

Abstract
Cryo-electron tomography (cryo-ET) is an emerging 3-D reconstruction technology that combines the principles of tomographic 3-D reconstruction with the unmatched structural preservation of biological material embedded in vitreous ice. Cryo-ET is particularly suited to investigating cell-biological samples and large macromolecular structures that are too polymorphic to be reconstructed by classical averaging-based 3-D reconstruction procedures. This unit aims to make cryo-ET accessible to newcomers and discusses the specialized equipment required, as well as the relevant advantages and hurdles associated with sample preparation by vitrification and cryo-ET. Protocols describe specimen preparation, data recording and 3-D data reconstruction for cryo-ET, with a special focus on macromolecular complexes. A step-by-step procedure for specimen vitrification by plunge freezing is provided, followed by the general practicalities of tilt-series acquisition for cryo-ET, including advice on how to select an area appropriate for acquiring a tilt series. A brief introduction to the underlying computational reconstruction principles applied in tomography is described, along with instructions for reconstructing a tomogram from cryo-tilt series data. Finally, a method is detailed for extracting small subvolumes containing identical macromolecular structures from tomograms for alignment and averaging as a means to increase the signal-to-noise ratio and eliminate missing wedge effects inherent in tomographic reconstructions.
Keywords: cryo-electron microscopy (cryo-EM), cryo-electron tomography (cryo-ET), Vitrification of macromolecules, tilt-series acquisition, sub-volume averaging
Cryo-electron Tomography for Structural Characterization of Macromolecular Complexes
Cryo-electron tomography (cryo-ET) is quickly becoming an accessible technique for investigating the structure of isolated macromolecular complexes, as well as studying the 3D structure and organization of cells. The advent of user-friendly technology, such as automated plunge freezers, cryo-specific specimen holders, automated tilt-series acquisition programs, and easy-to-follow software for tomogram reconstruction, has made cryo-ET available to nearly anyone with a primary interest in studying biological structure and function. For cryo-ET of macromolecular complexes, specimens are immobilized in a hydrated state by rapid freezing in a thin layer of vitreous ice. In the electron microscope, the sample is kept cold enough that ice crystals do not form and is tilted around an axis approximately from -60° to +60° while 2D projection images are collected typically every 1.5 degrees over this tilt range. The resulting tilt series, a stack of around 80 images, is then used to reconstruct the sample in 3D employing a mathematical algorithm. Macromolecular complexes can be analyzed directly in tomograms or studied further at higher resolution by averaging multiple copies of identical structures extracted from tomograms.
This unit describes the procedures for specimen preparation, data recording and 3D data reconstruction for cryo-ET as well as subsequent subvolume averaging and other analytical procedures. We begin with detailing the steps for plunge freezing, assuming that you already have a purified sample of the macromolecular complex, or preparation of small cells you wish to study (see Basic Protocol 1 and Alternate Protocol 1). Alternate Protocol 2 outlines the methods to prepare samples of large cells for cryo-ET by high pressure freezing and cryo-sectioning, though these techniques are not described here in any detail. The steps and considerations for transferring the sample into a cryo-holder and then into the electron microscope are described in Basic Protocol 2. This is followed by instructions for acquiring a tilt series, including advice on how to select an area appropriate for tilt-series acquisition, and how different imaging techniques may affect the final resolution of your data (see Basic Protocol 3). Reconstructing a tomogram from tilt series data is described in Basic Protocol 4. If your tomogram has multiple copies of a particular structure, small volumes containing the repeating structure can be selected as particles for alignment and averaging, as detailed in Basic Protocol 5. If the selected particles have an axis of symmetry, instructions for using symmetrization in combination with subvolume averaging are provided (see Supporting Protocol 1). A brief description on how to estimate the resolution of subvolume averages is given in Supporting Protocol 2.
Basic Protocol 1
PLUNGE FREEZING ISOLATED MACROMOLECULES USING A HOMEMADE PLUNGE FREEZER
For visualization in the electron microscope by cryo-ET, macromolecular complexes are embedded in vitreous ice that is generated by rapid plunge freezing of a very thin sample. The goal here is to freeze samples rapidly enough that water forms an amorphous solid called vitreous ice. This is important because ice crystals form quickly with slower freezing and severely damage the sample. This so-called “vitrification” preserves the specimen in a state that is very close to its native, hydrated state. The steps involved include: placing a droplet of your sample onto an electron microscopy grid, → blotting away excess fluid, → plunging the grid into a cryogenic liquid within seconds of blotting, and → transferring the frozen grid into a grid box for storage in liquid nitrogen until use.
For cryo-ET, grids coated with a holey carbon film are commonly used. This allows macromolecules to be imaged in the vitreous ice that covers the hole, avoiding the added noise that comes from a support film. An optimal sample concentration must be determined experimentally for each particular specimen, but is generally in the range of 0.1 μM - 4.0 μM.
An important step in preparing samples for cryo-ET is the addition of gold particles to the specimen. Gold particles are used to align the images in the tilt series for reconstruction of the tomogram as described in Basic Protocol 4. To facilitate good alignment, an even distribution of gold particles in the sample is highly desirable. Both 5 nm and 10 nm colloidal gold particles are suitable. However, if the specimen is not obscured by 10nm gold, these particles should be used as they are more visible in images taken at high tilts.
Most macromolecular complexes are sufficiently stable to be pre-formed in any way that is convenient. Where timing is critical, or aggregation must be prevented, multiple samples can be applied directly to a grid for freezing, with or without blotting in between. For example, decoration of microtubules by kinesin motor domains can be completed in a microfuge tube, then the motor-decorated microtubules adsorbed to grids for plunge freezing. In the case of kinesins known to cause microtubule bundling, which is not ideal for imaging, 5 μl of microtubules can be allowed to adsorb to the grid for 45 s, excess fluid blotted away, and then 5 μl of kinesin motor domains added to incubate with the pre-adsorbed microtubules on the grid.
Once excess fluid has been blotted away and the sample is left in a thin film ready for plunge freezing, ambient temperature and humidity are critical in specimen quality as samples can become dehydrated and cooled by evaporation. If the sample can tolerate low temperatures, plunge freezing in a walk-in 4°C cold room can be considered, as the humidity there is usually near saturation. However, while high humidity is desired before plunging, it is deleterious right after plunging, as it increases the build-up of frost in vessels containing liquid nitrogen. Frost contaminates the frozen-hydrated sample, so its accumulation must be avoided. It is often better to freeze samples in batches of only 8-12 grids, then defrost the freezer and allow it to dry completely before re-cooling it for another round of freezing.
Plunge frozen samples can be stored for months in liquid nitrogen. However, ice contamination will accumulate on the grids over time, which can interfere with image contrast and make tilt series acquisition more difficult.
Materials
Liquid nitrogen
Gaseous ethane or ethane/propane mixture with tubing and a pipette tip
Plunge freezing device (Figure 1)
Eye protection
Surgical facemask or face shield
Purified protein/macromolecule sample or thin cells
Suitable dilution buffer (if needed)
Glow discharger/plasma cleaner
Holey-carbon grids, 200 mesh (for example, C-flat™ or Quantifoil®)
Forceps
10 nm or 5 nm colloidal gold
Filter paper cut into strips (Whatman #1 or similar)
Grid storage boxes
Styrofoam box
50 ml conical tube
Figure 1.

(A) Setup of a homemade plunge-freezer. This basic freezing device that can easily be made by a local machine shop, has a height-adjustable metal plunger to which the forceps holding the grid can be attached and removed with a simple clip mechanism. Immediately after blotting excess fluid from the grid, the plunger is released by pushing the plunge controller. This injects the grid into a cup of ethane slush (magnified in B) that is kept cool by the liquid nitrogen in the outer foil-lined styrofoam box.
Set up the plunge freezer
Fill the outer container of the plunge freezer with liquid nitrogen (Figure 1).
Fill the inner container of the plunge freezer with ethane by blowing ethane gas through a hose with a pipette tip affixed to the end, taking care to touch the pipette tip to the bottom wall of the container. The ethane will condense on the cooled inner wall of the container and become liquid (Figure 1).
Ensure that you fill the container so that the grid will be completely submerged when plunged into the liquid ethane.
Liquid nitrogen and ethane can be dangerous and must be handled carefully. Work in a well-ventilated area and wear goggles to protect your eyes. We also recommend wearing a face shield to prevent formation of ice crystals from your breath.
Prepare sample for freezing
3. Plan whether the components of your specimen need to be incubated together in a separate tube or on the grid and calculate dilutions accordingly.
4. Dilute each component of the sample to the appropriate concentration.
Avoid buffers containing high concentrations of salt, sucrose, glycerol or DMSO because they can ruin the image contrast and promote beam damage. If these compounds have been added to specimens for storage or as cryo-protectant, dilute or dialyze them out as much as possible prior to plunge freezing.
Plunge freeze the specimen
5. Glow discharge the carbon film of the grids using a glow discharger or plasma cleaner. This makes the carbon film more hydrophilic and provides a more even adsorption of the specimen on to the grid (Aebi and Pollard, 1987).
It is recommended that grids be glow discharged as close as possible to the time of applying the sample and used within half an hour. If not used in that time, they can be glow-discharged again.
6. Mount the grid into the forceps you will be using for plunge freezing, with the carbon-coated side of the grid (the darker, shiny side) facing up.
Make sure to grip only the very edge of the grid with the forceps. The metal forceps cool slower than the sample in liquid ethane and the quality of freezing may be compromised if you grip too low down on the grid. Do not make any dents or bend the grid.
7. Apply 3-5 μl of the sample to a glow-discharged grid and allow it to adsorb for 10 s - 2 min.
8. Apply 1 μl of concentrated 10 nm colloidal gold (see Reagents and Solutions) and incubate for a further 30 s - 1 min.
1 μl of concentrated colloidal gold can also instead be mixed directly with the sample before applying it to the grid.
9. Mount the forceps with the sample onto the plunge freezing device.
This can also be done before step 7 if you prefer to apply the sample to the grid once it is already set in the freezing device.
10. Ensure the liquid ethane is at the correct temperature and consistency. If it is frozen, defrost the solid ethane by bubbling in ethane gas until it becomes slush. The ideal temperature for plunge freezing is when the walls of the ethane cup are coated by a layer of solid ethane while the center of the cup remains liquid.
Be aware that thawing frozen ethane can take more than two minutes and if the sample has already been applied to the grid, you run the risk of it drying out or time-sensitive reactions incubating on the grid for too long. It is helpful to defrost the ethane completely first and then wait for it to cool along the sides of the ethane cup while you apply your sample to the grid.
11. Using a strip of filter paper, blot away excess protein solution from the grid. Hold the filter paper flat against the front surface of the grid. Capillary forces will pull liquid away from the grid until the filter paper spontaneously releases. To ensure the sample is embedded in a thin enough layer of vitreous ice, it is important to wait for this spontaneous release, and not to pull the filter paper away beforehand.
12. Plunge the grid into the ethane slush immediately after blotting.
From this point on, everything that comes into contact with the grid must be at liquid nitrogen temperature and any exposure of the grid to humid air must be kept to an absolute minimum.
13. Transfer the frozen grid from the ethane slush into a grid storage box sitting in liquid nitrogen. Pull the grid slowly up out of the ethane so that excess ethane is drawn away by surface tension. Once the grid is three quarters out of the ethane, transfer it rapidly into the liquid nitrogen.
If there is a lot of ethane on the grid upon transfer into the liquid nitrogen, it can be wicked away with a piece of pre-cooled filter paper.
We keep our grids in grid boxes with slots for 4 grids and a screw top lid (Figure 2B). These are then stored in a 50ml conical tube in a liquid nitrogen Dewar. Drill two holes near the top of the conical tube to allow liquid nitrogen to flow in and put a small weight in the bottom to keep the conical tube low in the storage tank.
Figure 2.

(A) A cryo-holder seated in a transfer station. When filled with liquid nitrogen, the transfer station enables transfer of the grid from storage in a grid box onto the tip of the cryo-holder (area in the red box magnified in the image below it). A shutter is placed over the grid in the cryo-holder to protect it from contamination. The tip of the cryo-holder is cooled by liquid nitrogen from the Dewar, which keeps the specimen vitreous during insertion into and imaging in the electron microscope. (B-E) Common tools used for loading a sample into a cryo-holder. (B) Grids are stored in liquid nitrogen in a grid box (green) with slots for 4 grids that is kept closed with a screw-top lid (white). (C) A ladle used for transferring grid boxes from a liquid-nitrogen filled container into the transfer station so specimens remain submerged in liquid nitrogen throughout the transfer. (D) A tool that fits onto the grid box lid to open/close the grid box. (E) Forceps used to transfer the grid out of the grid box and into the tip of the cryo-holder. Ensure the tips of the forceps are straight to hold the grid securely and to prevent mechanical damage to the grid.
14. Image frozen grids immediately in the electron microscope, or store in liquid nitrogen.
If you wish to image your grids immediately after freezing, excess ethane will evaporate when the sample is inserted into the microscope, but this can crash the vacuum and also cause your sample to drift if the clip ring is clamped down over frozen ethane that evaporates upon insertion of the sample. To avoid this, grids can be stored in liquid nitrogen at least overnight before imaging so that excess ethane can dissolve away.
Alternate Protocol 1
PLUNGE FREEZING ISOLATED MACROMOLECULES USING A COMMERCIAL PLUNGE FREEZER
While homemade plunge freezing devices work very well for the majority of applications, results can vary based on differences in the ambient humidity and temperature, timing between the necessary steps and the competency of the user. If reproducibility is a concern, commercial automated plunge freezers, such as the Vitrobot™ by the FEI Company (Eindhoven, The Netherlands), the Leica EM GP (Wetzlar, Germany), and the Cryoplunge™3 from GATAN, Inc. (Pleasanton, CA) are available where all plunge freezing parameters can be precisely controlled.
The steps for plunge freezing using a commercial device are essentially identical to those described in Basic Protocol 1. The differences are that in an automated freezer, the specimen is kept in a chamber at a constant user-selected temperature and humidity between blotting and plunging, the force and time of blotting can be specified, and the time between blotting and plunging can be set. These parameters must be optimized for each sample. A typical setup for preparing isolated macromolecules such as kinesin-decorated microtubules and liposomes, is shown in Table 1. A thorough protocol for plunge freezing samples using the Vitrobot™ is described by Iancu et al., 2006.
Table 1.
Typical settings for preparing various macromolecular complexes using a Vitrobot™
| Intermediate Filaments | Kinesin-Decorated Microtubules | Membrane Proteins/Liposomes | |
|---|---|---|---|
| Cryogen | Ethane/propane mix | Ethane | Ethane/propane mix |
|
| |||
| Chamber Temperature | 22°C | 22°C | 22°C |
|
| |||
| Chamber Humidity | 85% | 90% | 70% |
|
| |||
| Blot Total | 1 | 1, 1 | 1 |
|
| |||
| Blot Force | 0 | 0, 0 | 1 |
|
| |||
| Blot Time (s) | 2 | 1.5, 3 | 3 |
|
| |||
| Drain Time (s) | 0 | 0, 0 | 0 |
|
| |||
| Wait Time (s) | 3-10 | 35, 60 | Manual |
|
| |||
| Special Considerations | •Apply 2 μl of sample to each side of grid | •Apply 5 μl of microtubules first, blot using 1st set of values listed. Then add 5 μl of motors and blot using 2nd set of values listed. | •Apply 2 μl of sample to each side of grid. |
| •No glow discharge prior to sample application. | |||
Alternate Protocol 2
HIGH PRESSURE FREEZING AND CRYO-SECTIONING FOR CRYO-ELECTRON TOMOGRAPHY
Cryo-ET is the method of choice to examine macromolecules in their native cellular environment, and if a complex exists in multiple copies, it can be investigated at higher resolution using subvolume averaging without the need to isolate it from the cell. As a method for sample preparation, however, plunge freezing is limited to small specimens such as isolated macromolecules, virus particles, and very small cells, all thinner than ~500 nm. Larger cells and chunks of tissue cannot be vitrified reliably by plunge freezing; their reliable cryo-immobilization requires high pressure freezing. A high pressure freezer shifts the freezing point of water by applying a high pressure (about 2000 bars) to the sample immediately before freezing, reducing nucleation and growth of ice crystals enough to vitrify samples up to 600 μm thick. Specimens prepared by high pressure freezing can then be transferred to a pre-cooled cryo-ultramicrotome chamber where they are cut into thin slices using a diamond knife by a process called cryo-sectioning. Ribbons of cryo-sections are then carefully transferred and pressed onto an electron microscope grid for storage in liquid nitrogen or direct imaging in the electron microscope.
High pressure freezing and subsequent cryo-sectioning are sophisticated procedures that are beyond the scope of this section, but are described in detail elsewhere. (Dubochet et al., 2007, Bouchet-Marquis and Fakan, 2009, Ladinsky, 2010, Bouchet-Marquis and Hoenger, 2011). The protocols explained in the remainder of this unit are applicable to both plunge frozen samples and cryo-sections unless specified otherwise.
Basic Protocol 2
SETTING UP THE MICROSCOPE FOR CRYO-ELECTRON TOMOGRAPHY
This section describes the steps necessary to transfer the frozen-hydrated sample from storage in liquid nitrogen into a cryo-holder and subsequently into the electron microscope. The two most important aspects of this process are to ensure that the grid remains at liquid nitrogen temperatures throughout, and that anything coming into contact with the grid must be pre-cooled by placing it into liquid nitrogen and waiting for the bubbling to stop. Transfers from storage into the cryo-holder, and insertion of the cryo-holder into the microscope should be done rapidly and if possible in a low-humidity environment. These steps may require some “dry-training” by learning how to use the equipment before trying it with an important specimen. It is recommended that you wear goggles to protect your eyes from spraying liquid nitrogen and a face shield to prevent ice crystal formation from your breath vapor. Use clean liquid nitrogen for all steps to reduce ice crystal contamination on your grid, and make sure that all tools are completely clean and dry before pre-cooling them in liquid nitrogen.
Materials
Styrofoam box
Goggles/eye protection
Surgical facemask or face shield
Liquid nitrogen
Forceps
Funnel with wire mesh filter
Cryo-holder (Figure 2A)
Transfer station (Figure 2A)
Ladle (Figure 2C)
Grid box lid opener (Figure 2D)
Electron microscope with cryo-capabilities (e.g. Tecnai F20 by the FEI Company)
Load a grid into the cryo-holder
Ensure that the cryo-holder has been pumped down on a vacuum station for at least an hour (but preferably overnight) and is completely dry and at room temperature before use.
Any water left on the holder’s tip or the shutter mechanism will freeze and may disrupt proper function of the holder.
2. Close the valve of the cryo-holder, vent the vacuum and remove the holder from the pumping station.
3. Check the o-ring on the rod of the cryo-holder under a dissecting microscope to make sure it is clean, free of dust and fibers, and not cracked.
4. Place the cryo-holder into the transfer station (Figure 2A).
5. Add clean liquid nitrogen to the Dewar of the cryo-holder and to the reservoir of the transfer station that holds the tip of the rod (Figure 2A).
Pour the liquid nitrogen through a small funnel with a wire mesh at the bottom to filter out any large ice crystals or other contaminating particles.
6. Wait until the nitrogen has finished bubbling and then fill up the Dewar of the cryo-holder with liquid nitrogen and fill up the reservoir to just slightly above the level of the cryo-holder tip.
7. Retrieve samples from storage in liquid nitrogen, and transfer them rapidly into a styrofoam container filled with liquid nitrogen.
8. Transfer the grid box containing the grid to be imaged from the styrofoam box into the reservoir of the transfer station using a pre-cooled “ladle” (Figure 2C) so the grid box is immersed in liquid nitrogen during the transfer.
9. Unscrew the lid of the grid box, keeping the lid under liquid nitrogen long enough to note any grids that may be stuck to the lid. Remove any grids stuck to the lid carefully with a pair of forceps and place them back into the grid box. Then set the grid box lid aside to stay cool in the liquid nitrogen in the styrofoam box.
10. Remove the clip ring from the tip of the cryo-holder, or open the slot that holds the grid by the mechanism specific to your cryo-holder.
11. Make sure to remove the grid that may still be in the cryo-holder from a previous user.
12. If necessary, top up the liquid nitrogen in the reservoir until the tip of the rod is covered.
13. Use pre-cooled forceps to carefully grab the edge of your grid and quickly transfer it through nitrogen vapor into the grid-slot at the tip of the cryo-holder.
14. Center the grid and then clamp it in place by replacing the clip ring or using the mechanism specific to your cryo-holder.
Make sure that the grid is firmly in place otherwise it will drift while imaging, making data collection extremely difficult.
15. Close the shutter on the cryo-holder and top up the transfer station reservoir with liquid nitrogen if necessary so that the tip of the cryo-holder is covered.
Transfer the cryo-holder to the electron microscope
16. Prepare the microscope for inserting the cryo-holder. This step will be specific to your microscope, but may include cooling the anti-contamination braids with liquid nitrogen, removing a room temperature holder that might be inserted to prevent dust from entering the goniometer, and prepumping the airlock.
The anti-contamination braids should be cooled for a minimum of 20 minutes prior to inserting the specimen.
17. Make sure that the anti-contamination blades on your microscope (if present) are inserted and centered over the specimen to avoid buildup of contamination on your sample. If necessary, test the holder settings in a dry run to make sure the holder tip does not touch the blades at high tilts.
18. Remove the cryo-holder from the transfer station and rapidly insert it into the microscope. If your holder is not of the free-hanging type, avoid smacking the crystal tip during insertion as they can break easily when cooled.
19. If you need to wait for the airlock to finish its pump time, then wait until it is complete before inserting the holder all the way into the column.
Airlock time should be set between 30s-60s.
20. If you had to pour out the liquid nitrogen from the Dewar of the cryo-holder during insertion, refill it immediately once the holder is fully inserted.
Fill the Dewar of the cryo-holder only up to, or just above the level of the rod (look inside the Dewar to see where the rod is). If the Dewar is filled completely, bubbling of liquid nitrogen in the neck of the Dewar can cause drift while imaging.
21. Wait for the column vacuum to come down and stabilize.
22. Open the shutter covering the grid on the cryo-holder. If the vacuum spikes, wait for it to stabilize before opening the column valves that protect the electron source.
Basic Protocol 3
TILT SERIES DATA COLLECTION
To collect a tilt series, the sample is tilted from approximately -60° to +60° (higher when possible) and a series of images of the area of interest is recorded according to a user-defined tilt increment. The images from this tilt series are then used to reconstruct the tomogram as discussed in Basic Protocol 4. For tilt series acquisition, the goal is to have every image in the series centered on the area of interest (the record area) and recorded at the same focus. When the sample is tilted, it changes its position both laterally and vertically, which necessitates corrections of position and focus. Drift in the sample also contributes to the position corrections necessary to keep the area of interest centered. Thus, the basic sequence of operations required is: tilting the sample to a new angle, → correcting the specimen’s position, → correcting the specimen’s focus, and → acquiring an image of the area of interest. Apart from SerialEM (Mastronarde, 2005), which is described in this section, there are several other tilt-series acquisition programs available to achieve the same basic steps. Examples include UCSF Tomo (Zheng et al., 2007), TOM Toolbox (Nickell et al., 2005) and Xplore3D (FEI Company, Eindhoven, The Netherlands).
The biggest problem to overcome in cryo-ET is the large number of images that must be taken of the particular sample under investigation. Frozen-hydrated biological material is very sensitive to the electron beam and deteriorates gradually upon increasing exposure to the beam. Hence, strict attention must be paid to the amount of dose applied to the sample. As a rule of thumb for cryo-ET, the volume to be reconstructed should not receive than a total dose of ≥100 e-/Å2 for the entire tilt series plus any other imaging (records, previews and views) combined. However, this is a rough guideline and some samples require a lot less electron exposure, especially when attempting to reconstruct high-resolution data. When working with a new specimen, it is useful to perform a dose-series by taking a set of low-dose images to determine the maximum dose that can be applied to a sample before it becomes too damaged. If the specimen has an internal order that creates a characteristic diffraction pattern with bright spots or layer lines according to the repetitive structure (such as helices or 2D crystalline arrays), beam damage can be assessed by the gradual disappearance of diffraction peaks at successively lower frequencies (i.e. the disappearance of progressively coarser structural detail). When bubbling occurs on the recording area the dose is at least a factor of two too high.
The extreme sensitivity of cryo-samples means that data collection for cryo-tomography differs greatly from that of plastic section tomography, particularly in the following ways:
Tracking and focusing must be done on an area separate from the record area (Figure 3).
The number of images and electron dose per image of each projection recorded in the tilt series.
The defocus at which the data are collected.
Figure 3.
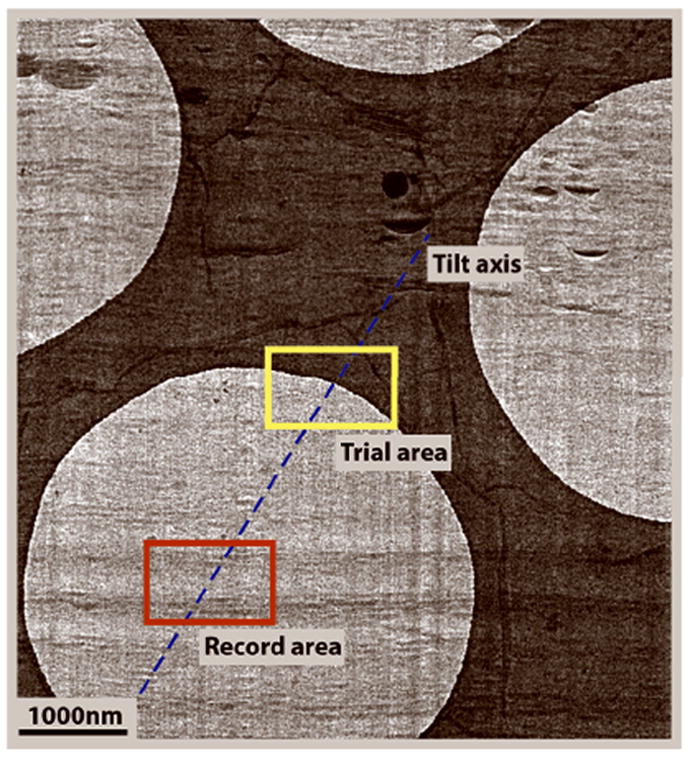
A cryo-section on a holey carbon grid showing the tracking area 2 μm away along the tilt axis from the record area, using the edge of the carbon hole as a high-contrast feature for cross-correlation. Image courtesy of Cédric Bouchet-Marquis, Univ. of Colorado at Boulder.
Each of these points is discussed below in more detail. Importantly, points ii) and iii) can affect the resolution of the data and should be considered carefully, keeping the ultimate goals of your research in mind.
-
Tracking:
The purpose of having a separate tracking area is to be able to focus and align the area of interest throughout the tilt series without exposing it to the additional dose required for these processes. The tracking area should be set about 1.5 - 2.5 μm away from the recording area and the beam must be adjusted so its application to the tracking area does not irradiate the area of interest or any other area from which you may want to collect data in the future. It is imperative that the tracking area be precisely aligned with the tilt axis to maintain accurate focus settings at the tilted positions. Most tilt series acquisition software allows for the tracking area to be selected on either side of the recording area along the tilt axis, so only two general tracking areas are possible. The tracking area must contain features that have enough contrast for cross-correlating, such as the edge of a carbon hole or a distinctive arrangement of biological material (Figure 3). Large chunks of ice contamination in the tracking area should be avoided as they may be at a different height (focus level) than the sample and may shift or disappear during the tilt series acquisition. Always track and focus at the same magnification at which you record, except if tracking becomes unreliable at high magnification where the image area is less than ~1 μm, then the magnification for tracking may be lowered to maintain a large enough field of view.
-
Image number and electron dose:
Tilt series acquisition requires a compromise between the number of images recorded (as defined by the chosen tilt-angle increment and tilt range), and the dose applied per image. Using a smaller tilt-increment provides a finer sampling of Fourier space and should therefore yield a better resolution. However, a smaller tilt increment implies recording more images so to stay below the tolerable dose limit, the dose per image must be decreased. A low dose per image (< 1 e-/Å2) will result “noisy” images (inadequate electron dose per pixel, so the counting statistics in the digital representation of the specimen provide a poor representation of the sample’s distribution of electron scattering). This situation may make the images in the tilt series impossible to align. For most cryo-applications, a 1.5° tilt increment with a 1 e-/Å2 dose per image is suitable. More complex schemes for acquiring tilt series have been developed to compensate for the increased thickness of the sample at high tilts. At 60° tilt, the beam has to penetrate twice as thick a sample as it does at zero tilt. Some acquisition programs allow the user to distribute the total dose in a manner such that a higher dose is given to images at high tilts. For example, by varying the dose by 1/cosine of the tilt angle, you can apply a dose of 1 e-/Å2 at zero tilt that increases proportionally with the tilt angle to a dose of 2 e-/Å2 at 60°.
The “Saxton Scheme” has been developed to distribute the tilt angles such that a smaller tilt increment is used at higher tilts to acquire more images where the sample is the thickest (Saxton et al., 1984). However, for amorphous structures we have found that varying the dose, instead of the tilt angle increment is more effective for achieving a better reconstruction. Sophisticated acquisition schemes might be beneficial, but be aware of the increased dose that may be applied to your sample and adjust the tilt series settings accordingly. Experimenting with several protocols will let you find the one that works best for your samples.
-
Defocus:
The advantage of cryo-ET is that samples are viewed in their native, hydrated state, without incorporation of the electron-dense stains sometimes used to generate amplitude contrast. The disadvantage is that unstained biological material has only about 1.2 times the electron density of vitreous ice so when the specimen is in focus, contrast is poor and detail is difficult to see. For cryo-electron microscopy (cryo-EM) in general, the sample may be imaged slightly below-focus to generate phase-contrast. This defocus makes biological material easier to visualize, but it comes at the cost of unfavorable phase modifications at higher frequencies and some loss of data due to the electron microscope’s contrast transfer function (CTF). The CTF is a complex function of defocus value, acceleration voltage and spherical aberration (Cs) of the lens that affects the way an image is formed at various levels of defocus and must be considered quantitatively for an accurate interpretation of defocused images. This means that you should examine the CTF of your particular microscope to see how defocus will affect your resolution. If you will use tomograms for averaging, it is generally better to stay closer to focus (-2 μm to -4 μm), but if you only want to identify large macromolecular structures without averaging, then you can use a higher defocus (-5 μm to -8 μm).
Considerations for choosing an area to acquire a tilt series are discussed under “Critical Parameters and Troubleshooting”.
Materials
State of the Art electron microscope providing an excellent vacuum environment. The microscope has to be equipped with a computer-controlled goniometer and a high-sensitivity charge-coupled device (CCD) camera. Typically phosphorus scintillators are providing higher sensitivity while Yttrium aluminium garnet (YAG) scintillators deliver better resolution (smaller point-spread function).
Automated tilt series acquisition software (e.g. SerialEM)
Take a low magnification map of the grid
If your image acquisition software has mapping and navigator capabilities, set the magnification to ~170 × and take a low magnification montage of the entire imaging area of the grid. With a 4096 × 4096 pixel camera, we collect low magnification maps with a binning of 4.
A low magnification map will enable you to easily see where the ice is of optimal thickness for imaging or where ribbons of sections are located on the grid. With a navigator function, it will enable you to mark areas on the map that have already been viewed to avoid imaging the same area multiple times and will allow you to move easily to or return to points of interest on the grid.
Use low dose imaging to find an area of interest
2. Go to an area of the grid without anything of interest for imaging, such as an area of intact carbon without any ice.
3. Complete any beam alignments.
In our facility, each user optimizes the gun tilt, gun shift, beam tilt, beam shift, rotation center, spot-size dependent gun shift and condenser and lens stigmation before every session. The alignments you need to complete will be dependent on your microscope and how your facility is managed.
4. Locate an open area (such as an empty hole) over which to acquire the gain reference.
If necessary you can use a focused beam to burn away ice to create an empty hole.
5. Set the stage to a height that gives eucentric tilting.
This is essential for collecting tilted images centered on the area of interest and to minimize changes in focus and magnification between different tilts.
6. Set up low dose imaging. Some typical low dose settings are shown in Table 2.
Table 2.
Typical settings for low dose imaging using SerialEM on a Tecnai F20 electron microscope operating at 200kV (FEI company, Eindhoven, The Netherlands).
| Magnification | Dose (e-/Å2) | Spot Size | Binning (4K×4K camera) | |
|---|---|---|---|---|
| View | 3000 × | 0.05 | 4 | 8 |
| Focus | 29 000 × | 3.50 | 6 | 2 |
| Trial | 29 000 × | 0.20 | 6 | 8 |
| Record | 29 000 × | 1.00 | 6 | 2 |
| Preview | 29 000 × | 0.05 | 6 | 6 |
Low dose imaging is used to minimize the exposure of the sample to the beam to prevent damaging it before or while recording a tilt series. It allows the user to independently set the parameters for viewing, trials (tracking), focusing, and recording. When setting up the low dose parameters it is important to have the beam centered and make sure it is wide enough to not see the edges of the beam in the image, but small enough to avoid exposing a lot of the surrounding area.
7. Set the defocus target.
Remember to consider how the contrast transfer function influences the effect of defocus on your images.
8. Image the sample by taking records at no more than 1 e-/Å2 until you find an area suitable for tilt series acquisition.
See “Critical Parameters and Troubleshooting” for further details on choosing a suitable area.
Tilt series acquisition
9. Take an image of the tracking area.
The tracking area should be set up about 1.5 – 2.5 μm away from the recording area and contain high contrast features for cross-correlating.
10. Refine the eucentricity of the microscope stage in the area of interest to ensure that the record area will be centered throughout the tilt series.
11. Tilt the microscope stage to the highest tilt on each side, usually ±60° - 70°.
This allows you to assess whether the sample will be obscured by a grid bar or piece of contamination at high tilt.
12. View the tracking area at high tilt to make sure the area of interest is still centered.
If not, the eucentric height may not be properly adjusted, or the grid is drifting.
13. Set up the tilt series parameters. Among others, these parameters include the starting and ending tilt angles and the tilt angle increment, the electron dose per image, and the specifications for autofocusing and tracking. An example of the tilt series controller in SerialEM is shown in Figure 4 with the parameters typical for imaging kinesin-microtubule complexes for tomogram reconstruction and subsequent volume averaging.
14. Acquire the tilt-series
Figure 4.
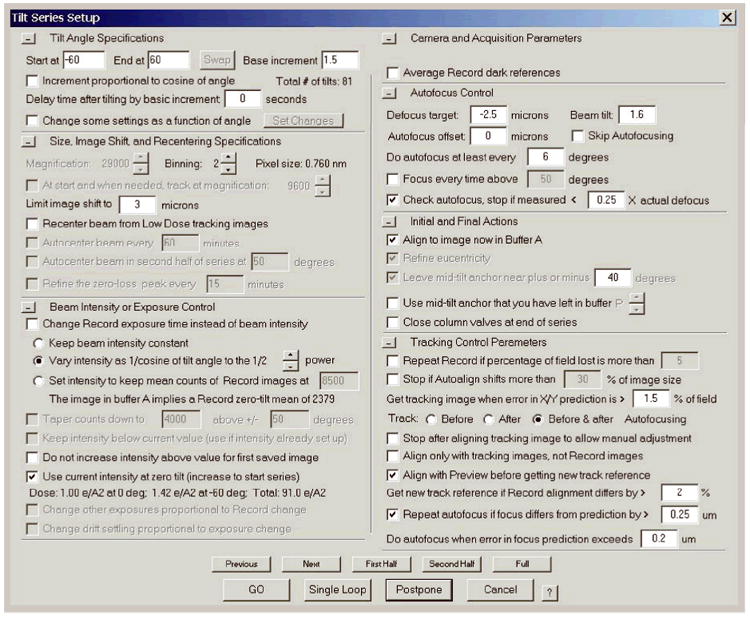
The tilt series controller in SerialEM with typical values for tilt series acquisition of kinesin-microtubules complexes on a 200 kV electron microscope.
Although this is automated, watch the first few cycles of the tilt series acquisition to make sure the area of interest remains centered. Cryo-holders can also be very sensitive to sound and noise should be kept to a minimum during tilt-series acquisition to prevent drift.
15. When the tilt-series is complete, return the microscope stage back to zero tilt.
16. Repeat steps 9-15 for every tilt series you wish to collect.
17. Transfer the data to the computer you will use for tomogram reconstruction and other image processing. It is a good idea to store your data with a safe backup system.
Basic Protocol 4
TOMOGRAM RECONSTRUCTION
The goal of tomogram reconstruction is to recover the 3D structure from which the tilt series images (2D projections) were collected. Unfortunately, in cryo-ET the 2D projections acquired in a tilt series are very noisy due to strict low-dose recording, and the tilt increments are chosen so only a limited number of projections are acquired. The process of reconstructing a tomogram involves aligning the images from the tilt series and then using a mathematical algorithm to generate, as accurately as possible, a 3D volume from the aligned tilt-series. Typically this is done with weighted back-projection of the aligned images through the reconstructed volume; it can also be done by using the 2D Fourier transforms of the individual images to fill in a common 3D Fourier space.
The image alignment step can greatly influence the quality of the final tomogram, because poor alignment produces blurry features. We typically do a two-step procedure where the 2D projections from the tilt-series are first shifted into translational alignment using cross-correlation. This coarse alignment is then refined with the help of gold particles that serve as fiducial markers. The fine alignment is carried out to correct small differences in image rotation, translations, variations in magnification, or non-uniform changes in the specimen that result from its interaction with the electron beam. Gold particles are ideal fiducial markers because they are electron dense and still visible at high tilts. To refine the alignment of the projections, the position of a selected group of gold particles are modeled and tracked through the tilt series. The coordinates of the gold particles are then fit to a set of projection equations that align the images in the tilt series consistently to one another. This fitting identifies gold particle positions that may not be accurately placed, so typically one modifies those positions where appropriate and repeats the fit. This process is iterated until no significant improvement in the alignment is seen.
In the case of cryo-sections or other samples where gold particles are unavailable, alignment must be achieved without the help of fiducial markers. In the past, this meant that alignment of images was calculated on cross-correlation alone, but improved methods have now become available for refining the alignment in the absence of fiducials, such as iterative area matching and geometry correction (Winkler and Taylor, 2006), Alignator (Castaño Díez et al., 2010), and patch tracking in eTomo. Patch tracking is similar to, but less sophisticated than the approach in Alignator; it uses cross-correlation to align small subregions of the image area in each of the views in the tilt series. The parameters can be adjusted until optimal tracking is obtained and a patch-fiducial model is generated that can be used as input for computing the fine-alignment just as a gold particle fiducial model would be. The resulting finely aligned stack is then used to reconstruct the tomogram.
A number of tomogram reconstruction methods are available. The most widely used is weighted back-projection (WBP) where the 2D projections are mathematically weighted before being projected back into the 3D reconstruction (Gilbert, 1972b). The weighting is applied to compensate for the oversampling of low frequencies in Fourier space by the images collected in a tilt series. This method results in a more accurate reconstruction than is achieved with simple back-projection alone. WBP is computationally fast and applicable to a wide range of data, generally without the need for optimization by the user. Two iterative algebraic reconstruction methods are also used in tomography: the Algebraic Reconstruction Technique (ART) (Gilbert, 1972a) and the Simultaneous Iterative Reconstruction Technique (SIRT) (Herman et al., 1973). ART-type and SIRT-type algorithms are more computationally intensive (thus slower) than WBP but are gaining popularity because of their different abilities to filter out noise, and because they can now be computed more rapidly with graphical processing units. The best choice of reconstruction method will depend on your sample and the application of the results. Further details of the mathematics and theory of these reconstruction algorithms can be found in Frank, 2006 and Penczek, 2010. Our lab uses WBP and occasionally SIRT to reconstruct tomograms with eTomo, part of the IMOD software package (Kremer et al., 1996), as described here. eTomo is operated through a simple graphic user interface with a flow chart of easy-to-follow steps and can be downloaded for free from the IMOD website (http://bio3d.colorado.edu/imod). Additional tomogram reconstruction packages available are TomoJ (Messaoudii et al., 2007), EM3D (Stanford University), TOM Toolbox (Nickell et al., 2005), and DigiECT (Digisens, France), among others.
Materials
A computer workstation (Linux, Macintosh or Windows with Cygwin)
Tomogram reconstruction software (e.g. eTomo, part of the IMOD software package)
Pre-process
Create a new directory for each tilt series you will reconstruct so all files generated during reconstruction will be easier to find. eTomo requires a new directory for each reconstruction.
Correct the effect of x-rays.
X-rays and other CCD defects appear in images as extremely dark or extremely bright pixels. These must be corrected because they ruin the dynamic range (contrast ratio) of the data and they can cause artifacts if included in the reconstruction. Such image anomalies can be identified as pixels that are higher than background by extreme amounts or that differ from adjacent pixels by extreme amounts. These extreme pixels should be replaced with an average value determined by interpolation from surrounding pixels. Detection and replacement of anomalous pixels in eTomo is described in detail in IMOD’s tomography guide, freely accessible on the IMOD website (http://bio3d.colorado.edu/imod).
Align the tilt-series images
3. Calculate the coarse alignment to translationally align images in the tilt series based on cross-correlation.
4. Fiducials available: In the zero-tilt image of the tilt-series, place a model point in the center of select gold particles to create a “seed model” for subsequent tracking and fine-alignment.
Only model gold particles that are lying inside the ice layer in a hole in the carbon. Gold particles lying on the carbon should not be included in the fiducial model as carbon interacts differently with the electron beam than the ice does and can negatively affect the alignment calculations. Similarly, gold particles that are very close to each other or in a clump should not be modeled for tracking and alignment.
Fiducials unavailable: Set up the parameters for and run patch tracking to create a patch-fiducial model. Skip steps 5-6 and use the patch-fiducial model as input for steps 7-11.
5. Complete a fiducial model of the gold particles in all images by automatically tracking the seed model through the tilt-series.
6. Manually fill in the gaps in the fiducial model on any views where the gold could not be tracked automatically. The alignment program can tolerate gaps in the model if a gold particle cannot be seen on every view.
Gold particles can also be detected and modeled automatically without creating and tracking a seed model by using software such as RAPTOR (Amat et al., 2008).
7. Compute the first round of fine-alignment. A notable output from the computation is a list of “residual errors”, which is based on the differences in positions between where each model point was placed in the model and where the point is predicted to be for a consistently aligned stack. A mean value of the residual error for all of the points, shown in the alignment summary, gives a representation for how well the model fits the alignment prediction.
8. Edit the fiducial model by examining the points with the largest residuals. If the predicted value moves the model point toward the center of the gold particle, then move the model point to the predicted location. Occasionally the computed alignment predicts that the point should be moved away from the gold particle. In this case, the model point should not be shifted to the predicted location, but should instead be centered on the gold particle.
9. Save the model with the corrected residuals and re-compute the alignment.
10. Examine the residual error mean value to see if it has improved (decreased).
11. Repeat steps 8-10 until little improvement in the residual error mean value is seen.
The residual error mean value should decrease with subsequent rounds of computing the alignment. In eTomo, this value is in pixels so the actual number will depend on the pixel size of the data. For example an error of 0.2 for a 10 Å pixel is the same as an error of 0.4 for a 5 Å pixel. If the pixel size is small, a high value does not necessarily indicate poor alignment.
12. Compute a binned version of the whole tomogram and use it to create a boundary model by outlining the edges of the sample area (i.e. the areas with biological material, or edges of a cryo-section) within the volume. The boundary model is used to position the tomogram so it will be as flat as possible and fit into the smallest volume, which makes the tomogram easier to analyze.
13. Calculate the final alignment.
Complete optional procedures using the final aligned stack
14. Optional: Correct the CTF
CTF correction is necessary if you collected data far from focus and want to recover information at resolutions where the contrast has been inverted by the microscope’s contrast transfer function (for CTF correction of tomographic data see: Xiong et al., 2009, Zanetti et al., 2009.
15. Optional: Erase gold
Gold fiducials generate strong ray artifacts in tomograms. These are particularly undesirable in cryo-ET due to the low contrast of the biological material and because they can obscure the specimen. Gold particles can be selected in the aligned stack of images and replaced with pixels of average gray value, effectively eliminating ray artifacts when the tomogram is reconstructed.
Reconstruct the tomogram and post process
16. Compute the tomogram from the final aligned (CTF-corrected, gold-erased) stack.
17. Trim the tomogram to include only areas with structure and material you wish to keep. This reduces the file size and make subsequent processing easier and quicker.
18. Scale the contrast to increase the dynamic range.
Gold particles are so dense that they occupy most of the dynamic range of the reconstruction, leaving little dynamic range for the biological material. This situation can be improved by scaling the data based on the pixel values in a region that contains biological material, but no gold particles.
19. Delete temporary and other files generated during the reconstruction that are no longer needed.
Basic Protocol 5
AVERAGING SUBVOLUMES OF IDENTICAL STRUCTURES FROM TOMOGRAMS
In many cases, macromolecular complexes can be identified and studied directly within cryo-tomograms, without the need for averaging, especially if the complex has a distinct shape. Microtubules (Bouchet-Marquis et al., 2007), actin filaments, ribosomes (Medalia et al., 2002), and the Golgi apparatus (Bouchet-Marquis et al., 2008), are just a few examples. The 26s proteasome has been identified in its native context because of its characteristic shape (Medalia et al., 2002) and ATP-synthase molecules have been clearly seen in cryo-tomograms of isolated mitochondria (Nicastro et al., 2000). However, because of the sensitivity of frozen-hydrated samples to the electron beam, the total dose on the specimen during tilt series acquisition is limited, resulting in a very low signal-to-noise ratio. In addition, being able to tilt a sample only from ±60°-70° results in a “missing wedge” of information that introduces smearing and other artifacts into the reconstruction. Thus, a method for aligning and averaging assumed identical subvolumes from tomograms has been developed as a way of increasing the signal-to-noise ratio and reducing missing wedge effects.
The steps for subvolume averaging include: modeling to select the particles to be averaged, → aligning these particles to a reference, → averaging aligned particles, and → iterating to improve the reference, alignment, and constructing a final average. We use 3dmod (Kremer et al., 1996), part of the IMOD software package for particle selection and PEET (Nicastro et al., 2006, Cope et al., 2010) for subvolume alignment and averaging. Because these packages are freely available and are easily downloaded and installed from our website (http://bio3d.colorado.edu/), this protocol describes the steps performed using this particular software. Additional methods and software for subvolume extraction and averaging are available (Beck et al., 2004, Al-Amoudi et al., 2007, Bartesaghi et al., 2008, Heymann et al., 2008).
Materials
Tomogram modeling software (e.g. 3dmod, part of the IMOD package)
Subvolume averaging software (e.g. PEET – Particle Estimation for Electron Tomography)
Select particles for subvolume averaging
Filter (denoise) the tomogram to enhance contrast for modeling. We use Frangakis and Hegerl’s algorithm for Nonlinear Anisotropic Diffusion with Edge Enhancing Diffusion (Frangakis and Hegerl, 2001).
The filtered tomogram should be used only for the modeling step to select particles. It should not be used as input for the volume averaging software.
-
2. Select individual subvolumes using a modeling program (e.g. 3dmod) by placing a model point in the center of each repeating unit. The 3dmod “slicer” window is ideal for locating the center of a particle in X, Y and Z for placement of a model point. The way that particles are modeled for selection is dataset specific. Here are a few examples for how we have modeled different types of macromolecules:
-
Isolated complexes scattered throughout a tomogram such as virus capsids (Figure 5A):
Place a model point in the center of each complex. For “spherical” assemblies, such as some viruses, this can be achieved by moving up and down through the tomogram and placing a model point at the Z-value at which the object appears widest.
If possible, select subvolumes from multiple tomograms to increase the number of particles available for averaging. Create a new model for each tomogram. This is recommended not only for isolated complexes, but for any type of structure being selected for averaging.
-
Structures that bind in between two filaments, such as kinesins forming “connections” in between microtubules (Figure 5C):
Use the slicer window to find the center of the connecting macromolecule, both in distance between the filaments, and in height of the connector itself. Place a model point at the center of every connection.
When choosing a particle size for averaging, described in step 15 below, we have achieved the best alignment if the particle volume is restricted to include all of the bridging protein, but very little of the flanking filaments.
-
Structures that repeat at a regular interval along the length of a filament.
Model the center of the filament. Starting near one end of the filament and working toward the other end, places points in the center of the filament. Place adjacent points sufficiently close to each other that the filament between them is essentially straight.
Run a program to automatically fill in points based on the initial model in step (C)i. For example, if the structure you want to average repeats every 8nm along the length of the filament, use the program “addModPts”, part of the PEET package, to automatically fill in points every 8nm along the filament after placing points by hand as described above. This is faster and more accurate than modeling points manually.
It is possible to select particles from filaments lying at multiple orientations within the same tomogram. If you are selecting particles with 3dmod, each filament must be modeled with a new contour.
-
Figure 5.
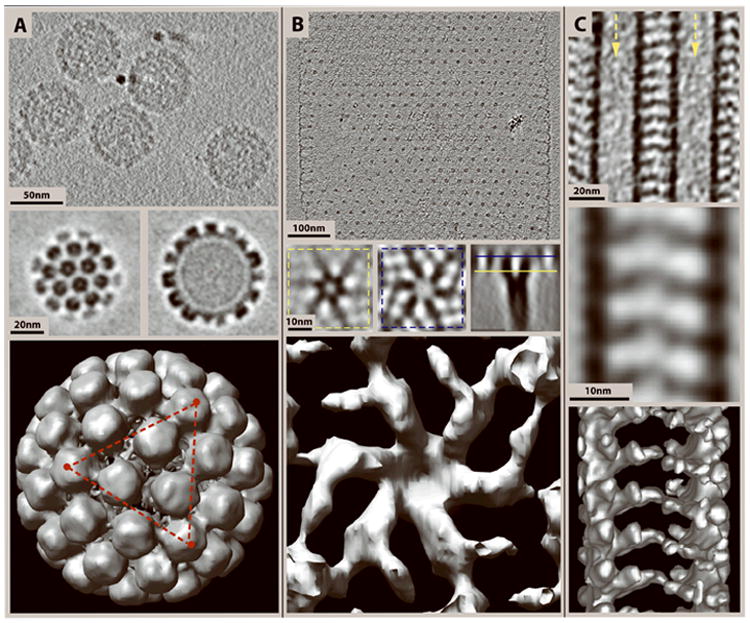
Different types of data studied by cryo-ET and subsequent subvolume averaging. In each column the top image shows a slice through the cryo-tomogram, the center panel shows slices through an average of subvolumes selected from tomograms, and the bottom image shows a surface rendering of the subvolume average. (A) Top: 4.5 nm slice through a cryo-tomogram of Bovine Papillomavirus (BPV) showing a side view of the capsomeres and packed DNA in the center. Center: Slices through the top (left) and center (right) of an average of 50 virus particles selected from cryo-tomograms, and “symmetrized” according to the 5-fold axis of symmetry. Bottom: Surface rendering of the averaged BPV capsid. The red triangle connects three of the twelve 5-fold axes of symmetry. (B) Top: 20 nm slice through a cryo-tomogram of an S-layer from the thermophilic archaeon Thermoproteus tenax. Center: 4 nm slices through various orientations of an average of 500 S-layer unit cell subvolumes extracted from tomograms. Bottom: Surface rendering of an S-layer unit cell from the average. (C) Top: 15 nm slice through a tomogram showing two adjacent microtubules decorated with the heterodimeric kinesin Kar3Vik1 that extends outwards forming a “bridge” in between the microtubules. Arrows point down the lumens of the two microtubules. Center: 15 nm slice through an average of 44 subvolumes. Bottom: A surface rendering of the average. BPV specimen courtesy of Prof. Robert Garcea, Univ. of Colorado at Boulder. S-layers from Thermoproteus tenax courtesy of Prof. Reinhard Rachel, Univ. of Regensburg, Germany.
Define the data to be averaged and select a reference volume
3. Create a new directory for each averaging run.
Many new files are created during averaging and this will make them easier to find later. In PEET, each averaging run must be in its own subdirectory.
4. Open the subvolume averaging program. While PEET is separate from the IMOD software, the PEET graphic user interface can be accessed from the eTomo start-up page.
5. Input the tomogram(s) and model(s) defining the centers of the subvolumes/particles to be aligned and averaged.
6. Select a reference volume. This can be either a single particle chosen from one of the input models, or it can be a volume from a separate file, such as the final reference from a previous round of averaging or from a different type of reconstruction method.
7. Optional: If applicable, generate an initial motive list for your data. This list has a comma separated value (.csv) file format and contains the rotations and translations necessary to align each particle in your model to the reference. If you have some idea of the orientation of the particles relative to the reference, the use of an initial motive list permits the use of smaller search angles, which speeds up the computation time of proper alignment parameters.
If no information on relative orientations is available, the volume averaging software will generate an initial motive list(s) for the first round of averaging. Motive lists are improved iteratively with each round of alignment.
8. Optional: If your data require it, select a suitable mask (such as a sphere, cylinder or other volume). For example, if you have variable background structures that could interfere with the particle alignment, a mask can be used to exclude these from the reference so they are not used in cross-correlation calculations and alignment will be performed using the structure of interest only.
9. Specify the tilt range of the tilt series from which the input tomogram was reconstructed. This is necessary only if you wish to use missing wedge compensation during alignment and averaging, which is recommended. With missing wedge compensation, weighted averaging is carried out in Fourier space so each particle contributes only the information that lies outside its missing wedge. The tilt range can be input from a tilt.log or a .tlt file generated during tomogram reconstruction.
Set up the search parameters to be used for each round of alignment and averaging
10. Define the first axis of rotation for the volume averaging software to use for the alignment search. In PEET, the default is to use the original Y-axis of the tomogram, but there is also an option to instead use the particle model points. In this case, the first rotation axis is the vector between two model points and is thus different for each particle. This option is especially useful for particles selected along a filament that does not lie in line with the Y-axis of the tomogram.
11. Complete the iteration table. The iteration table allows the user to specify values for the angular search range and search radius for each round (iteration) of subvolume alignment and averaging. The rotation angles are such that Phi is the rotation around the first axis defined in step 11, with the second (Theta) and third (Psi) axes determined using a right handed Cartesian coordinate system.
Appropriate values for the maximum search angle and the search angle increment vary greatly depending on the type of particle being averaged and thus search parameters must be optimized for your own data. This is discussed in more detail under “Critical Parameters and Troubleshooting” and typical values for two different examples are shown in Table 3 as a guide.
Table 3.
Iteration table with typical search parameters for aligning and averaging different types of samples using PEET. Search parameters are very dataset dependent and must be optimized for each specimen.
| Iteration Number | Angular search Range (degrees) | Search Radius (pixels) | ||||||
|---|---|---|---|---|---|---|---|---|
| SAMPLE | Phi | Theta | Psi | |||||
| Max | Incr. | Max | Incr. | Max | Incr. | |||
| Kinesin-decorated microtubules | 1 | 18.0 | 2.0 | 0.0 | 1.0 | 0.0 | 1.0 | 5 |
| 2 | 9.0 | 3.0 | 5.0 | 2.5 | 5.0 | 2.5 | 4 | |
| 3 | 4.5 | 1.5 | 3.2 | 1.6 | 3.2 | 1.6 | 2 | |
| 4 | 2.25 | 0.75 | 2.0 | 1.0 | 2.0 | 1.0 | 2 | |
| 5 | 1.125 | 0.375 | 1.2 | 0.6 | 1.2 | 0.6 | 2 | |
| Virus Particles | 1 | 36.0 | 9.0 | 36.0 | 9.0 | 36.0 | 9.0 | 3 |
| 2 | 12.0 | 3.0 | 12.0 | 3.0 | 12.0 | 3.0 | 2 | |
| 3 | 6.0 | 2.0 | 6.0 | 2.0 | 6.0 | 2.0 | 2 | |
| 4 | 3.0 | 1.0 | 3.0 | 1.0 | 3.0 | 1.0 | 2 | |
13. Optional: Set a low-pass filter to get rid of high frequency noise. Typically, we use a cutoff frequency of 0.30 with a Gaussian falloff (to avoid edge artifacts) standard deviation (sigma) of 0.05 for each iteration.
The appropriate filtering value will again be dependent on your specific data – if the images are noisy, better results may be obtained by using a cutoff frequency as low as 0.1 or 0.15.
14. Set a reference threshold. This controls the number of particles that are included to create the reference for a subsequent round of alignment and averaging. A general guide is to set the threshold at two-thirds the total number of particles to prevent “bad” particles from being incorporated into the reference.
Set the averaging parameters
15. Specify the particle volume. This represents the edge length, in pixels, of the cubic volume that the program will extract to obtain each particle from the tomogram(s) for averaging. The center of the particle volume will be the position, in X, Y and Z of the points in the particle selection model from step 2.
Note that for PEET, the minimum particle size must contain the volume of interest, plus twice the maximum search radius.
16. Input the number of particles to be included in the average. It is useful to generate a number of averages, each created from an increasing number of particles.
In some cases, incorporating more particles can actually decrease the quality of the average if they are too different in structure and/or image quality.
17. Compute the alignment and averages. Some examples of tomograms we have used for subvolume averaging, and the resulting averages are shown in Figure 5.
Supporting Protocol 1
APPLYING SYMMETRY WITH SUBVOLUME AVERAGING
When the initial subvolume average from Basic Protocol 5 indicates the presence of rotational and/or translational symmetry, or if the structure in question is known in advance to posses such symmetry, additional averaging based on symmetry operations can be highly advantageous. Examples include square or hexagonal symmetry in planar arrays, helical symmetry in 15- or 16-protofilament microtubules, and icosahedral symmetry in “spherical” viruses. By applying the symmetry operations, the effective number of particles to be averaged can be greatly increased (6-fold in the hexagonal case – or more if translational symmetry is used, 15-fold for 15-protofilament microtubules, and 60-fold for icosahedral viruses), with corresponding improvement in signal-to-noise ratio and reduction in missing wedge artifacts (Figure 6).
Figure 6.
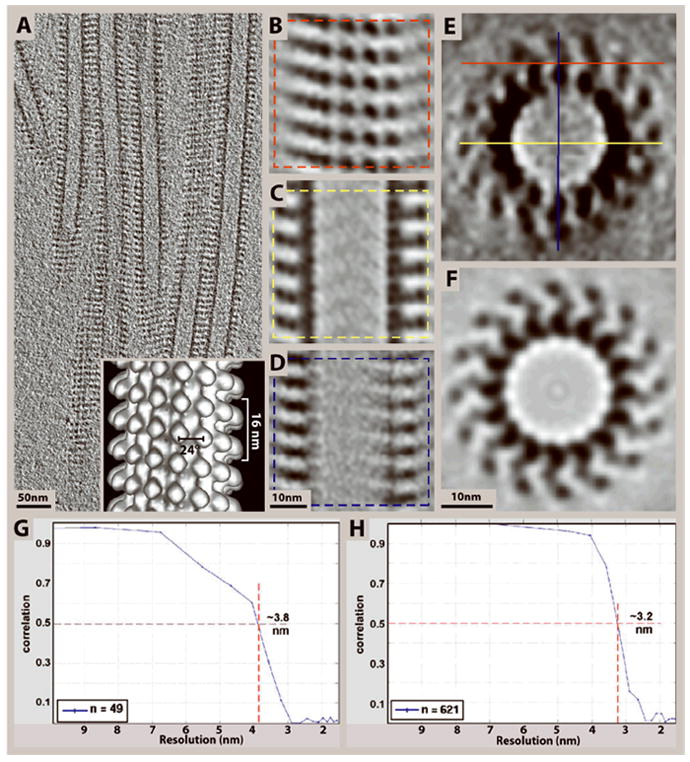
Incorporating symmetry with subtomogram averaging can effectively eliminate missing-wedge effects as demonstrated here on kinesin–microtubule complexes (Cope et al., 2010). (A) 10 nm slice through a cryo-tomogram of microtubules decorated with the heterodimeric kinesin Kar3Vik1. (B-D) 4 nm slices through the top and center of an average of 99 subvolumes selected from the tomogram in (A) according to the planes shown in (E). The central x-y slice in (C) is much less affected by the missing wedge, and shows well-defined densities corresponding to the α- and β- subunits of tubulin and the two globular domains of Kar3Vik1. (Only one of the two domains actually binds to the tubulin protofilaments). The central slice in (D) illustrates the loss of resolution along the Z-axis due to the missing wedge as compared to the x-y slice in (C). (E-F) A slice through the cross-section of the subvolume average before (E), and after rotational averaging (F), showing that rotational averaging can effectively eliminate missing-wedge effects. By averaging over all 16 protofilaments the asymmetric unit changes from axial slices along the microtubule to one αβ-tubulin–kinesin complex. Hence, the theoretical maximum number of asymmetric units included in the average is increased from 99 to 1584 (16 × 99). (Though our average is based on the 1242 particles with the best correlation to the reference because this subset gave the highest resolution). (G-H) For Fourier-shell correlation calculations, the datasets were split in half on a random basis and the two halves were correlated against each other. Fourier-shell correlation graphs obtained from the averages in E (G) and F (H) reveal resolution limits of 3.8 nm and 3.2 nm, respectively, based on the 50% correlation criterion. Inset in (A) shows a surface rendering of an average of a 15-protofilament microtubule after symmetrization as described in Supporting Protocol 1, step 3.
While the details depend on the specific case, the basic approach for using PEET to incorporate symmetry is the same. After an initial round of averaging, as described in Basic Protocol 5, PEET creates motive lists, one for each input tomogram, in comma-separated value (.csv) file format. The motive lists specify the rotations and translations applied to each particle to align it with a common reference. The basic steps involved in incorporating symmetry into a new round of averaging are: creating new motive lists to center the structure in a position optimal for applying symmetry information, → creating additional new motive lists applying symmetry operations to the centered structure, → generating a new reference based on the symmetrized motive lists, → refining the alignment using the new reference, and → calculating a symmetrized average based on the refined alignment.
PEET provides a program called “modifyMotiveList” which can be used to generate a new motive list by applying specified, common rotation and translation to all the particles in an existing motive list. By chaining such operations, the required transformations are readily accomplished.
Create new motive lists containing the rotations/translations for each symmetry group
1. Create a new directory in which the symmetrization will be performed.
We recommend keeping the original volume(s) and model(s) in a top level directory, with a subdirectory for each average. This structure prevents confusion and permits experimentation with alternate averaging strategies along with symmetrization without copying files.
2. Optional: From each final motive list produced in an initial round of averaging (described in Basic Protocol 5), create one or more new lists in which some or all of the symmetry operations are conveniently expressed.
For example, center a 15-protofilament microtubule within the average volume with its length parallel to the Y-axis, or for isolated particles such as icosahedral viruses, center the averaged virus within the subvolume.
3. From each motive list created in step 2 (or from Basic Protocol 5 if step 2 was omitted), create a new motive list corresponding to each unique member of the symmetry group. Newly created motive lists are treated as referring to independent particles, even though they are associated with the same volume as the originals, and may refer to different orientations and/or positions of the same subvolumes.
For example, a motive list created for a 15-protofilament helical microtubule would include rotations of 360*i / 15 = 24*i ° combined with translations of 16*i / 15 nm, where i = 0, 1, … 14, to bring a subunit from each protofilament to an equivalent position (Figure 6A, inset).
For an icosahedral virus that has twelve 5-fold axes of rotational symmetry, motive lists can be created in step 2 to rotate the original average in 12 different ways to produce 12 new volumes each with a different 5-fold axis rotated to lie on the Z-axis. Then in step 3, motive lists can be created that define the additional rotations of 0, 72, 144, 216, and 288 degrees to apply the 5-fold symmetry around the Z-axis for each of the 12 new volumes (Figure 5A, bottom).
Calculate a new average using the new motive lists
4. Generate a new reference by performing a normal PEET iteration with no search, using the motive lists from step 3 to specify the orientations and positions of each particle.
5. Perform a narrow range search over a few degrees and a few pixels to generate the refined alignment and “symmetrized” average. Use the motive lists from step 3 as the starting orientations and positions, and the newly generated reference from step 4 as the reference.
In principle, a symmetrized average can be created without a search. However, we recommend using a narrow range, refining search to minimize problems caused by missing data, less than perfect initial alignment, and reference bias. Depending on the initial search range and the final precision desired, from one to several iterations may be required.
Supporting Protocol 2
ESTIMATING RESOLUTION IN SUBVOLUME AVERAGES
Frequency response in subvolume averages can be estimated by computations over spherical shells in Fourier space. Comparing the average to the individual volumes leads to a method known as Spectral Signal-To-Noise Ratio or SSNR (Penczek, 2002). When many volumes are available, an alternative is to randomly split the data in half, compute averages over each half, and then calculate correlation between the averages over frequency shells of interest, resulting in the Fourier Shell Correlation or FSC (Harauz and van Heel, 1986). Typically, frequency response plots are used as relative indicators, for example, in choosing how many particles to use in the final average. In some cases, however, a single, absolute resolution number is desired. Choice of the FSC or SSNR cutoff value used to determine resolution in the latter case can be controversial (van Heel and Schatz, 2005). PEET provides programs to compute and plot both SSNR and FSC.
Reagents and Solutions
Concentrated colloidal gold
Place 1 ml of 5 nm or 10 nm colloidal gold in a microfuge tube
Spin at maximum speed in a bench top microfuge for 10 minutes to pellet the gold particles
Remove ~960 μl of the supernatant
Resuspend the gold pellet in the remaining 40 μl.
Add 1μl of concentrated colloidal gold to your sample before plunge freezing.
Software
See the respective website for each of these software packages for download/installation instructions and additional information including instruction manuals and tutorials.
SerialEM: http://bio3d.colorado.edu/SerialEM/
IMOD, including eTomo and 3dmod: http://bio3d.colorado.edu/imod/
Commentary
Background Information
The principle of electron tomography has been in use for many years but was first applied to biological material in the late 1960’s (De Rosier and Klug, 1968) and the improved computational possibilities that we have available today have given tomography a serious new boost (Lučić et al., 2005, Vanhecke et al., 2010). The principle is based on a mathematical projection theorem, which postulates that the Fourier transforms of 2D projections through an object at different tilts correspond to central slices of the object in 3D Fourier space. The 3D structure of a specimen can thus be reconstructed by collecting a series of 2D projections at various angles around the specimen. The projections, which aim to sample as much of Fourier space as possible, are then used to reconstruct the 3D volume by a mathematical reconstruction algorithm, such as weighted back-projection. The 3D reconstruction has the remarkable advantage of being able to be rotated and “sliced” computationally in any direction. Slices can be as thin as a single voxel (typically 0.5 - 1 nm3) and can be cut at any position or orientation through the tomogram providing views without interference from structures (or noise) above or below the selected slice somewhat similar to the effect of a confocal light microscope. However, due to the limited tilt capabilities of electron microscope stages (~±60°-70° instead of ±90°) the 3D Fourier space reconstruction lacks data that corresponds to a missing wedge (missing pyramid or cone, if multiple tilt axes were collected) and this is reflected in a loss of resolution along the Z-axis of the reconstruction as well as the presence of streaks and other artifacts.
For conventional electron tomography, samples are plunge frozen, high pressure frozen or chemically fixed and then dehydrated by replacing the water in the sample with an organic solvent, often in the presence of a fixative and/or osmium tetroxide. Dehydrated specimens are then embedded in a plastic resin and subsequently sectioned to be thin enough for good quality imaging in the microscope available (for example, 300 nm sections are imaged well in a microscope with an accelerating voltage of 300 kV). Often the sections are post-stained with lead and uranyl acetate to provide contrast of the biological material in the electron microscope. Plastic-section tomography has been used to unravel many scientific mysteries regarding the organization of cellular structures in various cell states, and still plays an important role in cell biology today (Marsh et al., 2001, O’Toole et al., 2003, Hoog et al., 2007 are just a few of many). However, the methods of sample preparation can lead to dehydration artifacts, and structural information is obtained implicitly through the use of an electron-dense stain, limiting the resolution that can be reached.
In the early 1980’s Dubochet and colleagues pioneered the technique of freezing samples rapidly enough (104 degrees/s) to prevent the specimen damage caused by ice crystal formation (Dubochet et al., 1981, Dubochet et al., 1988, Dubochet, 2007). While the properties, and particularly the formation, of vitreous (amorphous) ice are still somewhat elusive, the technique has been widely adopted as a means to preserve specimens as close to their native condition as possible. Rapid freezing allows for specimens to be immobilized and imaged in a fully-hydrated condition without the use of the dehydrating agents and contrast-generating stains used in plastic-section electron microscopy. This gives the advantages of superior specimen preservation and the potential to resolve much finer details (theoretically atomic detail, e.g. Gonen et al., 2005). Preparation of samples for cryo-ET is also much quicker than for conventional imaging, as samples can be frozen and imaged in the same day, while dehydrating by freeze-substitution and resin-embedding, depending on the protocol, can take anywhere from a full day to a week.
However, frozen-hydrated samples suffer from extreme sensitivity to the electron beam and as a consequence, the limit on the dose that can be applied to the specimen results in a low signal-to-noise ratio and rather poor image contrast. While specimen preparation for cryo-ET is quicker, it is technically more difficult, often requiring quite a bit of practice and more attention from the user during sample handling. In addition, the supporting equipment necessary for cryo-ET such as the cryo-holder and freezing device, is highly specialized and very expensive.
The routine collection of tilt-series data for cryo-ET only became practical after the development of software for automated tilt-series acquisition and dedicated imaging technology. The major “selling point” of cryo-ET is that it is uniquely suited to the study of complex cellular structures, because it does not initially rely on any kind of symmetry or averaging. If multiple copies of a structure are present, they can be further analyzed by subvolume averaging software to increase the image signal-to-noise ratio and reveal structures at higher resolution. A strong benefit here is that complexes can be averaged in their native environment without the need to purify them from the cell. Cryo-ET both with and without subvolume averaging is also a valuable tool for studying isolated complexes prepared in vitro because it permits the observation of non-symmetrical structures, such as microtubules that possess a lattice seam or are incompletely decorated by a kinesin motor (Cope et al., 2010).
Unfortunately, cryo-ET reconstructions suffer from missing wedge effects, which limit their resolution in the direction parallel to the electron beam typically by a factor of ~1.5-2 for a ±60° tilt range. The missing wedge effects can be eliminated by subvolume averaging if the subvolumes are randomly oriented in space. If they are not randomly oriented, as with linear tubular segments, particles picked from 2D crystalline arrays or particles fixed in a preferred orientation by surface effects, then symmetrization, where applicable, can effectively reduce the missing wedge problem (Figure 6).
Other image processing techniques for cryo-EM, such as electron crystallography, single particle or icosahedral reconstruction and helical reconstruction, are well-established methods for recovering 3D structural information from 2D projections and have yielded spectacular results to high resolution (Gonen et al., 2005, Sachse et al., 2007, Zhang et al., 2010 are a few of many). These methods rely strictly on averaging 2D projections containing various views of an identical structure and in some cases take advantage of an intrinsic symmetry of the macromolecule being studied. As such, identical, inflexible complexes, 2D crystalline arrays, perfect icosahedrons or perfect helices are required. However, such reproducible structures and/or regular arrays are more of an exception than a rule and most biological macromolecules and higher-order assemblies are too flexible and polymorphic to be subjected to averaging. In addition, these averaging methods are typically applied to isolated complexes that have been purified or prepared in vitro; the resulting complexes are studied outside of their native environment.
2D image-averaging techniques do have the advantage of being able to rapidly incorporate a very large number of particles and most of these approaches are not as affected by missing wedge/missing cone artifacts as those found in tomograms. Furthermore, averaging-based reconstructions are composed of data obtained from single projections, and therefore each image can be taken with much higher electron dose. As a result, the raw data may already reveal a much better signal-to-noise ratio than the individual projections that go into cryo-tomograms. In general, 2D image averaging based 3D reconstruction methods have the potential to reach a much higher resolution than cryo-ET, even when the latter is combined with subvolume averaging. Because of this, averaging based methods are not necessarily in direct competition with cryo-ET, but instead are rather complementary. Their application should be considered based on the desired outcomes of the experiment.
Recent improvements of cryo-EM and cryo-ET related technology and hardware, such as automated plunge freezers, cryo-specific specimen holders, more stable stages for tilting, better cameras and faster computation have resulted in a revival of ET in general and an initiation of cryo-ET, collectively constituting a strong push for what can be achieved in terms of resolution. User-friendly software and equipment have made the technique far more accessible, even to investigators who are not dedicated electron microscopists. The newest technology developments include high-sensitivity direct detection devices (such as CMOS-detectors) (Jin et al., 2008, Milazzo et al., 2010), and phase plates suitable for cryo-ET (Danev et al., 2010, Murata et al., 2010). Once leaving the experimental phase, these advances promise to improve the currently attainable image resolution and enhance the scope of biological questions that can be answered by cryo-ET in the future.
Critical Parameters and Troubleshooting
Grids
Holey carbon grids (grids with a thin film of carbon with holes 2 – 4 μm in diameter), or lacey carbon grids are the most popular choice for cryo-ET and can be homemade, or bought from a commercial vendor. While pricey, we prefer to purchase grids holey carbon grids because of the uniform size and spacing of the holes and have used C-Flat™ (Protochips Inc, Raleigh, NC) and Quantifoil® grids (Quantifoil Micro Tools GmbH, Jena, Germany) with equal success. A wide variety of holey carbon grids are available, differing in the size of their carbon holes, the spacing between the holes and the mesh number. For larger samples, such as whole-mount cells or cryo-sections, a wider hole should be used to facilitate imaging as much of the structure as possible in a single hole. For smaller samples, such as isolated protein complexes, a smaller hole-size may be favorable. Grids with a 200 mesh are preferred. 400 or higher mesh grids have an increased chance of having a grid bar come into view at high tilts which is not ideal for tilt-series acquisition.
The support films of manufactured grids are often hydrophobic. We therefore recommended glow discharging them immediately before use to obtain an even distribution of vitreous ice across the surface of the grid. However, some macromolecular complexes that are highly positively charged have been known to stick preferentially to the carbon and are excluded from the holes. To prevent this, glow discharging should be avoided. Instead, the grids can be rinsed in a droplet of chloroform and allowed to dry in a fume hood before use. Applying the sample to both sides of the grid and blotting from both sides has also been shown to help. Occasionally we receive grids that still have plastic (such as Formvar) over the holes left over from the manufacturing process (pseudo-holes). This can be cleaned up effectively by rinsing the grids in a droplet of ethylene dichloride or amyl-acetate before use. We have found that beam damage and non-uniform ice are more common with older grids, so grids should be purchased as needed and preferably not stored for longer than six months.
Blotting Parameters
The parameters used for blotting must be optimized for each specimen, and may vary even for the same sample depending on whether you are using a homemade or commercial plunge freezer. The most favorable blotting conditions are when specimens are routinely frozen with a layer of ice that is vitreous and of optimal thickness for your sample and for imaging. If ice is repeatedly not vitreous, make sure that the grid has been kept close to liquid nitrogen temperature at all times between freezing and insertion into the microscope. Increasing the humidity during blotting can also help to obtain vitreous ice. We have also successfully used a mixture of ethane and propane as described in Tivol et al., 2008 to improve freezing. If ice is repeatedly too thick, the blotting time should be increased and/or the volume of sample applied to the grid can be decreased. If many grid squares contain broken carbon, the blotting force should be reduced. Alternatively, grids can be blotted from the back, so the filter paper comes into contact with the side of the grid that is not coated with the carbon layer (the sample should still be applied to the carbon-coated side). If it seems that your sample is being blotted off onto the filter paper, increase the time that the sample has to adsorb to the carbon (the wait time). Blotting from the back of the grid or from the side can also help with this, though blotting from the side often results in ice that is too thick.
Electron Microscope
The choice of electron microscope for tilt series acquisition is an important one, as not all are equal. Firstly, data quality can vary depending on the electron source. For cryo-EM and cryo-ET a Field Emission Gun (FEG) is generally preferred over tungsten or lanthanum hexaboride thermal filaments because of the improved coherence of the beam that generates better phase contrast, and provides higher resolving power (though this is more important for high-resolution cryo-EM than for cryo-ET). Secondly, the accelerating voltage should be chosen on the basis of the type of sample being imaged. An accelerating voltage of 300 kV is recommended for thicker samples such as ~300 nm plastic sections as these energetic electrons penetrate the sample better. However, for thinner samples such as plunge frozen isolated macromolecules or vitrified sections, 200 kV is sufficient and often superior because of the reduced number of unscattered electrons that contribute to lower contrast. To improve contrast, particularly at 300 kV, electron microscopes may be fitted with an energy filter for so-called “zero-loss” imaging to eliminate inelastically scattered electrons that mostly contribute to background noise rather than image data.
To prevent accumulation of frost on the sample, electron microscopes used for cryo-EM and cryo-ET must be equipped with anti-contamination blades that are kept at liquid nitrogen temperature throughout the imaging session. A charge-coupled device (CCD) camera is essential for automated tilt series acquisition and the electron microscope must have a stable specimen stage that can be tilted eucentrically, so the imaging area remains centered throughout the tilt series. Also necessary are low-dose imaging capabilities, so focusing and specimen tracking during the tilt series can be done outside the area of interest, thereby minimizing beam damage to the specimen.
Magnification
The magnification at which data are collected determines the pixel size of tilt series images and thus has implications on the final resolution of the tomogram. In general, data should be collected at a magnification that gives a pixel size roughly one third of the desired resolution. For example, to resolve features in the ~2.5 nm range, we collect data on our Tecnai F20 FEG electron microscope (FEI Company, Eindhoven, The Netherlands) at 29 000x, which on our 4K ×4K camera corresponds to a pixel size of 7.6 Å when binned by 2. The higher the magnification, the more susceptible the specimen is to beam damage and drift during tilt series acquisition. Consequently, it is recommended to choose the lowest magnification suitable for the resolution you wish to achieve. Note that this will not affect the resolution produced by the lenses, but only the maximum sampling rate on the digitized image.
Tilt Series Acquisition Area
While there may be many areas on a grid with a good distribution of sample, not all these areas will be ideal for acquiring a tilt series. In addition to an optimal specimen, a number of parameters must be considered when choosing an area for tilt-series acquisition:
Ensure that the area of interest will not be obscured by a grid bar at high tilts. Also, the closer one remains to the center of the grid the lower the chance that the edge of the sample holder will come into view at high tilt.
The imaging area must be flat. If there is a dent in the grid near the area of interest, recording becomes suboptimal due to the focus gradient across the specimen and incorrect tilt readings from the goniometer.
The carbon layer should be intact within the grid square you are imaging. Samples imaged close to a tear in the carbon layer are susceptible to mechanical drift.
Biological material should be imaged in the ice that covers a hole, not over the carbon. The carbon layer contributes to background noise and reacts differently from ice under the electron beam making images more difficult to align.
Ice must be of optimal thickness for your sample. Ice that is too thick prevents electrons from penetrating the sample, and decreased contrast under low dose imaging conditions. Ice that is too thin may contain flattened or partially dehydrated samples.
Ice must be vitreous. If you are unsure whether the ice is vitreous, take a diffraction image near the area of interest. Spots in the diffraction pattern can indicate crystalline ice, while vitreous ice should only contain very diffuse rings at well-defined radii such as the 2Å water line (Bouchet-Marquis and Fakan, 2009).
The tracking area must contain distinctive features for cross-correlating, as described in Basic Protocol 3.
The number and distribution of gold fiducials in the imaging area must be considered. An even distribution of gold particles will serve as useful fiducials for achieving a good alignment of the tilt series images. Optimally a recording area should contain ~10-20 fiducials, evenly distributed over the record area without obscuring your sample.
Tilt Series Image Alignment
Proper image alignment is essential for a good tomographic reconstruction as blurring from poor alignment is very apparent in final reconstructions. The coarse alignment of tilt-series images by cross-correlation will rarely fail, but may be unsuccessful when aligning some pairs of cryo-images in a tilt series with very low contrast. Before proceeding with refining the alignment, check that the coarse alignment has been completed properly. If alignment has not been successful, errant images can be aligned manually. In eTomo, we use the program “midas” for this. Occasionally, a tilt series will contain images that are out of focus or suffer from drift. These views should be excluded from alignment calculations, and if necessary, can also be excluded from the final reconstruction.
If your dataset lacks gold fiducials, refine the alignment using patch-tracking or another fiducial-less alignment method, such as Alignator (Castaño Díez et al., 2010). Patch-tracking is most effective on areas with distinct structures. If the images have large empty areas without any biological material, a boundary model should be used to exclude these areas from the tracking process. If gold particles are available, the time and effort required to model and align using fiducials can be worthwhile to achieve a better alignment. Aim to select ~10-20 gold particles for a 2K × 2K image. Gold particles that are very close to each other or in a clump are not suitable for alignment. For cryo-ET, it is often the case that the number of gold particles is limited to less than 10 and various alignment parameters, such as grouping images, can be tested to achieve the best alignment of the tilt series. It is important to note that alignment using gold particles for cryo-ET is quite different from that of plastic section ET. Firstly, gold particles are on only one surface or distributed throughout the sample and not on two distinct surfaces as in plastic sections. Secondly, the plastic resin distorts differently under the electron beam depending on how the embedded material interacts with the beam, while it is thought that vitreous samples do not distort as much. Accordingly, parameters for solving the alignment with plastic section images will not necessarily give the best alignment for cryo-data. A useful way of assessing the alignment is to view the tomogram in the X-Z orientation. Here, strong features such as gold particles or electron dense material will looked slightly stretched in the Z orientation, but “banana” shaped features are indicative of poor alignment.
Search Parameters for Optimal Alignment of Subvolumes
A fundamental step in subvolume averaging using PEET is the correct choice of search parameters for alignment of the extracted subvolumes. Appropriate values for the maximum search angle and the search angle increment (step size) for each of the three rotations, Phi, Theta and Psi, vary greatly depending on the type of structure being averaged. There are two general rules that we have found apply well to our data. First, the initial iteration should have the largest angular search range, angular step size, and search radius. Once a rough approximation has been found, these parameters can be made smaller (and therefore more precise) through subsequent iterations. Second, multiple iterations with fewer angular steps are typically computationally quicker than a single iteration with many angular steps, particularly when searching around multiple axes. Often we set the maximum search angle to be three times that of the search angle increment. For example, a maximum angle of 6° with a step size of 2° would search from -6° to +6° at 2° intervals. With this pattern, both the maximum search angle and step size can be decreased by a factor of 2 for each subsequent iteration. When little is known about the particles’ relative orientations, a large search range such as 180°, can be used. This will search the entire space (-180° to +180°) around the chosen axis seeking the alignment that gives the highest cross-correlation with the reference. However, if a small step size is chosen in this case, the search will take an extremely long time. Conversely, using a large step size may result in not being able to find the best alignment. If it is necessary to search around the entire axis, a spherical or hemispherical search may be appropriate. Spherical searches search the space more efficiently by avoiding oversampling near the poles of the axes and are thus computationally faster than a corresponding full grid search. For particles repeating along a twisted filament, where the angular rotation/translation is known, a smaller search range can be used for the first iteration of averaging and fewer iterations are needed to attain precision in the alignment. Refer to Table 3 for example iteration tables for two different types of samples.
Anticipated Results
The results from cryo-ET should generally permit a reconstruction with the potential to resolve biological structures with recognizable features of ~3 nm on thin, plunge frozen macromolecules, and closer to ~4-5 nm on thicker specimens and cryo-sections. Smaller features are usually beyond the noise level and visualizing them would require further processing.
Results from subvolume averaging will vary substantially depending on the quality of the sample, the quality of the tomogram(s), the number of particles included in the average, the type of structure being averaged and whether some type of symmetry can be used during averaging.
Time Considerations
An experienced researcher can plunge freeze 8-12 grids in 2-3 hours depending on the complexity of sample preparation for freezing and incubation times required. Frozen samples can be stored for months in liquid nitrogen before transferring to the microscope for imaging. A skilled microscopist can collect 5-8 tilt series in an 8-hour microscope session if the sample quality is good. It is possible to remove a cryo-grid from the microscope and save it for future imaging, but this increases the chances of accumulating ice contamination and damage to the grid. It is better to plan ahead so that you have enough time to collect all the data you need in one session.
With today’s relatively fast processors, tomogram reconstruction will take an experienced operator less than one hour per tilt series. Post-reconstruction filtering takes about 10-20 minutes to set up, but it can take up to a few hours to run, depending on the computer’s ability to use parallel computing, processor number and speed, and the size of your tomogram.
Modeling to select particles for volume averaging can take anywhere between a few hours and a few days depending on the structure you are modeling, the number of datasets you will be selecting particles from, and whether you are able to automatically fill in model points or have to model each particle by hand. Using the graphic user interface to set up a PEET run will generally also only take 10-20 minutes, while computing the alignment and averaging will take hours or days to complete depending on processor number and speed and the number of particles being averaged. Bear in mind that you may need to carry out multiple rounds of averaging to optimize the search parameters for your data.
Acknowledgments
The authors would like to thank members of the Boulder 3-D lab; Eileen O’Toole, Cindi Schwartz, Mary Morphew, Cedric Bouchet-Marquis, Robert Kirmse, and David Mastronarde, as well as Dick McIntosh for critically reading the manuscript and providing valuable feedback. J. Heumann and A. Hoenger are supported by grant NIH/NCRR-P41-RR000592. J. Cope is supported by the National Institutes of Health / University of Colorado Molecular Biophysics Training Grant NIH T32 GM-065103.
Literature Cited
- Aebi U, Pollard TD. A glow discharge unit to render electron microscope grids and other surfaces hydrophilic. J Electron Microsc Tech. 1987;7:29–33. doi: 10.1002/jemt.1060070104. [DOI] [PubMed] [Google Scholar]
- Al-Amoudi A, Diez DC, Betts MJ, Frangakis AS. The molecular architecture of cadherins in native epidermal desmosomes. Nature. 2007;450:832–837. doi: 10.1038/nature05994. [DOI] [PubMed] [Google Scholar]
- Amat F, Moussavi F, Comolli LR, Elidan G, Downing KH, Horowitz M. Markov random field based automatic image alignment for electron tomography. J Struct Biol. 2008;161:260–275. doi: 10.1016/j.jsb.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Bartesaghi A, Sprechmann P, Liu J, Randall G, Sapiro G, Subramaniam S. Classification and 3D averaging with missing wedge correction in biological electron tomography. J Struct Biol. 2008;162:436–450. doi: 10.1016/j.jsb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Forster F, Ecke M, Plitzko JM, Melchior F, Gerisch G, Baumeister W, Medalia O. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science. 2004;306:1387–1390. doi: 10.1126/science.1104808. [DOI] [PubMed] [Google Scholar]
- Bouchet-Marquis C, Fakan S. Cryoelectron microscopy of vitreous sections: a step further towards the native state. Methods Mol Biol. 2009;464:425–439. doi: 10.1007/978-1-60327-461-6_24. [DOI] [PubMed] [Google Scholar]
- Bouchet-Marquis C, Hoenger A. Cryo-electron tomography on vitrified sections: a critical analysis of benefits and limitations for structural cell biology. Micron. 2011;42:152–162. doi: 10.1016/j.micron.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Bouchet-Marquis C, Starkuviene V, Grabenbauer M. Golgi apparatus studied in vitreous sections. J Microsc. 2008;230:308–316. doi: 10.1111/j.1365-2818.2008.01988.x. [DOI] [PubMed] [Google Scholar]
- Bouchet-Marquis C, Zuber B, Glynn AM, Eltsov M, Grabenbauer M, Goldie KN, Thomas D, Frangakis AS, Dubochet J, Chretien D. Visualization of cell microtubules in their native state. Biol Cell. 2007;99:45–53. doi: 10.1042/BC20060081. [DOI] [PubMed] [Google Scholar]
- Castaño Díez D, Scheffer M, Al-Amoudi A, Frangakis AS. Alignator: a GPU powered software package for robust fiducial-less alignment of cryo tilt-series. J Struct Biol. 2010;170:117–126. doi: 10.1016/j.jsb.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Cope J, Gilbert S, Rayment I, Mastronarde D, Hoenger A. Cryo-electron tomography of microtubule-kinesin motor complexes. J Struct Biol. 2010;170:257–265. doi: 10.1016/j.jsb.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Kanamaru S, Marko M, Nagayama K. Zernike phase contrast cryo-electron tomography. J Struct Biol. 2010;171:174–181. doi: 10.1016/j.jsb.2010.03.013. [DOI] [PubMed] [Google Scholar]
- De Rosier DJ, Klug A. Reconstruction of Three Dimensional Structures from Electron Micrographs. Nature. 1968;217:130–134. doi: 10.1038/217130a0. [DOI] [PubMed] [Google Scholar]
- Dubochet J. The physics of rapid cooling and its implications for cryoimmobilization of cells. Methods Cell Biol. 2007;79:7–21. doi: 10.1016/S0091-679X(06)79001-X. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, Mcdowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Booy FP, Freeman R, Jones AV, Walter CA. Low temperature electron microscopy. Annu Rev Biophys Bioeng. 1981;10:133–149. doi: 10.1146/annurev.bb.10.060181.001025. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Zuber B, Eltsov M, Bouchet-Marquis C, Al-Amoudi A, Livolant F. How to “read” a vitreous section. Methods Cell Biol. 2007;79:385–406. doi: 10.1016/S0091-679X(06)79015-X. [DOI] [PubMed] [Google Scholar]
- Frangakis AS, Hegerl R. Noise reduction in electron tomographic reconstructions using nonlinear anisotropic diffusion. J Struct Biol. 2001;135:239–250. doi: 10.1006/jsbi.2001.4406. [DOI] [PubMed] [Google Scholar]
- Frank J. Electron Tomography: Methods for Three-Dimensional Visialization of Structures in the Cell. Second Edition. Springer; New York, N.Y: 2006. [Google Scholar]
- Gilbert P. Iterative methods for the three-dimensional reconstruction of an object from projections. J Theor Biol. 1972a;36:105–117. doi: 10.1016/0022-5193(72)90180-4. [DOI] [PubMed] [Google Scholar]
- Gilbert PF. The reconstruction of a three-dimensional structure from projections and its application to electron microscopy. II. Direct methods. Proc R Soc Lond B Biol Sci. 1972b;182:89–102. doi: 10.1098/rspb.1972.0068. [DOI] [PubMed] [Google Scholar]
- Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- Herman GT, Lent A, Rowland SW. ART: mathematics and applications. A report on the mathematical foundations and on the applicability to real data of the algebraic reconstruction techniques. J Theor Biol. 1973;42:1–32. doi: 10.1016/0022-5193(73)90145-8. [DOI] [PubMed] [Google Scholar]
- Heymann JB, Cardone G, Winkler DC, Steven AC. Computational resources for cryo-electron tomography in Bsoft. J Struct Biol. 2008;161:232–242. doi: 10.1016/j.jsb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoog JL, Schwartz C, Noon AT, O’toole ET, Mastronarde DN, Mcintosh JR, Antony C. Organization of interphase microtubules in fission yeast analyzed by electron tomography. Dev Cell. 2007;12:349–361. doi: 10.1016/j.devcel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Iancu CV, Tivol WF, Schooler JB, Dias DP, Henderson GP, Murphy GE, Wright ER, Li Z, Yu Z, Briegel A, Gan L, He Y, Jensen GJ. Electron cryotomography sample preparation using the Vitrobot. Nat Protoc. 2006;1:2813–2819. doi: 10.1038/nprot.2006.432. [DOI] [PubMed] [Google Scholar]
- Jin L, Milazzo AC, Kleinfelder S, Li S, Leblanc P, Duttweiler F, Bouwer JC, Peltier ST, Ellisman MH, Xuong NH. Applications of direct detection device in transmission electron microscopy. J Struct Biol. 2008;161:352–358. doi: 10.1016/j.jsb.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, Mcintosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Ladinsky MS. Micromanipulator-assisted vitreous cryosectioning and sample preparation by high-pressure freezing. Methods Enzymol. 2010;481:165–194. doi: 10.1016/S0076-6879(10)81008-0. [DOI] [PubMed] [Google Scholar]
- Lučić V, Forster F, Baumeister W. Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem. 2005;74:833–865. doi: 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Marsh BJ, Mastronarde DN, Buttle KF, Howell KE, Mcintosh JR. Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc Natl Acad Sci U S A. 2001;98:2399–2406. doi: 10.1073/pnas.051631998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Medalia O, Weber I, Frangakis AS, Nicastro D, Gerisch G, Baumeister W. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science. 2002;298:1209–1213. doi: 10.1126/science.1076184. [DOI] [PubMed] [Google Scholar]
- Messaoudii C, Boudier T, Sanchez Sorzano CO, Marco S. TomoJ: tomography software for three-dimensional reconstruction in transmission electron microscopy. BMC Bioinformatics. 2007;8:288–296. doi: 10.1186/1471-2105-8-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milazzo AC, Moldovan G, Lanman J, Jin L, Bouwer JC, Klienfelder S, Peltier ST, Ellisman MH, Kirkland AI, Xuong NH. Characterization of a direct detection device imaging camera for transmission electron microscopy. Ultramicroscopy. 2010;110:744–747. doi: 10.1016/j.ultramic.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata K, Liu X, Danev R, Jakana J, Schmid MF, King J, Nagayama K, Chiu W. Zernike phase contrast cryo-electron microscopy and tomography for structure determination at nanometer and subnanometer resolutions. Structure. 2010;18:903–912. doi: 10.1016/j.str.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, Frangakis AS, Typke D, Baumeister W. Cryo-electron tomography of neurospora mitochondria. J Struct Biol. 2000;129:48–56. doi: 10.1006/jsbi.1999.4204. [DOI] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, Mcintosh JR. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- Nickell S, Forster F, Linaroudis A, Net WD, Beck F, Hegerl R, Baumeister W, Plitzko JM. TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol. 2005;149:227–234. doi: 10.1016/j.jsb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- O’Toole ET, Giddings TH, Mcintosh JR, Dutcher SK. Three-dimensional organization of basal bodies from wild-type and delta-tubulin deletion strains of Chlamydomonas reinhardtii. Mol Biol Cell. 2003;14:2999–3012. doi: 10.1091/mbc.E02-11-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penczek PA. Three-dimensional spectral signal-to-noise ratio for a class of reconstruction algorithms. J Struct Biol. 2002;138:34–46. doi: 10.1016/s1047-8477(02)00033-3. [DOI] [PubMed] [Google Scholar]
- Penczek PA. Fundamentals of three-dimensional reconstruction from projections. Methods Enzymol. 2010;482:1–33. doi: 10.1016/S0076-6879(10)82001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachse C, Chen JZ, Coureux PD, Stroupe ME, Fandrich M, Grigorieff N. High-resolution electron microscopy of helical specimens: a fresh look at tobacco mosaic virus. J Mol Biol. 2007;371:812–835. doi: 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton WO, Baumeister W, Hahn M. Three-dimensional reconstruction of imperfect two-dimensional crystals. Ultramicroscopy. 1984;13:57–70. doi: 10.1016/0304-3991(84)90057-3. [DOI] [PubMed] [Google Scholar]
- Tivol WF, Briegel A, Jensen GJ. An improved cryogen for plunge freezing. Microsc Microanal. 2008;14:375–379. doi: 10.1017/S1431927608080781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel M, Schatz M. Fourier shell correlation threshold criteria. J Struct Biol. 2005;151:250–262. doi: 10.1016/j.jsb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Vanhecke D, Asano S, Kochovski Z, Fernandez-Busnadiego R, Schrod N, Baumeister W, Lučić V. Cryo-electron tomography: methodology, developments and biological applications. J Microsc. 2010 doi: 10.1111/j.1365-2818.2010.03478.x. A current review discussing sample-preparation, data acquisition, data processing and interpretation and some recent examples of biological applications using cryo-ET. [DOI] [PubMed] [Google Scholar]
- Winkler H, Taylor KA. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy. 2006;106:240–254. doi: 10.1016/j.ultramic.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN. CTF determination and correction for low dose tomographic tilt series. J Struct Biol. 2009;168:378–387. doi: 10.1016/j.jsb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Riches JD, Fuller SD, Briggs JA. Contrast transfer function correction applied to cryo-electron tomography and sub-tomogram averaging. J Struct Biol. 2009;168:305–312. doi: 10.1016/j.jsb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell. 2010;141:472–482. doi: 10.1016/j.cell.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Keszthelyi B, Branlund E, Lyle JM, Braunfeld MB, Sedat JW, Agard DA. UCSF tomography: an integrated software suite for real-time electron microscopic tomographic data collection, alignment, and reconstruction. J Struct Biol. 2007;157:138–147. doi: 10.1016/j.jsb.2006.06.005. [DOI] [PubMed] [Google Scholar]