

Abstract
Objective
To explore the relationships between bactericidal antimicrobial treatment of sepsis, inflammatory response, severity of AKI, and outcomes.
Design
Controlled laboratory experiment.
Setting
University laboratory.
Interventions
Sepsis was induced by cecal ligation and puncture (CLP) in 52 rats and was treated with either bactericidal antibiotics (ampicillin/sulbactam) or placebo (saline). Serial blood specimens were obtained after CLP for serum creatinine, interleukin (IL)-6, and neutrophil gelatinase-associated lipocalin (NGAL) concentrations. RIFLE criteria were used to assess severity of AKI. All animals were observed for survival up to one week. In a separate experiment, 6 healthy animals were given antibiotics and renal function was assessed. Another 12 animals were sacrificed 2 days after laparotomy for kidney histology.
Measurements and Main Results
Survival in the placebo group was 50% compared to 81.8% in the antibiotic group (P <0.05). Most animals (93%) without antibiotics developed AKI, of which 39% exhibited greater than a 3-fold rise in serum creatinine (RIFLE-F). Furthermore survival decreased as AKI severity increased. Surprisingly, all antibiotic-treated animals developed AKI, of which 68.6% reached RIFLE-F. However, renal dysfunction was less persistent in these animals. Patterns of plasma IL-6 were similar to creatinine with higher concentrations seen earlier in antibiotic-treated animals but with faster resolution. IL-6 concentration at 24 hrs was independently associated with the development of RIFLE-F. Histological findings were consistent with functional parameters showing that antibiotics worsened AKI.
Conclusion
In polymicrobial sepsis bactericidal antibiotics resulted in more inflammation and more severe AKI. However, resolution of inflammation and AKI was faster with antibiotics and correlated best with survival. These results suggest that transient worsening of renal function may be an expected consequence of sepsis therapy. These findings also question the value of peak severity of AKI as a primary endpoint and suggest that resolution of AKI may be more appropriate.
Keywords: Acute kidney injury, acute renal failure, sepsis
Acute kidney injury (AKI) often complicates sepsis in critically ill patients (1, 2), and is also an important predictor for outcome even in non-critically ill patients (3). In general, sepsis and likelihood of death appear to correlate with severity of AKI (4, 5). However, observations in humans reflect both the degree of functional impairment as measured by serum creatinine as well as the severity of the underlying insult causing the impairment. It is assumed that measures that improve kidney function will reduce kidney injury or conversely, therapies which worsen kidney function acutely must also increase kidney injury. These assumptions were analogous to the way heart failure was approached twenty years ago, as interventions for inhibition of cardiac contractility were contraindicated in congestive heart failure. The introduction of beta blockers as a treatment for heart failure shattered the existing paradigm that only measures to increase cardiac function would be beneficial (6). Our current AKI paradigm is similarly constrained and we are evaluating treatments solely on the basis of the degree to which maximum functional impairment is affected. Under this paradigm, even therapies which shorten AKI duration but transiently result in worse function would be rejected.
Furthermore, the mechanisms of sepsis-induced AKI remain controversial (7-10). We have shown, in humans with community-acquired pneumonia, that AKI severity is associated with the degree of inflammatory response as measured by pro- and anti-inflammatory cytokine activation (11). If, in sepsis, AKI occurs as a result of the cytotoxic effects of cytokines and other inflammatory mediators, then experimental models of sepsis showing greater cytokine activation should result in worse AKI (12).
Bactericidal antibiotics are known to increase inflammation acutely by release of bacterial toxins (13-14). However, timing and appropriateness of antibiotics have also been shown to influence outcome in humans with sepsis (15) and are recommended as critical components of sepsis care bundles (15,16). We hypothesized that bactericidal antibiotics would result in increased inflammation and worse kidney function while still leading to improved survival and ultimately better organ function. This question is important because it asks whether AKI therapies can be solely evaluated on the basis of maximum functional impairment.
MATERIALS AND METHODS
Experimental Protocol
Following approval by the Animal Care and Use Committee of the University of Pittsburgh, we anesthetized 52 adult (24 to 28 weeks old, weight 400-600g), healthy, male, Sprague-Dawley rats with intraperitoneal injection of pentobarbital sodium (40 mg/kg). Cecal ligation and puncture (CLP) was performed with a predetermined 25% ligated length of cecum and 20-gauge needle: two punctures inferior to the ileocecal valve. This protocol is associated with a mortality of about 50-60% at Day 7 (17). The abdomen was closed and 20 ml/kg lactated Ringer’s solution was given subcutaneously for resuscitation. Topical anesthetic was applied to the surgical wound and rats were returned back to their cages and allowed food and water ad libitum.
Eighteen hours after CLP, animals were returned to the laboratory and assigned to either: group 1 (n=22) and given ampicillin/sulbactam (125mg/kg every 12 hrs) starting 18 hrs after CLP and continued for three days; or group 2 (n=30) which were given saline injections as placebo. A jugular vein catheter was also placed to draw blood and survival time was assessed up to 7 days. In order to exclude any possible effects of antibiotics on measures of renal function, we gave the same dose and courses of therapy to another 6 healthy (laparotomy but no CLP) animals as a control and obtained the same measurements. Another 12 animals (4 CLP, 4 CLP + antibiotics and 4 healthy +antibiotics) were sacrificed 2 days following laparotomy for kidney histology.
Measurements and Calculations
Blood (0.8ml) was drawn from a central venous catheter at 18 hrs, 24 hrs, 48 hrs, 72 hrs, 5 days and 7 days after CLP. Similar time points were obtained for healthy controls. In order to ensure that the maximum blood loss for each animal was controlled below the 20% total blood volume, we staggered sample collections so that only 4 time points were obtained in each animal. Therefore, data that were not collected for serum creatinine (Cr) were imputed by averaging the values before and after the missing value. The isolated plasma was kept at -80° for subsequent interleukin (IL)-6, neutrophil gelatinase-associated lipocalin (NGAL), and creatinine measurements. Survival time was recorded in days starting from CLP.
Plasma IL-6 was measured with an enzyme-linked immunosorbent assay (ELISA) (R & D Systems, Minneapolis, MN). Plasma NGAL was determined using an ELISA (BioPorto Diagnostics, Gentofte, Denmark). Plasma creatinine was detected with a creatinine enzymatic assay kit (BioVision Technologies, Mountain View, CA). The severity of AKI was assessed using the serum creatinine portion of the RIFLE criteria (18) which classified risk (R), injury (I), and failure (F), on the basis of maximum creatinine increase of 150%, 200% and 300% respectively over the 7 days following CLP.
Evaluation of kidney histology
Rat kidneys were fixed in 10% neutral buffered formalin, dehydrated in graded anhydrous absolute ethanol, and embedded in paraffin. Histological sections (5μm) of kidney were stained with hematoxylin-eosin and periodic acid schiff. We considered the morphological changes indicating acute tubular necrosis as the loss of brush border, the vacuolization of tubular epithelial cells, and the presence of intra-tubular debris.
In order to explore if the inflammatory response produces organ-specific effects, we also evaluated liver injury with alanine aminotransferase (ALT). ALT was determined using a LDH-NADH coupled assay (Pointe Scientific Inc, Canton, MI) from twenty animals (13 from antibiotics treated group, and 7 from placebo group).
Statistical Analysis
Descriptive data were expressed as means ± standard error (SE). The analysis of variance and unpaired student’s t-test were applied to compare the normally distributed variables within and between groups. Mann-Whitney U-test was used to compare the non-normally distributed data. Categorical variables were expressed as proportions and compared using the Chi-square test. The association between plasma IL-6 and severity of AKI was examined with logistic regression analysis. The survival analysis was assessed by Kaplan-Meier statistics and compared using Log rank test. A two sided P < 0.05 was considered statistically significant.
RESULTS
Antibiotics and AKI
Although antibiotics significantly improved 7-day survival compared to placebo (81.8% vs. 50% P = 0.01, Figure 1), antibiotics did not improve kidney function acutely. Most animals (93%) not receiving antibiotics and all animals treated with antibiotics developed AKI. However, maximum functional impairment was greater in animals receiving antibiotics. Of animals developing AKI, 68.6% of antibiotic-treated animals and 39% of placebo-treated animals reached (RIFLE-F) (Figure 2) (P <0.05). None of the healthy animals that received antibiotics developed AKI. Figure 3 shows the kidney histology using hematoxylin-eosin (HE) and periodic acid Schiff (PAS) stains under light microscopy with original magnification of X400. Loss of brush border was evident, as was mild dilation of the tubular lumen 48 hrs after CLP. Vacuolization was seen after CLP in almost all tubule cells (Figure 3A). These pathological changes were even worse at 48 hrs in the antibiotic-treated CLP animals (Figure 3B). There was no obvious kidney injury in healthy animals treated with antibiotics (Figure 3C).
Figure 1.

Kaplan-Meier Survival plots for all animals. CLP (n=30): animals with CLP treated with saline; CLP+Abs (n=22) animals with CLP treated with antibiotics (Abs); Abs (n=6): animals without CLP treated with antibiotics; P<0.05, CLP vs CLP+Abs
Figure 2.
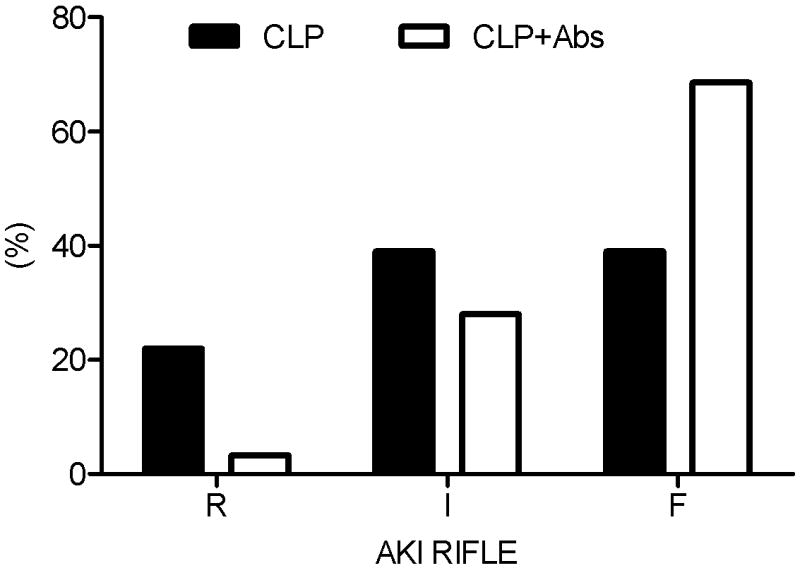
Distribution of severity of acute kidney injury. RIFLE categories, R, I, F = AKI RIFLE classes Risk, Injury and Failure. CLP (n=30): animals with CLP treated with saline; CLP+Abs (n=22): animals with CLP treated with antibiotics; P<0.05, CLP vs CLP+Abs regarding RIFLE-F.
Figure 3.

Kidney histology after 48 hrs of laparotomy. Histological sections (5μm) of kidney were stained with hematoxylin-eosin (HE) and periodic acid Schiff (PAS). A. CLP: animals with CLP treated with saline; B. CLP+Abs: animals with CLP treated with Abs; C. Abs: animals without CLP treated with Abs.
Outcomes by severity of AKI
When we compared the survival rates and severity of AKI, we found that mortality increased with increasing severity of AKI (Figure 4). However, 73% of animals with RIFLE-F died in the placebo group, compared to only 36.4% in the antibiotic-treated group. When we compared the creatinine concentration over time between the survivors and non-survivors, we found that the creatinine concentration increased with time in the non-survivors of both groups, while in all the survivors the creatinine concentration increased temporarily, and then gradually recovered (Figure 5).
Figure 4.
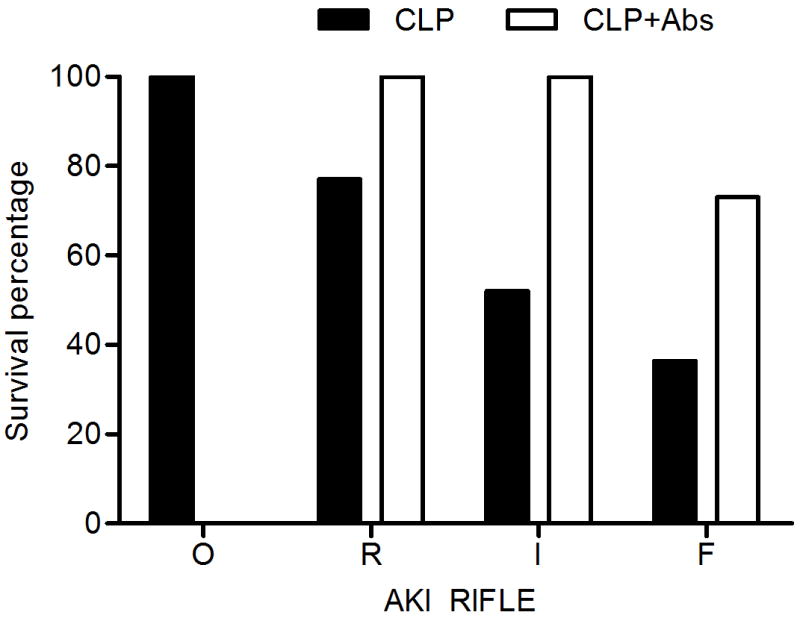
Survival of animals by severity of AKI (RIFLE class). O = no AKI; R,I,F = AKI RIFLE classes R, I and F.
Figure 5.

Serum creatinine (Cr) patterns over time for survivors and non-survivors in two groups (mean, mg/dl).
Biomarker patterns with time
Figure 6A shows the relationship between inflammatory response as measured by IL-6 over time and by group. IL-6 levels were not different prior to antibiotics or placebo in septic animals but in both groups levels were significantly increased compared to healthy controls at 18 hours. At 24 hours (6 hours after antibiotics were given), IL-6 levels peaked in the antibiotic-treated group and were significantly greater than placebo-treated animals (370.80 vs 561.46 pg/ml, P<0.05). While inflammation resolved quickly in antibiotic-treated animals, it persisted in placebo-treated animals such that the IL-6 levels were much greater in this group starting at 72hrs (336.79 vs 133.76 pg/ml, P<0.05) persisting throughout the 7 days.
Figure 6.

IL-6, plasma Cr and NGAL changes with time (mean ± SE). CLP (n=30): animals with CLP treated with saline; CLP+Abs (n=22): animals with CLP treated with Abs; Abs (n=6): animals without CLP treated with Abs; * p<0.05, CLP vs CLP+Abs. A: IL-6 (pg/ml); B: Cr (mg/dl); C: NGAL (IU/ml)
Changes in plasma Cr (Figure 6B) over time were similar to those of IL-6 but increased more slowly. Serum creatinine peaked at approximately 48 hrs in the antibiotic-treated CLP animals and then recovered gradually; however, it increased more slowly and remained elevated longer in placebo-treated animals. The peak serum creatinine concentration was observed 48-72 hrs after peak IL-6 concentrations, suggesting a temporal relationship between inflammation and onset of AKI. At 7 days, the Cr in the antibiotic-treated group was significantly lower than that of the placebo group (0.38 vs 0.62 mg/dl, p<0.05). There was no significant change in Cr with time in healthy animals treated with antibiotics.
Plasma NGAL increased with time in all groups as shown in Figure 6C. Eighteen hours after CLP, plasma NGAL had increased nearly 10 fold compared to healthy animals, reaching a peak (about 15 fold) at 24hrs, and remained significantly elevated for the entire week. While NGAL from antibiotic-treated animals gradually decreased after 48hrs, pNGAL concentrations remained significantly elevated in placebo-treated animals beginning at 96hrs and up to 7 days (Figure 6C, P<0.05).
Association between IL-6, AKI severity, and outcome
In logistic regression analysis, the plasma IL-6 concentration at 24 hrs of CLP was independently associated with AKI severity (RIFLE-F vs others). The odds ratio was 1.28 (95% confidential interval: 1.09-1.89, P<0.05). To determine the contributions of IL-6 to the predictive model for AKI severity, the areas under the ROC curve were measured for the logistic regression model based on the IL-6 concentration at 24 hrs after CLP. The area under the ROC curve was 0.79 (95% confidential interval: 0.61-0.91, P<0.05). The median IL-6 concentration at 24hrs after CLP in RIFLE-R, RIFLE-I and RIFLE-F were 270.75, 414.24 and 563.62 pg/ml respectively (P<0.05, RIFLE-F vs RIFLE-I; RIFLE-F vs RIFLE-R, Figure 7A). In all survivors, the median IL-6 levels decreased rapidly with time, while the levels were still high with time in all non-survivors (Figure 7B).
Figure 7.

Relationship between IL-6 levels, AKI severity and outcome.
A. Box-plot summaries of IL-6 level at 24 hrs of CLP in those with AKI RIFLE-R, RIFLE-I and RIFLE-F (pg/ml). P<0.05, RIFLE-F vs RIFLE-I, RIFLE-F vs RIFLE-R.
B. Median IL-6 (pg/ml) changes with time in all survivors and non-survivors.
Association between IL-6 and liver injury
In order to explore if the inflammatory response produces organ-specific effects, we analyzed the relationship between median IL-6 levels and median ALT levels. Figure 8 demonstrated that changes of ALT (Figure 8A) and IL-6 (Figure 8B) with time were consistent.
Figure 8.

Relationship between IL-6 and liver injury.
A. Median IL-6 (pg/ml) changes with time in twenty rats ((13 from antibiotics treated group, and 7 from placebo group).
B. Median ALT (IU/L) changes with time in twenty rats (13 from antibiotics treated group, and 7 from placebo group).
DISCUSSION
The main finding of this study was that while treatment with bactericidal antibiotics improved survival in CLP-induced sepsis, it neither prevented AKI nor limited its severity as measured by maximum RIFLE stage using serum creatinine. Indeed AKI severity was actually greater in antibiotic-treated animals though duration was attenuated. Although this finding seems paradoxical it is consistent with observations in humans. First, it has been known for many years that bactericidal antibiotics can release toxins as they kill bacteria causing inflammation and clinical symptoms (e.g. fever, chills). The classic Jarisch-Herxheimer reaction has been described with spirochetes, but similar reactions have been described with multiple other infections (13) and are probably quite common in sepsis (14). The downstream mechanism appears to be the activation of inflammatory cytokines especially IL-6, IL-8 and TNF (19). In our study we chose to measure only one cytokine to minimize blood loss and chose IL-6 as the representative cytokine (20, 21); and we found that IL-6 increased early after antibiotics. We chose ampicillin/sulbactam in our study as it is active against a wide range of bacterial groups, and used in various infections, including intra-abdominal, skin, lower respiratory tract, and gynecological infections. It is also a commonly-used bactericidal antibiotic. The “spike” in IL-6 induced by antimicrobials (ampicillin/sulbactam) was most likely related to rapid bacterial cell death resulting in endotoxin release (22,23), although it is also known that antimicrobials have immunomodulating properties (24) and macrophage activation with IL-6 release may also be a “side effect” of antimicrobials (25,26).
Second, we have recently shown a strong correlation between IL-6 expression and AKI severity in sepsis-induced AKI in humans (11). It is therefore not surprising that an increased acute inflammatory response should result in worse renal function. Although the mechanistic link between cytokine activation and AKI is not well understood, emerging evidence suggests that the inflammatory milieu may lead to renal cell dysfunction in a variety of ways (27). However, this inflammatory response is not organ-specific, as we also showed the consistent liver injury occurred with the increased IL-6. Finally, the duration of AKI appears to be a predictor of long term outcome in patients with AKI. Coca and colleagues reported that in diabetics with postoperative AKI the duration of renal impairment was independently associated with decreased survival and recovery (28). Our results are similar in many ways. As shown in figure 6, animals treated with antibiotics had a shorter duration of AKI by both RIFLE (serum creatinine) and plasma NGAL. Indeed, animals not receiving antibiotics had still not completely recovered by day 7. As shown in figure 5, surviving animals exhibited evidence of renal recovery relatively early (by 36-48hrs). Animals that did not resolve AKI early did not survive. Of course, unlike the clinical situation, our study design would have accentuated this effect because we did not provide renal replacement therapy. Nevertheless, our results show clear evidence that shorter duration of AKI is associated with survival as was seen by Coca et al (28).
Our results are also consistent with the vast majority of epidemiologic studies that found a relationship between severity of AKI and survival. In our study, like in clinical studies (1, 2), survival decreased as severity of AKI increased. However, our study was also able to extend these prior results because we could specifically control for resolution of infection. Our antibiotic-treated animals represent early and effective control of infection while non-treated animals represent the extreme case of inadequate control. In this way our results demonstrate the importance of adequate treatment of infection (appropriate antibiotics and source control) for the successful resolution (though not prevention) of AKI. These findings have potentially important implications for future studies of therapies for AKI, especially regarding endpoints. Like antibiotics, a drug might be effective in limiting the course of AKI and hence improving outcome, while simultaneously worsening renal function temporarily.
The results of our study also lend support to the concept that plasma NGAL is an early predictor for AKI (29). We found that NGAL increased prior to creatinine in animals developing AKI. However, given that most of our animals developed AKI, it is not possible to adequately test the discrimination of this biomarker. Importantly, NGAL levels began to decline after 48hrs in the antimicrobial-treated animals while they remained elevated in the placebo group. Even at day 7, there was still significantly increased plasma NGAL in placebo-treated animals whereas this biomarker had nearly normalized in the antimicrobial-treated group. To the extent that NGAL represents kidney damage, placebo-treated animals continued to manifest damage one week after CLP. Finally, although the onset of NGAL activation in the plasma appeared similar to that of IL-6, NGAL remained elevated in placebo-treated animals, as did Cr, even after IL-6 began to normalize. This suggests that NGAL may be a useful marker for resolution of AKI as we have recently shown in critically ill patients with sepsis-induced AKI (30).
Another interesting finding is that there was a small increase of NGAL without concomitant increase of IL-6 or Cr after antibiotic injection in the healthy animals. In order to better understand the mechanisms of this effect we injected saline into four healthy animals and obtained similar results (data not shown). Thus, it appears that this very small increase in plasma NGAL was simply due to the overall stress reaction. NGAL is also synthesized in peritoneal mesothelial cells, and is induced by the peritoneal and gut damage (31). Released by neutrophils upon activation, NGAL is also a marker of bacterial infection and systemic inflammation (32, 33).
There are important limitations to this study. First, this study was conducted in CLP induced septic rats. These animals only received antimicrobials and limited supportive care (fluid resuscitation). Surgical source control of infection, inotropes/vasopressors, mechanical ventilation and renal replacement therapies were not applied to these animals. Thus the course of sepsis and AKI may not be synonymous with the clinical scenario in humans. Second, we did not collect urine; therefore we lack urine output data to integrate into the RIFLE categories or to examine urine biomarkers. These factors may have contributed to a misclassification of some cases of AKI and may have influenced risk estimates of AKI. Urine NGAL may be a more specific marker of AKI compared to plasma NGAL (34). We studied only one time point (18hrs after CLP), for initiation of antibiotic therapy as this time point has been shown to correspond to the time when symptoms are fully manifest and therefore likely (35) simulates when a patient would seek medical attention. Furthermore, the concentrations of most mediators reach peek levels between 18-24 hrs (17, 36). We did not measure plasma endotoxin levels and furthermore, we tested only one of many possible antimicrobial treatments, and the antibiotic-induced endotoxin release may be dependent on the class of antibiotics used. For instance, Vianna et al have demonstrated in a similar model of sepsis that at 6h after CLP imipenem-treated animals showed endotoxin plasma concentrations higher than those observed in animals treated with ciprofloxacin plus clindamycin or the animals that received no antibiotic treatment (37).
In summary, this study demonstrates that bactericidal antibiotics transiently increase inflammation and do not prevent AKI although they significantly improved outcome. Resolution of inflammation and AKI, however, were more common and faster in animals receiving antibiotics. The induction of AKI during sepsis is strongly correlated and temporally related to activation of inflammatory mediators. Mortality was associated with failure to recover from, rather than development of, AKI in this sepsis model. Thus, these results question the value of peak severity of AKI as a primary endpoint and suggest that resolution of AKI may be more appropriate.
Acknowledgments
This study was funded, in part, by the National Institutes of Health.
Financial support: This work was funded in part by NIH R01HL080926 (JK and ZP). Additional funding to investigators came from R01DK070910 (JK), K08GM081459 (KS) and KL2 RR024154 (RM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, And Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of General Medical Science or the National Institutes of Health.
Footnotes
The authors have not disclosed any potential conflicts of interest.
References
- 1.Bagshaw SM, Uchino S, Bellomo R, et al. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol. 2007;2:431–439. doi: 10.2215/CJN.03681106. [DOI] [PubMed] [Google Scholar]
- 2.Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 3.Barrantes F, Feng Y, Ivanov O, et al. Acute kidney injury predicts outcomes of non-critically ill patients. Mayo Clin Proc. 2009;84:410–416. doi: 10.1016/S0025-6196(11)60559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagshaw SM, George C, Dinu I, et al. A Multi-Centre Evaluation of the Rifle Criteria forEarly Acute Kidney Injury in Critically Ill Patients. Nephrol Dial Transplant. 2008;23:1203–1210. doi: 10.1093/ndt/gfm744. [DOI] [PubMed] [Google Scholar]
- 5.Chen YC, Jenq CC, Tian YC, et al. Rifle classification for predicting in-hospital mortality in critically ill sepsis patients. Shock. 2009;31:139–145. doi: 10.1097/SHK.0b013e31817d419e. [DOI] [PubMed] [Google Scholar]
- 6.Hjalmarson A, Goldstein S, Fagerberg B, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA. 2000;283(10):1295–302. doi: 10.1001/jama.283.10.1295. [DOI] [PubMed] [Google Scholar]
- 7.Langenberg C, Bagshaw SM, May CN, et al. The histopathology of septic acute kidney injury: a systematic review. Crit Care. 2008;12:R38. doi: 10.1186/cc6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagshaw SM, Langenberg C, Wan L, et al. A systematic review of urinary findings in experimental septic acute renal failure. Crit Care Med. 2007;35:1592–1598. doi: 10.1097/01.CCM.0000266684.17500.2F. [DOI] [PubMed] [Google Scholar]
- 9.Kellum JA. Impaired renal blood flow and the ‘spicy food’ hypothesis of acute kidney injury. Crit Care Med. 2011;39:901–3. doi: 10.1097/CCM.0b013e31820f70bb. [DOI] [PubMed] [Google Scholar]
- 10.Mehta RL, Bouchard J, Soroko SB, Ikizler TA, Paganini EP, Chertow GM, Himmelfarb J Program to Improve Care in Acute Renal Disease (PICARD) Study Group. Sepsis as a cause and consequence of acute kidney injury: Program to Improve Care in Acute Renal Disease. Intensive Care Med. 2011;37:241–8. doi: 10.1007/s00134-010-2089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murugan R, Karajala-Subramanyam V, Lee M, et al. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010;77:527–535. doi: 10.1038/ki.2009.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chawla LS, Seneff MG, Nelson DR, et al. Elevated plasma concentrations of IL-6 and elevated APACHE II score predict acute kidney injury in patients with severe sepsis. Clin J Am Soc Nephrol. 2007;2:22–30. doi: 10.2215/CJN.02510706. [DOI] [PubMed] [Google Scholar]
- 13.Loscalzo J, Fauci AS, Braunwald E, et al. Harrison’s Principles of Internal Medicine. 17. Dubuque: McGraw-Hill Medical; 2008. pp. 1048–1067. [Google Scholar]
- 14.ALKharfy KM, Kellum JA, Matzke G. Unintended Immunomodulation: Part II. Effects of Pharmacological Agents on Cytokine Activity. Shock. 2000;13:346–360. doi: 10.1097/00024382-200005000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Garnacho-Montero J, Aldabo-Pallas T, Garnacho-Montero C, et al. Timing of adequate antibiotic therapy is a greater determinant of outcome than are TNF and IL-10 polymorphisms in patients with sepsis. Crit Care. 2006;10:R111. doi: 10.1186/cc4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vyas D, Javadi P, Dipasco PJ, et al. Early antibiotic administration but not antibodytherapy directed against IL-6 improves survival in septic mice predicted to die on basis of high IL-6 levels. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1048–1053. doi: 10.1152/ajpregu.00312.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng Z, Wang H, Carter M, et al. Hemoadsorption improves long-term survival after sepsis in the rats. Crit Care Med. 2008;36(12 Suppl):A1. [Google Scholar]
- 18.Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P Acute Dialysis Quality Initiative workgroup. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204–12. doi: 10.1186/cc2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vidal V, Scragg IG, Cutler SJ, et al. Variable major lipoprotein is a principal TNF-inducing factor of louse-borne relapsing fever. Nat Med. 1998;4:1416–1420. doi: 10.1038/4007. [DOI] [PubMed] [Google Scholar]
- 20.Kellum JA, Kong L, Fink MP, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis. Arch Intern Med. 2007;167:1655–1633. doi: 10.1001/archinte.167.15.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song M, Kellum JA. Interleukin-6. Crit Care Med. 2005;33:S463–S465. doi: 10.1097/01.ccm.0000186784.62662.a1. [DOI] [PubMed] [Google Scholar]
- 22.Gismondo MR, Chisari G, Lo-Bue AM. Effects of ampicillin and sulbactam/ampicillin on the immune system. J Int Med Res. 1991;(Suppl 1):24A–28A. [PubMed] [Google Scholar]
- 23.Van Den Berg C, de Neeling AJ, Schot CS, et al. Delayed antibiotic-induced lysis of Escherichia coli in vitro is correlated with enhancement of LPS release. Scand J Infect Dis. 1992;24:619–627. doi: 10.3109/00365549209054648. [DOI] [PubMed] [Google Scholar]
- 24.Van Vlem B, Vanholder R, De Paepe P, et al. Immunomodulating effects of antibiotics: literature review. Infection. 1996;24:275–291. doi: 10.1007/BF01743360. [DOI] [PubMed] [Google Scholar]
- 25.Holzheimer RG. The significance of endotoxin release in experimental and clinical sepsis in surgical patients--evidence for antibiotic-induced endotoxin release? Infection. 1998;26:77–84. doi: 10.1007/BF02767765. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto A, Sakai T, Ochiai M, et al. Augmenting effect of antibiotics on endotoxin activity may cause a safety problem. Microbiol Immunol. 2004;48:97–102. doi: 10.1111/j.1348-0421.2004.tb03494.x. [DOI] [PubMed] [Google Scholar]
- 27.Wen X, Murugan R, Peng Z, et al. Pathophysiology of acute kidney injury: a new perspective. Contrib Nephrol. 2010;165:39–45. doi: 10.1159/000313743. [DOI] [PubMed] [Google Scholar]
- 28.Coca SG, King JT, Jr, Rosenthal RA, et al. The duration of postoperative acute kidney injury is an additional parameter predicting long-term survival in diabetic veterans. Kidney Int. 2010;78:926–933. doi: 10.1038/ki.2010.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soni SS, Cruz D, Bobek I, et al. NGAL: a biomarker of acute kidney injury and other systemic conditions. Int Urol Nephrol. 2010;42:141–150. doi: 10.1007/s11255-009-9608-z. [DOI] [PubMed] [Google Scholar]
- 30.Srisawat N, Murugan R, Lee M, et al. Plasma NGAL predicts recovery from acute kidney injury following pneumonia. Kidney Int. 2011 doi: 10.1038/ki.2011.160. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung JC, Lam MF, Tang SC, et al. Roles of neutrophil gelatinase-associated lipocalin in continuous ambulatory peritoneal dialysis-related peritonitis. J Clin Immunol. 2009;29:365–378. doi: 10.1007/s10875-008-9271-7. [DOI] [PubMed] [Google Scholar]
- 32.Xu SY, Pauksen K, Venge P. Serum measurements of human neutrophil lipocalin (HNL) discriminate between acute bacterial and viral infections. Scand J Clin Lab Invest. 1995;55:125–131. doi: 10.3109/00365519509089604. [DOI] [PubMed] [Google Scholar]
- 33.Mårtensson J, Bell M, Oldner A, et al. Neutrophil gelatinase-associated lipocalin in adult septic patients with and without acute kidney injury. Intensive Care Med. 2010;36:1333–1340. doi: 10.1007/s00134-010-1887-4. [DOI] [PubMed] [Google Scholar]
- 34.Bennett M, Dent CL, Ma Q, et al. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol. 2008;3:665–673. doi: 10.2215/CJN.04010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hubbard WJ, Choudhry M, Schwacha MG, et al. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 36.Maier S, Traeger T, Entleutner M, et al. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock. 2004;21:505–511. doi: 10.1097/01.shk.0000126906.52367.dd. [DOI] [PubMed] [Google Scholar]
- 37.Vianna RC, Gomes RN, Bozza FA, et al. Antibiotic treatment in a murine model of sepsis: impact on cytokines and endotoxin release. Shock. 2004;21:115–120. doi: 10.1097/01.shk.0000111828.07309.21. [DOI] [PubMed] [Google Scholar]