 ATP (Roche), pH 7.3, adjusted to 275 mOsmol/kg with sucrose. For Kir2.3 recordings, MgCl2 was reduced to 1 mM to prevent channel rundown (Chuang et al., 1997). The standard bath solution contained 135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES-free acid, pH 7.4, 290 mOsmol/kg. In some experiments, a high-K+ bath was used that contained 90 mM NaCl, 50 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES-free acid, pH 7.4, 290 mM mOsmol/kg. Kir current recordings were collected as previously described (Bhave et al., 2011). After achieving stable whole-cell currents, VU573 was applied intermittently or continuously for 2–10 min, followed by application of 2 mM BaCl2. Data acquisition and analysis were performed using pClamp 9.2 software (Molecular Devices). All recordings were made at room temperature (20–23°C).
Test compound and stimulus plate preparation
Compound master antagonist plates were created by serial diluting compounds 1:3 from 10 mM stock in 100% DMSO using the BRAVO liquid handler (Agilent Technologies, Santa Clara, CA, USA). Assay daughter plates were created using the ECHO 555 liquid hander (Labcyte, Sunnyvale, CA, USA), transferring 240 nL from the master plate to the daughter plate for each well followed by addition of 40 μL of assay buffer resulting in antagonist compound concentration–response curves (CRC) starting at 60 μM (2× final concentration). Glutamate was diluted in Tl+ buffer [125 mM sodium bicarbonate (added fresh the morning of the experiment), 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM Tl+ sulfate, and 10 mM HEPES, pH 7.3] at 5× the final concentration to be assayed.
Kinetic imaging, data analysis, and statistics
Cells were loaded with the Tl+ sensitive fluorescent dye FluoZin-2 and plated in clear-bottom 384-well plates essentially as described previously (Lewis et al., 2009; Bhave et al., 2011). Cell plates and daughter compound plates were loaded onto a kinetic imaging plate reader (FDSS 6000; Hamamatsu Corporation, Bridgewater, NJ, USA). All recordings were made at room temperature (20–23°C). Appropriate baseline readings were taken (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm) and 20 μL test compounds were added followed by 50 images at 1 Hz additional baseline. Following a 20-min incubation period, baseline readings were taken for 30 s followed by addition of 10 μL Glutamate at an EC80 concentration. An additional 170 images were taken at 1 Hz. Glutamate EC80 concentration was determined the day of the assay.
Data were analyzed using Excel (Microsoft Corp, Redmond, WA, USA). Raw data were opened in Excel and each data point in a given trace was divided by the first data point from that trace (static ratio). For experiments in which antagonists were added, data were again normalized by dividing each point by the fluorescence value immediately before the glutamate addition to correct for any subtle differences in the baseline traces after the compound incubation period. The slope of the fluorescence increase beginning 5 s after Tl+/glutamate addition and ending 15 s after Tl+/glutamate addition was calculated. The data were then plotted in Prism software (GraphPad Software, San Diego, CA, USA) to generate CRC after correcting for the slope values determined for baseline waveforms generated in the presence of vehicle controls. Potencies were calculated from fits using a four-parameter logistic equation.
Chemical synthesis
General
All NMR spectra were recorded on a 400-MHz AMX Bruker NMR spectrometer. 1H chemical shifts are reported in δ values in part per million downfield with the deuterated solvent as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). Low resolution mass spectra were obtained on an Agilent 1200 series 6130 mass spectrometer with electrospray ionization. High-resolution mass spectra were recorded on a Waters Q-TOF API-US plus Acquity system with electrospray ionization. Analytical thin layer chromatography was performed on EM Reagent 0.25 mm silica gel 60-F plates. Samples were analyzed for ≥95% purity using LC-UV/Vis-MS. Analytical HPLC was performed on an Agilent 1200 series with UV detection at 214 and 254 nm along with ELSD detection. LC/MS: method 1 = J-Sphere80-C18, 3.0 mm × 50 mm, 4.1 min gradient, 5%[0.05%TFA/CH3CN]:95%[0.05%TFA/H2O] to 100%[0.05%TFA/CH3CN]; method 2 = Phenomenex-C18, 2.1 mm × 30 mm, 2 min gradient, 7%[0.1%TFA/CH3CN]:93%[0.1%TFA/H2O] to 100%[0.1%TFA/CH3CN]; method 3 = Phenomenex-C18, 2.1 mm × 30 mm, 1 min gradient, 7%[0.1%TFA/CH3CN]:93%[0.1%TFA/H2O] to 95%[0.1%TFA/CH3CN]. Preparative purification was performed on a custom HP1100 purification system (Leister et al., 2003) with collection triggered by mass detection. Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification.
Reagents
Substituted phenols were purchased from various commercial sources, 2-Amino-1-benzylbenzimidazole and 2-Amino-1-methylbenzimidazole were purchased from Acros Organics. 1-(4-Chlorophenylmethyl)-2-aminobenzimidazole was made using published procedures (Caroti et al., 1986).
General synthetic procedure
The desired phenol (2 mmol) was dissolved in 0.4 mL NaOH (5 M) and then treated with epichlorohydrin (0.32 mL, 3.7 mmol; Scheme 1). The mixture was heated in a Biotage Initiator microwave reactor at 100°C for a 15-min period. The reaction was cooled to ambient temperature and diluted with dichloromethane. The aqueous layer was removed and the organic layer extracted with water. The mixture was dried and solvent removed under reduced pressure. The crude mixture was then treated with the desired 2-aminobenzimidazole (3 mL, 0.2 mmol, 0.7 M MeOH) dissolved in methanol Scheme 1). The mixture was allowed to stir for 4 days at ambient temperature. The solvent was removed under reduced pressure and the residue was diluted with dichloromethane. The mixture was extracted with water, dried, and concentrated under reduced pressure. The crude was purified using reverse phase chromatography (C18, Acetonitrile/H2O 0.1% TFA).
Scheme 1.

Synthesis of theVU573 scaffold.
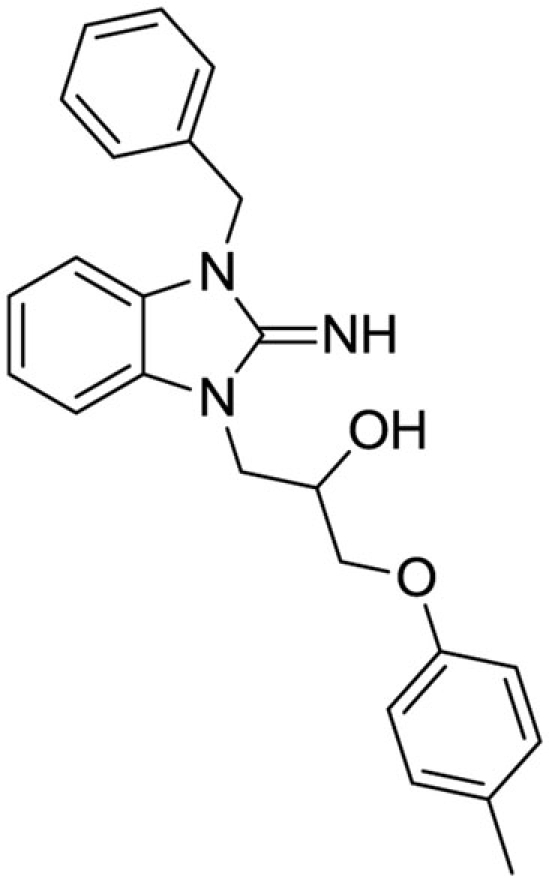
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(p-tolyloxy)propan-2-ol (1). Purchased from ChemBridge.
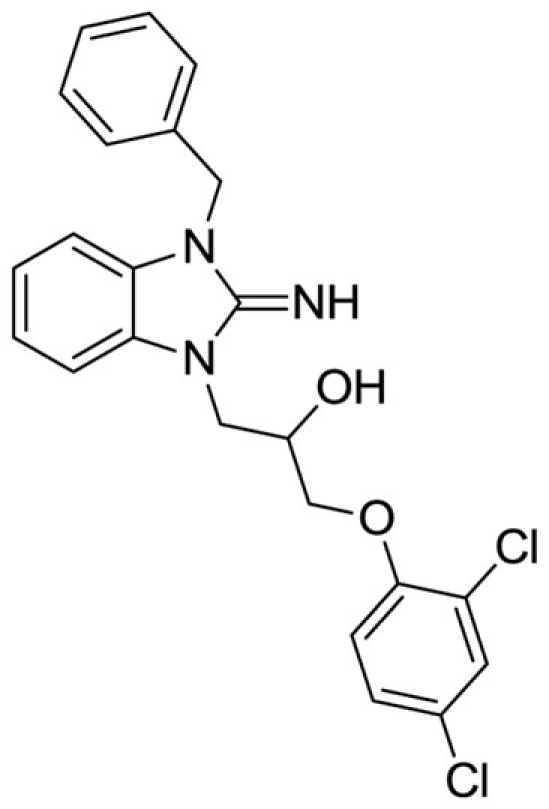
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(2,4-dichlorophenoxy)propan-2-ol (2).
LCMS: Rt = 1.41 min., >98%, m/z = 443 [M + H].
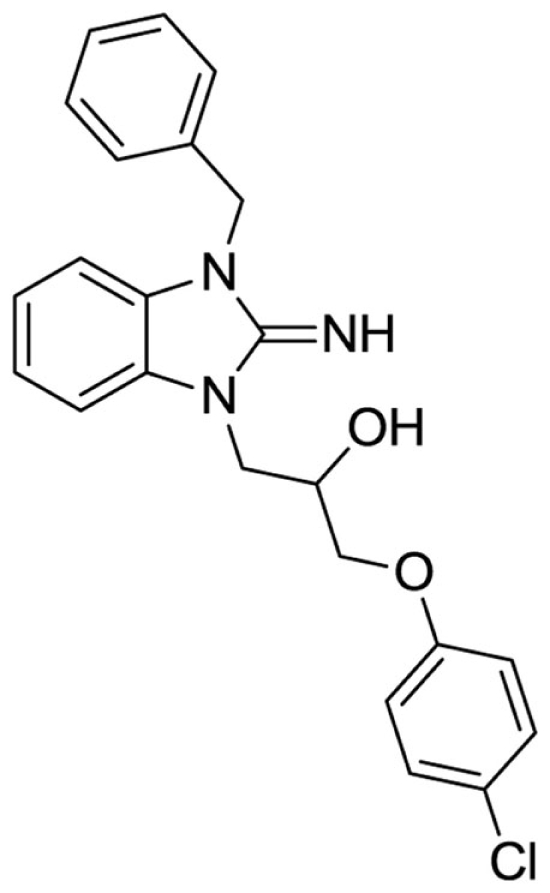
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-chlorophenoxy)propan-2-ol (3).
LCMS: Rt = 1.38 min., >98%, m/z = 408 [M + H].
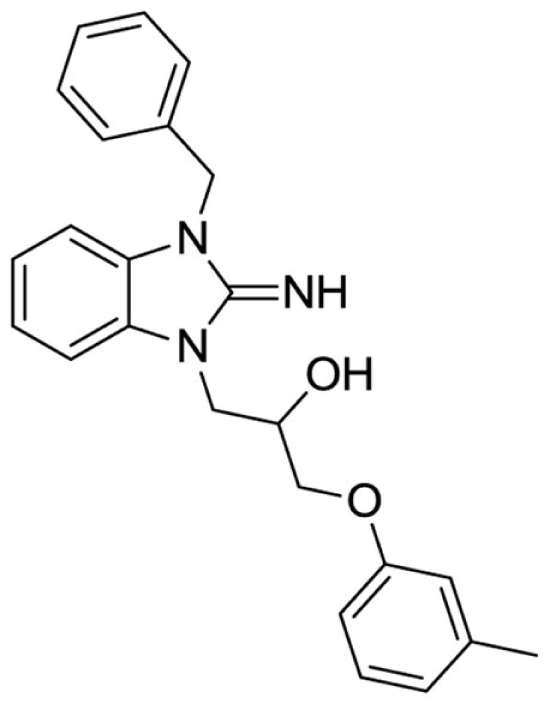
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(m-tolyloxy)propan-2-ol (4).
Purchased from InterBioScreen.
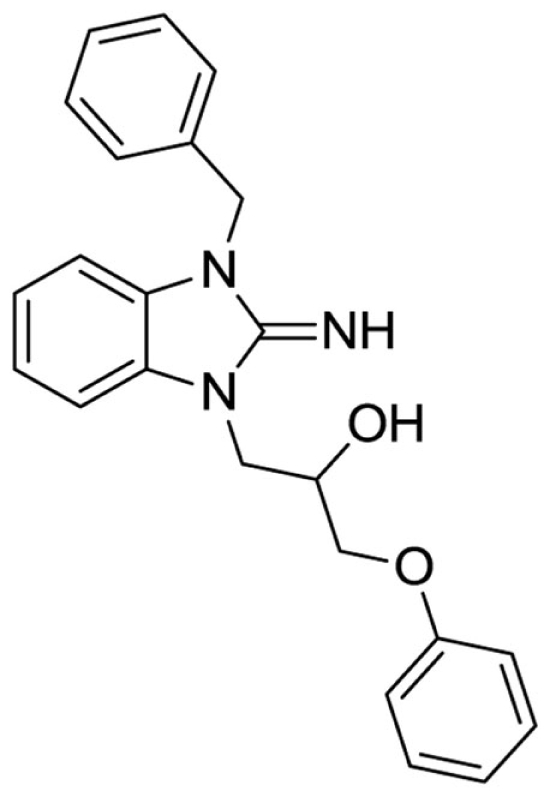
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-phenoxypropan-2-ol (5).
Purchased from ChemBridge.
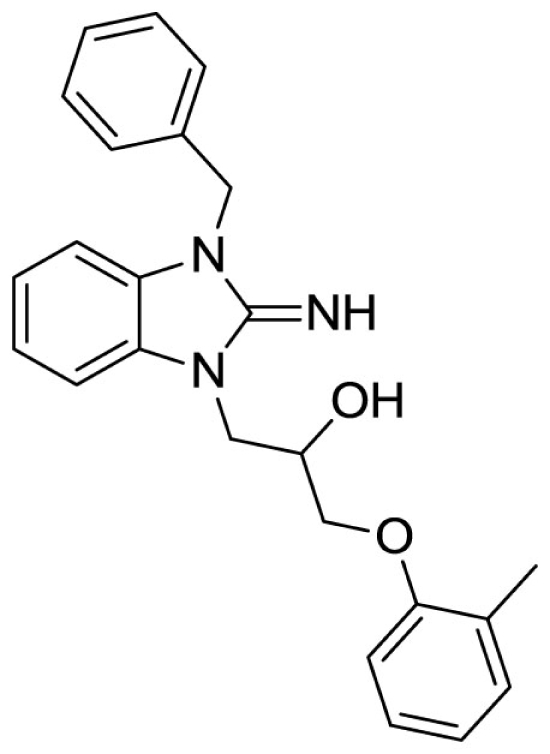
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(o-tolyloxy)propan-2-ol (6).
Purchased from ChemBridge.
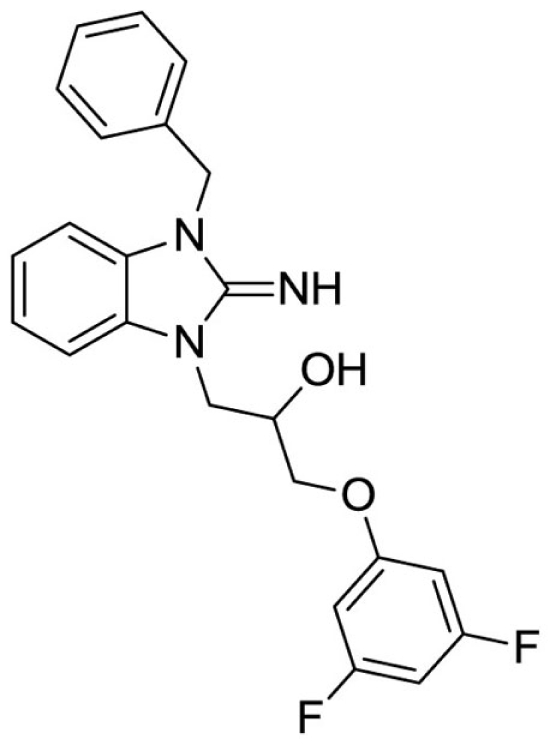
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(3,5-difluorophenoxy)propan-2-ol (7).
LCMS: Rt = 1.05 min., >98%, m/z = 410 [M + H].
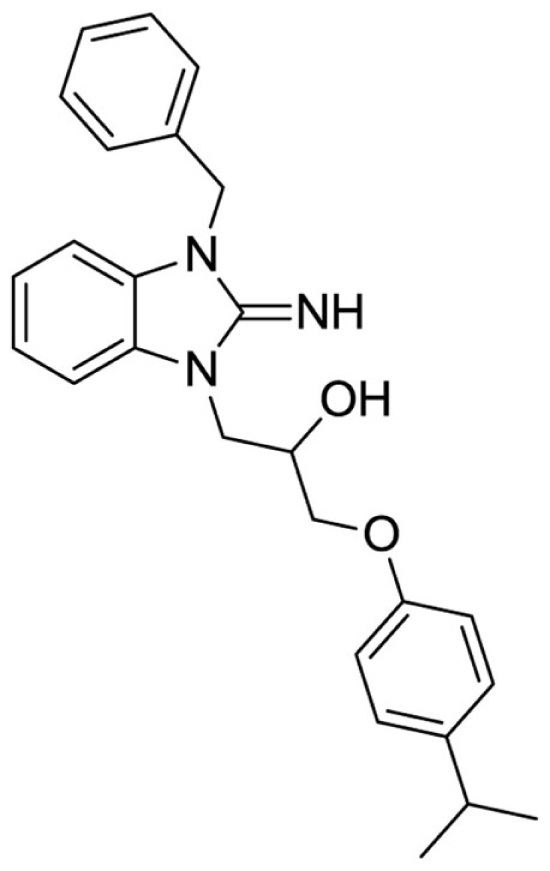
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-isopropylphenoxy)propan-2-ol (8).
LCMS: Rt = 1.14 min., >98%, m/z = 416 [M + H].
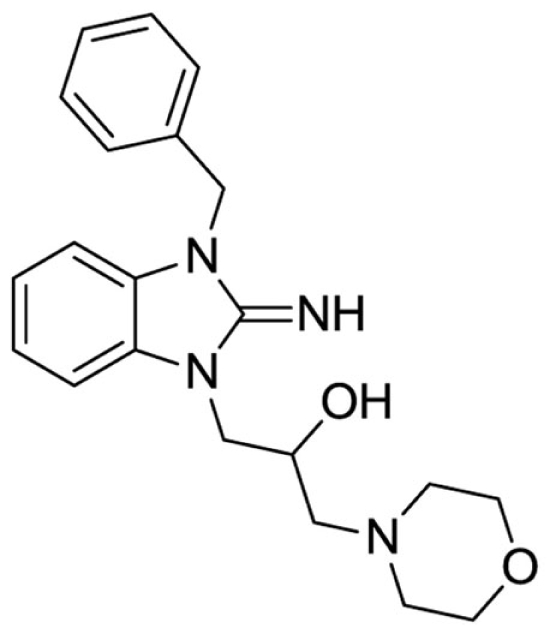
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-morpholinopropan-2-ol (9).
LCMS: Rt = 0.67 min., >98%, m/z = 367 [M + H].
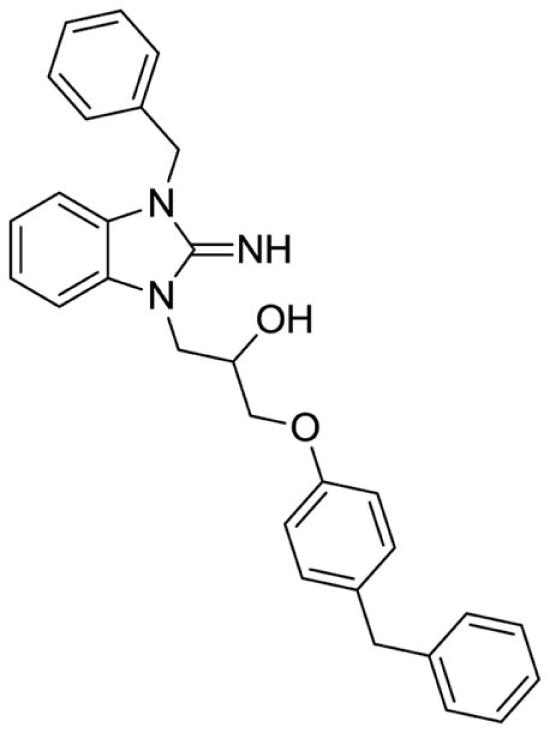
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-benzylphenoxy)propan-2-ol (10).
LCMS: Rt = 1.19 min., >98%, m/z = 464 [M + H].
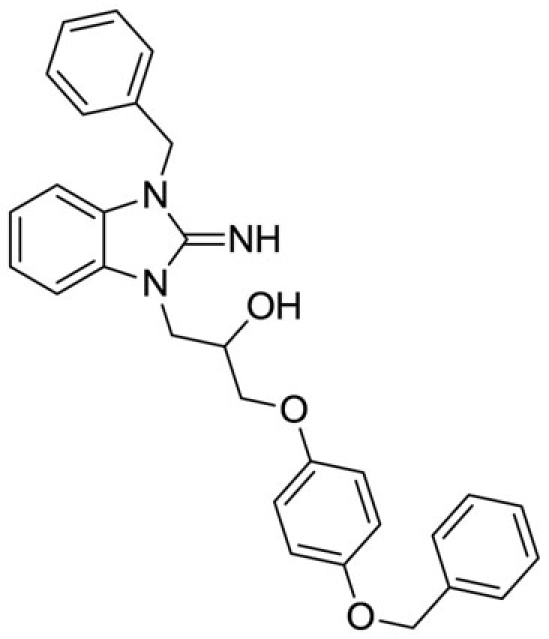
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-(benzyloxy)phenoxy)propan-2-ol (11).
LCMS: Rt = 1.16 min., >98%, m/z = 480 [M + H].
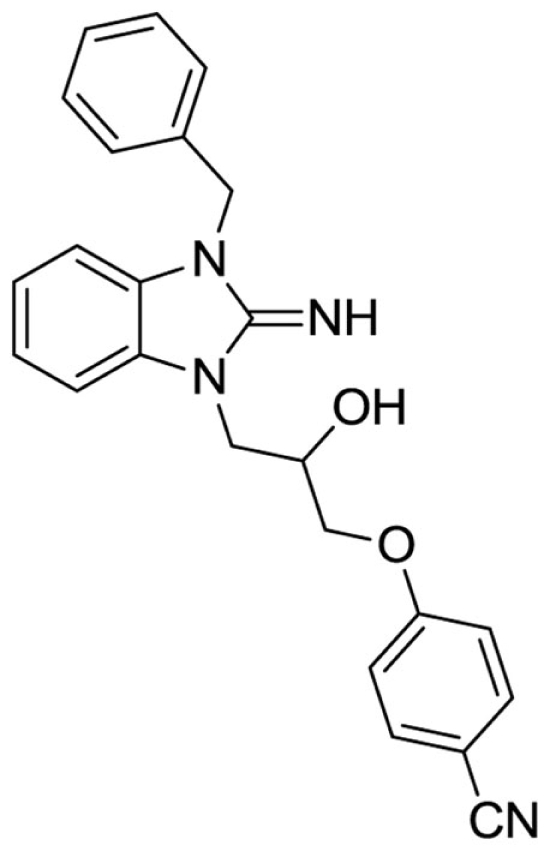
4-(3-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-2-hydroxypropoxy)benzonitrile (12).
LCMS: Rt = 1.00 min., >98%, m/z = 399 [M + H].
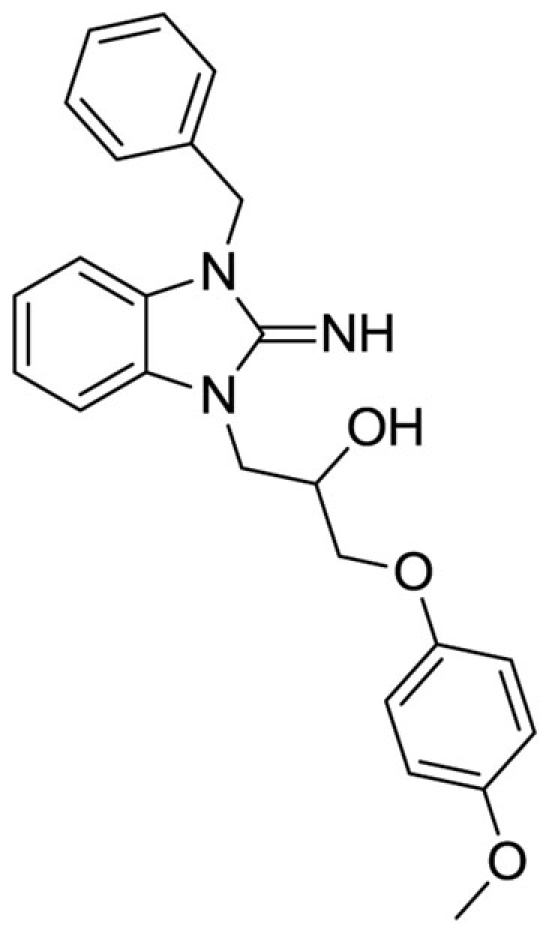
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-methoxyphenoxy)propan-2-ol (13).
LCMS: Rt = 1.00 min., >98%, m/z = 404 [M + H].
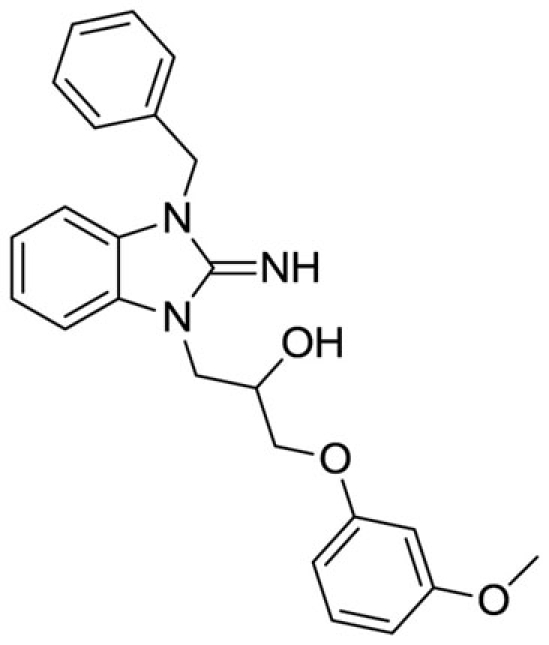
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(3-methoxyphenoxy)propan-2-ol (14).
LCMS: Rt = 1.01 min., >98%, m/z = 404 [M + H].
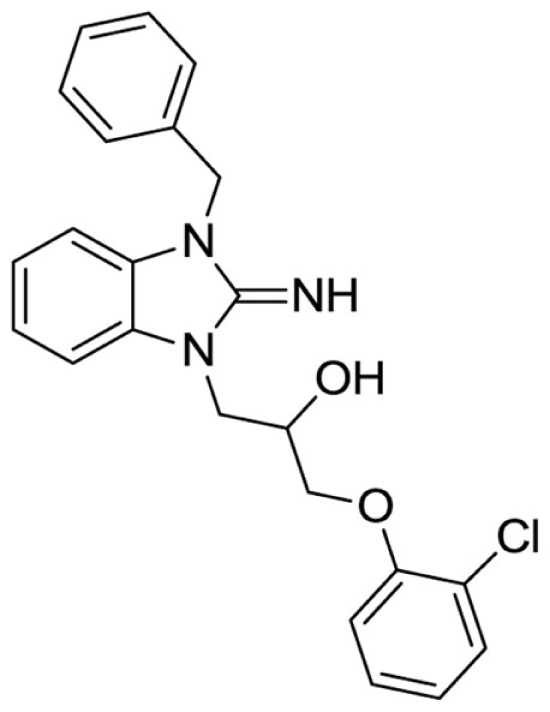
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(2-chlorophenoxy)propan-2-ol (15).
LCMS: Rt = 1.08 min., 90%, m/z = 408 [M + H].
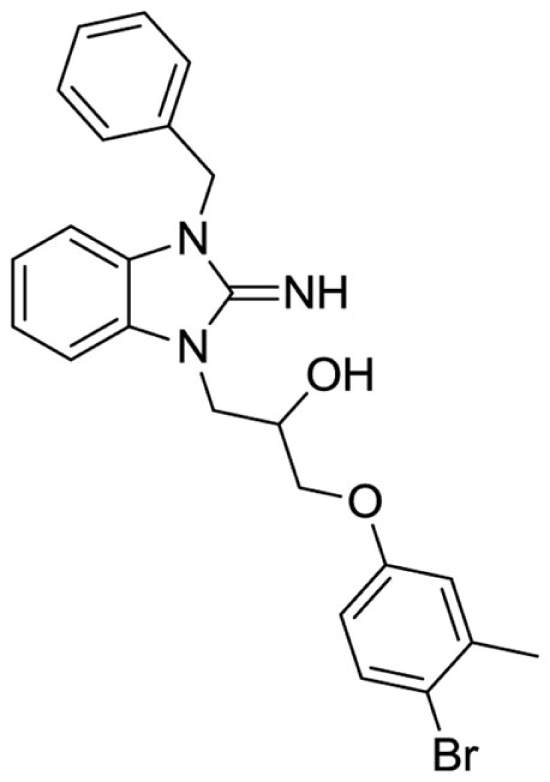
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-bromo-3-methylphenoxy)propan-2-ol (16).
LCMS: Rt = 1.13 min., >98%, m/z = 466 [M + H].
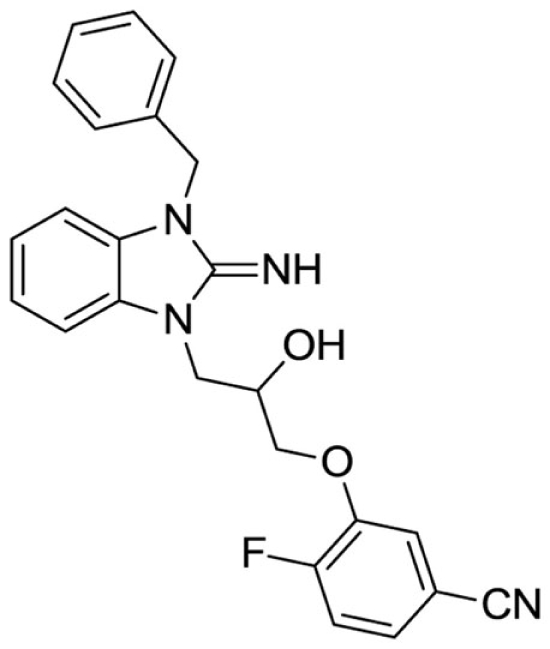
3-(3-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-2-hydroxypropoxy)-4-fluorobenzonitrile (17).
LCMS: Rt = 0.99 min., >98%, m/z = 417 [M + H].
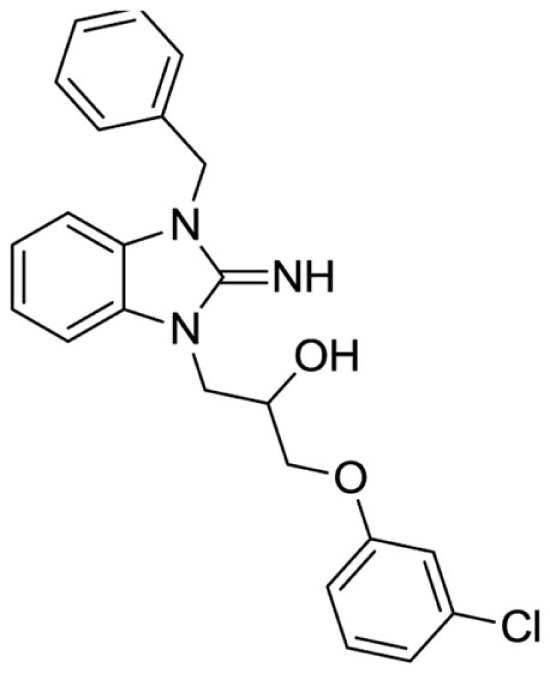
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(3-chlorophenoxy)propan-2-ol (18).
LCMS: Rt = 1.06 min., >98%, m/z = 408 [M + H].
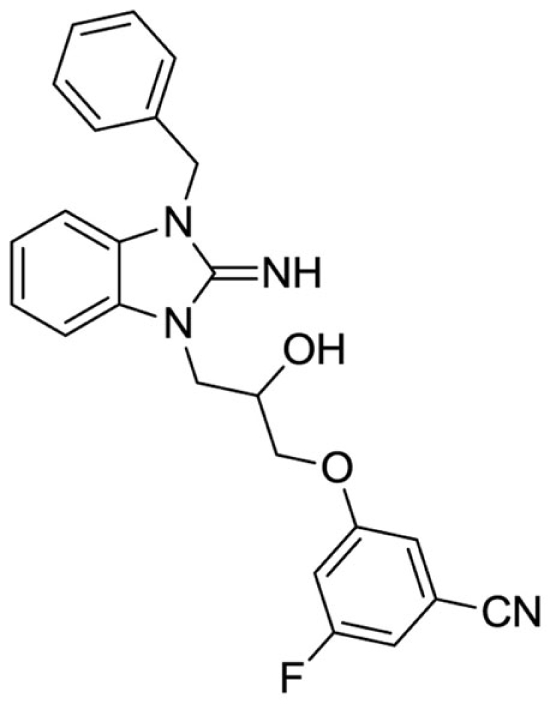
3-(3-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-2-hydroxypropoxy)-5-fluorobenzonitrile (19).
LCMS: Rt = 1.00 min., 90%, m/z = 417 [M + H].
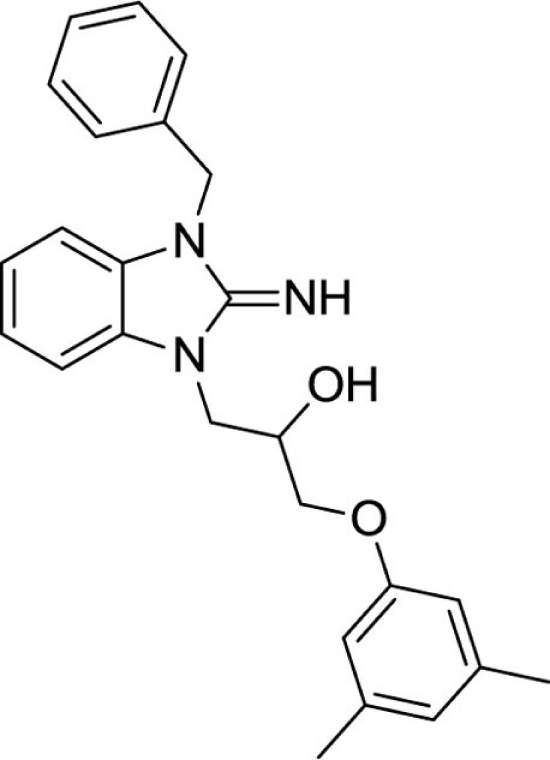
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(3,5-dimethylphenoxy)propan-2-ol (20).
LCMS: Rt = 1.10 min., 93%, m/z = 402 [M + H].
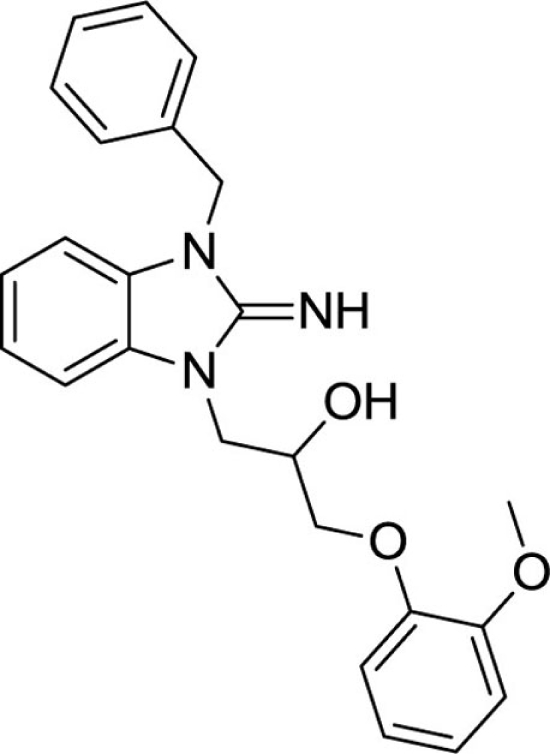
1-(3-benzyl-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(2-methoxyphenoxy)propan-2-ol (21).
LCMS: Rt = 0.98 min., >98%, m/z = 404 [M + H].
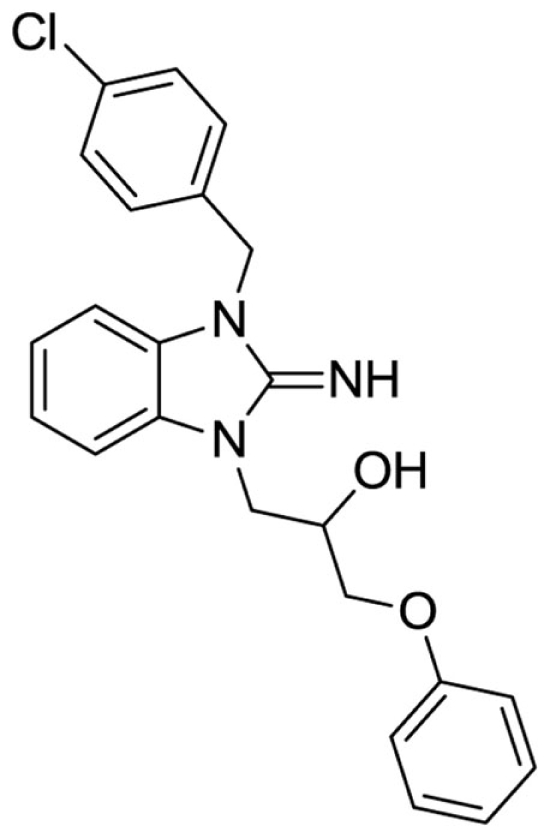
1-(3-(4-chlorobenzyl)-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-phenoxypropan-2-ol (22).
LCMS: Rt = 1.33 min., 87%, m/z = 408 [M + H].
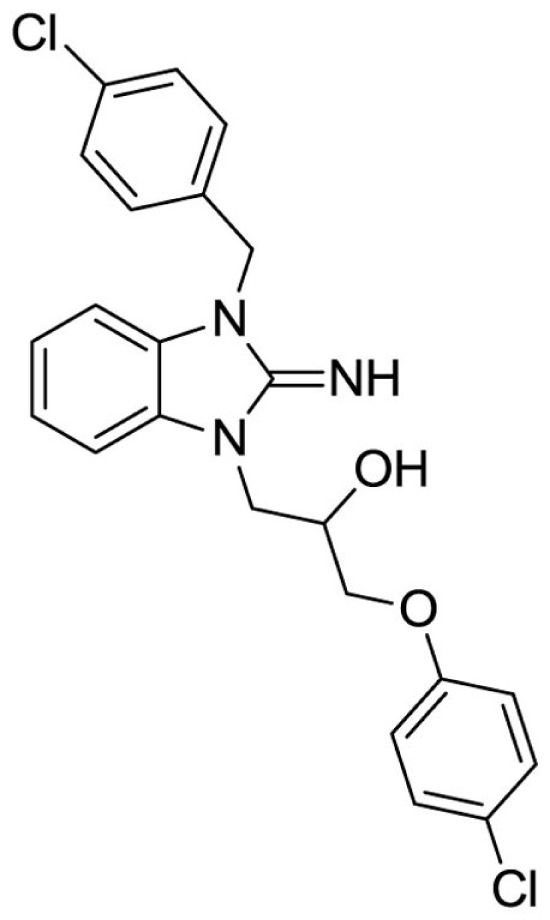
1-(3-(4-chlorobenzyl)-2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-chlorophenoxy)propan-2-ol (23).
LCMS: Rt = 1.38 min., >98%, m/z = 442 [M + H].
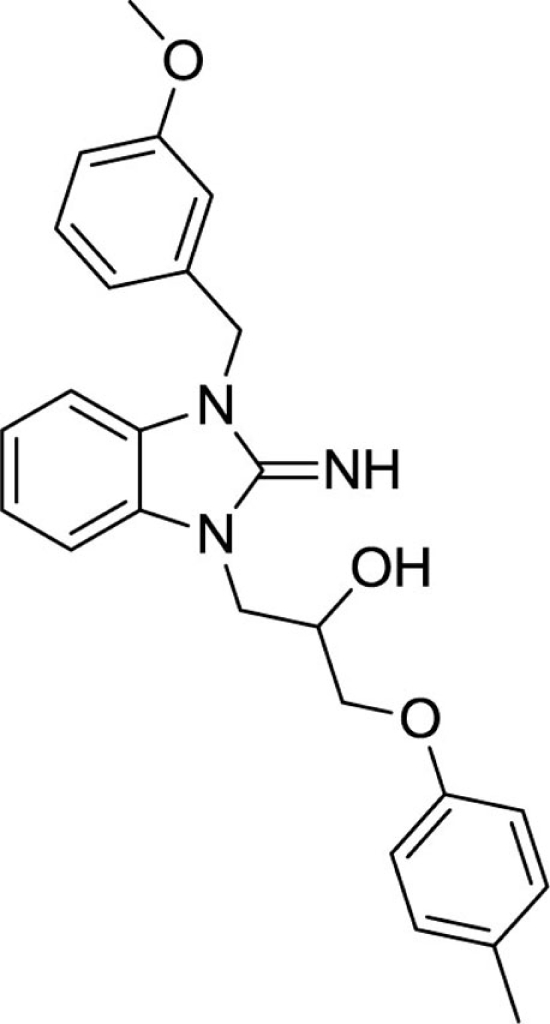
1-(2-imino-3-(3-methoxybenzyl)-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(p-tolyloxy)propan-2-ol (24).
LCMS: Rt = 1.05 min., >98%, m/z = 418 [M + H].
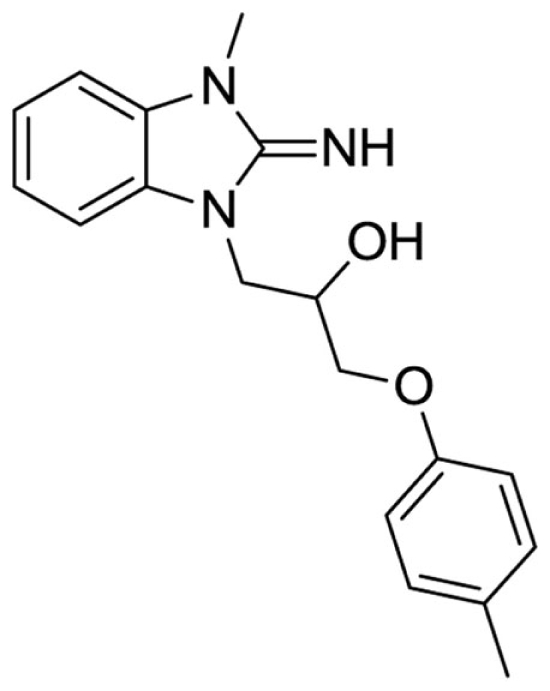
1-(2-imino-3-methyl-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(p-tolyloxy)propan-2-ol (25).
LCMS: Rt = 0.91 min., >98%, m/z = 312 [M + H].
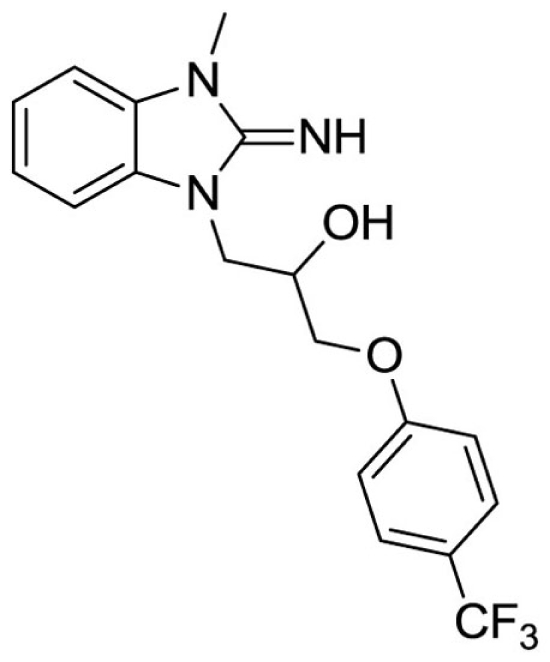
1-(2-imino-3-methyl-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-3-(4-(trifluoromethyl)phenoxy)propan-2-ol (26).
LCMS: Rt = 1.11 min., >98%, m/z = 442 [M + H].
Results
VU573: A weak Kir1.1 inhibitor that preferentially inhibits GIRK
We recently performed a high-throughput screen (HTS) for small-molecule modulators of Kir1.1 (Lewis et al., 2009), the founding member of the inward rectifier potassium (Kir) channel family (Ho et al., 1993; Zhou et al., 1994) and putative diuretic target (Simon et al., 1996; Ji et al., 2008; Pasternak et al., 2010). A focused library of reproducibly active inhibitors was assembled from the primary screen, with the goal of mining the library for antagonists of other inward rectifiers. As noted in the Introduction, a growing body of evidence suggests that GIRK channels expressed in atrial cardiomyocytes may represent a viable drug target for the treatment of atrial fibrillation. Given the paucity of potent and selective small-molecule inhibitors of GIRK and therapeutic interest in the channel, we screened the focus library for GIRK modulators using a Tl+ flux-based fluorescence assay developed recently at Vanderbilt (Niswender et al., 2008). The assay reports Tl+ flux through heteromeric Kir3.1/3.2 channels co-expressed in HEK-293 cells with the group III metabotropic glutamate receptor mGluR8. Intracellular Tl+ concentration is reported using the commercially available Tl+-sensitive fluorescent dye FluoZin-2. As shown in Figure 1A, glutamate addition dose-dependently increased Tl+ flux as GIRK channels are activated by mGluR8 stimulation. The focused library was screened at a single concentration of 10 μM using an 80% maximally effective concentration (EC80) of glutamate. Several compounds were identified that preferentially inhibited GIRK over Kir1.1 (data not shown). One of these compounds, termed VU573, was selected for further study.
Figure 1.

VU573 inhibits mGluR8-activated Kir3.1/3.2 channel activity in thallium flux assays. (A) Representative FluoZin-2 fluorescence traces recorded from HEK-293 cells stably expressing mGluR8 and Kir3.1/3.2 before and after co-application of thallium and different doses of glutamate (shaded box). From an 11-point glutamate concentration–response curve (not shown), the glutamate concentration evoking approximately 80% (EC80) of the maximal response (ECmax) was determined and used for subsequent experiments. (B) Representative traces for changes of Tl+-induced FluoZin-2 fluorescence following 20 min pre-treatment of cells with the indicated concentrations of VU573 and subsequent thallium and glutamate-EC80 addition (shaded box). (C) Mean ± SEM concentration–response curve for VU573-dependent inhibition of Kir3.1/3.2 (n = 3). The chemical structure of VU573 is shown in the inset. (D) Representative FluoZin-2 fluorescence traces recorded from monoclonal Kir1.1-S44D expressing cells cultured overnight in absence (−Tet) or presence (+Tet) of Tetracycline. (E) Representative traces for changes of Tl+-induced FluoZin-2 fluorescence following pre-treatment of cells with the indicated concentrations of VU573 and then exposed to thallium (shaded box). (F) Mean ± SEM concentration–response curve for VU573-dependent inhibition of Kir1.1-S44D (n = 3).
The chemical structure of VU573 is shown in the inset of Figure 1C. The potency of VU573 was evaluated in 11-point CRC using an EC80 of glutamate to activate GIRK. As shown in Figure 1B, VU573 inhibited GIRK-dependent Tl+ flux in a dose-dependent manner with a 50% inhibition concentration (IC50) of approximately 1.9 μM (Figure 1C).
To verify that VU573 is a more potent inhibitor of GIRK than Kir1.1, a full VU573 CRC was generated using an established Tl+ flux assay for Kir1.1 (Lewis et al., 2009; Bhave et al., 2011). The assay employs an inducible system in which Kir1.1 is expressed from a tetracycline-regulatable promoter. As shown in Figure 1D, extracellular Tl+ addition produced a dramatic increase in FluoZin-2 fluorescence in cells cultured with tetracycline, but not in uninduced cells. Thus, most of the Tl+ flux in tetracycline-induced cells occurs through Kir1.1. VU573 exhibited weak yet dose-dependent inhibition of Kir1.1 (Figure 1F), with an IC50 of approximately 19 μM. Whole-cell patch clamp experiments confirmed that 20 μM VU573 inhibited Kir1.1 by 49.5 ± 0.03% (n = 5). Thus, VU573 is a preferential inhibitor of GIRK over Kir1.1.
VU573 shows preference for Kir2.3, GIRK, and Kir7.1 over Kir1.1 and Kir2.1
To further assess the selectivity of VU573, several members of the Kir channel family were expressed in Xenopus oocytes and screened for VU573 sensitivity using the TEVC technique. We first tested whether VU573 discriminates between Kir3.1/3.2 and Kir3.1/3.4 channels, the predominant heterotetrameric forms of GIRK found in the nervous system and heart, respectively (Hibino et al., 2010). Figure 2A illustrates a typical experiment performed on an oocyte co-injected with cRNA encoding Kir3.1 and Kir3.2. Current recorded at −80 mV is shown as a function of time. The oocyte was initially bathed in a potassium-free (0K) solution and then switched to one containing 90 mM K+ (90K) to activate GIRK. The dramatic increase in inward current in the presence of 90K buffer was not observed in water-injected oocytes (data not shown) and therefore largely reflects current through Kir3.1/3.2 channels. Bath application of 10 μM VU573 inhibited GIRK current by approximately 50%, which was significantly lower than expected from Tl+ flux experiments (Figure 1C). The residual current was blocked by Ba2+. Figure 2F shows mean ± SEM percentage block of Kir3.1/3.2 and Kir3.1/3.4 channels by 1, 10, and 25 μM VU573 or Ba2+. The mean ± SEM time constant (τ) for inhibition of Kir3.1/3.2 and Kir3.1/3.4 currents by 25 μM VU573 were 28 ± 4 s (n = 6) and 27 ± 5 s (n = 6), respectively. There were no significant (P > 0.05) differences in the degree of VU573-dependent block of Kir3.1/3.2 and Kir3.1/3.4 channels at any of the concentrations.
Figure 2.

Effect of VU573 on Kir channels expressed in oocytes. (A) Representative Kir3.1/3.2 current traces recorded from an oocyte using the two-electrode voltage-clamp technique. Oocytes were initially bathed in a potassium-free (0K) solution and then switched to one containing 90 mM potassium (90K) to activate Kir3.1/3.2. After reaching a steady-state, the oocyte was exposed to 10 μM VU573 (in 90K) bath to inhibit Kir3.1/3.2. Residual Kir3.1/3.2 currents were inhibited with 2 mM barium (Ba2+). A final switch back to 0K was used to measure leak current at the end of each experiment. Representative whole-cell current traces recorded from oocytes expressing respectively (B) Kir1.1, (C) Kir2.1, (D) Kir2.3, and (E) Kir7.1 before and after application of 50 μM VU573. Residual Kir currents were inhibited with 2 mM barium (Ba2+). (F) Mean ± SEM percent inhibition of current evoked by Kir3.1/3.2, Kir3.1/3.4, Kir1.1, Kir2.1, Kir2.3, and Kir7.1 with the indicated concentrations of VU573 ( ) or Ba2+ (■) n = 4–6).
) or Ba2+ (■) n = 4–6).
GIRK channels are co-expressed with other members of the Kir channel family in a tissue- and cell type-specific manner. As noted in the Introduction, there is growing evidence that inhibitors of cardiac GIRK channels may be therapeutically beneficial in patients with atrial fibrillation. However, Kir2.1 and Kir2.3 are also expressed in the heart (Hibino et al., 2010) and therefore represent potential off-targets for VU573 actions. GIRK, Kir2.1, and Kir2.3 are also co-expressed in various brain regions, as is one of the newest Kir channel family members, Kir7.1 (Krapivinsky et al., 1998). We therefore determined whether VU573 inhibits any of these channels expressed in Xenopus oocytes. Kir1.1 was also expressed to confirm the results from Tl+ flux and patch clamp experiments.
Representative TEVC recordings from oocytes expressing Kir1.1, Kir2.1, Kir2.3, or Kir7.1 are shown in Figures 2B–E. The oocytes were voltage-clamped at −75 mV and stepped for 200 ms every 5 s to −120 mV to evoke inward current. The current amplitude at −120 mV is shown as a function of time. Barium was again used at the end of each experiment as a control blocker. Consistent with the Tl+ flux data showing greater potency toward GIRK than Kir1.1, 50 μM VU573 inhibited Kir1.1 by only 4.3 ± 1.7% with a time constant of 95 ± 15 s (n = 8). Kir2.1 was also relatively insensitive to VU573, and the time constant was very slow (τ = 735 ± 85 s; n = 4; Figure 2C). After 25 min of constant bath perfusion, 50 μM VU573 inhibited Kir2.1 by only 30.2 ± 9.4%. In contrast, Kir2.3 was inhibited comparatively quickly (τ = 154 ± 32 s) by 80.9 ± 5.0% (n = 7). Similarly, Kir7.1 was inhibited by 66.0 ± 6% with a rapid time constant of 38 ± 6 s (n = 8).
Patch clamp analysis of VU573 potency in mammalian cells
GIRK is inhibited by VU573 with an IC50 of approximately 2 μM in Tl+ flux assays in HEK-293 (Figure 1) cells and 10 μM in TEVC experiments in oocytes (Figure 2). To determine if the discrepancy is due to the expression system or assay type, whole-cell patch clamp techniques were used to assess the potency of VU573 toward Kir3.1/3.2 GIRK channels expressed in HEK-293 cells. The cells were voltage-clamped at a holding potential of −75 mV and then stepped to −120 mV for 200 ms, after which the membrane potential was ramped between −120 and 120 mV at a rate of 2.4 mV/ms. As shown in Figure 3B, GIRK activity was low in the presence of 5 mM extracellular K+ (5K), but increased upon elevation of bath K+ to 50 mM (50K), paralleling the behavior of GIRK observed in oocytes. Consistent with Tl+ flux data (Figure 1C), bath application of 30 μM VU573 led to a near complete inhibition of GIRK. As shown in the voltage ramp experiments in Figure 3A, VU573 inhibited GIRK across all potentials tested. The residual current was inhibited by barium (Ba2+; Figure 3B). The mean ± SEM time constant of inhibition was 16 ± 1 s (n = 5). A fit of the CRC data (Figure 3C) from patch clamp experiments yielded an IC50 of 1.3 μM, which is very near that derived from Tl+ flux assays (Figure 1C).
Figure 3.

VU573-dependent inhibition of Kir3.1/3.2, Kir2.3, and Kir7.1 channel activity expressed in HEK-293 cells. (A,D,G) Representative whole-cell Kir3.1/3.2, Kir2.3, and Kir7.1 current traces recorded from transfected HEK-293 cells. The cell was voltage ramped every 5 s between −120 and 120 mV for 200 ms from a holding potential of −75 mV. Normalized Kir3.1/3.2, Kir2.3, and Kir7.1 currents recorded before (black line) and after reaching steady-state inhibition by 30 μM VU573 (gray line) are shown. (B,E,H) Representative time course traces of VU573-dependent inhibition of Kir3.1/3.2, Kir2.3, and Kir7.1 by 30 μM VU573 using the protocol described above. After achieving whole-cell access, the current was allowed to stabilize before adding 30 μM VU573 and then 2 mM barium (Ba2+) to inhibit channel activity. (C,F,I) Mean ± SEM concentration–response curves for Kir3.1/3.2 (n = 4–7), Kir2.3 (n = 4–6), and Kir7.1 (n = 4–7), respectively.
We extended our patch clamp experiments to include Kir2.3 and Kir7.1 due to their strong inhibition by VU573 in oocytes (Figure 2F). A monoclonal stable T-REx-HEK-293 cell line expressing Kir2.3 under the control of a tetracycline-inducible promoter was patch clamped following overnight induction with tetracycline. Kir7.1 was studied in transiently transfected HEK-293 cells (see Materials and Methods). The cells were subjected to the same voltage ramp protocol described above for GIRK. Steady-state Kir2.3 and Kir7.1 currents recorded before (black line) and after (gray line) bath application of 30 μM VU573 are shown in Figures 3D,G, respectively. Both channels were inhibited with a rapid time course (Figures 3E,H) and IC50 values of approximately 1 μM (Figures 3F,I). The mean ± SEM time constants for inhibition of Kir2.3 and Kir7.1 were 23 ± 2 and 14 ± 2 s, respectively (n = 4 each).
Development of Tl+ flux assays for Kir2.3 and Kir7.1-M125R
We next set out to employ medicinal chemistry in an effort to improve the potency and selectivity of VU573 toward GIRK, Kir2.3, and Kir7.1. Conventional electrophysiological methods are too slow and labor intensive to support a robust medicinal chemistry campaign, leading us to establish high-throughput Tl+ flux assays for Kir2.3 and Kir7.1.As shown in Figure 4A, robust tetracycline-inducible Tl+ flux was observed in monoclonal stable T-REx-HEK-293 expressing Kir2.3. VU573 dose-dependently inhibited Tl+ flux through Kir2.3 with an IC50 of 4.7 μM (Figures 4B,C), a value that is reasonably close to that derived from patch clamp experiments (Figure 3F).
Figure 4.

Development of a thallium flux assay for Kir2.3. (A) Thallium flux-dependent FluoZin-2 fluorescence recorded from monoclonal Kir2.3 T-REx-HEK-293 cells cultured overnight in absence (−Tet) or presence (+Tet) of Tetracycline. The fluorescence emission was recorded before and after the addition of extracellular thallium (shaded box). (B) Representative traces for changes of Tl+-induced FluoZin-2 fluorescence following 20 min pre-treatment of cells with the indicated concentrations of VU573. (C) CRC for VU573-dependent inhibition of Kir2.3 activity. Values are mean ± SEM (n = 3). A fit of the CRC with a single-site four-parameter logistic function yielded IC50 of 4.7.
In contrast, despite numerous attempts on multiple polyclonal and monoclonal cell lines, we were unable to detect Tl+ flux through Kir7.1. Western blot analysis revealed that this was not due to lack of channel protein expression (data not shown), suggesting that other channel properties were responsible. Kir7.1 is unique among Kir channels in that it has a very small unitary conductance, which has been estimated from noise analysis to be on the order of 50 femptoSiemens (fS; Krapivinsky et al., 1998). We reasoned that this could keep Tl+ flux through Kir7.1 below the limit of detection of FluoZin-2. The small conductance of the channel is due at least in part to a non-conserved Methionine (M) residue at position 125 located in the pore. Mutation of the residue to Arginine (R),which occupies the homologous position in all other Kir channels (not shown), has no effect on protein expression or targeting to the plasma membrane, but increases the single-channel conductance by approximately 20-fold (Krapivinsky et al., 1998). We therefore wondered if the M125R mutation could enable measurement of Tl+ flux using FluoZin-2. As shown in Figure 5B, tetracycline-induced robust Tl+ flux in polyclonal stable T-REx-HEK-293 cells expressing Kir7.1-M125R. VU573 induced concentration-dependent inhibition of Tl+ flux with an IC50 of 4.9 μM (Figures 5C,D). The good correlation between IC50 values from patch clamp and Tl+ flux experiments and the fact that the M125R mutation had no effect on the sensitivity to VU573 in patch clamp experiments (Figure 5A) indicated that the Kir7.1-M125R assays could be used as a screening tool to support medicinal chemistry efforts for VU573.
Figure 5.

Development of a thallium flux assay for Kir7.1 (M125R). (A) Mean ± SEM % inhibition of wild type (closed bars; n = 6–7) or M125R mutant (open bars; n = 4–6) Kir7.1 by the indicated concentration of VU573.Note that the wild type data are reproduced from Figure 3. (B) Thallium flux-dependent FluoZin-2 fluorescence recorded from polyclonalKir7.1 (M125R) T-REx-HEK-293 cells cultured overnight in absence (−Tet) or presence (+Tet) of Tetracycline. The fluorescence emission was recorded before and after the addition of extracellular thallium (shaded box). (C) Representative traces for changes of Tl+-induced FluoZin-2 fluorescence following 20 min pre-treatment of cells with the indicated concentrations of VU573. (D) CRC for VU573-dependent inhibition of Kir2.3 activity. Values are mean ± SEM (n = 3). A fit of the CRC with a single-site four-parameter logistic function yielded IC50 of 4.9 μM.
Synthesis and structure–activity relationships of VU573 analogs
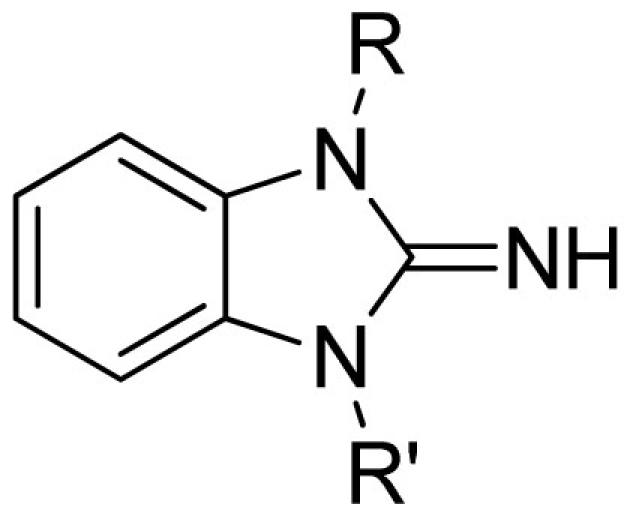
Lead optimization efforts directed at VU573 to improve its potency and selectivity toward Kir2.3, GIRK, and Kir7.1 were initiated by conserving the benzyl moiety and substituting the R group (Table 1). Structure–activity relationships (SAR) revealed few analogs with channel selectivity. Replacement of the phenoxy moiety with a morpholino group to the VU573 scaffold resulted in compound (9), which lost activity (IC50 values > 100 μM) for Kir7.1 and Kir1.1. The IC50 values for Kir2.3 and GIRK were 24 and 15.3 μM, respectively. Substitution of the R group of the VU573 scaffold with a phenoxy benzyloxy functional group led to an analog (11), which was equipotent for Kir2.3 and GIRK, but loss activity toward Kir7.1 (IC50 values > 100 μM). An alternate functional group for the VU573 scaffold, a 4-cyanophenyl (12), led to an analog compound with moderate selectivity for GIRK (IC50 = 5.27 μM) and weak inhibition of Kir2.3 and Kir7.1, with IC50 values of 11.3 and 19.7 μM, respectively. All other analogs in which substitutions were made at both R and R′ groups led to similar potency among the Kir2.3, Kir7.1, and GIRK channels as compared to VU573, but with weak inhibition for Kir1.1. The development of two VU573 analogs, R70 and R5C (Table 1), that retain activity toward GIRK and Kir2.3 but have lost activity toward Kir7.1 suggest that chemically optimized analogs based on the VU573 scaffold can be developed to increase their selectivity.
Table 1.
Structure–activity relationships and lead optimization summary.
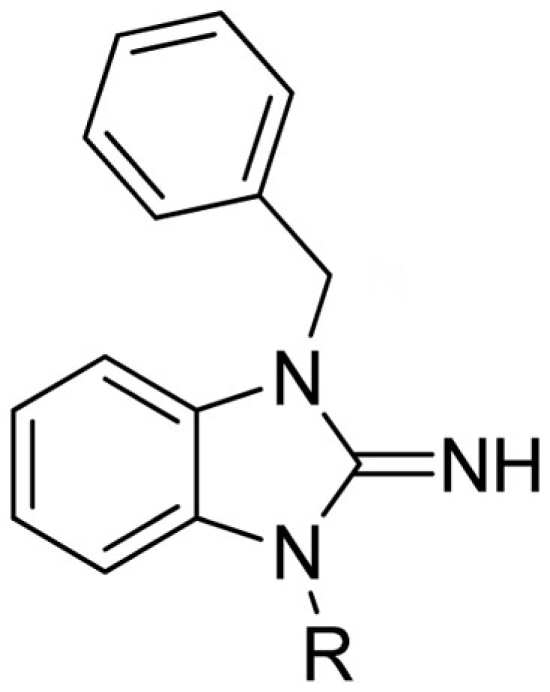 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R | VU#/Barcode | IC50 (μM) |
||||
| Kir2.3 | Kir7.1 | GIRK | Kir1.1 | ||||
| 1 | 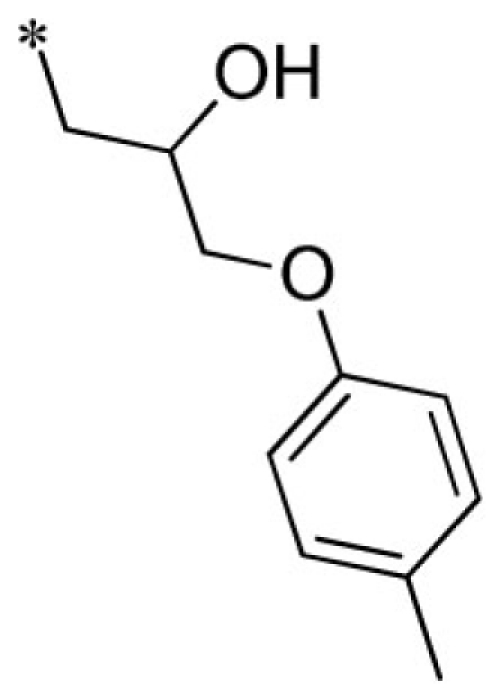 |
VU0160573-1/IC4X | 4.15 ± 0.35 | 4.77 ± 2.89 | 2.17 ± 0.40 | 10.8 ± 3.8 | |
| 2 | 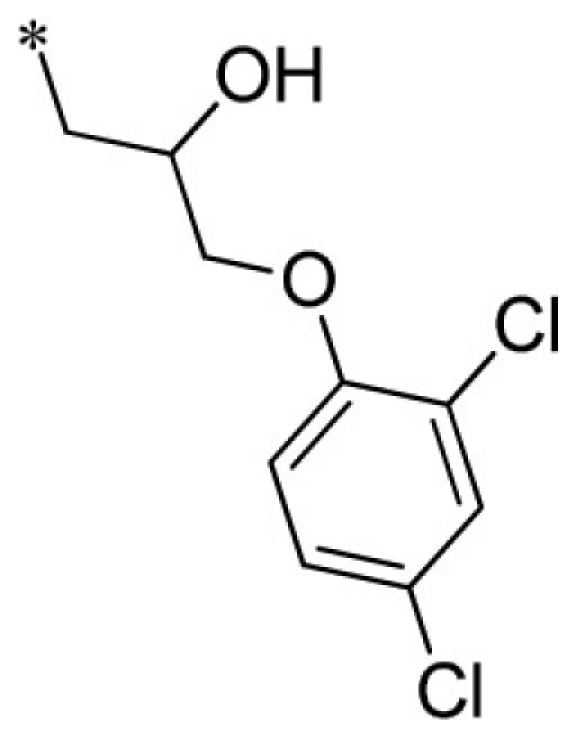 |
VU0403134-1/IC3Y | 5.33 ± 2.08 | 4.73 ± 2.57 | 3.20 ± 0.71 | 7.07 ± 2.30 | |
| 3 | 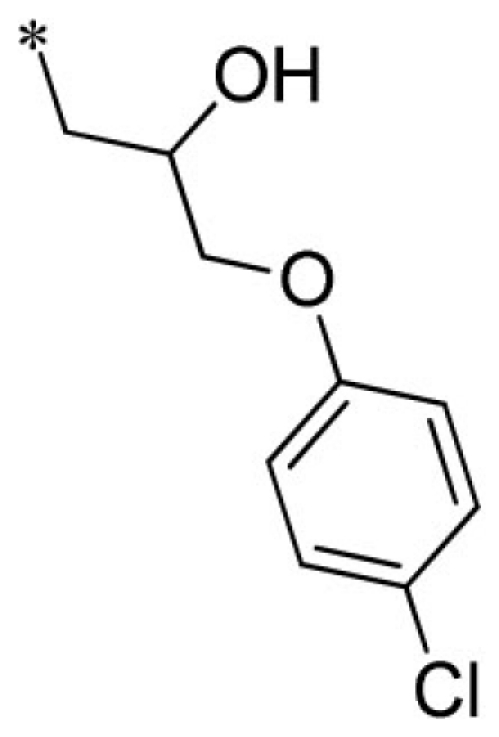 |
VU0403131-1/IC58 | 4.20 ± 1.25 | 5.17 ± 1.89 | 2.90 ± 1.10 | 6.57 ± 3.80 | |
| 4 | 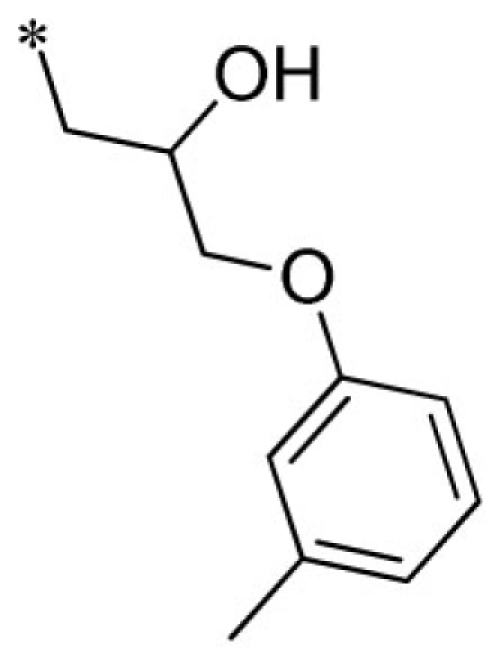 |
VU0340260-1/IC38 | 7.53 ± 2.34 | 8.00 ± 3.86 | 4.95 ± 1.80 | 11.2 ± 2.8 | |
| 5 | 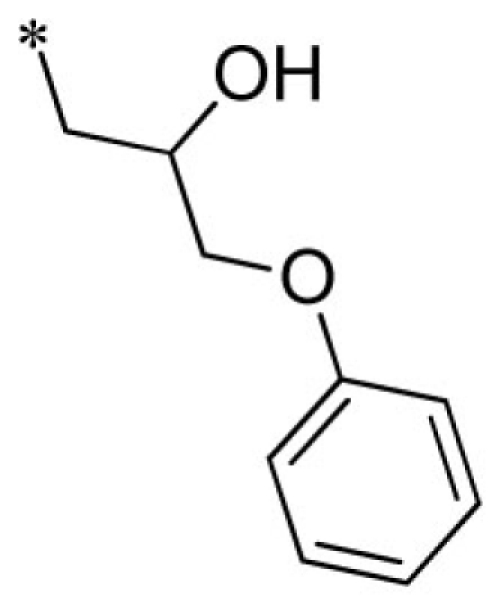 |
VU0026784-1/IC3L | 3.43 ± 0.67 | 4.47 ± 2.40 | 1.95 ± 0.49 | 11.7 ± 4.1 | |
| 6 | 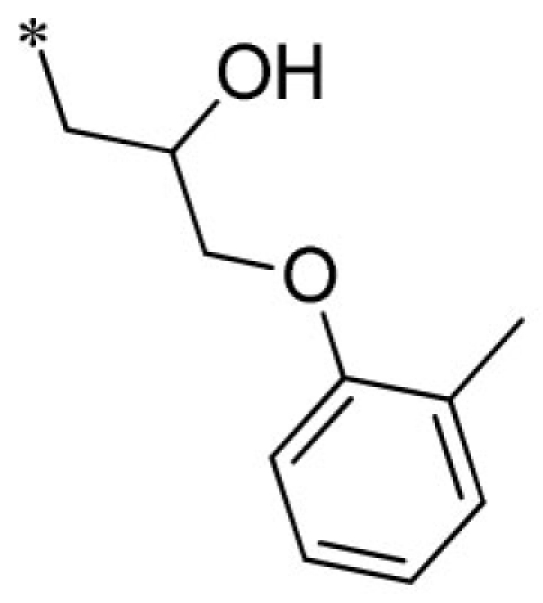 |
VU0288495-1/IC39 | 5.33 ± 1.70 | 4.10 ± 1.93 | 1.95 ± 0.07 | 11.9 ± 2.2 | |
| 7 | 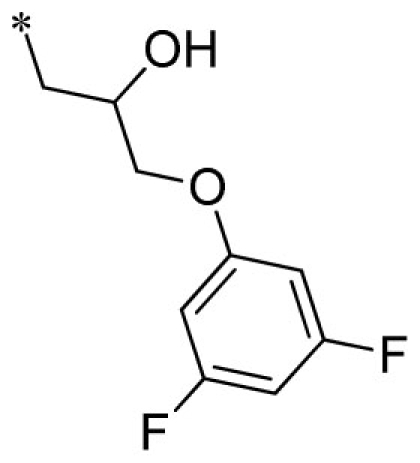 |
VU0451348-1/R6P | 3.53 ± 1.33 | 4.20 ± 0.95 | 1.40 ± 0.35 | 7.57 ± 2.20 | |
| 8 | 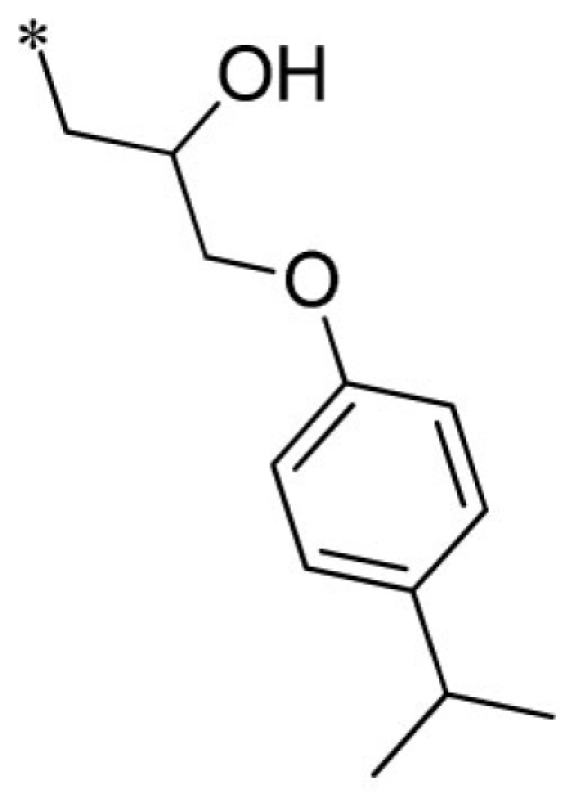 |
VU0451344-1/R80 | 5.03 ± 1.96 | 6.03 ± 1.60 | 3.37 ± 0.70 | 6.77 ± 1.70 | |
| 9 | 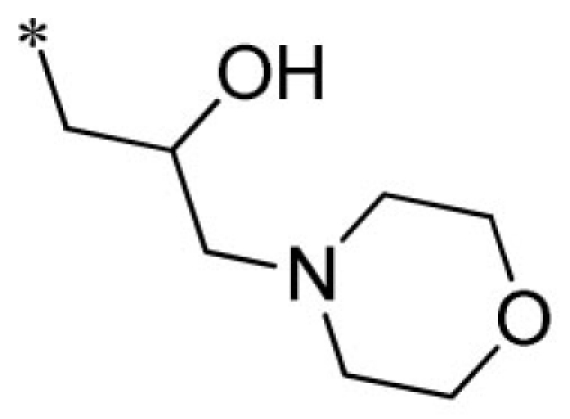 |
VU0451342-1/R70 | 24.0 ± 10.4 | >100 | 15.3 ± 0.6 | >100 | |
| 10 | 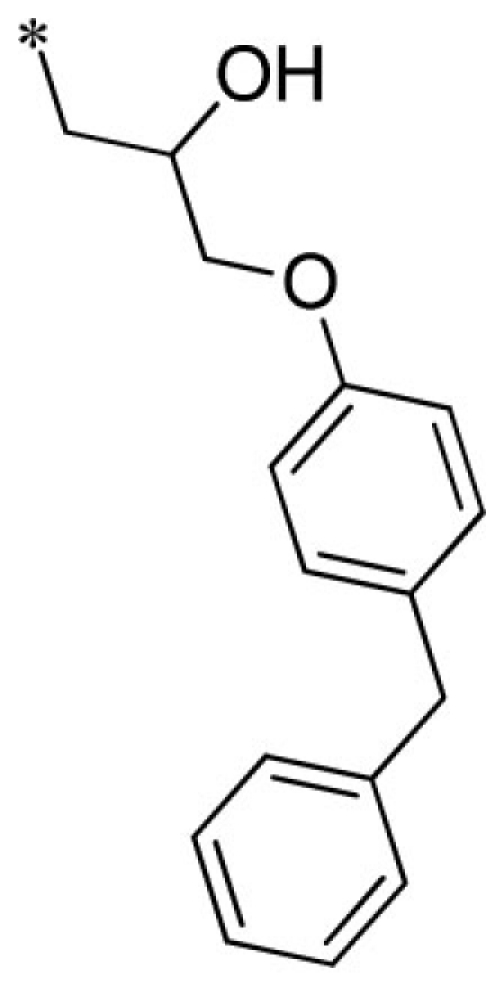 |
VU0451341-1/R7M | 2.80 ± 0.99 | 6.07 ± 2.71 | 3.83 ± 1.10 | 6.13 ± 0.23 | |
| 11 | 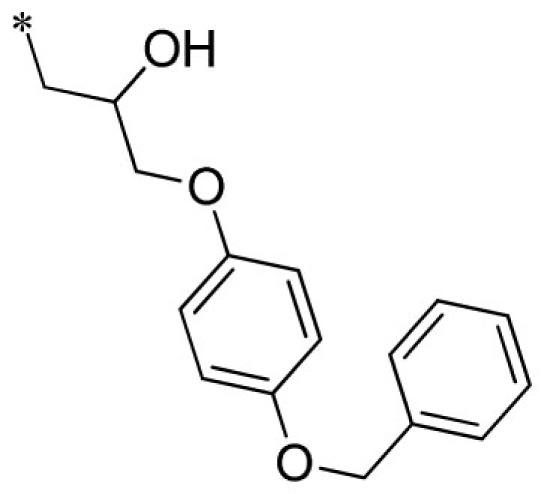 |
VU0451340-1/R5C | 2.33 ± 0.68 | >100 | 3.17 ± 0.86 | 5.23 ± 0.59 | |
| 12 | 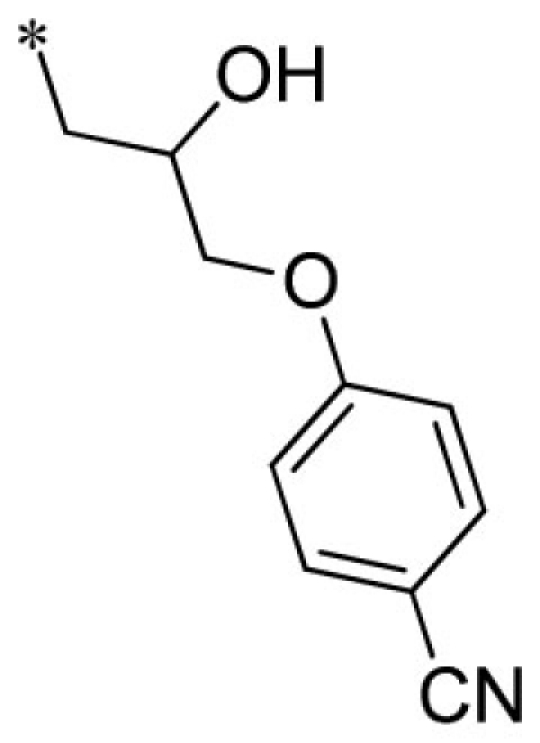 |
VU0451339-1/R56 | 11.3 ± 1.2 | 19.7 ± 7.4 | 5.27 ± 0.40 | 15.7 ± 2.1 | |
| 13 | 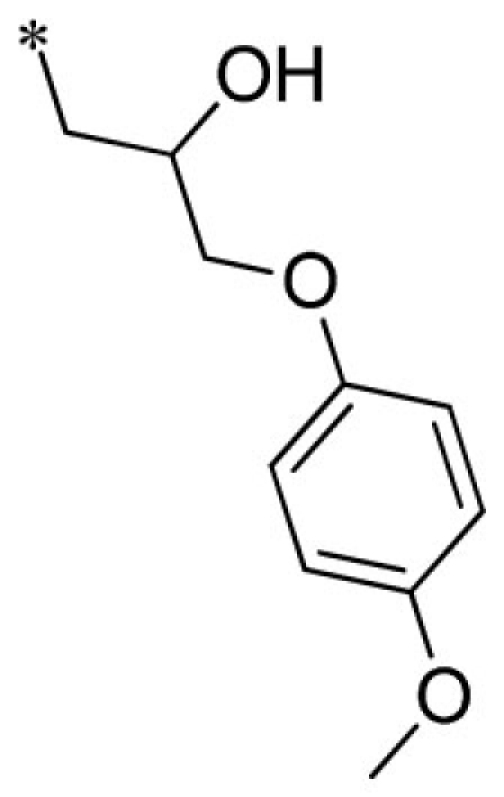 |
VU0066224-6/R7N | 3.87 ± 1.40 | 6.70 ± 1.39 | 1.50 ± 0.44 | 12.7 ± 1.2 | |
| 14 | 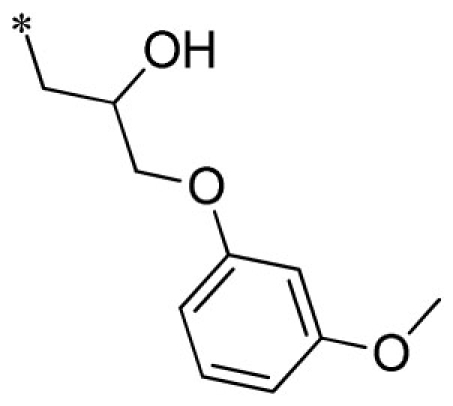 |
VU0451336-1/R5B | 3.13 ± 1.45 | 3.47 ± 0.67 | 1.43 ± 0.25 | 10.1 ± 2.9 | |
| 15 | 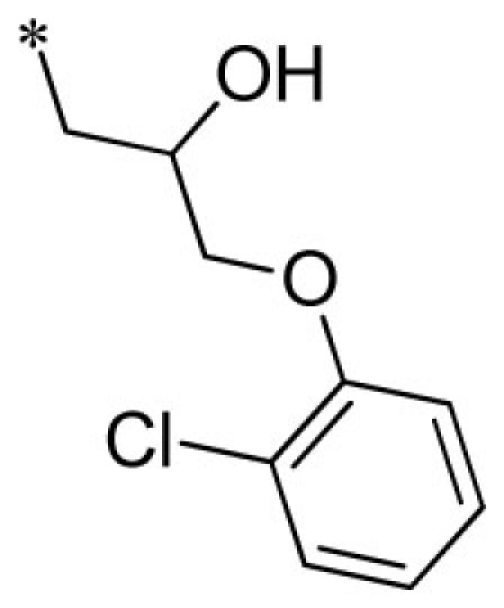 |
VU0451333-1/R5X | 4.95 ± 1.34 | 5.33 ± 2.91 | 2.77 ± 0.60 | 8.60 ± 1.20 | |
| 16 | 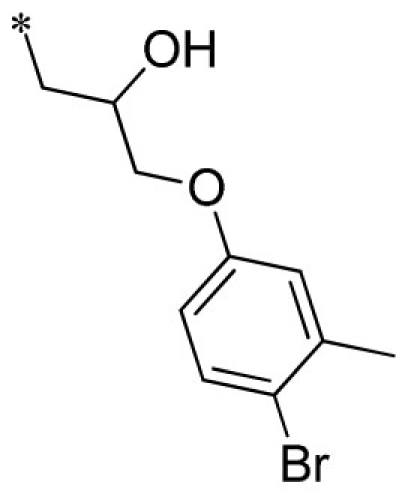 |
VU0451337-1/R5K | 5.10 ± 1.27 | 6.10 ± 3.12 | 3.43 ± 0.21 | 8.53 ± 1.70 | |
| 17 | 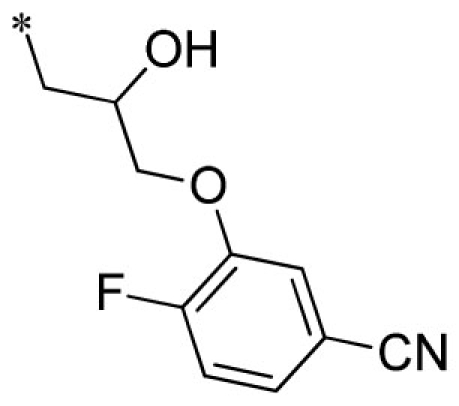 |
VU0451332-1/R7B | 5.30 ± 1.56 | 4.33 ± 1.33 | 1.40 ± 0.30 | 8.27 ± 0.29 | |
| 18 | 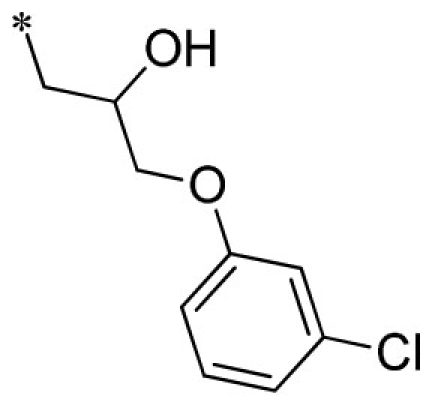 |
VU0451338-1/R71 | 4.45 ± 0.92 | 5.63 ± 2.24 | 2.83 ± 0.42 | 11.7 ± 2.1 | |
| 19 | 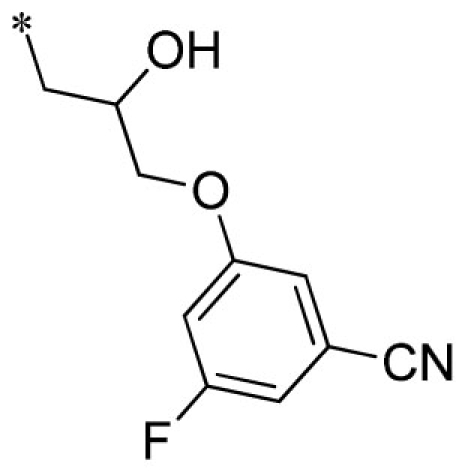 |
VU0451330-1/R6J | 7.15 ± 0.71 | 8.27 ± 2.37 | 1.93 ± 0.21 | 14.3 ± 1.5 | |
| 20 | 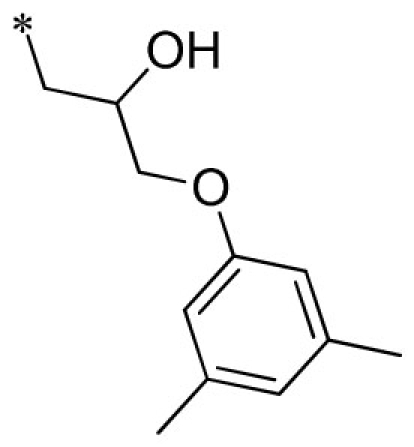 |
VU0451331-1/RKN | 3.65 ± 0.21 | 3.03 ± 0.68 | 4.33 ± 0.68 | 16.0 ± 2.0 | |
| 21 | 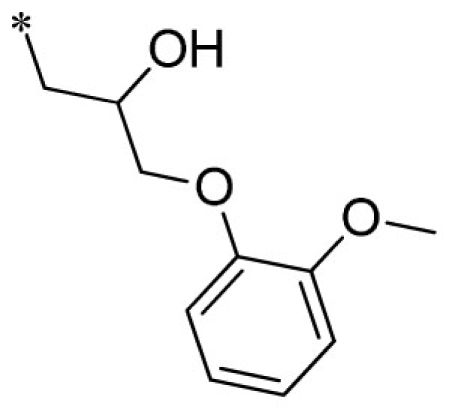 |
VU0451846-2/R87 | 3.60 ± 0.28 | 8.50 ± 2.18 | 5.13 ± 1.60 | 19.3 ± 1.5 | |
 | |||||||
| Cmpd | R | R′ | VU#/Barcode |
IC50 (μM) |
|||
| Kir2.3 | Kir7.1 | GIRK | Kir1.1 | ||||
| 22 | 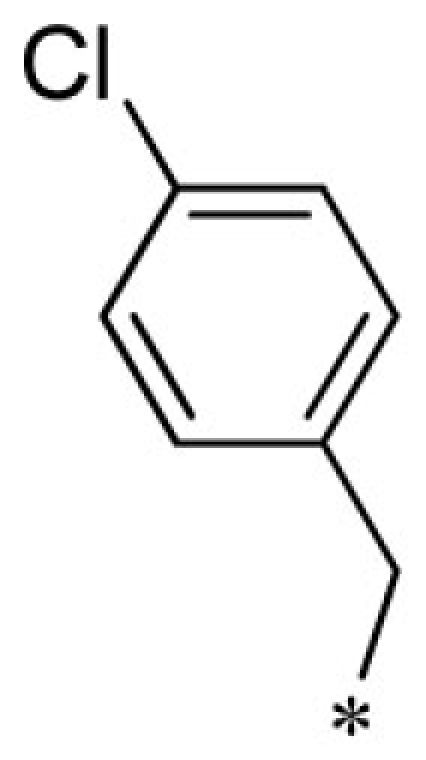 |
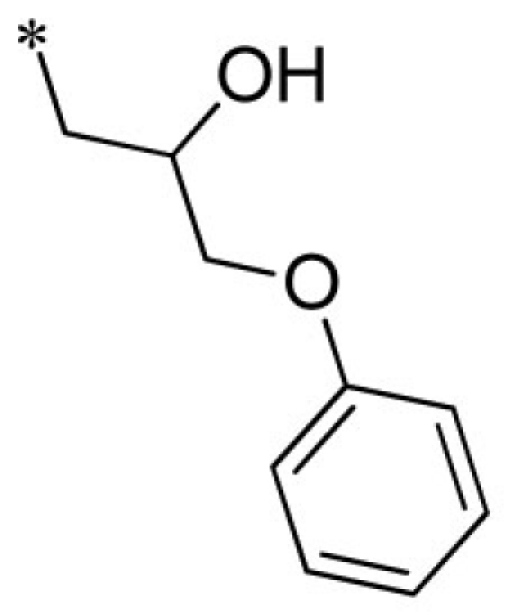 |
VU0401333-1/IC4L | 7.20 ± 1.73 | 8.93 ± 3.53 | 4.80 ± 1.10 | 14.7 ± 2.1 |
| 23 | 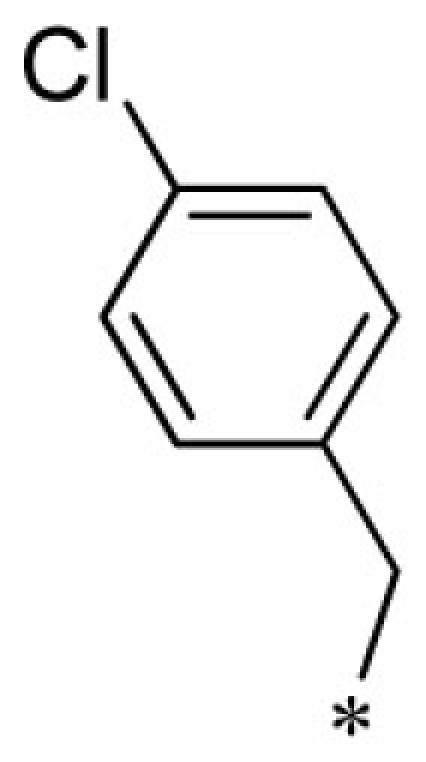 |
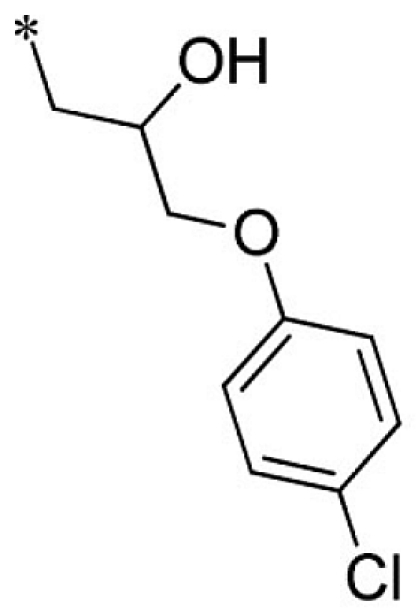 |
VU0403132-1/IC48 | 5.70 ± 3.18 | 19.5 ± 28.2 | 3.35 ± 0.92 | 6.53 ± 3.90 |
| 24 | 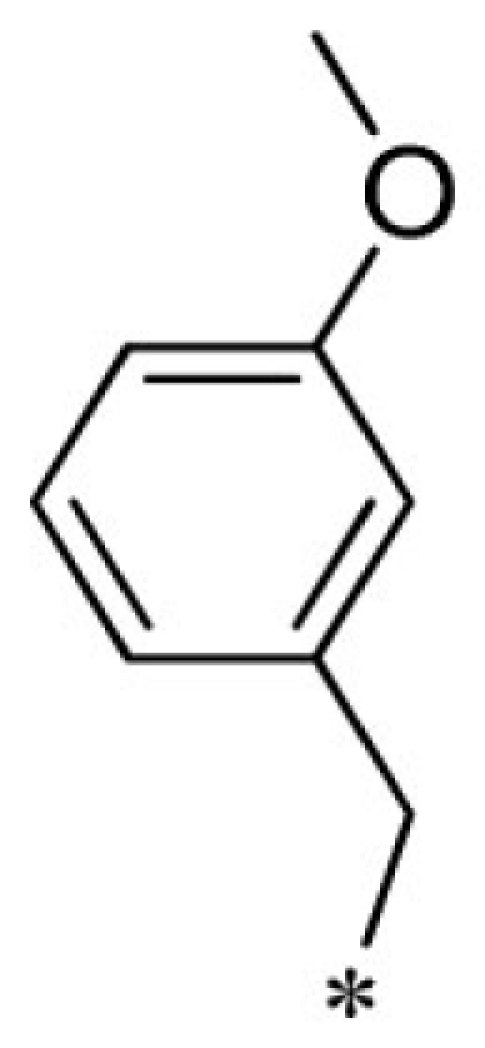 |
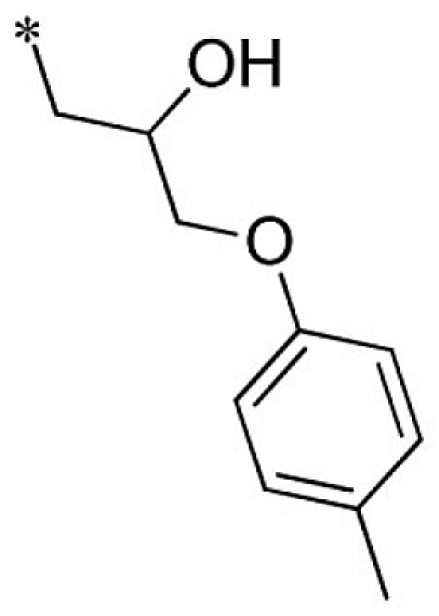 |
VU0451343-1/RJ4 | 2.80 ± 0.89 | 3.00 ± 0.87 | 4.20 ± 0.69 | 13.7 ± 4.4 |
| 25 |  |
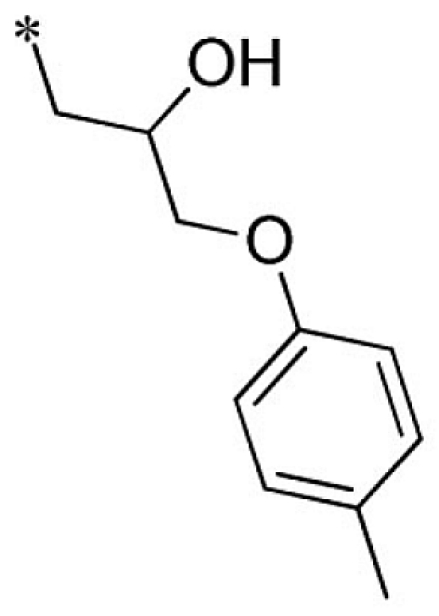 |
VU0451335-1/R5P | 16.5 ± 0.7 | 25.3 ± 8.5 | 12.3 ± 2.5 | 13.0 ± 2.6 |
| 26 |  |
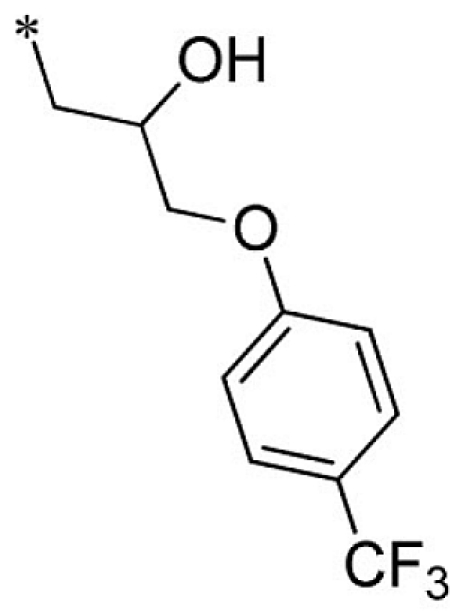 |
VU0451334-1/R7L | 4.90 ± 0.14 | 5.20 ± 1.82 | 2.87 ± 0.31 | 6.97 ± 2.00 |
Summary of structure–activity relationships of VU573 analogs. Reported IC50 values were measured by Tl+ flux assay. Values are mean ± SEM (n = 3).
To determine if the apparent loss of activity is an artifact of the Tl+ flux assay or the use of the M125R mutant, we examined the effects of the VU573 analog R70 on wild type Kir7.1 currents in whole-cell patch clamp experiments. The cells were voltage-clamped between −120 and 120 mV in 20 mV increments from a −75-mV holding potential (Figure 6E). As illustrated in the average current traces recorded before (Figure 6A) or during bath application of 10 μM R70 (Figure 6B), the analog had no effect on wild typeKir7.1 activity. In contrast, subsequent addition of 10 μM VU573 led to a near complete inhibition of the channel (Figures 6C). The mean ± SEM current–voltage relationships recorded in the three conditions are shown in Figure 6D.
Figure 6.

Lack of effect of R70 on wild type Kir7.1 activity. Average Kir7.1 current traces (n = 4 each) recorded in the absence (A) or presence of (B) 10 μM R70 or (C) 10 μM VU573. (D) Mean ± SEM (n = 4) current–voltage Kir7.1 relationships recorded in the indicated conditions using the voltage-clamp protocol shown in (E).
Discussion
The Tl+ flux assay developed by Weaver et al. (2004) and subsequently commercialized by Invitrogen under the FluxOR© label has had a major impact on high-throughput screening for potassium channels. The assay takes advantage of the fact that Tl+ readily permeates most K+ channel pores. With the exception of wild type Kir7.1, the assay seems to be particularly well suited for inward rectifier K+ channels due to their high open probability near the resting membrane potential of a cell. This obviates the need for depolarizing the cell with a high-potassium step to open voltage-gated K+ channels. To date, Tl+ flux assays have been reported for the inward rectifiers Kir1.1 (Lewis et al., 2009), Kir2.1 (Wang et al., 2010), Kir2.3 (this study), Kir3.1/3.2 (Niswender et al., 2008), and Kir7.1-M125R (this study). Tl+ flux assays have also been developed for some ligand- and voltage-gated potassium channels, including SK3 and KCNQ2 (Weaver et al., 2004), KCNQ4 (Li et al., 2011), and hERG (Titus et al., 2009; Bridal et al., 2010; Huang et al., 2010; Schmalhofer et al., 2010; Zou et al., 2010). A Tl+ flux assay was used to screen for modulators of the potassium-chloride co-transporter KCC2 (Delpire et al., 2009). This is notable because KCC2 is electroneutral and therefore cannot be screened using voltage-sensitive dyes. Thus, the Tl+ flux assay appears to be broadly adaptable to both electrogenic and electroneutral K+ transport proteins.
In the present study, Tl+ flux- and electrophysiological-based counterscreens revealed a preferential inhibitor of GIRK, Kir2.3, and Kir7.1. It is unclear why IC50 values are lower in oocytes compared to mammalian cells, but likely reflects differences in membrane properties or intracellular factors between the two cell types. VU573 inhibits GIRK independent of GPCR stimulation and does not discriminate between cardiac (Kir3.1/3.4) and neuronal (Kir3.1/3.2) forms of the channel. The latter observation suggests that the VU573 binding site is located within the Kir3.1 subunit, which is common to both heteromeric forms, or is shared between all three subunits. Pharmacology experiments on homotetrameric Kir3.1, Kir3.2, and Kir3.4 channels should help resolve the issue. Given the broad tissue distribution and important physiological functions of GIRK channels in multiple organ systems, the development of subtype selective modulators may be important for developing, for example, cardiac-specific drugs to treat atrial fibrillation without toxic side effects on the nervous system. The availability of high-resolution crystal structures of the GIRK cytoplasmic domain (Nishida and Mackinnon, 2002), GIRK-bacterial Kir channel chimeras (Nishida et al., 2007) and now full-length mammalian Kir2.2 (Tao et al., 2009) should be helpful in guiding and interpreting experiments to define the molecular binding site for VU573 and other GIRK antagonists. The relatively flat SAR of VU573 against GIRK, Kir2.3, and Kir7.1 suggests that the VU573 scaffold may be amenable to the addition of diazirine or other light-reactive moieties for photoaffinity labeling of the channels for identification of the binding site using mass spectrometry. A detailed understanding of both unique and overlapping binding sites will provide novel insights into the molecular architecture of GIRK and should facilitate the development of subtype-specific inhibitors.
Members of the Kir2.X subfamily, including Kir2.1, Kir2.2, Kir2.3, Kir2.4, and Kir2.6, are broadly expressed in neurons, cardiac, skeletal and smooth muscle cells, endothelial cells, macrophages, and epithelial cells (De Boer et al., 2010a). Much of our understanding of the physiology of Kir2.X channels comes from studies of disease-causing mutations in humans (Plaster et al., 2001; Ryan et al., 2010) and knockout mice (Zaritsky et al., 2001). While it is clear from this work that Kir2.X channels are essential in many organ systems, pinpointing their individual roles has been difficult due to their overlapping expression profiles, propensity to form Kir2.X heterotetramers and poorly developed pharmacology. The development of subtype selective Kir2.X inhibitors would undoubtedly bolster efforts to understand the physiology and druggability of Kir2.X homo- and heteromeric channels. To date, however, the inhibitor pharmacology of Kir2.X channels is limited to barium, cesium, and a handful of non-specific drugs such as chloroquine (Rodriguez-Menchaca et al., 2008), mefloquine (Lopez-Izquierdo et al., 2011b), quinacrine (Lopez-Izquierdo et al., 2011a), carvedilol (Ferrer et al., 2011), tamoxifen (Ponce-Balbuena et al., 2009), and pentamidine (De Boer et al., 2010b). Pregnenolone sulfate (Kobayashi et al., 2009) and flecainide (Caballero et al., 2010) are activators of Kir2.3 and Kir2.1, respectively. Like VU573 (this study), mefloquine, quinacrine, tamoxifen, and carvedilol preferentially inhibit Kir2.3 over Kir2.1. Electrophysiological analysis of mutagenized channels suggests that chloroquine and pentamidine are cytoplasmic pore blockers, whereas mefloquine and carvedilol appear to disrupt channel interactions with the activating membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2). There is evidence that quinacrine has a complex mechanism of action involving both pore block and disruption of channel-PIP2 interactions. Because VU573 is also a preferential inhibitor of Kir2.3, we are currently testing if it shares a common mechanism of action and binding site(s) with these drugs. Looking ahead, it will be important to determine whether VU573 or any of the aforementioned drugs show preference for homomeric versus heteromeric Kir2.X channels. These studies may help inform medicinal chemistry efforts to develop subtype-specific inhibitors of Kir2.X channels.
To our knowledge, VU573 is only the second and most potent Kir7.1 inhibitor available. We recently reported that the ∼300 nM Kir1.1 inhibitor 7,13-bis[(4-nitrophenyl)methyl]-1,4,10-trioxa-7,13-diazacyclopentadecanedihydrochloride, or VU590, inhibits Kir7.1 with an IC50 of approximately 8 μM, making VU590 the first publically disclosed small-molecule inhibitor of both Kir1.1 and Kir7.1 (Lewis et al., 2009). In the present study, we found that VU573 preferentially inhibits Kir7.1 (IC50 ∼ 1 μM) over Kir1.1 (IC50 ∼ 19 μM).
Kir7.1 is widely expressed in the brain, retinal pigment epithelial cells of the eye, the choroid plexus, and epithelial cells of the intestine, nephron, and inner ear (Krapivinsky et al., 1998; Nakamura et al., 1999; Ookata et al., 2000; Pondugula et al., 2006; Yang et al., 2008). Very little is known about the physiology of Kir7.1 due in part to the unusually small (i.e., ∼50 fS) unitary conductance of the channel. This precludes the use of single-channel recording techniques to study its activity in native cell types, where other inward rectifiers of larger unitary conductances are often co-expressed. Whole-cell patch clamp recordings could conceivably be used to dissect out the relative contributions of Kir7.1 and other Kir channels to the macroscopic current provided that specific blockers were available. This scenario highlights the need for better pharmacological modulators of Kir7.1 and other members of the Kir channel family. The discovery of VU573 and its inactive analogs represent important steps toward filling this gap. Furthermore, and importantly, the development of a robust Tl+ flux assay using the Kir7.1-M125R mutant will enable HTS for chemically diverse modulators of the channel.
Interestingly, heritable mutations in the Kir7.1-encoding gene KCNJ13 were recently found in patients with two forms of retinal disease (Hejtmancik et al., 2008; Sergouniotis et al., 2011). These findings confirm the importance of Kir7.1 in retinal pigmented epithelia of the eye and raise questions regarding the detailed physiological roles of Kir7.1 in the eye and other organ systems. The development of potent and selective Kir7.1 antagonists will greatly facilitate those efforts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
HEK-293 cells stably expressing GIRK 1, GIRK 2, and the human M4 muscarinic receptor (HEK/GIRK cells) were generously provided by Drs Huai Hu Chang and Lily Jan (University of California San Francisco, San Francisco, CA, USA). The authors would like to thank the B. H. Robbins Scholars Fellowship Program (Daniel F. Lonergan), NIH 1R01DK082884 (Jerod S. Denton), T32GM07628 (Thuy T. Nguyen), and NIH/MLPCN (5U54MH084659-02) for support of this research. Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
References
- Bhave G., Chauder B. A., Liu W., Dawson E. S., Kadakia R., Nguyen T. T., Lewis L. M., Meiler J., Weaver C. D., Satlin L. M., Lindsley C. W., Denton J. S. (2011). Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol. Pharmacol. 79, 42–50 10.1124/mol.110.066928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G., Lonergan D., Chauder B. A., Denton J. S. (2010). Small-molecule modulators of inward rectifier K+ channels: recent advances and future possibilities. Future Med. Chem. 2, 757–774 10.4155/fmc.10.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridal T. R., Margulis M., Wang X., Donio M., Sorota S. (2010). Comparison of human Ether-a-go-go related gene screening assays based on IonWorks Quattro and thallium flux. Assay Drug Dev. Technol. 8, 755–765 10.1089/adt.2010.0267 [DOI] [PubMed] [Google Scholar]
- Caballero R., Dolz-Gaiton P., Gomez R., Amoros I., Barana A., Gonzalez De La Fuente M., Osuna L., Duarte J., Lopez-Izquierdo A., Moraleda I., Galvez E., Sanchez-Chapula J. A., Tamargo J., Delpon E. (2010). Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc. Natl. Acad. Sci. U.S.A. 107, 15631–15636 10.1073/pnas.1004021107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroti P., Ceccotti C., Da Settimo A., Palla F., Primofiore G. (1986). A facile synthesis of 5,7-dihydro-5-oxopyrido[3′,2′:5,6]pyrimido[1,2-a]benzimidazoles. A new heterocyclic ring system. J. Heterocycl. Chem. 82, 1833–1836 10.1002/jhet.5570230645 [DOI] [Google Scholar]
- Chuang H. H., Jan Y. N., Jan L. Y. (1997). Regulation of IRK3 inward rectifier K+ channel by m1 acetylcholine receptor and intracellular magnesium. Cell 89, 1121–1132 10.1016/S0092-8674(00)80299-8 [DOI] [PubMed] [Google Scholar]
- De Boer T. P., Houtman M. J., Compier M., Van Der Heyden M. A. (2010a). The mammalian K(IR)2.x inward rectifier ion channel family: expression pattern and pathophysiology. Acta Physiol. (Oxf.) 199, 243–256 [DOI] [PubMed] [Google Scholar]
- De Boer T. P., Nalos L., Stary A., Kok B., Houtman M. J., Antoons G., Van Veen T. A., Beekman J. D., De Groot B. L., Opthof T., Rook M. B., Vos M. A., Van Der Heyden M. A. (2010b). The anti-protozoal drug pentamidine blocks KIR2.x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel. Br. J. Pharmacol. 159, 1532–1541 10.1111/j.1476-5381.2010.00658.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E., Days E., Lewis L. M., Mi D., Kim K., Lindsley C. W., Weaver C. D. (2009). Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 106, 5383–5388 10.1073/pnas.0812756106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D., Friedrich A., Voigt N., Jost N., Wettwer E., Christ T., Knaut M., Ravens U. (2005). The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706 10.1161/CIRCULATIONAHA.105.575332 [DOI] [PubMed] [Google Scholar]
- Ehrlich J. R. (2008). Inward rectifier potassium currents as a target for atrial fibrillation therapy. J. Cardiovasc. Pharmacol. 52, 129–135 10.1097/FJC.0b013e31816c4325 [DOI] [PubMed] [Google Scholar]
- Fallen K., Banerjee S., Sheehan J., Addison D., Lewis L. M., Meiler J., Denton J. S. (2009). The Kir channel immunoglobulin domain is essential for Kir1.1 (ROMK) thermodynamic stability, trafficking and gating. Channels (Austin) 3, 57–68 10.4161/chan.3.1.7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer T., Ponce-Balbuena D., Lopez-Izquierdo A., Arechiga-Figueroa I. A., De Boer T. P., Van Der Heyden M. A., Sanchez-Chapula J. A. (2011). Carvedilol inhibits Kir2.3 channels by interference with PIP2-channel interaction. Eur. J. Pharmacol. 668, 72–77 10.1016/j.ejphar.2011.05.067 [DOI] [PubMed] [Google Scholar]
- Hashimoto N., Yamashita T., Tsuruzoe N. (2006). Tertiapin, a selective IKACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol. Res. 54, 136–141 10.1016/j.phrs.2006.03.021 [DOI] [PubMed] [Google Scholar]
- Hebert S. C., Desir G., Giebisch G., Wang W. (2005). Molecular diversity and regulation of renal potassium channels. Physiol. Rev. 85, 319–371 10.1152/physrev.00051.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejtmancik J. F., Jiao X., Li A., Sergeev Y. V., Ding X., Sharma A. K., Chan C. C., Medina I., Edwards A. O. (2008). Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am. J. Hum. Genet. 82, 174–180 10.1016/j.ajhg.2007.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 10.1152/physrev.00021.2009 [DOI] [PubMed] [Google Scholar]
- Ho K., Nichols C. G., Lederer W. J., Lytton J., Vassilev P. M., Kanazirska M. V., Hebert S. C. (1993). Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362, 31–38 10.1038/362031a0 [DOI] [PubMed] [Google Scholar]
- Huang X. P., Mangano T., Hufeisen S., Setola V., Roth B. L. (2010). Identification of human Ether-a-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol. 8, 727–742 10.1089/adt.2010.0331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W., Foo J. N., O’Roak B. J., Zhao H., Larson M. G., Simon D. B., Newton-Cheh C., State M. W., Levy D., Lifton R. P. (2008). Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 40, 592–599 10.1038/ng.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W., Lu Z. (1998). A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 37, 13291–13299 10.1021/bi980031v [DOI] [PubMed] [Google Scholar]
- Kitamura H., Yokoyama M., Akita H., Matsushita K., Kurachi Y., Yamada M. (2000). Tertiapin potently and selectively blocks muscarinic K+ channels in rabbit cardiac myocytes. J. Pharmacol. Exp. Ther. 293, 196–205 [PubMed] [Google Scholar]
- Kobayashi T., Washiyama K., Ikeda K. (2009). Pregnenolone sulfate potentiates the inwardly rectifying K channel Kir2.3. PLoS ONE 4, e6311. 10.1371/journal.pone.0006311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G., Medina I., Eng L., Krapivinsky L., Yang Y., Clapham D. E. (1998). A novel inward rectifier K+ channel with unique pore properties. Neuron 20, 995–1005 10.1016/S0896-6273(00)80480-8 [DOI] [PubMed] [Google Scholar]
- Leister W., Strauss K., Wisnoski D., Zhao Z., Lindsley C. (2003). Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries. J. Comb. Chem. 5, 322–329 10.1021/cc0201041 [DOI] [PubMed] [Google Scholar]
- Lewis L. M., Bhave G., Chauder B. A., Banerjee S., Lornsen K. A., Redha R., Fallen K., Lindsley C. W., Weaver C. D., Denton J. S. (2009). High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol. Pharmacol. 76, 1094–1103 10.1124/mol.109.059840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Rottlander M., Xu M., Christoffersen C. T., Frederiksen K., Wang M. W., Jensen H. S. (2011). Identification of novel KCNQ4 openers by a high-throughput fluorescence-based thallium flux assay. Anal. Biochem. 418, 66–72 10.1016/j.ab.2011.06.040 [DOI] [PubMed] [Google Scholar]
- Lopez-Izquierdo A., Arechiga-Figueroa I. A., Moreno-Galindo E. G., Ponce-Balbuena D., Rodriguez-Martinez M., Ferrer-Villada T., Rodriguez-Menchaca A. A., Van Der Heyden M. A., Sanchez-Chapula J. A. (2011a). Mechanisms for Kir channel inhibition by quinacrine: acute pore block of Kir2.x channels and interference in PIP2 interaction with Kir2.x and Kir6.2 channels. Pflugers Arch. 462, 505–517 10.1007/s00424-011-0995-5 [DOI] [PubMed] [Google Scholar]
- Lopez-Izquierdo A., Ponce-Balbuena D., Moreno-Galindo E. G., Arechiga-Figueroa I. A., Rodriguez-Martinez M., Ferrer T., Rodriguez-Menchaca A. A., Sanchez-Chapula J. A. (2011b). The antimalarial drug mefloquine inhibits cardiac inward rectifier K+ channels: evidence for interference in PIP2-channel interaction. J. Cardiovasc. Pharmacol. 57, 407–415 10.1097/FJC.0b013e31820b7c03 [DOI] [PubMed] [Google Scholar]
- Machida T., Hashimoto N., Kuwahara I., Ogino Y., Matsuura J., Yamamoto W., Itano Y., Zamma A., Matsumoto R., Kamon J., Kobayashi T., Ishiwata N., Yamashita T., Ogura T., Nakaya H. (2011). Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ. Arrhythm. Electrophysiol. 4, 94–102 10.1161/CIRCEP.110.951608 [DOI] [PubMed] [Google Scholar]
- Makary S., Voigt N., Maguy A., Wakili R., Nishida K., Harada M., Dobrev D., Nattel S. (2011). Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ. Res. 109, 1031–1043 10.1161/CIRCRESAHA.111.253120 [DOI] [PubMed] [Google Scholar]
- Matsuda T., Ito M., Ishimaru S., Tsuruoka N., Saito T., Iida-Tanaka N., Hashimoto N., Yamashita T., Tsuruzoe N., Tanaka H., Shigenobu K. (2006). Blockade by NIP-142, an antiarrhythmic agent, of carbachol-induced atrial action potential shortening and GIRK1/4 channel. J. Pharmacol. Sci. 101, 303–310 10.1254/jphs.FP0060324 [DOI] [PubMed] [Google Scholar]
- Nakamura N., Suzuki Y., Sakuta H., Ookata K., Kawahara K., Hirose S. (1999). Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+, K+-ATPase. Biochem. J. 342(Pt 2), 329–336 10.1042/0264-6021:3420329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M., Cadene M., Chait B. T., Mackinnon R. (2007). Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26, 4005–4015 10.1038/sj.emboj.7601828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M., Mackinnon R. (2002). Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111, 957–965 10.1016/S0092-8674(02)01227-8 [DOI] [PubMed] [Google Scholar]
- Niswender C. M., Johnson K. A., Luo Q., Ayala J. E., Kim C., Conn P. J., Weaver C. D. (2008). A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 73, 1213–1224 10.1124/mol.107.041053 [DOI] [PubMed] [Google Scholar]
- Ookata K., Tojo A., Suzuki Y., Nakamura N., Kimura K., Wilcox C. S., Hirose S. (2000). Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J. Am. Soc. Nephrol. 11, 1987–1994 [DOI] [PubMed] [Google Scholar]
- Pasternak A. P., Shahipour A., Tang H., Teumelsan N. H., Yang L., Zhu Y., Walsh S. P. (2010). Inhibitors of the renal outer medullary potassium channel. US patent number: US20100286123.
- Plaster N. M., Tawil R., Tristani-Firouzi M., Canun S., Bendahhou S., Tsunoda A., Donaldson M. R., Iannaccone S. T., Brunt E., Barohn R., Clark J., Deymeer F., George A. L., Jr., Fish F. A., Hahn A., Nitu A., Ozdemir C., Serdaroglu P., Subramony S. H., Wolfe G., Fu Y. H., Ptacek L. J. (2001). Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105, 511–519 10.1016/S0092-8674(01)00342-7 [DOI] [PubMed] [Google Scholar]
- Ponce-Balbuena D., Lopez-Izquierdo A., Ferrer T., Rodriguez-Menchaca A. A., Arechiga-Figueroa I. A., Sanchez-Chapula J. A. (2009). Tamoxifen inhibits inward rectifier K+ 2.x family of inward rectifier channels by interfering with phosphatidylinositol 4,5-bisphosphate-channel interactions. J. Pharmacol. Exp. Ther. 331, 563–573 10.1124/jpet.109.156075 [DOI] [PubMed] [Google Scholar]
- Pondugula S. R., Raveendran N. N., Ergonul Z., Deng Y., Chen J., Sanneman J. D., Palmer L. G., Marcus D. C. (2006). Glucocorticoid regulation of genes in the amiloride-sensitive sodium transport pathway by semicircular canal duct epithelium of neonatal rat. Physiol. Genomics 24, 114–123 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Menchaca A. A., Navarro-Polanco R. A., Ferrer-Villada T., Rupp J., Sachse F. B., Tristani-Firouzi M., Sanchez-Chapula J. A. (2008). The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. U.S.A. 105, 1364–1368 10.1073/pnas.0708153105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D. P., Da Silva M. R., Soong T. W., Fontaine B., Donaldson M. R., Kung A. W., Jongjaroenprasert W., Liang M. C., Khoo D. H., Cheah J. S., Ho S. C., Bernstein H. S., Maciel R. M., Brown R. H., Jr., Ptacek L. J. (2010). Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 140, 88–98 10.1016/j.cell.2009.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalhofer W. A., Swensen A. M., Thomas B. S., Felix J. P., Haedo R. J., Solly K., Kiss L., Kaczorowski G. J., Garcia M. L. (2010). A pharmacologically validated, high-capacity, functional thallium flux assay for the human Ether-a-go-go related gene potassium channel. Assay Drug Dev. Technol. 8, 714–726 10.1089/adt.2010.0351 [DOI] [PubMed] [Google Scholar]
- Sergouniotis P. I., Davidson A. E., Mackay D. S., Li Z., Yang X., Plagnol V., Moore A. T., Webster A. R. (2011). Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 89, 183–190 10.1016/j.ajhg.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D. B., Karet F. E., Rodriguez-Soriano J., Hamdan J. H., Dipietro A., Trachtman H., Sanjad S. A., Lifton R. P. (1996). Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 14, 152–156 10.1038/ng1096-152 [DOI] [PubMed] [Google Scholar]
- Tanaka H., Hashimoto N. (2007). A multiple ion channel blocker, NIP-142, for the treatment of atrial fibrillation. Cardiovasc. Drug Rev. 25, 342–356 10.1111/j.1527-3466.2007.00025.x [DOI] [PubMed] [Google Scholar]
- Tao X., Avalos J. L., Chen J., Mackinnon R. (2009). Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326, 1668–1674 10.1126/science.1180310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus S. A., Beacham D., Shahane S. A., Southall N., Xia M., Huang R., Hooten E., Zhao Y., Shou L., Austin C. P., Zheng W. (2009). A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel. Anal. Biochem. 394, 30–38 10.1016/j.ab.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt N., Friedrich A., Bock M., Wettwer E., Christ T., Knaut M., Strasser R. H., Ravens U., Dobrev D. (2007). Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK,ACh channels in patients with chronic atrial fibrillation. Cardiovasc. Res. 74, 426–437 10.1016/j.cardiores.2007.02.009 [DOI] [PubMed] [Google Scholar]
- Wakili R., Voigt N., Kaab S., Dobrev D., Nattel S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968 10.1172/JCI46315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K. B. (2010). A real-time screening assay for GIRK1/4 channel blockers. J. Biomol. Screen 15, 1229–1237 10.1177/1087057110381384 [DOI] [PubMed] [Google Scholar]
- Wang H. R., Wu M., Yu H., Long S., Stevens A., Engers D. W., Sackin H., Daniels J. S., Dawson E. S., Hopkins C. R., Lindsley C. W., Li M., Mcmanus O. B. (2010). Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem. Biol. 6, 845–856 10.1021/cb200146a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver C. D., Harden D., Dworetzky S. I., Robertson B., Knox R. J. (2004). A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J. Biomol. Screen 9, 671–677 10.1177/1087057104268749 [DOI] [PubMed] [Google Scholar]
- Welling P. A., Ho K. (2009). A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am. J. Physiol. Renal Physiol. 297, F849–F863 10.1152/ajprenal.00181.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Zhang X., Hughes B. A. (2008). Expression of inwardly rectifying potassium channel subunits in native human retinal pigment epithelium. Exp. Eye Res. 87, 176–183 10.1016/j.exer.2008.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaritsky J. J., Redell J. B., Tempel B. L., Schwarz T. L. (2001). The consequences of disrupting cardiac inwardly rectifying K+ current (IK1) as revealed by the targeted deletion of the murine Kir2.1 and Kir2.2 genes. J. Physiol. 533, 697–710 10.1111/j.1469-7793.2001.t01-1-00697.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Tate S. S., Palmer L. G. (1994). Primary structure and functional properties of an epithelial K channel. Am. J. Physiol. 266, C809–C824 [DOI] [PubMed] [Google Scholar]
- Zou B., Yu H., Babcock J. J., Chanda P., Bader J. S., Mcmanus O. B., Li M. (2010). Profiling diverse compounds by flux- and electrophysiology-based primary screens for inhibition of human Ether-a-go-go related gene potassium channels. Assay Drug Dev. Technol. 8, 743–754 10.1089/adt.2010.0339 [DOI] [PMC free article] [PubMed] [Google Scholar]