

Abstract
Indian hedgehog (Ihh) is indispensable for osteoblast differentiation during embryonic development of the endochondral skeleton. In the absence of Ihh, cells of the osteoblast lineage fail to activate the expression of Runx2, a transcription factor integral to osteoblast differentiation. However, it is hitherto unclear whether the lack of Runx2 expression is solely responsible for the failure of osteoblast formation in Ihh-null embryos. Here, by creating a mouse allele that expresses Runx2 in a Cre-dependent manner, we show that force-expression of Runx2 in the skeletogenic cells restores bone formation in the Runx2-null, but not in the Ihh-null embryo. Thus, the mechanism through which Ihh induces osteoblast differentiation requires other effectors in addition to Runx2.
Keywords: Indian hedgehog, Ihh, Runx2, osteoblast, bone
Introduction
Most elements of the mammalian skeleton originate from a cartilage intermediate through endochondrial ossification (Kronenberg, 2003). In this process, the skeletogenic mesenchymal cells condense to form a cartilage anlage composed of chondrocytes and several layers of surrounding fibroblastic cells that constitute the perichondrium. Following the initial phase of proliferation, chondrocytes located at the center of the anlage exit the cell cycle and undergo hypertrophy (increase in cell size). It is at this time that the bone-forming osteoblasts differentiate from the perichondrium adjacent to the hypertrophic chondrocytes. Thus, osteoblast differentiation during endochondral ossification is tightly coupled with chondrocyte development.
Indian hedgehog (Ihh) is a key signal emanating form the chondrocytes to induce osteoblast differentiation. Among the three mammalian Hedgehog proteins, Ihh is uniquely expressed by chondrocytes transitioning to the fully hypertrophic state, commonly known as the prehypertrophic and early hypertrophic chondrocytes (Lanske et al., 1996; St-Jacques et al., 1999; Vortkamp et al., 1996). Genetic deletion of Ihh in the mouse resulted in a complete lack of osteoblasts in the endochondral skeleton (St-Jacques et al., 1999). Similarly, ectopic induction of osteoblast differentiation by precocious hypertrophic chondrocytes in a chimeric mouse model also required Ihh (Chung et al., 2001). Moreover, studies of Smoothened (Smo), which encodes a 7-pass transmembrane protein indispensable for Hh signaling in the receiving cell, demonstrated a cell-autonomous requirement for Smo in the perichondrium for osteoblast differentiation (Long et al., 2004). These studies support a direct role for Ihh signaling in osteoblastogenesis.
The mechanism through which Ihh induces osteoblast differentiation is not well understood. Analyses of the Ihh−/− embryo have revealed that the perichondrium is severely hypoplastic, and that none of known markers for the osteoblast lineage is detectable, indicating that the differentiation process is arrested at a very early stage (Hu et al., 2005; St-Jacques et al., 1999). Although our previous work has shown that Ihh exerts its osteogenic effect through both Gli3 suppression and Gli2 activation (Hilton et al., 2005; Joeng and Long, 2009), the relevant target genes for either Gli2 or Gli3 are not known.
Runx2, a runt-domain transcription factor, is an attractive candidate as an important mediator for the osteogenic activity of Ihh. Molecular and genetic studies have established the essential role of Runx2 in osteoblast differentiation (Ducy et al., 1997; Lee et al., 1997; Mundlos et al., 1997; Otto et al., 1997). Importantly, similar to Ihh removal, deletion of Runx2 in the mouse leads to no osteoblasts, and hypoplasia of the perichondrium (Komori et al., 1997; Otto et al., 1997). Moreover, Runx2 expression in the perichondrium was abolished in the Ihh−/− embryo. These findings raise the possibility that force-expression of Runx2 in the perichondrium may be sufficient to restore osteoblast differentiation in Ihh−/− embryos. Here we test this possibility by genetic means.
Results
Generation of a mouse strain expressing Runx2 in a Cre-dependent manner
To create a versatile tool to express Runx2 in a tissue-specific manner, we modified the Rosa26 genomic locus through homologous recombination so that Runx2 expression can be achieved following Cre-mediated recombination (Fig. 1A). The modified allele was termed R26Runx2. As expected, mice carrying either one or two copies of the allele (genotypes designated R26Runx2/+ or R26Runx2/Runx2, respectively) were completely normal. When these mice were crossed with a Col2-Cre transgenic line that targets both chondrocyte and osteoblast lineages in the endochondral skeleton, they produced progenies with the genotype of Col2-Cre; R26Runx2/+ (or C2Cre; R26Runx2/+) that were viable and possessed a relatively normal skeleton at E18.5 (Fig. 1B1-B2).
Fig. 1.
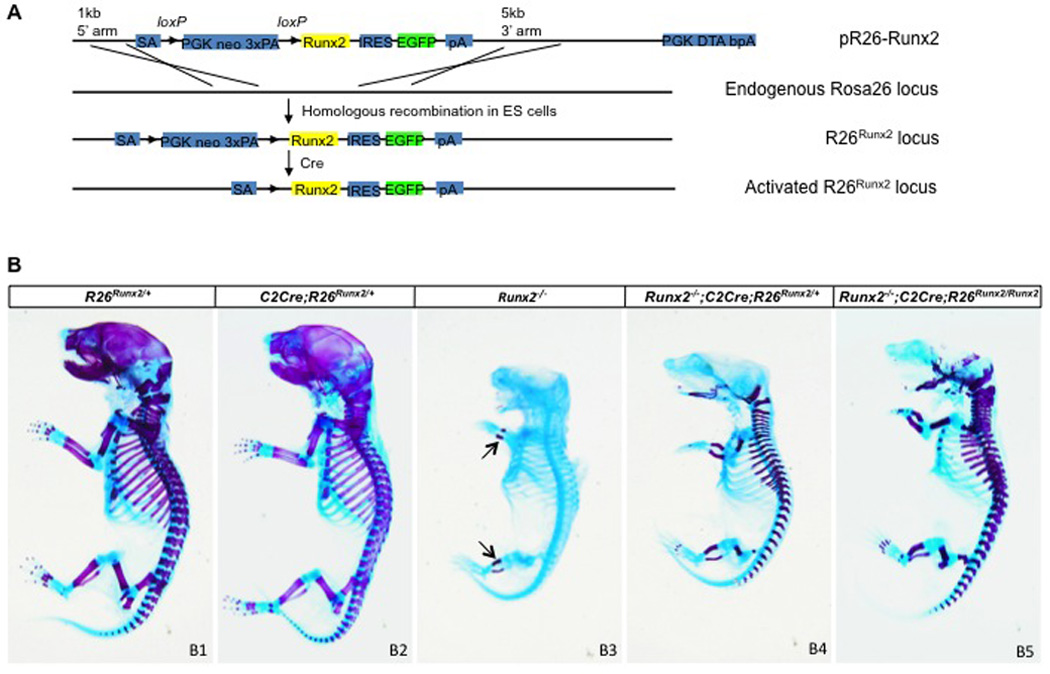
(A) Diagram for generation of the R26Runx2 allele. (B) Whole-mount skeletal staining of E18.5 mouse embryos. Arrows denote mineralized cartilage.
Forced expression of Runx2 rescues bone formation in Runx2-null mice
To determine the efficacy of Runx2 expressed from the R26Runx2 allele, we tested whether activation of R26Runx2 allele in the skeletogenic cells could functionally replace the endogenous Runx2 alleles in the embryonic skeleton. Specifically, we generated Runx2-null embryos (Runx2−/−), and those that also carried the Col2-Cre transgene, and one or two R26Runx2 alleles (Runx2−/−; C2Cre; R26Runx2/+, or Runx2−/−; C2Cre; R26Runx2/Runx2, respectively). When analyzed at E18.5 by whole-mount skeletal staining, the Runx2−/− embryos, exhibited no alizarin red staining throughout the body except for the zeugopod (Fig. 1B3). This staining pattern was consistent with the previous reports that the Runx2−/− embryos possessed no bone and only a small amount of mineralized cartilage in the zeugopod. Importantly, the Runx2−/− embryos expressing one or two R26Runx2 alleles exhibited progressively more alizarin red staining characteristic of bone (distinguishable from mineralized cartilage by a more red color) (Fig. 1B4-B5). The restoration of bone formation in the endochondral skeleton but not the skull was consistent with the specific targeting of Col2-Cre to the former. Thus, activation of the R26Runx2 allele in the endochondral skeleton is sufficient to restore bone formation in the Runx2-null embryo.
To corroborate the findings above, we conducted further analyses of the tibia. H&E staining of longitudinal sections from E18.5 embryos confirmed that activation of the R26Runx2 allele in the wild-type background (C2Cre; R26Runx2/+) did not overtly alter the morphology of the cartilage, the bone collar or the marrow (Fig. 2A-B). On the other hand, the Runx2−/− tibia lacked a bone collar or a marrow cavity, but possessed an elongated region of hypertrophic chondrocytes (Fig. 2C). In contrast, the Runx2−/− embryos expressing one or two R26Runx2 alleles formed a bone collar (Fig. 2D-E). Surprisingly, a marrow cavity was only observed in the embryos expressing two R26Runx2 alleles, revealing a dependence of marrow formation on Runx2 dosage (see Discussion). Moreover, no trabecular bone was observed within the marrow cavity of the Runx2−/− embryos expressing two R26Runx2 alleles. Nonetheless, these results confirm that activation of a single R26Runx2 allele is sufficient to induce osteoblast differentiation within the perichondrium in the absence of endogenous Runx2.
Fig. 2.
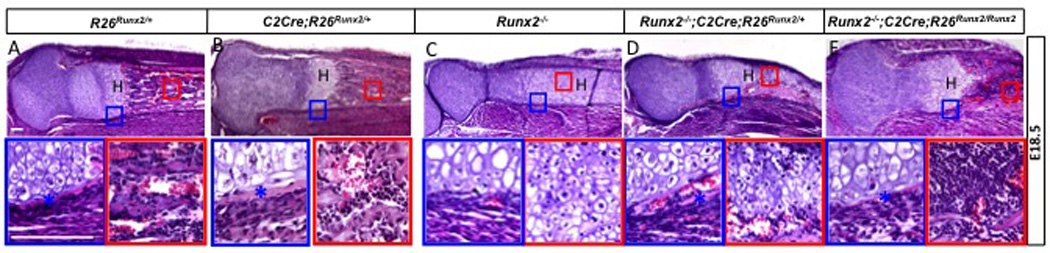
H&E staining of the tibia from E18.5 embryos. Proximal ends to the left. Boxed regions shown at a higher magnification below. Scale bar: 100 µm. Asterisks denote bone collar. H: hypertrophic zone.
The efficacy of the R26Runx2 allele was further demonstrated by molecular analyses of the tibia in E18.5 embryos. In situ hybridization confirmed that Runx2 was normally expressed in the perichondrium and the primary spongiosa, and at a lower level in the prehypertrophic and early hypertrophic cartilage (Fig. 3A1). Notably, prominent Runx2 signals were also detected in the perichondrium and the cartilage of the Runx2−/− embryo (which did not possess a primary spongiosa)(Fig. 3A2), indicating that the Runx2-null allele produced a mutant mRNA sufficiently stable to be detected by the in situ probe (see Methods). Moreover, activation of either one or two R26Runx2 alleles did not noticeably increase the overall Runx2 signal, indicating a relatively low abundance of the exogenous Runx2 (Fig. 3A3-A4). Further analyses of other osteoblast-lineage markers revealed that the Runx2−/− tibia expressed alkaline phosphatase (AP) and bone sialoprotein (Bsp) in the perichondrium, but no osterix (Osx) or osteocalcin (OC) (Fig. 3B2-E2). On the other hand, activation of either one or two R26Runx2 alleles induced the expression of both Osx and OC in the Runx2−/− embryos (Fig. 3D3-E3, D4-E4). Thus, molecular analyses further corroborated that activation of the R26Runx2 allele restored the entire osteogenic program in the perichondrial cells in the absence of endogenous Runx2.
Fig. 3.
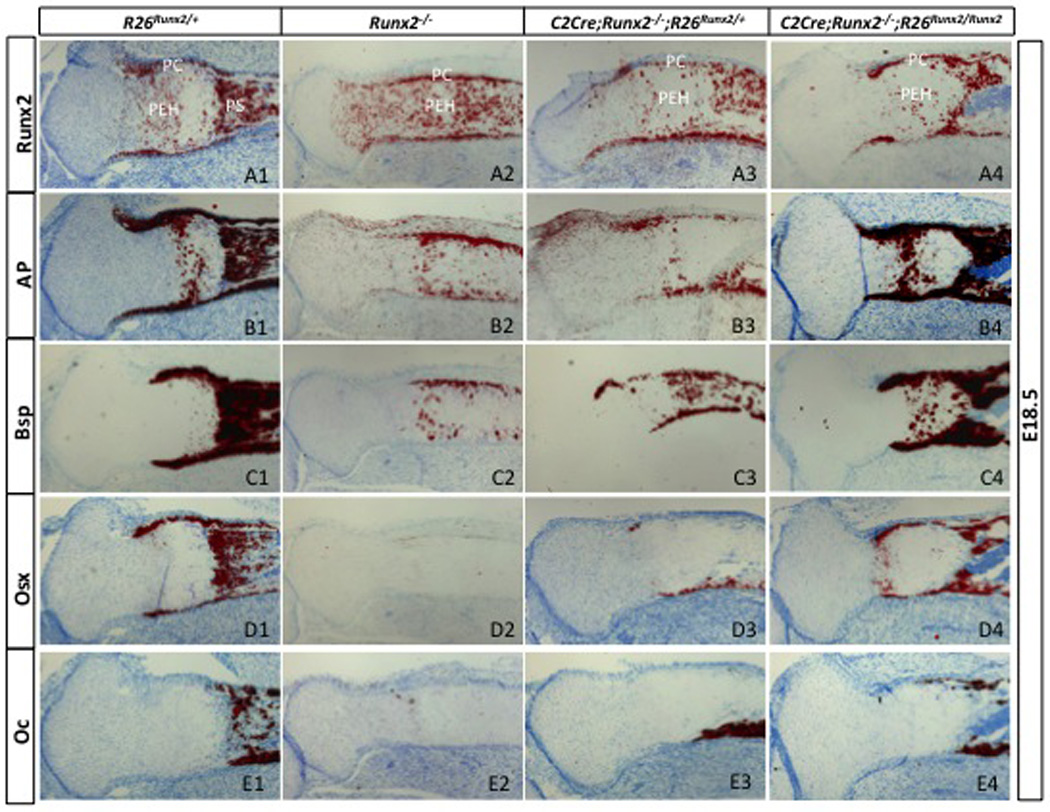
In situ hybridization of osteoblast markers. Shown are longitudinal sections through the tibia of E18.5 embryos, with proximal ends to the left. Signals shown in red. PC: perichondrium; PEH: prehypertrophic and early hypertrophic region; PS: primary spongiosa.
Forced expression of Runx2 promotes chondrocyte hypertrophy in Runx2-null mice
Activation of the R26Runx2 allele also accelerated chondrocyte hypertrophy in the Runx2−/− embryo. Apart from the lack of mature osteoblasts, Runx2 deletion was known to cause a marked delay in cartilage hypertrophy and subsequent mineralization throughout the endochondral skeleton. This deficiency was already evident from the lack of alizarin red staining in most of the skeleton at E18.5 (Fig. 1B3), and was confirmed here by analyses of the femur. Indeed, in contrast to the wild-type humerus in which the central hypertrophic region had been replaced by a marrow cavity, the Runx2−/− sample showed little sign of cellular hypertrophy (identifiable by a lighter H&E staining) (Fig. 4B1), and only minimal expression of Col10a1, a molecular marker for early and mid-stage hypertrophic chondrocytes (Fig. 4B2). Importantly, activation of one or two R26Runx2 alleles in the Runx2-null background induced chondrocyte hypertrophy, although only two alleles led to the formation of a nascent marrow cavity (Fig. 4C1-C2, D1-D2). All together, the data so far demonstrate that exogenous Runx2 expression from the R26Runx2 allele is sufficient to cause both osteoblast differentiation and cartilage hypertrophy in the absence of endogenous Runx2.
Fig. 4.

Analyses of humeri at E18.5. Proximal ends to the left. (A1-D1) H&E staining. (A2-D2) In situ hybridization of Col10a1. Red lines denote hypertrophic zone. BM: bone marrow.
Runx2 does not restore osteoblast differentiation in Ihh-null embryo
Having established that activation of a single R26Runx2 allele was sufficient to restore osteoblastogenesis in Runx2−/− embryos, we next tested whether it could do the same in the Ihh−/− embryo. To this end, we generated littermate embryos that were either Ihh−/− only, or Ihh−/− but also carried the Col2-Cre transgene and one R26Runx2 allele (Ihh−/−; C2Cre; R26Runx2/+). Whole-mount staining at E18.5 revealed that the skeletons of the two embryos were very similar, except that the Runx2-expressing embryo displayed more intense alizarin red staining (reflecting more cartilage mineralization, see below) throughout the endochondral skeleton (Fig. 5A2-A3). Likewise, histological analyses showed that R26Runx2 expression did not alter the limb skeletal morphology characteristic of the Ihh−/− embryo (Fig. 5C1-D1). However, a closer examination revealed that Runx2 expression further reduced the zone of nonhypertrophic chondrocytes in the Ihh−/− embryo (Fig. 5C2-D2). Moreover, in situ hybridization demonstrated that Runx2 expression accelerated the progression of hypertrophic chondrocytes from the Col10a1- (early and mid-stage hypertrophy) (Fig. 6A1-C1) to the Mmp13-expressing (late hypertrophy) stage (Fig. 6A2-C2); this acceleration is expected to increase mineralization that could account for the more intense alizarin red staining described above. Thus, force-expression of Runx2 further expedited chondrocyte hypertrophy in the absence of Ihh.
Fig. 5.
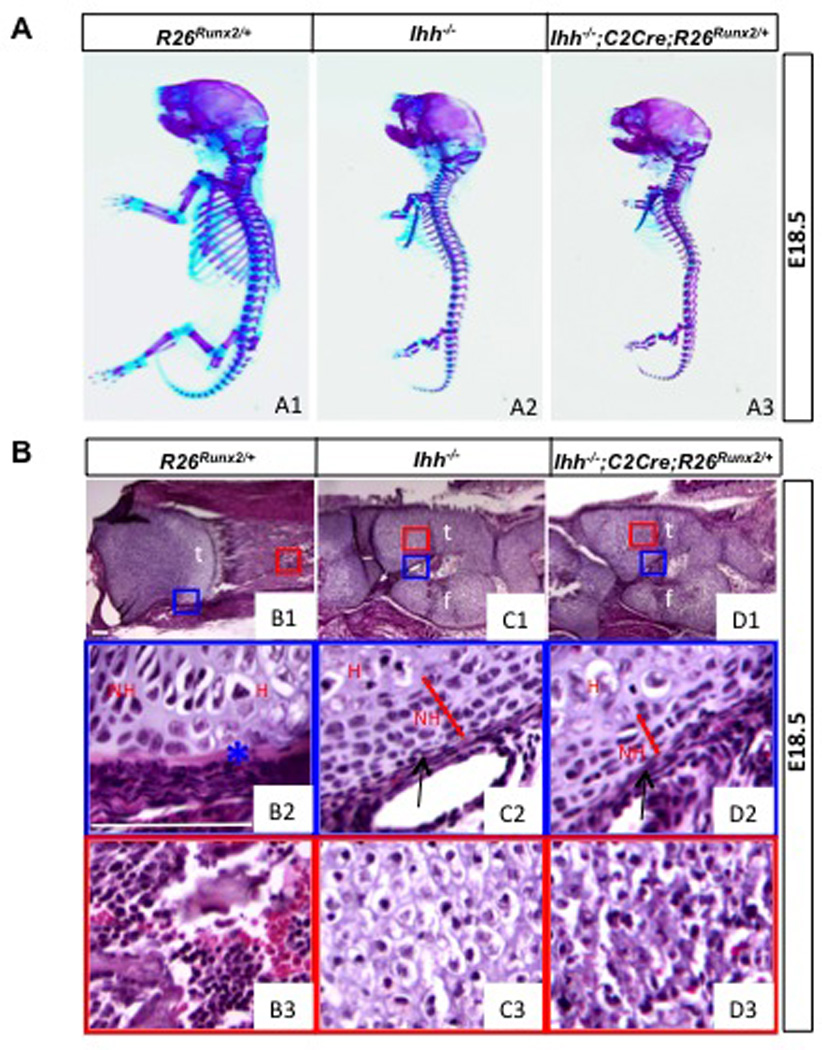
Morphological analyses of Ihh−/− embryos expressing Runx2. (A) Whole-mount skeletal staining. (B) H&E staining of longitudinal sections of the tibia. Proximal ends to the left. Boxed areas in B1-D1 shown at a higher magnification in B2-D2 and B3-D3. Scale bar: 100 µm. t: tibia; f: fibula; NH: nonhypertrophic chondrocytes; H: hypertrophic chondrocytes. Arrows denote hypoplastic perichondrium. Asterisk denotes bone collar.
Fig. 6.
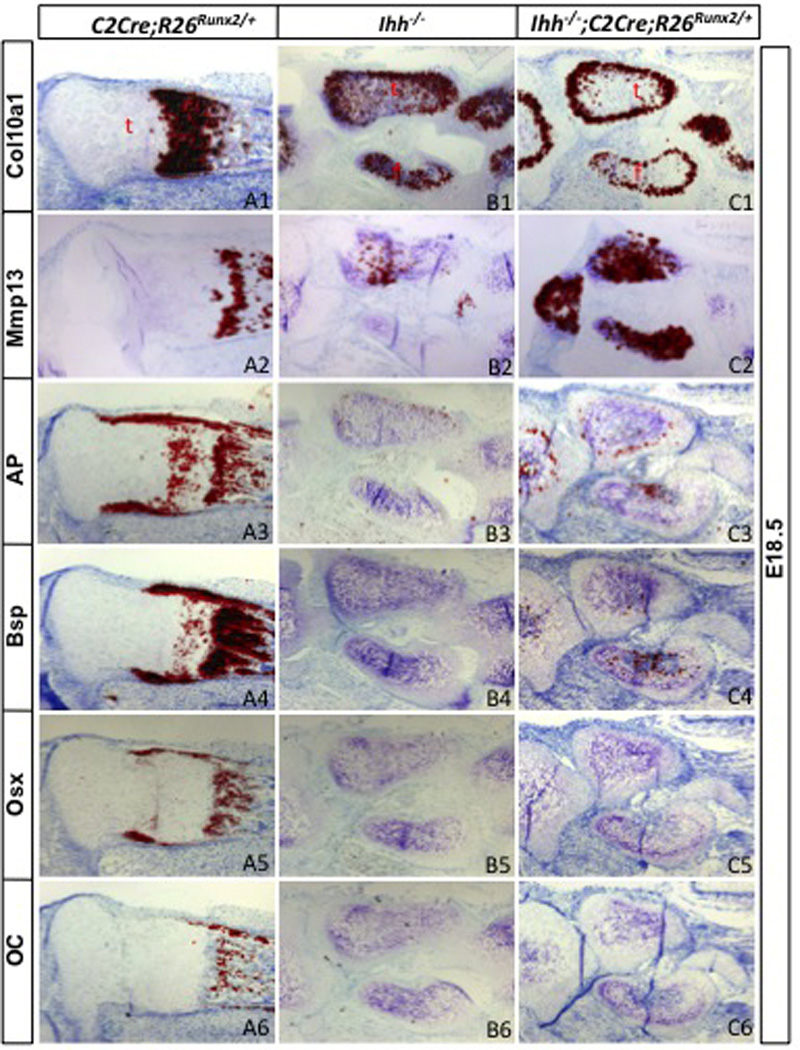
Molecular analyses of Ihh−/− embryos expressing Runx2. Shown are in situ hybridization results from longitudinal sections through the hindlimb. Signals in red. t: tibia; f: fibula.
However, Runx2 expression did not lead to bone formation in the Ihh−/− embryo. Indeed, the perichondrium from which the bone collar normally forms stayed thin regardless of Runx2 expression (arrows, Fig. 5C2-D2). Molecular analyses confirmed that AP, Bsp, Osx and Oc were not induced in the perichondrium by Runx2 expression (Fig. 6A3-C6). Thus, force-expression of Runx2 failed to rescue osteoblast differentiation in the absence of Ihh.
Discussion
We have genetically tested the hypothesis that Runx2 alone mediates the function of Ihh in osteoblast differentiation. By force-expressing Runx2 in the skeletogenic cells, we show that Runx2 is sufficient to rescue osteoblastogenesis in the Runx2−/− but not the Ihh−/− embryo. Thus, induction of osteoblast differentiation by Ihh most likely requires other effectors besides Runx2.
We cannot exclude the possibility that a higher level of Runx2 expression might restore osteoblast differentiation in the absence of Ihh. Indeed, the level of Runx2 from the R26Runx2 allele was likely to be low as in situ hybridization (with a probe detecting transcripts from both the Runx2-null and the R26Runx2 allele) did not detect an obvious increase in overall Runx2 mRNA when the R26Runx2 allele was activated (Fig. 3A3-A4). Moreover, a dose-dependent effect was observed in the experiments with Runx2−/− embryos (Fig. 1B4- B5). We have attempted to generate Ihh−/− embryos expressing two R26Runx2 alleles without success, due to unexplained early embryonic lethality. Nonetheless, because a single R26Runx2 allele was sufficient to restore osteoblast differentiation in the absence of endogenous Runx2, we conclude that the defect caused by Ihh removal was greater than the loss of Runx2.
Notably, restoration of bone formation in the Runx2−/− embryo was restricted to the diaphyseal region where a bone collar would normally form. This was so even though the Col2-Cre transgene that was used to activate Runx2 expression targeted all chondrocytes and perichondrial cells (Long et al., 2001b). Similarly, overexpression of Runx2 in the wild type background did not cause any ectopic osteoblast differentiation. Thus, Runx2 alone appeared to be insufficient to induce osteoblast differentiation from all perichondrial cells. This conclusion is in agreement with a previous report that viral expression of Runx2 in the chick limb bud did not induce ectopic osteoblast differentiation (Stricker et al., 2002). On the other hand, targeted overexpression of Runx2 in osteoblasts was previously shown to inhibit their maturation and to cause osteopenia in postnatal mice (Liu et al., 2001). It will be of interest to determine in the future whether overexpression of Runx2 from the R26Runx2 allele has a similar postnatal bone phenotype.
We have confirmed the positive role of Runx2 in chondrocyte hypertrophy. This was most evident in the experiments with the Runx2−/− embryos, wherein force-expression of Runx2 restored chondrocyte hypertrophy that was otherwise absent in most of the endochondral cartilage elements at E18.5. Similarly, Runx2 force-expression further expedited the hypertrophic program in the Ihh−/− embryo. These results are consistent with the previous finding that direct expression of Runx2 from a Col2a1 promoter restored chondrocyte hypertrophy in the Runx2−/− embryo (Takeda et al., 2001).
It is worth noting that in either Runx2−/− or Ihh−/− background, activation of a single R26Runx2 allele did not restore vascularization of the hypertrophic cartilage, even though hypertrophy appeared to have reached an advanced stage as judged by Mmp13 expression (Fig. 6C2). This could be explained partly by a dose-dependence on Runx2, as activation of two R26Runx2 alleles led to partial vascularization of the hypertrophic region (Fig. 2F). Because Runx2 was shown to induce Vegfa expression in hypertrophic chondrocytes (Zelzer et al., 2001), it is conceivable that a threshold level of Vegfa could only be achieved by a higher level of Runx2. On the other hand, the results could mean that spatial and temporal regulation of Runx2 expression (which is absent in the R26Runx2 allele) is critical for proper vascularization of the cartilage. Finally, in the case of Ihh−/− embryos, the failure of cartilage vascularization may be due to reasons independent of Runx2; this view is consistent with the relatively normal expression of Runx2 in the hypertrophic chondrocytes of Ihh−/− embryos (Hu et al., 2005; St-Jacques et al., 1999). In summary, the present study calls for future studies to discover the critical mediators of Ihh function in osteoblast differentiation.
Methods
Mouse strains
The Runx2+/−, Ihh+/− and Col2-Cre (line 3) mouse lines are as previously described. The Runx2+/− mouse was generously provided by Dr. Gerard Karsenty (Columbia University, NY). To generate the R26Runx2 mouse, the mouse Runx2 cDNA encoding the MASNS isoform (kindly provided by Dr. Gerard Karsenty) was sequentially cloned into pCIG (Megason and McMahon, 2002), pBIG-T (Srinivas et al., 2001) and pRosa-PAS (Soriano, 1999; Srinivas et al., 2001) to produce the final construct pR26-Runx2. Specifically, a DNA fragment containing 123 nt of the 5’UTR and the entire coding sequence for MASNS Runx2 was released from “pBS Cbfa1 sh” by XhoI and XbaI, and cloned into XhoI and EcoRV sites of pCIG. The resultant plasmid was then digested with SalI and the relevant piece cloned into the same site in pBigT. Finally, the Runx2-containing fragment was cloned into the PacI/AscI sites of pRosa-PAS. The final construct was then linearized and electroporated into RW-4 ES cells (Murine Embryonic Stem Cell Core, Siteman Cancer Center, Washington University School of Medicine). The correctly targeted ES clone was identified by Southern analyses with a previously described probe (Kisseberth et al., 1999). Chimeric mice were generated by injecting ES cells into C57BL6 blastocytes (Tg/KO Micro-injection Core, Washington University School of Medicine). PCR with ear biopsy samples were performed to determine germline transmission and for subsequent genotyping (Soriano, 1999). All animal studies were approved by Animal Studies Committee at Washington University.
Analyses of Mouse Embryos
Whole mount skeletal staining was performed with alizarin red and alcian blue as previously described (Long et al., 2001a). Embryonic limbs were sectioned after fixation in 10% buffered formalin overnight at room temperature, and embedded in paraffin. Limbs from E18.5 embryos were decalcified in 14 % EDTA/PBS (pH7.4) for 48 hours after fixation and before processing. In situ hybridization was performed by using 35S-labeled riboprobes as previously described (Hilton et al., 2005; Hu et al., 2005; Long et al., 2004; Long et al., 2001b). The Runx2 probe includes 221 nt of the 5’UTR and much of the first coding exon of the transcript producing the MASNS isoform, and recognizes the transcripts from both the null allele (which replaces part of the second coding exon and the rest of the gene with a LacZ expression cassette) and the R26Runx2 allele (which includes 123 nt of the 5’UTR and the entire coding sequence for MASNS).
Highlights.
Generation of a mouse strain expressing Runx2 from the Rosa26 locus in a Cre-dependent manner
Forced expression of Runx2 rescues bone formation in Runx2−/− embryo
Forced expression of Runx2 expedites chondrocyte maturation
Forced expression of Runx2 does not rescue bone formation in Ihh−/− embryo
Acknowledgement
The work was supported by an NIH grant DK065789 (FL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chung UI, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. The Journal of clinical investigation. 2001;107:295–304. doi: 10.1172/JCI11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339–4351. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- Joeng KS, Long F. The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development. 2009;136:4177–4185. doi: 10.1242/dev.041624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseberth WC, Brettingen NT, Lohse JK, Sandgren EP. Ubiquitous expression of marker transgenes in mice and rats. Dev Biol. 1999;214:128–138. doi: 10.1006/dbio.1999.9417. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LH, Ho C, Mulligan RC, Abou-Samra AB, Juppner H, Segre GV, Kronenberg HM. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–166. doi: 10.1083/jcb.200105052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development. 2004;131:1309–1318. doi: 10.1242/dev.01006. [DOI] [PubMed] [Google Scholar]
- Long F, Schipani E, Asahara H, Kronenberg H, Montminy M. The CREB family of activators is required for endochondral bone development. Development. 2001a;128:541–550. doi: 10.1242/dev.128.4.541. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001b;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker S, Fundele R, Vortkamp A, Mundlos S. Role of Runx genes in chondrocyte differentiation. Dev Biol. 2002;245:95–108. doi: 10.1006/dbio.2002.0640. [DOI] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Glotzer DJ, Hartmann C, Thomas D, Fukai N, Soker S, Olsen BR. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106:97–106. doi: 10.1016/s0925-4773(01)00428-2. [DOI] [PubMed] [Google Scholar]