

Abstract
The stereoselective synthesis of novel photoreactive γ-secretase inhibitors 2 and 3 has been achieved. Key steps of the strategy involve preparation of α-N-Boc-epoxide 8 and formation of lactone 14 in a practical and stereo-controlled fashion. Compounds 2 and 3 are potent γ-secretase inhibitors and directly interact with presenilin-1, a catalytic subunit of γ-secretase.
γ-Secretase cleaves the amyloid precursor protein (APP) to generate β-amyloid (Aβ) peptides, which are believed to play a causative role in the pathogenesis of Alzheimer Disease (AD).1 γ-Secretase is an aspartyl protease composed of at least four proteins, including presenilin, nicastrin, APH and Pen2.2 Genetic and biochemical studies have indicated that presenilin is the catalytic core of γ-secretase3–5 and as such, familial mutations of presenilin have been associated with early on-set of AD6 through alteration of the specificity of γ-secretase. Furthermore, γ-secretase represents a novel class of protease that hydrolyzes the scissile bond within the transmembrane domain of substrate.7, 8
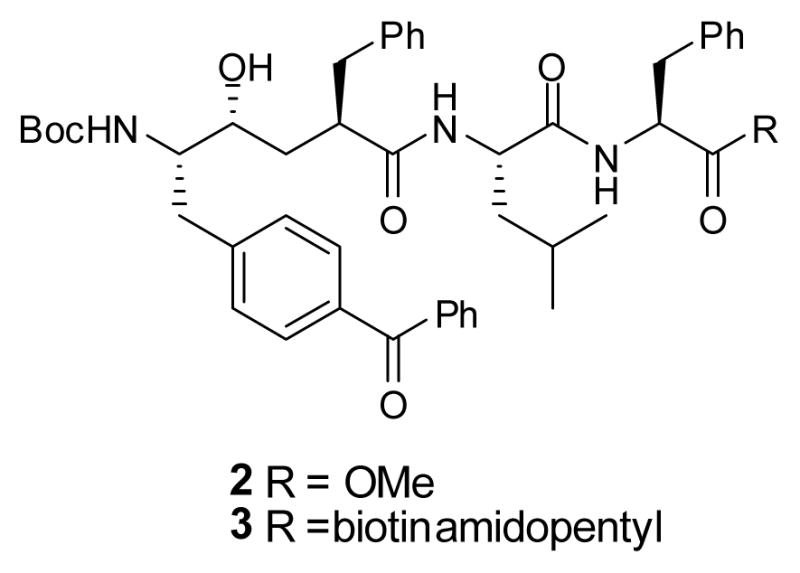
L-685,458 (1) (Figure 1), a potent γ-secretase inhibitor9 that contains a hydroxyethylene isostere, can be modified into a photoreactive compound by replacing an unsubstituted phenyl with a benzophenone (BP). These substitutions at P2, P1′ and P3′ have been synthesized and utilized to study γ-secretase.4, 10 However, synthesis of a photoreactive dipeptide isostere at the P1 position has not yet been achieved. In the current study, we describe the stereo-controlled synthesis of two new analogs of L-685,458 (2 and 3, Chart 1) with BPA (benzophenone alanine) at the P1 position and demonstrate that they directly interact with presenilin, the catalytic subunit of γ-secretase. Moreover, this novel BPA-Phe isostere could be useful as a functional unit to synthesize active site directed inhibitors for profiling aspartyl proteases.
Figure 1.
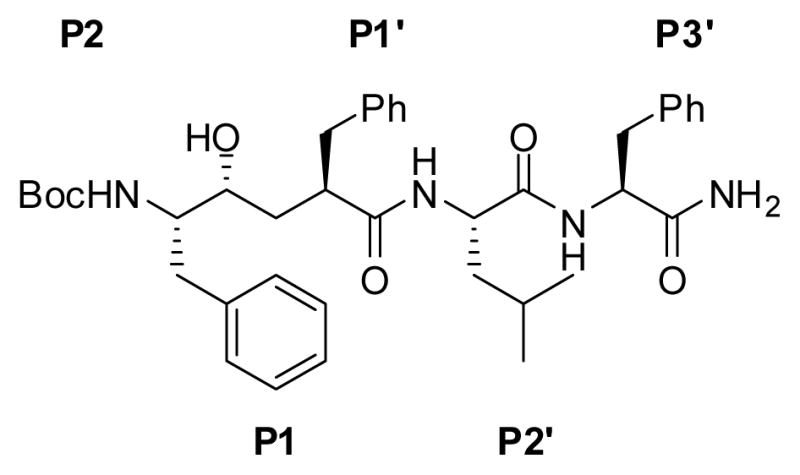
Structure of L-685,458 (1). The side chains corresponding to the P and P′ sites are marked.
Chart 1.
The synthesis of 2 and 3 started with the preparation of epoxide 8 using a modified Barrish-Polniaszek’s method11 (Scheme 1). Methylation of Boc-p-Bz-Phe-OH (4) with TMSCHN2 in methanol12 provided methyl ester 5.
SCHEME 1. Synthesis of Epoxide 8.

Reagents and conditions: (a) TMSCHN2, MeOH, 0 °C to rt, 18 h, 90%; (b) NaBH4, MeOH, −60 °C, 86%; (c) TBSCl, CH2Cl2, imidazole, rt, 95%; (d) 4 equiv. CH2ICl, 5 equiv. LDA, THF, −78 °C; (e) 4 equiv. NaBH4, MeOH, −78 °C to 0 °C, 65%; (f) KOH, EtOH, 0 °C to rt, 95%.
However, an attempt that followed the same synthetic route for preparation of Phe-BPA isostere10 to protect benzophenone 5 as a dioxolane using ethylene glycol, p-TsOH and benzene at reflux for 2 days failed to generate any product. Thus, we changed our strategy by reducing ketone to an alcohol. We intended to find conditions that allow for the stereo- and regioselective reduction the ketone of benzophenone. Initially, we treated 5 with NaBH4 at 0 °C13 with favorable stereoselectivity (85:15 dr) and 70% yield, but this condition also led to the formation of a small amount of reduced methyl ester. However, when we performed the same reaction at −60 °C, we obtained the stereoselective product (85:15 dr) in 86% yield without reducing the methyl ester. Silylation of the resulting secondary alcohol produced 6, which led to the generation of a chiral center at the benzylic carbon. The configuration of 6 is assigned by X-ray crystallographic analysis of intermediate 12 (Scheme 2) as described later in Figure 2. Treatment of methyl ester 7 with excess LDA/CH2ICl provided an α-chloroketone, which was reduced with NaBH4 to give chlorohydrin 7 (9:1 dr) in favor of the desired stereoisomer as demonstrated by X-Ray analysis. The two diastereoisomers of 7 were separated by column chromatography (65% yield of the desired compound, based on recovered starting material 6). Cyclization of chlorohydrin 7 produced epoxide 8 in 95% yield.14
SCHEME 2. Synthesis of Acid 15.

Reagents and conditions: (a) CH2(CO2Et)2, NaOEt, EtOH, rt, 70%; (b) LiOH/DME-H2O, 50 °C, 6 h; (c) toluene, reflux, 8 h, 60%; (d) LDA, PhCHO, THF, −78 °C; (e) Ac2O, Et3N, 120 °C, 80% for 2 steps; (f) H2, 10% Pd/C, EtOAc, rt, 6 h, 90%; (g) HF·Py, THF, 18 h, 83%; (h) MnO2, CH2Cl2, 18 h, 76%; (i) (ia) LiOH/DME-H2O, rt; (ib) TBSCl, imidazole, DMF, rt; (ic) MeOH, 79% for 3 steps.
Figure 2.

X-Ray Crystallographic Structure of 12: C37H49NSiO5, M 615.86, orthorhombic P212121 (No. 19), a = 5.9239(12) Å, b = 12.923(3) Å, c = 46.419(9) Å, V = 3553.0(12) Å3, Dc (Z = 4) = 1.151 g/cm3, T = 100 K, μ = 0.106 cm−1. The final R value is 0.2116 for 3442 independent reflections with I > 2σI and 398 parameters. (The crystal structure of 12 has been deposited at the Cambridge Crystallographic Data Centre with the deposition number: CCDC 710680)
Treatment of epoxide 8 with the sodium salt of diethyl malonate directly provided lactone 9 as a mixture of stereoisomers (Scheme 2).15 Hydrolysis of 9 with aqueous LiOH, followed by decarboxylation gave lactone 10 in 60% yield. Aldol condensation of 10 with benzaldehyde followed by dehydration with acetic anhydride-triethylamine at 120 °C gave the α,β-unsaturated lactone 11 in 80% yield.16 Hydrogenation of 11 with 10% Pd/C (1 atm, 6 h) provided lactone 12 as the sole product. The assignment of three chiral centers, as indicated in Scheme 2, was confirmed by the X-ray crystallographic analysis of 12 (Figure 2). Removal of the silyl group in lactone 12 with n-Bu4NF (TBAF) led to epimerization at the α-lactone position, perhaps due to the basicity of the TBAF reagent. However, we were able to find that treatment of 12 with pyridine/HF overnight successfully removed the silyl protecting group to give 13 without any epimerization.17 Oxidation of the benzylic alcohol with MnO218 gave benzophenone 14 in 76% yield.19 Hydrolysis of lactone 14 with LiOH and silylation of the resulting hydroxy acid produced 15 in 79% yield.
Esterification of Leu-Phe-OH with TMSCl in MeOH,20 followed by coupling of the resulting amine with acid 15, and deprotection of the resulting silyl ether with TBAF, produced the desired compound 2 in reasonable yield (Scheme 3).21 In order to facilitate the purification of the labeled proteins or fragments thereof, biotinylated compound 3 was prepared (Scheme 3). Mild saponification of the methyl ester in 2 led to the corresponding carboxylic acid, which was coupled with 5-(biotinamido)pentylamine in the presence of EDC and HOBt and resulted in compound 3.
SCHEME 3. Synthesis of Compounds 2 and 3.

Reagents and conditions: (a) TMSCl, MeOH, 0 °C to rt, 18 h, 80%; (b) 15, EDC, HOBt, i-Pr2NEt, DMF, 57% (c) n-Bu4NF, THF, rt, 85%; (d) LiOH, THF/H2O, rt, 90%; (e) 5-biotinamido pentylamine, EDC, HOBt, DMF, rt, 50%.
We next examined the biological activities of 2 and 3. First, we determined their inhibitory potency against γ-secretase using an in vitro assay.22 The IC50 values of 2 and 3 are 0.7 nM and 0.6 nM, respectively (Fig. 3A), which is similar to the parent compound, L-685,458 (1). These findings have demonstrated that incorporating BPA into the P1 position and attaching a biotin tag at the C-terminus do not affect their potency for inhibition of γ-secretase. Second, we tested whether 3 was capable of photo-crosslinking to γ-secretase. HeLa cell membranes were incubated with 3 at a final concentration of 10 nM in the absence and the presence of 2 μM of L-685,458 for 2.5 h. Then samples were irradiated with UV light (> 350 nm) and the labeled proteins were solubilized and isolated with streptavidin beads.4
Figure 3.

Both 2 and 3 are potent γ-secretase inhibitors that directly bind to Presenilin-1. A) Inhibitory potencies of compounds 2 and 3 against γ-secretase. B) Scheme of photoaffinity labeling procedure. After photo-crosslinking, the biotinylated proteins were captured, eluted and analyzed by Western analysis. C) Analysis of photolabeled proteins. The photo-crosslinked proteins were resolved by SDS-PAGE and probed with PS1-NTF (N-terminal fragment) antibody.
The biotinylated proteins were eluted and analyzed by Western blotting with antibodies against presenilin-1 (PS-1). Inhibitor 3 directly photolabels PS-1 (Fig. 3C). Moreover, an excess of L-685,458 is able to block photoinsertion of this probe into presenilin-1. Taken together, these results have demonstrated that compounds 2 and 3 are potent γ-secretase inhibitors that can specifically label the catalytic core of γ-secretase. Therefore, compounds 2 and 3 should be valuable probes for mapping the active site of γ-secretase.
Acknowledgments
This work was supported by NIH grant AG026660 (YML), the Alzheimer’s Association (Zenith Fellows Award to YML), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics. CCS is supported by an NIH NRSA predoctoral fellowship 5F31NS053218-02. We thank Dr. George Sukenick, Ms. Sylvi Rusli (NMR Core Facility, Sloan-Kettering Institute) for mass spectral analyses and Dr. Louis Todaro (Hunter College New York) for X-ray structure analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hardy J, Allsop D. Trends Pharmacol Sci. 1991;12:383. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 2.De Strooper B. Neuron. 2003;38:9. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 3.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Nature. 1999;398:513. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 4.Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ. Nature. 2000;405:689. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 5.Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai JY, Rahmati T, Xia W, Selkoe DJ, Wolfe MS. Nat Cell Biol. 2000;2:428. doi: 10.1038/35017062. [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Nature. 1995;375:754. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 7.Wolfe MS, Kopan R. Science. 2004;305:1119. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- 8.Brown MS, Ye J, Rawson RB, Goldstein JL. Cell. 2000;100:391. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 9.Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL. Biochemistry. 2000;39:8698. doi: 10.1021/bi0005456. [DOI] [PubMed] [Google Scholar]
- 10.Chun J, Yin YI, Yang G, Tarassishin L, Li YM. J Org Chem. 2004;69:7344. doi: 10.1021/jo0486948. [DOI] [PubMed] [Google Scholar]
- 11.Chen P, Cheng PTW, Spergel SH, Zahler R, Wang XB, Thottathil J, Barrish JC, Polniaszek RP. Tetrahedron Lett. 1997;38:3175. [Google Scholar]
- 12.Chai W, Murray WV. Tetrahedron Lett. 1999;40:7185. [Google Scholar]
- 13.Orito K, Sato S, Suginome H. J Chem Soc Perkin Trans 1. 1995;1:63. [Google Scholar]
- 14.Preparion of 8. Tert-Butyl (S)-2-(4-(S)-tert-butyldimethylsilyloxy phenylmethylphenyl)-1-((S)-oxiran-2-yl)ethylcarbamate (8). To an ice-cold solution of 7 (1.25 g, 2.41 mmol) in EtOH (30 mL) was added KOH (163 mg, 2.9 mmol), and the mixture was stirred at rt. for 2 h. The mixture was concentrated under reduced pressure, and the residue was partitioned between EtOAc (200 mL) and H2O (50 mL). The organic layer was washed with saturated NH4Cl solution, H2O, and brine, dried with Na2SO4, and concentrated under reduced pressure. Purification of the residue by column chromatography (20 % EtOAc in Hexane) gave 8 (1.11 g, 95 %) as a yellow syrup: [α]25D 9.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): 7.38 (d, J = 7.4 Hz, 2H), 7.33 (m, 4H), 7.22 (t, J = 7.1 Hz, 1H), 7.17 (d, J = 8.0 Hz, 2H), 5.75 (s, 1H), 4.47 (brs, 1H), 3.70 (brs, 1H), 3.93 (m, 2H), 2.80 (m, 3H), 1.38 (s, 9H), 0.94 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H); 13C NMR: 155.2, 145.1, 143.7, 135.3, 129.3, 128.1, 126.9, 126.5, 126.2, 79.5, 76.4, 53.2, 46.8, 37.2, 28.2, 25.8, 18.3, −4.8; EIMS: 506.3 [M + Na+], HRMS (ESI) calcd for C28H41NSiO4Na: 506.2703, found 506.2698.
- 15.Evans BE, Rittle KE, Homnick CF, Springer JP, Hirshfield J, Veber DF. J Org Chem. 1985;50:4615. [Google Scholar]
- 16.Nadin A, Lopez JMS, Neduvelil JG, Thomas SR. Tetrahedron. 2001;57:1861. [Google Scholar]
- 17.Preparation of 13. Tert-Butyl (S)-1-((2R,4R)-4-benzyl-5-oxotetrahydrofuran-2-yl)-2-(4-(S)-hydroxyphenyl methylphenyl) ethyl carbamate (13). A solution of silyl ether 12 (390 mg, 0.63 mmol) in THF (10 mL) was transferred to a polyethylene vial and Py·HF (70% HF, 1.5 mL) was dropwise added at 0 °C. The mixture was stirred at rt. overnight. The reaction mixture was quenched with saturated NaHCO3 solution, and extracted with EtOAc (3 × 50 mL). The combined organic layer was washed with brine, dried with Na2SO4. The residue was purified by column chromatography (50 % EtOAc in Hexane) to give 13 (264 mg, 83 %) as a white solid: mp 59–60 °C; [α]25D −58.8 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3): 7.35–7.21 (m, 10 H), 7.15 (d, J = 7.2 Hz, 2H), 7.10 (d, J = 7.9 Hz, 2H), 5.77 (d, J = 2.7 Hz, 1H), 4.43 (br, 1H), 4.09 (br, 1H), 3.85 (br, 1H), 3.24 (dd, J = 4.0, 13.8 Hz, 1H), 2.87–2.68 (m, 4H), 2.63 (d, J = 3.3 Hz, 1H, OH), 2.18 (m, 1H), 1.75 (m, 1H), 1.31 (s, 9H); 13C NMR: 177.6, 155.3, 143.9, 142.5, 138.4, 135.9, 129.6, 128.8, 128.7, 128.5, 128.4, 127.5, 127.3, 127.2, 126.8, 126.7, 126.5, 79.8, 78.8, 75.9, 54.3, 43.0, 36.1, 31.6, 28.2; EIMS: 524.1 [M + Na+], HRMS (ESI) calcd for C31H35NO5Na: 524.2413, found 524.2402.
- 18.Fauq AH, Cherif-Ziani C, Richelson E. Tetrahedron: Asymmetry. 1998;9:2333. [Google Scholar]
- 19.Preparation of 14. Tert-Butyl (S)-2-(4-benzoylphenyl)-1-((2R,4R)-4-benzyl-5-oxotetrahydrofuran-2-yl) ethyl carbamate (14). To an ice-cold solution of 13 (240 mg, 0.478 mmol) in CH2Cl2 (10 mL) was added MnO2 (415 mg, 4.78 mmol). The suspension was stirred at rt. overnight. The reaction mixture was filtered through celite, washed with EtOAc. The combined organic layer was concentrated, the residue was purified by column chromatography (40 % EtOAc in Hexane) to give 14 (185 mg, 76 %) as a white solid: mp 59–60 °C; [α]25 D −68.7 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3): 7.74 (dd, J = 7.2, 11.9 Hz, 4H), 7.57 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 7.28 (m, 4 H), 7.17 (m, 3H), 4.68 (d, J = 9.4 Hz, 1H), 4.32 (br, 1H), 3.93 (br, 1H), 3.26 (dd, J = 4.1, 13.9 Hz, 1H), 3.03–2.73 (4H), 2.30 (m, 1H), 1.86 (m, 1H), 1.34 (s, 9H); 13 C NMR: 196.4, 177.6, 155.3, 142.3, 138.4, 137.7, 136.1, 132.5, 130.4, 130.0, 129.5, 128.9, 128.8, 128.4, 126.9, 80.0, 79.2, 54.6, 42.5, 36.5, 36.2, 31.4, 28.3; EIMS: 522.2 [M + Na+], HRMS (ESI) calcd for C31H33NO5Na: 522.2256, found 522.2250.
- 20.Kokotos G, Padrón JM, Martín T, Gibbons WA, Martín VS. J Org Chem. 1998;63:3741. [Google Scholar]
- 21.Preparation of 2. {1S-Benzoyl-phenyl-4R-[1-(1S-methyl-oxycarbonyl-2-phenyl-ethylcarbamoyl)-3-(1S)-methyl-butylcarbamoyl]-2R-hydroxy-5-phenyl-pentyl}-carbamic acid tert-butyl ester (2). A solution of 15 (20 mg, 0.0316 mmol), Leu-Phe-OMe (16 mg, 0.054 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (11 mg, 0.057 mmol) and 1-hydroxylbenzotriazole (8 mg, 0.057 mmol) in DMF (2 mL) was stirred at rt. The reaction mixture was diluted with EtOAc, and the organic layer was washed with aqueous citric acid, saturated NaHCO3 solution, and brine, dried with Na2SO4, and concentrated under reduced pressure. Purification of the residue by column chromatography (50 % EtOAc in hexane) gave 16 (20 mg, 70%) as a white foam: 1H NMR (400 MHz, CDCl3): 7.76 (d, J = 7.1 Hz, 2H), 7.68 (d, J = 8.0 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.48 (d, J = 7.7 Hz, 2H), 7.27 (m, 4H), 7.20 (m, 4H), 7.11 (d, J = 7.0 Hz, 2H), 6.96 (d, J = 8.0 Hz, 2H), 6.49 (t, J =8.05 Hz, 2H), 4.78 (m, 1H), 4.55 (d, J = 8.0 Hz, 1H), 4.43 (m, 1H), 3.84 (m, 1H), 3.69 (s, 3H), 3.65 (m, 1H), 3.12–3.00 (m, 2H), 2.65 (dd, J = 5.7, 13.6 Hz, 1H), 2.59 (m, 1H), 2.45 (m, 1H), 1.87 (m, 1H), 1.79–1.58 (m, 3H), 1.37 (s, 9H), 0.91 (s, 9H), 0.86 (dd, J = 6.7, 11.1 Hz, 6H), 0.07 (s, 3H), 0.06 (s, 3H); 13 C NMR: 196.6, 174.9, 171.9, 171.4, 155.7, 144.2, 139.5, 137.9, 136.1, 135.9, 132.4, 130.5, 130.2, 129.5, 129.4, 129.2, 128.8, 128.8, 128.5, 127.3, 126.7, 79.9, 72.5, 54.9, 53.5, 52.5, 51.9, 45.5, 40.8, 38.9, 38.1, 36.0, 33.9, 28.6, 26.1, 25.0, 22.7, 22.6, 18.2, −4.2, −4.7; EIMS: 928.2 [M + Na+], calcd for C53H71N3O8Si: 905.50.To an ice-cold solution of 16 (20 mg, 0.022 mmol) in THF (2 mL) was added a solution of TBAF in THF (1.0 M, 0.2 mL). The mixture was stirred at rt. overnight. The reaction mixture was diluted with EtOAc and washed with citric acid and brine, dried with Na2SO4, and concentrated under reduced pressure. Purification of the residue by column chromatography (5% MeOH in CH2Cl2 )gave 2 (17 mg, 85%) as a white solid: mp 147–148 °C; [α]25D −12.1 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3, a few drop of d4-MeOH): 7.77 (d, J = 7.3 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.7 Hz, 2H), 7.33–7.17 (m, 6H), 7.13 (t, J = 7.5 Hz, 2H), 6.69 (d, J = 7.1 Hz, 1H), 6.17 (d, J = 7.2 Hz, 1H), 4.78 (q, J = 7.3 Hz, 1H), 4.71 (d, J = 8.5 Hz, 1H), 4.26 (q, J = 7.3 Hz, 1H), 3.70 (m+s, 6H), 3.09 (m, 2H), 2.93 (dd, J = 7.2, 13.5 Hz, 1H), 2.86–2.62 (m, 4H), 1.83 (m, 1H), 1.74 (m, 1H), 1.56 (m, 1H), 1.46 (m, 2H), 1.39 (s, 9H), 0.84 (dd, J = 6.3, 8.5 Hz, 6H); 13 C NMR: 196.6, 175.8, 172.2, 171.8, 156.2, 143.6, 139.0, 137.9, 136.0, 135.9, 132.5, 130.5, 130.1, 129.5, 129.16, 129.0, 128.8, 128.8, 128.7, 128.4, 127.3, 126.8, 79.9, 73.3, 56.6, 53.5, 52.5, 47.0, 40.8, 39.3, 38.1, 35.7, 35.5, 29.9, 28.4, 26.0, 24.8, 23.0, 22.1; HRMS (ESI) calcd for C47H58N3O8: 792.4224, found 792.4229.
- 22.Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Proc Natl Acad Sci USA. 2000;97:6138. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]