

Graphical abstract

Abbreviations: 6-ECDCA, 6-ethylchenodeoxycholic acid; AF, activation function; BSEP, bile salt export pump; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; DMEM, Dulbecco’s Modified Eagle’s Medium; EF, enrichment factor; FBS, fetal bovine serum; FXR, farnesoid X receptor; H-bond, hydrogen bond; HEK-293, human embryonic kidney-293; IBABP, intestinal bile acid-binding protein; LCA, lithocholic acid; LRH-1, liver receptor homolog 1; LXR, liver X receptor; NCI, National Cancer Institute; PDB, Protein Data Bank; RXR, 9-cis-retionic acid receptor; SD file, structure-data file; SHP-1, small heterodimer partner 1; WDI, World Drug Index
Keywords: Farnesoid X receptor, Molecular modeling, Virtual screening, Lead identification, FXR agonist
Abstract
The farnesoid X receptor (FXR) is involved in glucose and lipid metabolism regulation, which makes it an attractive target for the metabolic syndrome, dyslipidemia, atherosclerosis, and type 2 diabetes. In order to find novel FXR agonists, a structure-based pharmacophore model collection was developed and theoretically evaluated against virtual databases including the ChEMBL database. The most suitable models were used to screen the National Cancer Institute (NCI) database. Biological evaluation of virtual hits led to the discovery of a novel FXR agonist with a piperazine scaffold (compound 19) that shows comparable activity as the endogenous FXR agonist chenodeoxycholic acid (CDCA, compound 2).
1. Introduction
The farnesoid X receptor (FXR, NR1H4), a ligand-dependent transcription factor, is a member of the nuclear hormone receptor superfamily comprising a highly conserved DNA-binding domain in the N-terminal region and a moderately conserved ligand-binding domain in the C-terminal region.1 Two activation functions, one ligand-independent (AF-1) and one ligand-dependent (AF-2), are situated in the N-terminal and C-terminal regions, respectively.1 Two cysteine-co-ordinated Zink fingers located in the DNA-binding domain are crucial for DNA binding and dimerization.1 For the regulation of gene expression the nuclear receptor FXR forms either a heterodimer with RXR, the 9-cis-retionic acid receptor, or a homodimer.2 FXR, as a sensor for bile acids, is strongly involved in the regulation of target gene expression which is crucial for bile acid biosynthesis and transport.3 Upon bile acid binding, the nuclear receptor FXR undergoes conformational changes and induces the expression of the small heterodimer partner 1 (SHP-1), an atypical member of the nuclear receptor family that lacks a DNA-binding domain.4 SHP-1 represses the expression of CYP7A1, the rate-limiting enzyme in the conversion of cholesterol into bile acids, by inhibiting the activity of the liver X receptor (LXR) and the liver receptor homolog 1 (LRH-1; NR5A2).4–6 Moreover, the activated FXR regulates genes encoding the intestinal bile acid-binding protein (IBABP) and the bile salt export pump (BSEP).5,7,8 FXR, with its central function in the physiological maintenance of bile acid homeostasis, is expressed in liver, intestine, kidney, and other cholesterol-rich tissues such as adrenal glands.9
Key bile acids in humans comprise the group of primary bile acids with cholic acid (CA, 1), chenodeoxycholic acid (CDCA, 2), as well as their glycine and taurine conjugates, and the group of secondary bile acids with deoxycholic acid (DCA, 3) and lithocholic acid (LCA, 4) (Chart 1).9
Chart 1.

Examples for endogenous (1–4) and synthetic (5–10) FXR ligands.
Several synthetic FXR ligands (Chart 1), that is, GW-4064 (5), 6-ethylchenodeoxycholic acid (6-ECDCA, 6), AGN-29 (7), and AGN-31 (8) have been generated for biomolecular investigations.10–12 It could already be shown that synthetic FXR agonists protect against the development of aortic plaques formation in murine models characterized by a pro-atherogenic lipoprotein profile and accelerated atherosclerosis.13 In addition to these and other synthetic compounds14–18 several natural products have been found to act as potent FXR ligands and modulators, respectively. These compounds include E- and Z-guggulsterone (9),19 stigmasterol, and its 3-acetate (10, Chart 1).20
Its regulative role in glucose and lipid metabolism in addition to its complex architecture renders the nuclear receptor FXR an attractive target for the discovery of novel bioactive compounds with the ability to control endogenous pathways in close relation to diseases, including the metabolic syndrome, dyslipidemia, atherosclerosis, and type 2 diabetes.21,22 In order to identify novel FXR agonists, a virtual screening workflow based on pharmacophore modeling was established.
Structure- and ligand-based pharmacophore modeling and subsequent virtual screening is a well-established workflow to identify new lead structures from compound libraries.23–25 The most critical step hereby is the theoretically sound validation of the screening models. Data sets for model validation, so-called test sets, can be manually collected from literature or readily taken from bioactivity databases. Until recently, several data sets containing thousands of bioactive compounds with activity data from literature, for example, the Derwent World Drug Index (WDI, www.thomsonreuters.com), the WOMBAT database (www.sunsetmolecular.com), and the MDDR database (www.symyx.com) have only been commercially available. With the start of the PubChem project26 the tide shifted and a large amount of biological data became available at no charge to the scientific community. Currently, bioactivities for about 1000 targets (mostly HTS data) are included in the PubChem BioAssay database. For these pharmaceutical targets, the PubChem contains a lot of useful information. A big advantage is the availability of large sets of confirmed inactive compounds which enable a thorough validation of virtual screening predictions. However, many targets are not contained in the PubChem. Additionally, often only data from cell-based assays (e.g., the overall effect on the growth rate of a tumor cell line is measured) or qualitative data from high-throughput screening assays are published. For many highly interesting targets, for which hundreds of compounds are found in recent literature, only a handful compounds are included in PubChem, for example, for 11-β-hydroxysteroid-dehydrogenase type 1, where just seven compounds are found in the PubChem. For molecular modeling studies, the knowledge about the molecular mode of action and the potency of compounds is essential. Therefore, the exploitation of the PubChem data for virtual screening studies is limited for many pharmaceutical targets.
Recently, the ChEMBL database, which is the successor of the Starlite database, was made publicly available.27 It is an open access source for compounds with their associated biological data. This project is funded by the Wellcome Trust, the EMBL member states, the EU Innovative Medicines Initiative, and the EU Framework 7 Program. The ChEMBL is an online database of 622,824 bioactive, drug-like, small molecules (version 02, November 2009). The database contains 2D structures, calculated properties such as molecular weight and Lipinski parameters, and information on bioactivities (e.g., binding constants, inhibition constants, and ADMET data). These data are extracted from the primary literature including some of the most important medicinal chemistry journals (e.g., J Med Chem, Bioorg Med Chem, Bioorg Med Chem Lett) and other leading publications (e.g., Nature and Science). From this point of view, the ChEMBL very much resembles the WOMBAT database, but contains a larger amount of validated information. Recently, PubChem now also contains the ChEMBL bioactivity data sets, which makes it convenient to search for activity data using just one search portal. In this study, the ChEMBL was analyzed for its composition and separated in target-specific subsets. Additionally, it was used besides other data sets to validate the generated FXR pharmacophore models.
No 3D pharmacophore-based virtual screening study on FXR has been published before. In this study, we systematically developed a structure-based pharmacophore model collection, theoretically validated it on active FXR ligands and decoys from the ChEMBL, and used it to virtually screen the National Cancer Institute (NCI) database.28 Selected virtual hits from the NCI were biologically evaluated, which led to the identification of a novel scaffold that could serve as lead structure for FXR agonist design.
2. Results and discussion
2.1. Data sets
2.1.1. ChEMBL database
The ChEMBL database is readily available as downloadable structure-data (SD) file (www.ebi.ac.uk/chembldb/). However, this file does not include bioactivity information. Accordingly, structure and bioactivity data were merged as described in the experimental section. The data merging and filtering process resulted in a database with 302,924 (version 02) unique entries, all with annotated biological activity. For the majority of compounds, one or two activities on biological targets are included. However, there is also a considerable number of compounds with more than ten indicated bioactivities (Fig. 1).
Figure 1.

Number of targets reported per ChEMBL compound.
The ChEMBL reports data on 3,485 differently annotated pharmacological targets. However, some targets are present more than once, with similar names; for example, 11-β-hydroxysteroid-dehydrogenase-1/11-beta-hydroxysteroid-dehydrogenase type 1, alpha-l-fucosidase-1/alpha-l-fucosidase-I, or carbonic-anhydrase-1/carbonic-anhydrase-I, to name a few.
2.1.2. FXR-actives
The set of active FXR ligands, that will further be referred to as ‘FXR-actives’ was extracted from the ChEMBL database using the workflow described in the experimental section. In the ChEMBL, FXR ligands are annotated as ‘bile acid receptor FXR’ ligands. Out of the initially 326 FXR ligands from ChEMBL, only unique ligands that were reported as active (EC50 <100 μM) were kept. As this was the pilot study using the ChEMBL database, nothing was known about the error rate regarding the reported molecular structures and bioactivities. Therefore, the 2D structures and the biological data were manually compared to the values from the original literature (Supplementary data Refs. 1–16). Remarkably, all remaining 224 2D structures and reported biological activities in the FXR-actives ChEMBL subset perfectly matched the data reported in literature. Where more than one FXR activity value was reported for a ligand, the least active one was found in the ChEMBL. The final 3D multiconformational FXR-actives data set comprised 221 compounds.
2.1.3. FXR-decoys
The FXR-decoy set was extracted from the ChEMBL database. Compounds with similar physicochemical properties like the FXR-actives (molecular weight, c log P, number of hydrogen bond donors, number of hydrogen bond acceptors, and number of aromatic rings) were selected. The final FXR-decoy database comprised 5598 entries.
2.1.4. DrugBank
The DrugBank is a publicly available database that links drug information with target data.29 Currently, the DrugBank contains about 4,800 small molecule entries including over 1,350 FDA-approved small molecules and over 3,240 experimental drugs, besides smaller compound groups.
2.1.5. Virtual DB
Previously, we have generated a virtual database using the program ilib diverse (www.inteligand.com) that contains 12,775 presumably inactive decoy molecules.30 This data set – the so-called Virtual DB – has been designed to include nonreactive, drug-like compounds and is frequently used in our group to assess a model’s ability to discard inactive compounds in a database screen.
2.1.6. NCI database
The compound database of the NCI is part of the Developmental Therapeutic Program. As a part of the whole concept, it offers small molecule compounds to research institutions free of charge. The database is available as SD file which can be downloaded and used for virtual screening experiments.
2.2. Analysis of the ChEMBL database and comparison with other databases
In order to evaluate the suitability of the ChEMBL database as benchmarking data set for virtual screening aiming at lead discovery, the physicochemical property distribution of the ChEMBL compounds was calculated, analyzed, and compared to marketed and investigational drugs from the DrugBank. In order to additionally get insights into the physicochemical property distribution our Virtual DB, that we frequently use as decoy set for model validation, we also compared this data sets with the ChEMBL and the DrugBank compounds.
The generated ChEMBL database contains 302,924 unique chemical structures. About two third – 196,381 compounds – match Lipinski’s Rule of Five criteria for drug-likeness.31 With respect to the definition of Irwin et al.32 56,086 compounds are lead-like possessing a molecular weight between 150 and 350, a maximum number of hydrogen bond (H-bond) donors of 3, at most 6 H-bond acceptors, and a c log P lower than 4. Finally, 10,085 structures are fragment-like with a molecular weight less than 250, a number of H-bond donors less than 3, a number of H-bond acceptors less than 6, a c log P between −2 and 3, and a number of rotatable bonds less than 3.33 A detailed distribution of molecular properties and the number of violations of Lipinski’s Rule of Five are shown in Figure 2.
Figure 2.

Molecular property distribution of the ChEMBL database compounds compared with the DrugBank and Virtual DB.
When comparing the DrugBank, the ChEMBL, and the Virtual DB, the numbers of H-bond acceptors and donors, the c log P values, the number of rotatable bonds, and the number of Lipinski Rule of Five violations are similarly distributed in the respective databases. Remarkably, the DrugBank contains the largest fraction of very rigid compounds bearing no or only 1 and 2 rotatable bonds. The most pronounced differences can be seen in the molecular weight distribution, where the ChEMBL includes more compounds with a high molecular weight. The DrugBank includes the highest fraction of small compounds among the three databases. The Virtual DB was optimized in molecular property distribution during database generation following guidelines for druglikeness30 and therefore it is not surprising that it follows a smooth Gaussian-like curve in molecular weight distribution.
2.3. Analysis of FXR ligand binding modes
FXR is known to accommodate a flexible ligand binding domain, where ligand binding changes the position of the AF-12 helix and thus the recruitment of co-activator or -repressor proteins. For pharmacophore model generation, a systematic understanding of ligand binding and the elucidation of essential amino acids participating in the process is crucial. Therefore, the published X-ray crystal structures of FXR-ligand complexes available at the time of this study were extracted from the Protein Data Bank34 (PDB, entries 1osh,35 3bej,36 3dct,37 3dcu,37 3fli,38 3fxv,39 3gd2,40 3hc5,41 and 3hc6;41 Fig. 3) and analyzed for their protein–ligand interactions (Table 1).
Figure 3.

FXR agonists that have been co-crystallized with human FXR and served for binding mode analysis and training compounds for structure-based pharmacophore model generation. The corresponding PDB code is given for each compound and chemical interactions are shown in 2D.
Table 1.
Protein–ligand interactions observed in FXR–agonist complexes from the PDB. Hydrophobic interactions are indicated as H, H-bond acceptors/donors as HBA/D, and negatively ionizable groups as NI
| Amino acid | 1osh⁎ | 3fli⁎ | 3bej | 3dct | 3dcu | 3fxv⁎ | 3gd2 | 3hc5 | 3hc6 |
|---|---|---|---|---|---|---|---|---|---|
| Met265 | H | H | HBA, H | HBA, H | H | HBA, H | H | H | |
| Ile269 | H | ||||||||
| Thr270 | HBA, H | H | H | H | H | H | |||
| Ile273 | H | H | |||||||
| Phe284 | H | H | H | H | H | H | H | H | H |
| Leu287 | H | H | H | H | H | H | H | H | H |
| Thr288 | HBA/D, H | H | H | H | H | H | H | ||
| Met290 | H | H | H | H | H | H | H | H | H |
| Ala291 | H | H | H | H | H | H | H | H | H |
| His294 | HBA | HBA | |||||||
| Val325 | H | H | H | H | H | ||||
| Met328 | H | H | H | H | H | H | H | H | H |
| Phe329 | H | H | H | H | H | H | H | H | |
| Arg331 | H | NI, HBA | NI, HBA | NI, HBA, H | NI, HBA, H | NI, HBA, H | NI, HBA, H | NI, HBA, H | |
| Phe333 | HBA | ||||||||
| Ile335 | H | H | H | H | H | H | H | H | H |
| Phe336 | H | H | |||||||
| Leu340 | H | ||||||||
| Ser342 | H | ||||||||
| Leu348 | H | H | H | H | H | H | H | ||
| Leu352 | H | H | H | H | H | ||||
| Ile352 | H | H | H | H | |||||
| Ile357 | H | H | H | H | H | H | H | H | |
| Tyr361 | H | ||||||||
| Ile362 | H | ||||||||
| Met365 | H | H | H | H | H | H | H | H | |
| Phe366 | H | ||||||||
| Tyr369 | HBA, H | ||||||||
| His447 | HBA | ||||||||
| Met450 | H | H | H | H | H | ||||
| Leu451 | H | ||||||||
| Trp454 | H | H | H | H | H | H | |||
| Phe461 | H | H | H | ||||||
| Trp469 | H | H | H | H | H | H |
The nine PDB entries represent four structural classes: y-shaped hydrophobic compounds (1osh), tricyclic heteroaromatic compounds (3fli), steroids (3bej), and derivatives of compound 5 (3dcu = compound 5, derivatives: 3dct, 3gd2, 3fxv, 3hc5, and 3hc6). The latter share very similar binding modes (Figs. 3 and 4, Table 1); thus, these structures were represented by a single model, while for the other PDB entries, one model was created for each structure.
Figure 4.

Comparison of FXR ligand binding modes and conformational changes of the binding pocket. (A) Binding modes of 5 (3dct) and its derivatives 14 and 16–18. Hydrophobic areas on the ligands are indicated in yellow, the charged interaction with Arg331 is depicted as red star, and H-bonds with Met265 and Arg331 are highlighted by red arrows. (B) Comparison of binding modes for 11 (black) and 15 using the 3fxv binding pocket. Compound 11 sterically clashes with the binding site in the 3fxv conformation. (C) Compound 13 fitted into the 3dct binding pocket, sharing a large part of it. The acidic functions differ in location and observed interactions. (D) Compound 12 fitted into the 3fxv binding pocket. The difluorophenyl moiety of 12 sterically clashes with the binding pocket conformation from 1fxv.
For 1osh and 3fli, conformational changes of the binding pocket and the observation of different interaction patterns between the ligand and the protein were observed. In addition, a 15 amino acid long flexible loop between helices 1 and 3 is completely distorted and therefore missing in the X-ray structure 1osh. Accordingly, 1osh and 3fli have to be treated separately for model building. The steroidal ligand from 3bej fits well into the compound 5-formed binding site. However, the location of the acidic function differs so that it establishes other interactions with the protein. In addition, 5-based ligands occupy a larger part of the binding site and thus form interactions that are not observed for 3bej. Therefore, 3bej was also subject of separate model generation.
2.4. Pharmacophore model generation and validation
Pharmacophore models based on the PDB entries 1osh, 3bej, 3fli, and 3dct, respectively, were generated using LigandScout software (www.inteligand.com). From each entry, several models were built that differed in the number and composition of chemical features, so that distinctive binding modes were reflected by the models. The resulting models were exported into DiscoveryStudio for optimization and virtual screening. For each model, one version containing a shape restriction was generated. All pharmacophore models were validated by calculating enrichment factors (EFs) using the FXR-actives data set and the complete ChEMBL as decoy set (best models see Table 2). For comparison, EFs using the FXR-decoys, the Virtual DB, and the DrugBank as decoy sets were calculated as well.
Table 2.
Performance of the generated pharmacophore models in screening the FXR-actives dataset and the validation DBs ChEMBL, DrugBank, and the Virtual DB. The model quality was assessed by EF calculations
| Model | Hits FXR-actives (n = 221) | Hits ChEMBL (n = 304,052 w/o FXR-actives) | EF ChEMBL/model rank | Hits DrugBank (n = 4,722) | EF DrugBank/model rank | Hits Virtual DB (n = 12,775) | EF Virtual DB/model rank | Hits FXR decoys (n = 5,598) | EF FXR decoys/model rank |
|---|---|---|---|---|---|---|---|---|---|
| 1osh-1 | 51 | 10,139 | 6.9/9 | 73 | 9.2/9 | 99 | 19.2/8 | 526 | 2.3/10 |
| 1osh-1-s | 34 | 350 | 121.9/3 | 2 | 21.1/4 | 5 | 51.3/6 | 17 | 17.6/4 |
| 1osh-2 | 69 | 2688 | 34.5/6 | 13 | 18.8/5 | 32 | 40.2/7 | 159 | 8.0/6 |
| 1osh-2-s | 44 | 114 | 383.4/2 | 2 | 21.4/3 | 2 | 56.2/5 | 7 | 22.7/2 |
| 3bej-1 | 7 | 777 | 12.3/8 | 5 | 13.0/8 | 0 | 58.8/1 | 27 | 5.4/8 |
| 3bej-1-s | 7 | 648 | 14.7/7 | 4 | 14.2/6 | 0 | 58.8/1 | 24 | 5.9/7 |
| 3bej-2 | 119 | 33280 | 4.9/10 | 316 | 6.1/10 | 429 | 12.8/9 | 982 | 2.8/9 |
| 3bej-2-s | 100 | 1552 | 83.3/5 | 66 | 13.5/7 | 415 | 11.4/10 | 57 | 16.8/5 |
| 3dct | 57 | 734 | 99.2/4 | 2 | 21.6/2 | 2 | 56.8/4 | 23 | 18.8/3 |
| 3dct-s | 43 | 12 | 1076.4/1 | 0 | 22.4/1 | 0 | 58.8/1 | 1 | 25.7/1 |
| 3fli | 143 | 61,788 | 3.2/12 | 427 | 5.6/12 | 1572 | 4.9/11 | 2473 | 1.4/11 |
| 3fli-s | 135 | 55,965 | 3.3/11 | 381 | 5.9/11 | 1542 | 4.7/12 | 2396 | 1.4/12 |
| All | 194 | 82,006 | 3.2 | 654 | 5.1 | 1895 | 5.5 | 2927 | 1.6 |
| All without 3fli-models | 162 | 40,979 | 5.4 | 374 | 6.8 | 531 | 13.7 | 1389 | 2.8 |
2.5. Description of the generated pharmacophore models
2.5.1. 1osh-1(-s) and 1osh-2-(s)
The first pharmacophore model derived from 1osh (1osh-1) consisted of five hydrophobic features, one H-bond acceptor with His294, and 27 exclusion volume spheres (Fig. 5A). Due to the missing loop between helices 1 and 3, no exclusion volume spheres could be placed on this part of the binding site. However, as this region is considered as highly flexible, it could adapt to the size of a ligand and thereby not be a strict sterical restriction for ligand binding. The model mainly recognized fexaramine analogs from the FXR-actives data set. However, also one steroidal hit with an EC50 of 11.6 μM returned from the FXR-actives set. According to literature data, hydrophobic contacts are established between the methyl ester group and the FXR binding pocket. Thus, a model containing this additional feature was created. However, this model was very restrictive. In order to provide a less restrictive model, the hydrophobic feature on the dimethylamine group was deleted. The resulting model (1osh-2) performed well in the theoretical validation. Although the 1osh-1 and 1osh-2 models are very similar to each other, their FXR-actives hitlists differed from each other. Eight out of 51 FXR-actives hits from the 1osh-1 model were not recognized by 1osh-2. Vice versa, 26 out of 69 FXR-actives hits from the 1osh-2 model did not fit into the 1osh-1 model. Thus, both models were kept in the final model collection. The comparison of the shape-containing models revealed similar results (eight out of 34 and 18 out of 44 unique hits from the 1osh-1-s and the 1osh-2-s-based FXR-actives screenings, respectively). The shape-containing models (1osh-1-s and 1osh-2-s) outperformed the initial models with up to 18-fold higher EFs (Table 2).
Figure 5.

Pharmacophore models for FXR ligands. Chemical features are color-coded: yellow – hydrophobic, red arrow – H-bond acceptor. (A) Model derived from the binding of 11 to FXR. His294 is shown in ball-and-stick style. (B) Compound 13-based model. His294 and Arg331 are shown in ball-and-stick-style. (C) Compound 5-scaffold model. (D) Compound 12-based model. Tyr369, the H-bonding partner, lies behind the molecule in this perspective; therefore, the H-bond acceptor is not visible. For showing how much the FXR binding pocket changes upon ligand binding compared to 5, His294 and Arg331 are also depicted in ball-and-stick-style.
2.5.2. 3bej-1(-s) and 3bej-2(-s)
The initial pharmacophore model based on the PDB entry 3bej (3bej-1) included three hydrophobic features, two H-bond acceptors anchoring the ligand with His294 and Thr288, a negatively ionizable feature representing the interaction with Arg331, and 25 exclusion volume spheres (Fig. 5B). In the database search of the FXR-actives set, the 3bej-1 only returned compound 5-related hits. This reflects the binding similarities between compounds 5 and 13, although they belong to completely different chemical scaffolds. Because 3bej-1 was very restrictive, a model variation was created that only contained the chemical features on the steroidal core and the neighboring atoms. The hydrophobic feature and the H-bond acceptor on the p-hydroxyphenyl ring were deleted resulting in model 3bej-2. This reduced model correctly identified FXR ligands from diverse chemical scaffolds including steroids; however, it also returned more decoys leading to a diminished EF. The addition of a shape to the 3bej-2 model (3bej-2-s) restricted the virtual hits to those compounds that also fulfilled the geometric requirements for FXR binding, which led to a considerable EF improvement (Table 2).
2.5.3. 3dct(-s)
The model built from the PDB entry 3dct was not only based on ligand binding information from this structure, it represented the observed binding interactions of all compound 5 derivatives that have been co-crystallized with FXR. The model 3dct consisted of five hydrophobic features, one H-bond acceptor with His294, one negatively ionizable group, and 29 exclusion volume spheres (Fig. 5C). From the FXR-actives set, the model retrieved only derivatives of compound 5. Due to the strong focus on a relatively high MW compound class and the high number of chemical features, the model was very restrictive. The shape-containing version of 3dct (3dct-s) retrieved the highest EFs in this study (Table 2).
2.5.4. 3fli(-s)
Based on the PDB entry 3fli, which shows a distinct FXR ligand binding domain conformation compared with the other entries, a relatively unrestrictive 3D pharmacophore model was created. The positively charged side chain of Arg331 points away from the ligand, therefore allowing only hydrophobic interactions. His294 is not engaged in direct protein–ligand interactions. As a particularity of this structure, the H-bond with Tyr369 was kept for the model, along with four hydrophobic features and 28 exclusion volume spheres (Fig. 5D). Due to the comparably few chemical features that formed this model, many hits were retrieved from the FXR-actives set including compound 5-like compounds, steroids, and derivatives of compound 13. However, 3fli also returned the largest number of inactive decoys from the Virtual DB which led to a comparably low EF. (Table 2).
2.6. Theoretical Evaluation of the FXR model collection using the ChEMBL, FXR decoys, DrugBank, and Virtual DB
In order to assess the overall performance of the model collection, the individual hit lists from the FXR-actives, FXR-decoys, ChEMBL, DrugBank, and Virtual DB were merged for each model, respectively, and cleared from duplicate entries. In the final file, all hits from a database, for example, all FXR-actives that were retrieved by any of our models, were present once. The model collection was able to retrieve 194 out of 221 (87.8%) FXR-actives. EFs for the parallel screening of the databases were 5.5 or lower (Table 2). The impact of low quality models was assessed by discarding the 3fli(-s) models and re-evaluating the respective EFs. Still, 162 FXR-actives (73.3%) from different chemical scaffolds were correctly recognized. The rates of false positive hits decreased over 50% thereby improving EFs for all four databases (Table 2). By additionally eliminating the unrestrictive model 2bej-2, still 122 (55.2%) of FXR-actives were retained while raising the EF for the ChEMBL up to 11.5. Obviously, the inclusion of models with low restrictivity and low EF into a model collection can considerably impair the predictive power of the entire system. Accordingly, the advantage of using multiple restrictive pharmacophore models representing different binding modes for one target compared to using one quite general model covering most active ligands is a way to avoid a high number of false positive hits.
As can be seen from the EFs calculated for the four different databases (Table 2), EFs cannot be directly compared in order to get an idea how predictive a model is. The reason lies in EF calculation, which is based on the distribution between active and (putative) inactive molecules within the validation database. The maximum EF, which is reached when only active and no inactive compound are returned from database screening, is for the ChEMBL 1376.8, for the FXR-decoys 26.33, for the DrugBank 22.4, and for the Virtual DB 58.8. Therefore, a model ranking based on EFs was performed (Table 2). For most models, the obtained rank was equal or similar (±1) in the compared rankings. The best-ranked model was 3dct-s in all three rankings, the lowest ranks were assigned to the 3fli(-s) models.
EF differences were most pronounced in the ChEMBL, which allowed for a clearer ranking of models. For example, 1osh-2-s and 3dct show very close EFs in the DrugBank (21.4 vs 21.6) and in the Virtual DB (56.2 vs 56.8), respectively. Only the ChEMBL-based EF calculation revealed a considerable better performance of the 1osh-2-s model (EF 383.4 vs 99.2 for 3dct). Another big advantage of the ChEMBL is that all compounds include bioactivity information. Thus, the user is able to study retrieved hit lists in more detail and evaluate whether false positive hits belong to related targets and may therefore be active.
The most challenging data set for discriminating FXR-active from putatively inactive molecules was the FXR-decoy database. In comparison to the DrugBank-based validation (which has a similar maximum EF as the FXR-decoys), most EFs were lower when using the FXR-decoy set. However, some models were still able to considerably enrich active compounds in the FXR-active versus FXR-decoy validation experiments (Table 2). EFs of around 17 and up were achieved with the models 1osh-1-s, 1osh-2-s, 3bej-2-s, and 3dct(-s), pointing towards a highly favorable impact of the shape restriction to the models performance.
2.7. Model collection evaluation with FXR agonists from recent literature
The ChEMBL dataset in its used version (02) did not contain the most recent ligands from literature. For a theoretical model evaluation that was not relied on the ChEMBL database, FXR agonists were collected from the most recent literature, starting December 2009. Only two papers have been published at the time of this study: Iguchi et al.42 reported on steroidal, bile acid-related FXR agonists, while Lundquist et al.43 presented tetrahydroazepinoindole derivatives (Supplementary data, Chart S-1). The 15 most active compounds from both papers were prepared for virtual screening and submitted to pharmacophoric profiling by the FXR pharmacophore model collection. All but one out of 15 compounds were correctly retrieved by at least one of the models. These results further confirmed the accuracy of the pharmacophore model collection.
2.8. Selection of virtual hits for biological testing
In order to experimentally verify the quality of high-quality models, virtual hits were selected for biological testing. The 3dct(-s) models were not able to find compounds classically considered as drug-like according to their physicochemical properties (compare Fig. 2). The reason for this lies in the high number of chemical features forming the model, making it very specialized for compound 5-like molecules. Although it can’t be ruled out that the 3dct(-s) models find interesting hits in compound databases, that was not the case for the NCI database. The 3fli(s) models and 3bej-2 showed the lowest EFs for all validation databases (Table 2) and were therefore also discarded for further virtual screening studies. Of the remaining models, those with the best EFs (compare Table 2), that is, the shape-containing models, and a high restrictivity were employed to select virtual hits from the NCI database. 1osh-1-s returned 126 hits, 1osh-2-s 59 hits, and 3bej-1-s 32 hits. Among the 217 total hits, 170 were very flexible with more than 10 rotatable bonds. These compounds were excluded from further investigations. The remaining 47 hits were visually inspected for their predicted binding pose. Based on these results and the current compound availability at the NCI, eight structurally diverse compounds were submitted to in vitro evaluation of their agonistic effect on human FXR. Four compounds were taken from the 1osh-1-s hitlist, the other four from the 3bej-1-s hitlist (Chart 2). All compounds that were selected to validate hits from the 1osh-2-s hitlist were not available at the time of our request.
Chart 2.

Virtual hits from the models 1osh-1-s and 3bej-1-s that were tested for biological activity in the FXR transactivation assay.
2.9. Results of the biological evaluation of virtual hits
Biological evaluation of the substances with respect to their ability to activate FXR was performed in HEK293 cells transiently transfected with elements of the FXR reporter assay system comprising the ECRE-Luc reporter plasmid containing quintuple RXR:FXR binding site and the respective expression plasmids for full length murine FXR and RXRα. The cells were treated with increasing concentrations (0–100 μM) of the specific FXR ligand 2, or with the selected virtual hits 19–26, respectively. A dose–dependent FXR-agonism was observed for compounds 19 and 20, whereas 21–26 were inactive. Activation of FXR by the respective ligand reached statistical significance between 1 and 10 μM, at concentration of 100 μM the system was saturated. The estimated EC50 values of the novel agonists were with 3.15 μM for 19 and 6.17 μM for 20 more active than substance 2 (16.8 μM) as shown in Figure 6. Compound 19 represents a so-far novel chemical scaffold for FXR agonists (see ‘novelty assessment of compound 19’ in Section 4). Therefore, it can serve as a lead structure for developing a new series of more potent FXR agonists.
Figure 6.
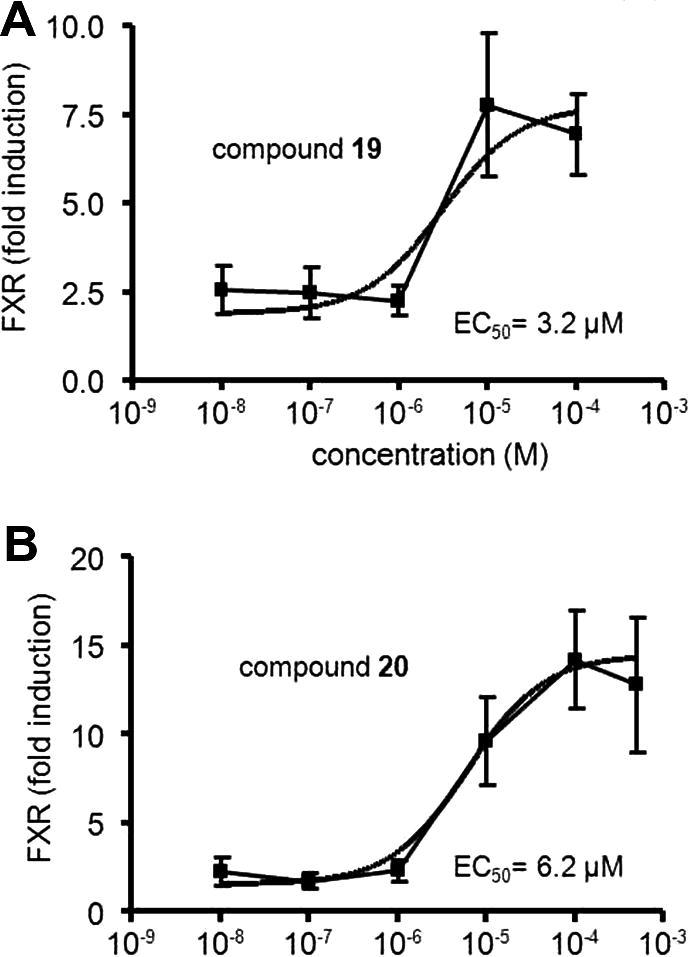
Reporter gene assay for FXR activation. Application of increasing concentrations (10 nM–100 μM) of the virtual hits 19 (A) and 20 (B), respectively, leads to dose dependent signal increase. FXR activity is expressed as fold difference of relative luciferase units (RLU) represented by mean values ± SEM resulting from triplicate determinations of three independent experiments (∗P <0.05, ∗∗P <0.01).
In order to obtain an independent evidence of the ability of 19 and 20 to activate FXR, we tested their effects on the expression of a gene known to be downstream of FXR. CYP7A1 expression is down-regulated by FXR via a well-characterized molecular mechanism involving SHP and LRH-1, and is widely used to monitor activity of endogenously expressed FXR.44 In full agreement with our promoter–reporter data, the two compounds significantly decreased levels of CYP7A1 mRNA; the degree of inhibition was similar to that induced by the classical FXR agonist 2 (Fig. 7).
Figure 7.

Expression of CYP7A1 mRNA. HepG2 cells treated with 19 and 20 (50 μM) for 6 h in DMEM containing 10% FCS; data expressed as fold of control cells treated with medium/0.1% DMSO (∗P <0.05, ∗∗P <0.01, ∗∗∗P <0.001).
In addition, it was investigated if there have already been reports on the biological properties of 19. In the literature, this compound has never been reported before. The enhanced NCI database browser (129.43.27.140/ncidb2/), where biological data from in-house screens by the NCI are reported, no experimental results were given for compound 19. Additionally, the structural topology of 19 was compared to known bioactive molecules from the WOMBAT and MDDR databases using the publicly available similarity search portal at sea.docking.org.45 Finally, the 1-(4-methylpiperazin-1-yl)-3-phenoxypropan-2-ol core structure was used as substructure search query in the online version of the ChEMBL DB (version 09). All 371 compounds that returned from this search were inspected for any reported activities on nuclear receptors. Also in these experiments, the compound or related molecules from this scaffold were not associated with FXR or any other nuclear receptor, underscoring the novelty of this chemical scaffold for FXR agonist design.
2.10. Mapping of compound 19 into the FXR binding pocket
The mapping conformation of compound 19 was fitted and energetically minimized into the FXR binding site. The resulting binding pose suggested a similar interaction pattern compared to the co-crystallized ligand compound 11, however, additional hydrogen bonds and fewer hydrophobic interactions were predicted (Fig. 8). Compound 19 was observed to be anchored within the receptor by additional hydrogen bonds to Ser332 and Tyr369 compared to the binding mode of compound 11. Although the piperazine ethylene moiety of 19 was mapped into a hydrophobic feature in the pharmacophore-based search, no such interactions could be observed in the minimized binding pose prediction. In fact, piperazine rather increases the hydrophilicity of a compound due to its basic character. The piperazine moiety can therefore be seen as a spacer linking two hydrophobic parts of the molecule, which enhances the compound’s solubility.
Figure 8.

Mapping of compound 19 (black) into the FXR ligand binding site. In comparison to the original ligand, compound 11 (white), 19 established additional hydrogen bonds (arrows) between the hydroxy group and Ser332/Tyr369. The hydrogen bond acceptor (red arrow) with His294 was observed in both compounds 11 and 19, respectively. Hydrogen bond-forming amino acids are depicted in ball-and-stick style. Three hydrophobic interactions (yellow spheres) were predicted to be identical for compounds 11 and 19. The piperazine moiety was not observed to form direct interactions with the receptor and could therefore be regarded as a solubility-enhancing spacer.
The generated pharmacophore models have been successfully applied to screen our in-house Chinese Herbal Medicine (CHM) database46 comprising 10,216 compounds, which are related to natural products used in traditional Chinese medicine. This in silico approach led to the identification of lanostane-type triterpenes from the mushroom Ganoderma lucidum as active FXR ligands. A complete description of this work is provided in part II of this study.47
3. Conclusions
The present work shows how a model collection for FXR can be assembled and thoroughly theoretically validated. In comparison to smaller DBs, the ChEMBL returns more distinct EFs from validation screenings. The developed 6FXR agonist models ranked by EF calculations using the ChEMBL DB were successfully applied to identify novel active compounds (19 and 20) from the NCI database. By the application of a promoter–reporter study and analysis of the mRNA levels of the FXR-regulated gene CYP7A1, the observed FXR agonistic activity was confirmed.
4. Experimental
4.1. Preparation of the ChEMBL database
The ChEMBL database was downloaded from the ChEMBL Group homepage (www.ebi.ac.uk/chembldb/) and imported into a MySQL (Sun Microsystems GmbH, Kirchheim-Heimstetten, Germany) database. A MySQL query for all compounds comprising canonical smiles, names, literature, target annotation, and published binding affinity (Ki) or biological activity (IC50/EC50) data resulted in a tab-delimited file containing 1,057,661 single entries. Subsequently, a PipelinePilot (www.accelrys.com) script was applied to divide the data by biological target and to generate 3D structures for the compounds.
This PipelinePilot script consisted of two subprotocols. The first subprotocol was responsible for splitting the ChEMBL data by their target annotation. It was based on a list of all ChEMBL target names and a filter component. This filter subprotocol retrieved only compound data annotated with the first target of the list and stored these data into a tab-delimited file. Subsequently, a loop caused the repetition of the whole subprotocol code using the next target on the list as filter query. After the loop was executed for all targets, the resulting tab-delimited files served as input for the second subprotocol which translated the canonical smiles code for each compound from each target file into a 3D structure, chemically standardized the molecules, generated all possible stereoisomers and tautomers, calculated the protonation state at physiological pH, energetically minimized the molecules using the Clean force field, and stored the results as MDL SD-file. Also this subprotocol included a loop which caused the repeatedly execution of the code until all tab-delimited files were converted into SD-files containing 3D structures, names, literature, and activity data for all compounds for each target. This automated procedure resulted in 3485 SD-files with unique target annotation. On the one hand, these subsets are readily available for molecular modeling experiments. On the other hand, the whole ChEMBL database may be useful to determine retrieval rates and EFs of virtual screening experiments. For this purpose, the ChEMBL including all 302,924 unique compounds was converted into a searchable 3D multiconformational database using DiscoveryStudio 2.5 (www.accelrys.com). The Build 3D Database protocol was employed for these calculations (FAST conformer generation algorithm with a maximum of 250 conformers per molecule and a maximal energy range of 20 kcal/mol above the calculated energy minimum).
4.2. Preparation of the DrugBank database
The DrugBank small molecules subset was downloaded from www.drugbank.ca/downloads and submitted to conformational analysis using DiscoveryStudio 2.5, using the Build 3D Database protocol with identical settings as described for the ChEMBL database.
4.3. Preparation of the FXR-actives dataset
The set of active FXR ligands was extracted from the ChEMBL database (www.ebi.ac.uk/chembldb/). Out of 326 compounds that were included in the ChEMBL subset linked with FXR activity, only ligands with a reported EC50 <100 μM were kept. After removing duplicate structures (among them, for example, the endogenous ligand CDCA) and calculating standard 3D structures using Omega2 (www.openeye.com), 221 entries remained in the FXR-actives data set. For the modeling studies, these compounds were submitted to conformational model analysis using DiscoveryStudio’s FAST conformer generation algorithm employing the settings described for the ChEMBL.
4.4. Preparation of the FXR-decoys database
The FXR decoys were extracted from the ChEMBL version 09. Only compounds with indicated bioactivities (IC50, EC50, Ki) were kept. From the over 390,000 remaining entries, those with similar physicochemical properties compared to the FXR-active dataset were kept. All decoys fulfilled the following criteria in comparison to one of the FXR-active molecules: molecular weight ±50, c log P ±0.1, equal number of aromatic rings, hydrogen bond acceptors, and hydrogen bond donors. Five thousand six hundred and thirteen molecules passed this filtering process. Conformational analysis was performed as described for the ChEMBL database. For 15 entries, the conformer generation was not successful, so the final FXR-decoys database comprised 5598 compounds.
4.5. Preparation of the Virtural DB
A previously generated virtual database that contains 12,775 drug-like, presumably inactive decoy molecules.30 This data set – the so-called Virtual DB – was used to assess the model’s ability to discard inactive compounds in a database screen. For an accurate comparison of screening results, the Virtual DB was re-submitted to conformational analysis using the same settings as for the ChEMBL data set.
4.6. Preparation of the NCI database
The NCI database was retrieved from the NCI download site (dtp.nci.nih.gov/docs/3d_database/Structural_information/structural_data.html) and converted into a 3D database using Catalyst 4.11 software (www.accelrys.com). The 3D multiconformational database included 247.041 compounds.
4.7. Analyses of binding modes
Ligand binding modes were analyzed using LigandScout 3 software (www.inteligand.com). LigandScout derives protein and ligand atom co-ordinates from the PDB and translates them into chemical structures according to the distances and geometries of neighboring atoms. Ligands can be aligned using either their chemical properties represented by pharmacophoric features48 or so-called reference points which reflect the position of the ligand in the protein context. The latter alignment option was used in this study for the binding mode analysis of FXR ligands.
4.8. Pharmacophore model generation and theoretical validation
Structure-based pharmacophore models were generated within LigandScout (www.inteligand.com). This program automatically analyzes interactions between the ligand and the protein by evaluating the electrostatic and geometric (distances, size, and angles) properties of areas where binding takes place. As a result, the user is provided with an interaction network that can consist of hydrophobic areas, H-bond acceptors/donors, positively or negatively ionizable/charged groups, and aromatic rings involved in aromatic interactions. The generated models can be used to align small sets of molecules to the model or be exported in file formats compatible with other molecular modeling software such as DiscoveryStudio (www.accelrys.com), MOE (www.chemcomp.com), or Phase (www.schrodinger.com). In this study, the model was adapted to DiscoveryStudio’s requirements – only one H-bonding or charged feature per ligand atom – and exported in hypoedit format. Using the hypoedit tool that comes with DiscoveryStudio, the model was transformed into a chm pharmacophore file. The resulting model was checked for validity via mapping of the bioactive ligand conformation it was derived from. This step is crucial because different virtual screening programs do not use identical chemical feature definitions, which can influence the mapping of a compound. In addition, active FXR ligands from the FXR-actives dataset were fitted into the model. In order to create sufficiently representative models, they were optimized to find a set of active compounds while keeping the number of decoys in the hitlist low. As a quantitative measure of model quality, EFs were calculated using the FXR-actives and VirtualDB, DrugBank, and ChEMBL data sets. The EF is calculated using the equation49:
where TP is the number of true positive hits in the hitlist (hits FXR-actives), n is the number of hits from virtual screening of both the active and decoy data sets (hits FXR-actives and Virtual DB/DrugBank/ChEMBL), A is the number of active ligands in the validation database (FXR-actives, n = 221), and N is the number of all compounds in the validation database (221 FXR-actives plus 12,775 in the Virtual DB gives 12,996; 221 plus 4722 from the DrugBank gives 4943, and the ChEMBL database consists of 304,273 entries that already include the FXR-actives). Database screening for EF calculations were performed within DiscoveryStudio 2.5 using the Search 3D Database protocol. Default settings were used except for the minimum interfeature distance, which was set to 0.00001.
The FXR ligand structures from recent literature42,43 were created using ChemBioDraw Ultra 11.0 (www.cambridgesoft.com) and converted into 3D multiconformational SD files using the FAST conformer generation algorithm of DiscoveryStudio 2.5. The calculation settings were equal to the database calculation settings described above for the ChEMBL database.
4.9. Biological evaluation of virtual hits
4.9.1. Cell culture, plasmids, and reagents
Human embryonic kidney-293 cells (HEK-293) from American Type Culture Collection (Manassas, VA, USA) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1% l-glutamine. The luciferase reporter plasmid (ECRE)5TK-Luc, the expression vector for mouse FXR, the expression vector for mouse RXR were kindly provided by Professor Glass CK (UCSD, La Jolla, CA). The vector pSV40-renillaLuc was obtained from Promega (Madison, WI, USA), the pcDNA3.1 from Invitrogen (Carlsbad, CA, USA). DMSO and 2 were purchased from Sigma–Aldrich (St. Louis, MO).
4.9.2. Reporter gene assay
Activation of FXR was tested in HEK-293 cells seeded in 48 well plates (NUNC) and transiently transfected with the indicated plasmids (total DNA 0.3 ng/well). Transient transfections were performed with the calcium phosphate technique as described.50 For monitoring transfection efficiency pSV40-renillaLuc was cotransfected. To normalize the amount of DNA transfected, pcDNA3.1 vector was added where appropriate. Cells were stimulated for 18 h with vehicle (DMSO) or with ligands at 25 μM concentration.
Luciferase activity was determined from the cell lysates using Dual-Luciferase Kit (Promega, Madison, WI, USA), measured with Victor2 multilabel counter (Wallac, Finland). The firefly luciferase values were normalized with the renilla luciferase value measured for the respective sample (relative luciferase units). FXR induction is expressed as fold difference of relative luciferase units (RLU) comparing values of stimulated samples with the vehicle treated control (mean values ± SEM) of triplicate determinations from at least three independent experiments. Statistical significance of FXR induction was assessed by ANOVA-multiple comparison with Bonferroni post-test, whereby P values of less than 0.05 were regarded as statistically significant. The EC50 values were estimated by nonlinear regression analysis using the equation for the sigmoidal dose response of GraphPad Prism 4.0 (GraphPad Software Inc., La Jolla, CA).
4.9.3. Analysis of expression of FXR downstream genes by qPCR
HepG2 cells were grown in 24-well dishes (NUNC) in DMEM supplemented with antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, 25 μg/ml amphotericin B), 1% glutamine and 10% FBS. The cells were stimulated in the same medium with the analyzed compounds or positive control (2) dissolved in DMSO (final concentration 0.1%) at a concentration of 50 μM for 6 h. The incubation was terminated and RNA was isolated using TriFast reagent (PeqLab, Erlangen, Germany). A GeneAmp RNA-PCR kit and oligo d(T)16 primers (Applied Biosystems, Foster City, CA) were used for cDNA synthesis from 900 ng of total RNA. Quantitative real-time PCR (qPCR) was performed using the Step One Plus (Applied Biosystems), FastStart SYBR Green Master Mix (Roche Diagnostics, Indianapolis, IN), and specific primers for amplification of CYP7A1 (Qiagen, Venlo, The Netherlands). CYP7A1 expression was normalized to the expression levels of 2-microglobulin using primers obtained from the same company.
4.10. Novelty assessment of compound 19
Apart from literature search using SciFinder and investigation of the data provided by the NCI, compound 19 was topologically compared with bioactive compounds reported in the WOMBAT and MDDR database. For this experiment, the sea.docking.org45 online portal was used. Scitegic’s ECFP4 fingerprinting algorithm was used for identifying compounds that are structurally related to compound 19. The search returned targets on which related compounds have reported to be active and ranked them according to the significance of the resulting similarity scores. From the results, compound 19 has not been associated with FXR before.
4.11. Identity and purity determination
Compounds 19 and 20 fulfilled the identity and purity criteria (⩾95%) as determined by HPLC.
4.12. Mapping of compound 19 into the FXR binding site
The 1osh-1-s model-mapping conformation of compound 19 was copied into the X-ray crystal structure co-ordinates of FXR (PDB entry 1osh). In order to optimize the fitting of the compound into the binding site, it was minimized using the MMFF94 force field implemented in LigandScout 3.02. Protein–ligand interactions were automatically analyzed using LigandScout and visually inspected.
Acknowledgment
This work was funded by the NFN-project ‘Drugs from Nature Targeting Inflammation – DNTI’, Subprojects-No. S10702/10711, S10703, and S10709/10713 from the Austrian Science Fund (FWF). D.S. is grateful for a young talents grant and the Erika Cremer Habilitations Program from the University of Innsbruck. We thank the National Cancer Institute for providing the FXR test compounds free of charge.
In memoriam Univ.-Prof. Dr. Bernd R. Binder
Footnotes
Supplementary data associated (The literature sources of the FXR ligands from the ChEMBL and the 2D structures of the FXR ligands from recent literature (Chart S-1).) with this article can be found, in the online version, at doi:10.1016/j.bmc.2011.09.056. These data include MOL files and InChiKeys of the most important compounds described in this article.
Supplementary data
The literature sources of the FXR ligands from the ChEMBL and the 2D structures of the FXR ligands from recent literature (Chart S-1).
MOL files
The following ZIP file contains the MOL files of the most important compounds referred to in this article.
ZIP file containing the MOL files of the most important compounds in this article.
References and notes
- 1.Pellicciari R., Costantino G., Fiorucci S. J. Med. Chem. 2005;48:5383. doi: 10.1021/jm0582221. [DOI] [PubMed] [Google Scholar]
- 2.Zhang M.-Z., Xu J., Yao B., Yin H., Cai Q., Shrubsole M.J., Chen X., Kon V., Zheng W., Pozzi A., Harris R.C. J. Clin. Invest. 2009;119:876. doi: 10.1172/JCI37398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X., Lin Y., Gilson M.K. J. Comb. Chem. High Throughput Screen. 2001;4:719. doi: 10.2174/1386207013330670. [DOI] [PubMed] [Google Scholar]
- 4.Goodwin B., Jones S.A., Price R.R., Watson M.A., McKee D.D., Moore L.B., Galardi C., Wilson J.G., Lewis M.C., Roth M.E., Maloney P.R., Willson T.M., Kliewer S.A. Mol. Cell. 2000;6:517. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 5.Makishima M., Okamoto A.Y., Repa J.J., Tu H., Learned R.M., Luk A., Hull M.V., Lustig K.D., Mangelsdorf D.J., Shan B. Science (New York, NY) 1999;284:1362. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 6.Lee F.Y., Lee H., Hubbert M.L., Edwards P.A., Zhang Y. Trends Biochem. Sci. 2006;31:572. doi: 10.1016/j.tibs.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Lew J.-L., Zhao A., Yu J., Huang L., de Pedro N., Pelaez F., Wright S.D., Cui J. J. Biol. Chem. 2004;279:8856. doi: 10.1074/jbc.M306422200. [DOI] [PubMed] [Google Scholar]
- 8.Willson T.M., Jones S.A., Moore J.T., Kliewer S.A. Med. Res. Rev. 2001;21:513. doi: 10.1002/med.1023. [DOI] [PubMed] [Google Scholar]
- 9.Fiorucci S., Mencarelli A., Palladino G., Cipriani S. Trends Pharmacol. Sci. 2009;30:570. doi: 10.1016/j.tips.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Pellicciari R., Fiorucci S., Camaioni E., Clerici C., Costantino G., Maloney P.R., Morelli A., Parks D.J., Willson T.M. J. Med. Chem. 2002;45:3569. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 11.Costantino G., Macchiarulo A., Entrena-Guadix A., Camaioni E., Pellicciari R. Bioorg. Med. Chem. Lett. 2003;13:1865. doi: 10.1016/s0960-894x(03)00281-6. [DOI] [PubMed] [Google Scholar]
- 12.Synold T.W., Dussault I., Forman B.M. Nat. Med. 2001;7:584. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 13.Mencarelli, A.; Fiorucci, S. J. Cell. Mol. Med.2010, E-pub ahead of print.
- 14.Bass J.Y., Caravella J.A., Chen L., Creech K.L., Deaton D.N., Madauss K.P., Marr H.B., McFadyen R.B., Miller A.B., Mills W.Y., Navas F., III, Parks D.J., Smalley T.L., Jr., Spearing P.K., Todd D., Williams S.P., Wisely G.B. Bioorg. Med. Chem. Lett. 2011;21:1206. doi: 10.1016/j.bmcl.2010.12.089. [DOI] [PubMed] [Google Scholar]
- 15.Richter H.G.F., Benson G.M., Bleicher K.H., Blum D., Chaput E., Clemann N., Feng S., Gardes C., Grether U., Hartman P., Kuhn B., Martin R.E., Plancher J.-M., Rudolph M.G., Schuler F., Taylor S. Bioorg. Med. Chem. Lett. 2011;21:1134. doi: 10.1016/j.bmcl.2010.12.123. [DOI] [PubMed] [Google Scholar]
- 16.Richter H.G.F., Benson G.M., Blum D., Chaput E., Feng S., Gardes C., Grether U., Hartman P., Kuhn B., Martin R.E., Plancher J.-M., Rudolph M.G., Schuler F., Taylor S., Bleicher K.H. Bioorg. Med. Chem. Lett. 2011;21:191. doi: 10.1016/j.bmcl.2010.12.123. [DOI] [PubMed] [Google Scholar]
- 17.Abel U., Schlüter T., Schulz A., Hambruch E., Steeneck C., Hornberger M., Hoffmann T., Perovic-Ottstadt S., Kinzel O., Burnet M., Deuschle U., Kremoser K. Bioorg. Med. Chem. Lett. 2010;20:4911. doi: 10.1016/j.bmcl.2010.06.084. [DOI] [PubMed] [Google Scholar]
- 18.Mehlmann J.F., Crawley M.L., Lundquist IV J.T., Unwalla R.J., Harnish D.C., Evans M.J., Kim C.Y., Wrobel J.E., Mahaney P.E. Bioorg. Med. Chem. Lett. 2009;19:5289. doi: 10.1016/j.bmcl.2009.07.148. [DOI] [PubMed] [Google Scholar]
- 19.Wu J., Xia C., Meier J., Li S., Hu X., Lala D.S. Mol. Endocrinol. 2002;16:1590. doi: 10.1210/mend.16.7.0894. [DOI] [PubMed] [Google Scholar]
- 20.Carter B.A., Taylor O.A., Prendergast D.R., Zimmerman T.L., von Furstenberg R., Moore D.D., Karpen S.J. Pediatr. Res. 2007;62:301. doi: 10.1203/PDR.0b013e3181256492. [DOI] [PubMed] [Google Scholar]
- 21.Lin H.-R., Abraham Donald J. Bioorg. Med. Chem. Lett. 2006;16:4178. doi: 10.1016/j.bmcl.2006.05.084. [DOI] [PubMed] [Google Scholar]
- 22.Tobin J.F., Freedman L.P. Trends Endocrinol. Metab. 2006;17:284. doi: 10.1016/j.tem.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Langer T., Hoffmann R.D. Wiley-VCH; Weinheim: 2006. Pharmacophores and Pharmacophore Searches. [Google Scholar]
- 24.Leach A.R., Gillet V.J., Lewis R.A., Taylor R. J. Med. Chem. 2010;53:539. doi: 10.1021/jm900817u. [DOI] [PubMed] [Google Scholar]
- 25.Kim K.-H., Kim N.D., Seong B.-L. Exp. Opin. Drug Discov. 2010;5:205. doi: 10.1517/17460441003592072. [DOI] [PubMed] [Google Scholar]
- 26.Bolton E.E., Wang Y., Thiessen P.A., Bryant S.H. Ann. Rep. Comput. Chem. 2008;4:217. [Google Scholar]
- 27.Warr W.A. J. Comput. Aided Mol. Des. 2009;23:195. doi: 10.1007/s10822-009-9310-3. [DOI] [PubMed] [Google Scholar]
- 28.Farese S., Kruse A., Pasch A., Dick B., Frey B.M., Uehlinger D.E., Frey F.J. Kidney Int. 2009;76:877. doi: 10.1038/ki.2009.269. [DOI] [PubMed] [Google Scholar]
- 29.Wishart D.S., Knox C., Guo A.C., Cheng D., Shrivastava S., Tzur D., Gautam B., Hassanali M. Nucleic Acids Res. 2008;36:D901. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuster D., Palusczak A., Hartmann R.W., Langer T. J. Chem. Inf. Model. 2006;46:1301. doi: 10.1021/ci050237k. [DOI] [PubMed] [Google Scholar]
- 31.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Adv. Drug Deliv. Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 32.Irwin J.J., Shoichet B.K. J. Chem. Inf. Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verdonk M.L., Cole J.C., Hartshorn M.J., Murray C.W., Taylor R.D. Proteins. 2003;52:609. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 34.Berman H., Henrick K., Nakamura H. Nat. Struct. Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 35.Downes M., Verdecia M.A., Roecker A.J., Hughes R., Hogenesch J.B., Kast-Woelbern H.R., Bowman M.E., Ferrer J.-L., Anisfeld A.M., Edwards P.A., Rosenfeld J.M., Alvarez J.G.A., Noel J.P., Nicolaou K.C., Evans R.M. Mol. Cell. 2003;11:1079. doi: 10.1016/s1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soisson S.M., Parthasarathy G., Adams A.D., Sahoo S., Sitlani A., Sparrow C., Cui J., Becker J.W. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5337. doi: 10.1073/pnas.0710981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akwabi-Ameyaw A., Bass J.Y., Caldwell R.D., Caravella J.A., Chen L., Creech K.L., Deaton D.N., Jones S.A., Kaldor I., Liu Y., Madauss K.P., Marr H.B., McFadyen R.B., Miller A.B., Navas F., III, Parks D.J., Spearing P.K., Todd D., Williams S.P., Wisely G.B. Bioorg. Med. Chem. Lett. 2008;18:4339. doi: 10.1016/j.bmcl.2008.06.073. [DOI] [PubMed] [Google Scholar]
- 38.Flatt B., Martin R., Wang T.-L., Mahaney P., Murphy B., Gu X.-H., Foster P., Li J., Pircher P., Petrowski M., Schulman I., Westin S., Wrobel J., Yan G., Bischoff E., Daige C., Mohan R. J. Med. Chem. 2009;52:904. doi: 10.1021/jm8014124. [DOI] [PubMed] [Google Scholar]
- 39.Feng S., Yang M., Zhang Z., Wang Z., Hong D., Richter H., Benson G.M., Bleicher K., Grether U., Martin R.E., Plancher J.-M., Kuhn B., Rudolph M.G., Chen L. Bioorg. Med. Chem. Lett. 2009;19:2595. doi: 10.1016/j.bmcl.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 40.Bass J.Y., Caldwell R.D., Caravella J.A., Chen L., Creech K.L., Deaton D.N., Madauss K.P., Marr H.B., McFadyen R.B., Miller A.B., Parks D.J., Todd D., Williams S.P., Wisely G.B. Bioorg. Med. Chem. Lett. 2009;19:2969. doi: 10.1016/j.bmcl.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 41.Akwabi-Ameyaw A., Bass J.Y., Caldwell R.D., Caravella J.A., Chen L., Creech K.L., Deaton D.N., Madauss K.P., Marr H.B., McFadyen R.B., Miller A.B., Navas F., III, Parks D.J., Spearing P.K., Todd D., Williams S.P., Wisely G.B. Bioorg. Med. Chem. Lett. 2009;19:4733. doi: 10.1016/j.bmcl.2009.06.062. [DOI] [PubMed] [Google Scholar]
- 42.Stauffer S.R., Coletta C.J., Tesdesco R., Nishiguchi G., Carlson K., Sun J., Katzenellenbogen B.S., Katzenellenbogen J.A. J. Med. Chem. 2000;43:4934. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 43.Lundquist IV J.T., Harnish D.C., Kim C.Y., Mehlmann J.F., Unwalla R.J., Phipps K.M., Crawley M.L., Commons T., Green D.M., Xu W., Hum W.-T., Eta J.E., Feingold I., Patel V., Evans M.J., Lai K., Borges-Marcucci L., Mahaney P.E., Wrobel J.E. J. Med. Chem. 2010;53:1774. doi: 10.1021/jm901650u. [DOI] [PubMed] [Google Scholar]
- 44.Penning T.M. J. Steroid Biochem. Mol. Biol. 2011;125:46. doi: 10.1016/j.jsbmb.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keiser M.J., Roth B.L., Armbruster B.N., Ernsberger P., Irwin J.J., Shoichet B.K. Nat. Biotechnol. 2007;25:197. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 46.Fakhrudin N., Ladurner A., Heiss E.H., Kramer M., Baumgartner L., Joa H., Schuster D., Markt P., Wolber G., Ellmerer E.P., Rollinger J.M., Atanasov A.G., Stuppner H., Dirsch V.M. Mol. Pharmacol. 2010;77:559. doi: 10.1124/mol.109.062141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grienke U., Mihály-Bison J., Schuster D., Afonyushkin T., Binder M., Guan S.-h., Chen C.-r., Wolber G., Stuppner H., Guo D.-a., Bochkov V.N., Rollinger J.M. Bioorg. Med. Chem. 2011 doi: 10.1016/j.bmc.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolber G., Dornhofer A., Langer T. J. Comput. Aided Mol. Des. 2006;20:773. doi: 10.1007/s10822-006-9078-7. [DOI] [PubMed] [Google Scholar]
- 49.Schuster D., Wolber G. Curr. Pharm. Des. 2010;16:1666. doi: 10.2174/138161210791164072. [DOI] [PubMed] [Google Scholar]
- 50.Perkins N.D., Edwards N.L., Duckett C.S., Agranoff A.B., Schmid R.M., Nabel G.J. EMBO J. 1993;12:3551. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The literature sources of the FXR ligands from the ChEMBL and the 2D structures of the FXR ligands from recent literature (Chart S-1).
ZIP file containing the MOL files of the most important compounds in this article.