

Abstract
A new concept for cancer screening has been preliminarily investigated. A cancer targeting agent loaded with a near-infrared (NIR) dye was topically applied on the tissue to highlight cancer-suspect locations and guide optical coherence tomography (OCT) imaging, which was used to further investigate tissue morphology at the micron scale. A pilot study on ApcMin mice has been performed to preliminarily test this new cancer screening approach. As a cancer-targeting agent, poly(epsilon-caprolactone) microparticles (PCLMPs), labeled with a NIR dye and functionalized with an RGD (argenine-glycine-aspartic acid) peptide, were used. This agent recognizes the ανβ3 integrin receptor (ABIR), which is over-expressed by epithelial cancer cells. The contrast agent was administered topically in vivo in mouse colon. After incubation, the animals were sacrificed and fluorescence-guided high resolution optical coherence tomography (OCT) imaging was used to visualize colon morphology. The preliminary results show preferential staining of the abnormal tissue, as indicated by both microscopy and laser-induced fluorescence imaging, and OCT’s capability to differentiate between normal mucosal areas, early dysplasia, and adenocarcinoma. Although very preliminary, the results of this study suggest that fluorescence-guided OCT imaging might be a suitable approach for cancer screening. If successful, this approach could be used by clinicians to more reliably diagnose early stage cancers in vivo.
OCIS codes: (170.0170) Medical optics and biotechnology, (170.4580) Optical diagnostics for medicine, (170.4500) Optical coherence tomography, (170.6935) Tissue characterization
1. Introduction
Standard clinical diagnosis of gastrointestinal cancers relies almost exclusively on endoscopic examination followed by pathological evaluation of tissue biopsies collected from suspected locations [1,2]. However, endoscopy does not provide the clinician with sufficient resolution to detect early-stage, pre-invasive high grade dysplasia or carcinoma in situ in small exophytic or flat adenomas. As a result, early stage cancers might be missed in high risk patients [3,4]. The sensitivity of traditional white-light endoscopy for cancer identification ranges from 47% to 53% [3]. If cancer is detected at later stages when it has already spread to regional lymph nodes (stage III), the cure rate degrades significantly [5,6]. Therefore, there is a clear need for novel technologies capable of detecting cancers at earlier stages, when they are easier to manage.
Ideally, an improved endoscopic technology that would maintain the existing visualization approach, while adding a more sensitive imaging capability and simultaneously a quantitative method for non-invasive assessment of tissue morphology in the cancer-suspect areas could prove to be a more powerful technique for detection of early stage cancers. Although such a technology does not exist yet, important steps have been already taken by researchers to address this need. For example, to increase imaging sensitivity, new strategies based on targeting molecular signatures of cancers were launched in recent years [7–11]. Nanoscale or microscale entities are functionalized to recognize molecular signatures and provide sensitivity enhancement in either radiological or optical imaging to allow for cancer localization [11,12]. With regard to visualization of tissue morphology in cancer suspect areas, high-resolution imaging technologies, such as confocal microscopy (CM) and optical coherence tomography (OCT) were tested. They both are capable of revealing tissue structure alterations at the sub-micron and micron scale, respectively [13,14]. However, OCT has the competitive advantage of providing up to 1.5 mm penetration depth, while still providing micron scale resolution. This is essential in determining cancer depth spreading. Studies in various organs have demonstrated the ability of OCT to differentiate between normal and pre-malignant conditions. For example, ovarian cysts, masses, and abnormal tissue have been successfully identified with laparoscopic OCT [15]. Laryngeal dysplasia and malignancy have been successfully imaged with OCT probes during laryngoscopy [16]. Intestinal metaplasia and dysplastic Barrett’s esophagus have been evidenced with endoscopic OCT [17,18]. Early investigations of OCT in the pancreas have also demonstrated the ability of this technology to distinguish between the layers of main duct epithelium in inflammatory and malignant diseases and to differentiate between benign and malignant cysts [19,20]. Unfortunately, OCT has a relatively small field of view (FOV), on the order of several mm, and thus its use in screening large size organs without a guidance tool is not a practical approach.
We investigated the potential for combining molecular markers with fluorescence imaging to guide OCT as a means to detect neoplastic changes in colon mucosal layer. Our goal was to demonstrate that the combined imaging modalities could improve our ability to detect pre-malignant and early malignant lesions that are not identifiable with endoscopy. In this paper we demonstrate for the first time that a fluorophore-loaded cancer targeting agent, topically applied on colon tissue, is capable of highlighting high grade lesions relative to the surrounding non-neoplastic tissue. Thus, fluorescence imaging can be used to guide OCT explorations of tissue and detect early cancer markers. The preliminary testing of this approach in a mouse model of colon cancer is discussed.
2. Materials and methods
2.1. Contrast agent
A contrast agent comprised of Poly(epsilon caprolactone) (PCL) microspheres, loaded with a fluorescent dye and functionalized with an argenine-glycine-aspartic acid (RGD) peptide to target cancer receptors, was designed and fabricated by us. The microspheres were designed to have a diameter of 1- 2 μm in order to reduce the probability for the contrast agent to be taken up by the vascular system and induce systemic toxicity. It has been shown before that micron-size particles (MPs) cannot easily penetrate the lympho-endoreticular system [21].
The detailed fabrication steps of this contrast agent are presented elsewhere [22]. Briefly, the PCLMPs were surface functionalized with RGD with a linear sequence ACD/CRG/DCF/CGG/GGG/COOH, which was synthesized at the research core facility, Tufts University, Boston, Massachusetts, USA. This peptide specifically recognizes the alpha-V-beta-3 (ανβ3) integrin receptor overexpressed by the pre-cancerous epithelial cells [23–26]. Although other sequences were also tested, this one demonstrated the highest affinity to various colon cancer cell lines, e.g., HT-29, SW-480, etc. A key issue we addressed was optimization of the RGD peptide concentration on the MP surface in order to maximize cell binding. To this end, we used gold nanobeads (10 to 20 nm in size) as a linker between RGD and MP surface. The uniform decoration of the MPs with nanometer size gold nanobeads turned out to be a very efficient method to achieve high strength binding between the peptide and the MP surface. To make these particles “visible” during the endoscopy, we encapsulated a near-infrared (NIR) dye (D-12731, Invitrogen USA) in the MP core, which becomes fluorescent when illuminated with 785 nm laser radiation. The choice for the NIR dye reduces the influence of tissue autofluorescence and improves imaging depth (the attenuation coefficient of tissue is significantly lower in the NIR than in the visible spectrum). Our approach to perform dye encapsulation and topical administration of the contrast, were two important measures to reduce contrast agent toxicity by limiting its spread into the body.
After fabrication, the PCLMPs were characterized for size and size distribution using a Multisizer 3, Beckman Coulter (Fullerton CA, USA). The mean size and standard deviation were 1.6 μm and 0.5 μm, respectively. A Scanning Electron Microscopy image of the PCLMPs is shown in Fig. 1 . A Hitachi S-4800 (Hitachi High-Technologies Europe, UK) field emission scanning microscope at an acceleration voltage of 20 kV was used. The gold coated surface morphology of the freeze-dried PCLMPs was observed in the reflectance mode.
Fig. 1.

Scanning electron microscope images of the PCLMPs. Gold coverage with 10-20 nm nanobeads can be observed in the left side higher resolution image.
2.2. Instrumentation
A multimodal imaging instrument was built to test this new approach for cancer screening. It is based on a swept-source OCT system, which was modified to add fluorescence and visible imaging capabilities. OCT, fluorescence, and visible channels were combined within the same optical path to offer perfect co-registration of the images. The schematic and the photographs of the instrument are shown in Fig. 2 . The instrument consists of three subsystems: OCT system, imaging probe, and system control unit.
Fig. 2.
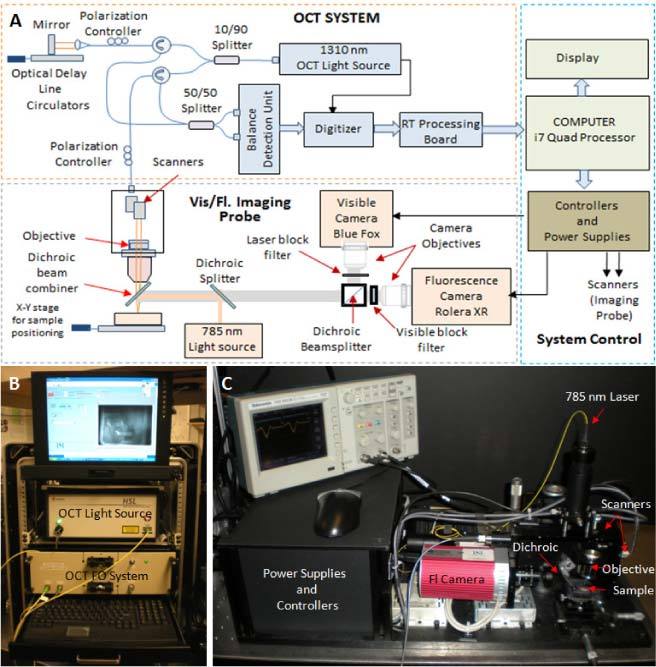
Schematic (A) and photographs (B,C) of the SS OCT system and imaging probe.
The OCT system uses a sweeping-wavelength light source (Model HS-2000, Santec, USA) with a bandwidth of 135 nm, which allows for an axial imaging resolution in tissue of about 6 μm. The other elements of this system are the fiber-optic interferometer, the balance detection unit, the signal digitizer, and a customized real-time data acquisition and processing board, which was specially designed by PSI to allow for fast-rate imaging and display of OCT data with a standard PC. Detailed design of this board was presented elsewhere [27]. The OCT imaging frame rate of this instrument is 20 to 50 frames/sec (with 1024 or 512 A-lines/frame), while the fluorescence imaging frame rate is 5 frames /sec (200 ms integration time is needed to provide good SNR in the fluorescence mode). Visible imaging was performed at video rates. A LabVIEW-based software program was used for some image processing, to display the processed OCT images, for configuration and communication with the real time board, and other functionality related to user interface. The OCT system was integrated into a 19-inch rack for portability at the animal facility. To test the fluorescence-guided OCT imaging concept we designed a combined OCT-fluorescence imaging probe with a field of view (FOV) of 20 mm x 20 mm in fluorescence mode and 5 mm x 5 mm in OCT mode. Our approach was to examine samples of admixed tissue (normal and tumor) and guide the OCT beam to suspect areas indicated by the combined bright-field/fluorescence images. A NIR enhanced response camera (Rolera XR, Q-Imaging) was used for fluorescence imaging, while a 785 nm laser diode was used for fluorophore excitation. A dichroic filter was used to combine the OCT and fluorescence excitation beams in the same optical path, while a custom barrier filter with an 800 nm to 850 nm pass-band (Chroma Technology Corp.) was placed in front of the camera lens to block the 785 nm excitation light. A visible camera was used to ensure proper placement of the sample under the imaging objective. The multimodal imaging probe has been designed to provide the same working distance (7.5 mm) for all three imaging modalities.
A side-looking catheter-type probe was designed and built to test feasibility of in vivo imaging of colon wall morphology. This was an important step taken by us towards in vivo testing of this technology. The outer diameter of this probe is ~640 μm. Probe scanning was performed with a custom-designed scanning engine, which provides a high-speed (50 Hz) axial scan and a low-speed angular scan. This scanning engine allows for an imaging linearity of 98%. The combination of the axial and rotary scan offers high-resolution 3D imaging capabilities. A lateral resolution of 15 μm and a maximum axial imaging range of 1.5 mm were possible with this probe.
2.3. Imaging protocols
Fluorescence microscopy was first performed on two cancer cell lines (HT-29 and SW-480) to test and optimize the contrast agent. Combined fluorescence-OCT imaging was then performed on an animal model of colon cancer (C57BL/6J-ApcMin mice) to preliminarily test the proposed fluorescence-guided OCT approach for cancer screening. BALB/c wild-type mice were used as controls in this study.
The cells lines were cultured and planted in chamber slides (Lab-TekTM, Thermo Fisher Scientific, USA), which have the bottom side made of a microscope cover glass No #1.5. A 200 μm coverslip was placed on the top of the chamber. The MPs (Au PCL-DiR-RGD) in PBS solution (~10 μM concentration) were added and the cells were placed back in the incubator for 1 to 2 hours. After incubation the cells were vigorously washed with PBS (3 to 5 times), while Dulbecco's Modified Eagle Medium (DMEM) was added before being transferred to the microscope for imaging. A fully motorized Olympus IX81 microscope was used to study cell binding efficacy. Bright field (visible) and fluorescence images were collected from the same field of view and correlated.
The animal study was performed on 15 ApcMin mice and 6 wild-type mice. Four week old ApcMin mice were purchased from Jackson Laboratories (Bar Harbor, ME) and aged for up to 16 weeks in the animal facility at Northeastern University NEU). Aging was necessary because colorectal lesions in these animals start to develop after 8 weeks of age. Wild mice, existent in the NEU animal facility, were used as controls. The mice were divided in 3 groups, each group containing 5 APC mice and 2 wild mice. Group 1 was investigated at the age of 8 weeks, group 2 at the age of 12 weeks, and group 3 at the age of 16 weeks. Since a microcatheter containing fibers for both OCT and fluorescence imaging was not available for the study, 3 APC mice from each group were used for ex vivo fluorescence-guide OCT imaging, while 2 APC mice were used for in vivo OCT imaging only. In vivo OCT imaging was performed to determine the feasibility of OCT use for differentiating between various pathologies of the colon in live animals.
Before imaging, the contrast agent was administered in vivo in the animals selected for the ex vivo study using a Teflon® tube-based microcatheter with a 1.5 mm outer diameter. Mice were anesthetized a few minutes in advance with 2.0% Avertin, administered intraperitoneally. The Teflon® tube was lubricated with a water-based lubricant and inserted into the anus with the mouse in the dorsal supine position. Then 2 mg PCLMPs mixed up with 50 ml of PBS were topically applied (sprayed in the colon). After 2 hrs of incubation, the colon was washed three times with PBS to remove non-binding MPs. Subsequently, all the mice planned for the ex vivo study were sacrificed by CO2 inhalation and the colon of each mouse was dissected longitudinally. Fluorescence imaging was first performed and then the tissue was fixed in 10% formalin for histological examination.
For the in vivo OCT study, images were collected at eight rotation angles separated by 45 deg. and 4 depths (25, 20, 15, and 10 mm) with respect to the tissue boundary. After imaging the animals from each group were sacrificed with CO2 inhalation and the colon tissue was dissected out longitudinally. Multiple longitudinal slices were taken for each imaging depth of the catheter and fixed in 10% formalin in order to correlate OCT findings with histology.
3. Results
3.1. Contrast agent binding study
HT-29 and SW-480 colon cancer cell lines were used in our experiments to test contrast agent binding sensitivity and specificity. We have incubated over 20 slides for each type of cell line and carefully examined microparticle attachment to the cells. As a control we used blank (non-functionalized particles). Representative images showing the attachment of the MPs to the HT-29 cells and no attachment of the blank MPs are presented in Fig. 3 .
Fig. 3.
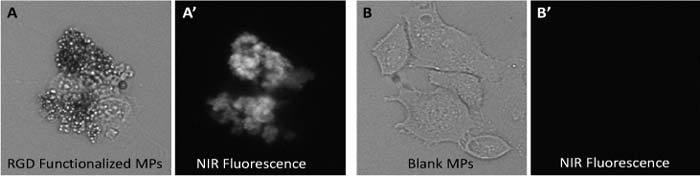
Bright field and fluorescence images of HT-29 cells incubated with PCLMPs. A, A’ RGD-functionalized PCLMPs; B, B’ Blank PCLMPs. Magnification 20X.
To determine binding efficiency and optimize incubation time we studied binding statistics for incubation times between 30 minutes and 3 hours. We took both bright field and fluorescence images and counted the fraction of the cells that were labeled with the contrast agent. The fraction of cells that were stained with non-functionalized MPs was negligible compared to the number of cells that were stained with functionalized MPs. A statistical analysis on over 20 slides (each cell type) showed that over 75% of the cells were labeled with MPs. The optimum incubation time was determined to be about 1.5 hrs (Fig. 4 ). These results demonstrate the successful functionalization of the MPs. More details about the testing of MPs on cell cultures and optimization approach for this contrast agent were presented elsewhere [22].
Fig. 4.
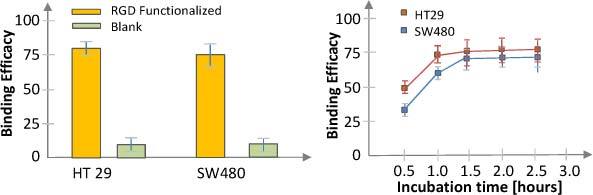
Binding statistics and incubation time for the HT-29 and SW-480 cell lines
3.2. Ex vivo fluorescence-OCT imaging study
The goal of this study was to preliminarily test the multimodal imaging approach, as well as the capability of OCT for differentiating between normal and abnormal colon tissue. At least two segments of the descending colon, about 10 mm in length, were analyzed from each animal. Besides the 785 nm fluorescence excitation and 1310 nm OCT imaging beams, an 835 nm aiming beam was launched into the OCT channel of the imaging probe to make the OCT scan visible within the fluorescence image. An offset signal was generated for the OCT galvanometers to move the OCT scan to any region-of-interest (ROI) within the fluorescence image. The OCT images of each tissue specimen were finally correlated with histological findings by identifying India ink marks, which were placed during OCT imaging.
The colon wall of the 6 BALB/c wild-type (control) mice and of 3 of the ApcMin mice, which did not show any sign of disease, was not notably stained with the contrast agent. However, consistent staining was observed in the remaining 12 ApcMin aged mice, with higher prevalence on the aged animals from the 2nd and 3rd group. The stained areas correlated very well with changes within colon wall morphology. Representative cases are presented in detail bellow.
Normal colon: A representative case of normal colon is shown in Fig. 5 . The fluorescence image shows some fluorescence spots, indicating that a very small fraction of the MPs got attached to tissue. The OCT image shows the normal appearance of the mucosa (M), submucosa (SM), and muscularis propria (MP) layers. The SM layer was measured to be very thin in some specimens, and not always very well resolved by OCT. However, most of tissue boundaries remained clear and distinct at all depths within the colon. Due to the small fraction of the MPs attached to the normal colon, the OCT images did not show any area of increased scattering.
Fig. 5.
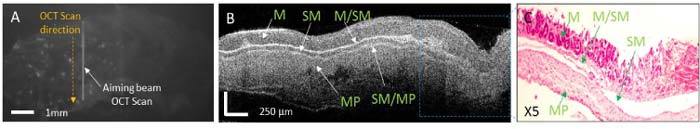
(A) Fluorescence image of the flattened colon showing very low attachment of the PCLMPs, as indicated by random bright spots. The vertical line indicates the position of the OCT scan, while the dashed line indicates the direction of this scan. (B) Cross-sectional OCT image showing the morphology of the colon layers; The OCT image displays the normal mucosa (M), the thin submucosal (SM) layer, and the muscularis propria (MP). The boundaries between these layers are well demarcated (see M/SM and SM/MP). (C) Colon histology within the ROI indicated in B.
To further investigate if the same morphology is present on other areas of the tissue specimens, relatively dense raster scans (1024 x 256 pixels within a 2.5 mm x 1.75 mm area) were taken to better visualize morphologic details of the colon wall. Indeed, as shown in Fig. 6 , the two frames selected from Media 1 (1.4MB, MOV) show the same morphology as it was observed in Fig. 5. Furthermore, similar morphology can be observed in all the frames from the raster scan.
Fig. 6.

(Media 1 (1.4MB, MOV) ). Fly-through video of both fluorescence and OCT measurements. (A) Fluorescence imaging showing the OCT raster scan, as indicated by the aiming beam. (B) Combined enface and cross-sectional OCT imaging. The en face view was taken at a depth indicated by the line from the cross-sectional image. The raw fluorescence video was acquired at 5 fps, while the raw OCT video was acquired at 20 fps. OCT scale bar = 250 μm.
Very similar findings were observed in all 6 wild-type mice, as well as in 2 of the 8 weeks old ApcMin mice and one 12 weeks old ApcMin mouse, which did not show any sign of adenomatous polyps. The absence of adenomatous polyps or carcinomas in ApcMin mice was noted by other investigators and explained by the fact that this animal model usually develops small intestine carcinomas, rather than colon adenomas or carcinomas [28].
Colon adenoma: The fluorescence images of the ApcMin mice that developed colonic adenomas showed substantially higher attachment of the MPs, especially in areas indicated by OCT as having changes in the morphology of the mucosal layer. These changes consisted of mucosal thickening over time (larger in older mice), or even growth of large polyps, and in some cases the disappearance of the mucosa/submucosa boundary.
Figure 7 shows a representative case of a 12 weeks old ApcMin mouse, where the MPs attached preferentially (see red circled areas in (A)) in the two areas with increased thickness of the mucosal layer of the colon, as shown in the OCT image (B) and confirmed by histology (C).
Fig. 7.
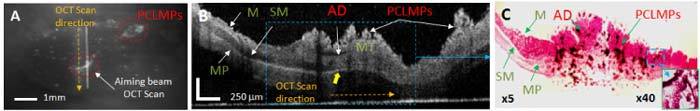
(A) Fluorescence, (B) OCT, and (C) histology of adenoma (AD) in mouse upper colon. The adenoma almost triples the thickness of the mucosal layer in the inflamed area (mucosal thickening-MT). The adjacent normal mucosa (M), submucosa (SM), and muscularis propria (MP) are labeled. Accumulation of PCL microparticles (PCLMPs) in the adenoma area is visible in the OCT image and confirmed by histology. Individual PCLMPs are discernable in the magnified (x40) histology area. OCT scale bar: 250 μm; Histology magnification: 5x and 40x, respectively
A raster scan over a larger area (~4 mm x 3mm) of the same tissue specimen shows indeed a good correlation between the brighter fluorescence areas and MP binding to tissue (Fig. 8 ). The en face image of the tissue surface shows the enhanced scattering from the MPs, while the cross-sectional images in areas indicated by the dotted lines show significant changes in the morphology of the mucosal layer (thickening of the mucosal layer, which is specific to adenomas).
Fig. 8.
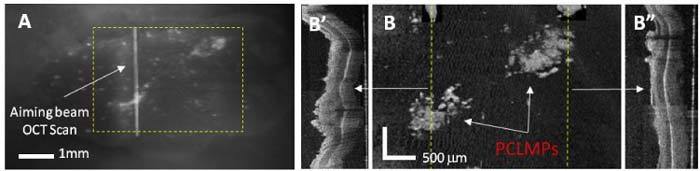
(A) Fluorescence image of a colon tissue specimen, (B) En face OCT image of the ROI indicated in A by a dotted rectangle. Cross-sectional images (B′ & B″) along the dotted lines from B show evident changes in the morphology of the mucosal layer, which are characteristic to adenomas.
Besides mucosal thickening, adenomatous polyps, as large as 2 to 3 mm in diameter, were found in several mice with the age of 12 weeks or higher. These colonic polyps are overgrowths of the colonic mucosa that carry a small risk (< 1%) of becoming malignant. Such lesions are classified histologically as tubular adenomas, tubulovillous adenomas (villoglandular polyps), or villous adenomas. The likelihood of cancer in an adenomatous polyp at the time of discovery is related to size, histologic type, and degree of dysplasia.
A representative case of an adenomatous polyp in a 12 weeks old ApcMin mouse is shown in Fig. 9 . The polyp invades the SM layer of the colon. The magnified histology image shows the disappearance of the lobular structure of the mucosal layer where the polyp is located. The presence of the MPs can be observed in both histology (darker spots) and OCT image (brighter spots on the top of the polyp), demonstrating the presence of the cancer receptors on these abnormal areas of the mouse colon.
Fig. 9.
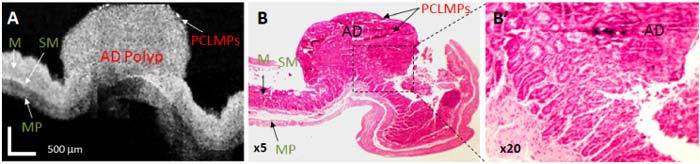
OCT image and histopathology of adenomatous polyp. (A). OCT image of adenoma displays a markedly expanded mucosa (development of a large polyp) and not very well defined tissue boundaries underlying the lesion. B. Corresponding histopathology; B’. Magnified histology on the polyp area confirms the presence of adenoma. M- mucosa; SM-submucosa; MP-muscularis propria; AD: adenoma; OCT scale bar: 500 μm; Histology magnification: 5x and 20x, respectively.
Colorectal adeno-carcinoma: The colorectal cancer, commonly known as bowel cancer, is a cancer caused by uncontrolled cell growth (neoplasia), in the colon, rectum, or vermiform appendix. Adenocarcinoma is a malignant epithelial tumor, originating from glandular epithelium of the colorectal mucosa. It invades the wall, infiltrating the MM, the SM[REMOVED HYPERLINK FIELD] and even the MP. Tumor cells describe irregular tubular structures, harboring pluristratification, multiple lumens, reduced stroma. Sometimes, tumor cells secrete mucus, which invades the interstitium producing large pools of mucus/colloid (optically “empty” spaces).
Signs of colorectal adenocarcinoma were identified in only 2 ApcMin mice, 16 weeks old. The adenocarcinoma was invading and replacing all three layers of the rectum, as shown in Fig. 10 . The OCT images showed the loss of tissue texture (absence of normal layers) and presence of pockets of uneven echoic or dark areas, which are caused by high signal absorption in the necrotic tissue or by the presence of the pools of mucus.
Fig. 10.
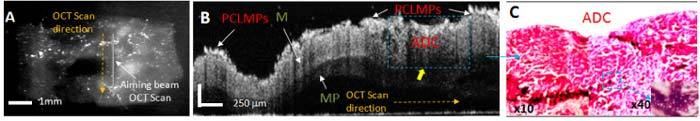
Fluorescence (A), OCT image (B). and histopathology (C) of adenocarcinoma in mouse colorectal region. As observed, adenocarcinoma (ADC) was invading and replacing the all three layers: mucosa (M), Submicosa (SM) and muscularis propria (MP). OCT scale bar: 250 μm; Histology magnification: x10 and x40, respectively.
OCT raster scans (2.25 mm x 1.75 mm) were taken as well on the tissue specimens from the two ApcMin mice identified with colon adenocarcinoma. Very strong attachment of the MPs was observed in some areas, as shown in the two frames selected from Media 2 (1.8MB, MOV) (see Fig. 11 ). The OCT images showed the loss of tissue texture (absence of normal layers). However, the presence of pockets of uneven echoic or dark areas was less prominent in this case, suggesting that cancer was not as advanced like it was observed in the previous case.
Fig. 11.

(Media 2 (1.8MB, MOV) ). Fly-through video of both fluorescence and OCT measurements. (A) Fluorescence imaging showing the OCT raster scan, as indicated by the aiming beam. (B) Combined enface and cross-sectional OCT imaging. The en face view was taken at a depth indicated by the line from the cross-sectional image. The raw fluorescence video was acquired at 10 fps, while the raw OCT video was acquired at 20 fps. OCT scale bar: 250 μm.
To further determine the correlation between tissue pathology and the integrated intensity of the fluorescence signal, a detailed analysis was performed on several tissue specimens that were assigned by a pathologist to one of the next classes: normal colon, adenomas, and adenocarcinomas. To account for the differences within specimen size, the contour of each of each tissue specimen was generated from the visible image and superimposed on the corresponding fluorescence image, as shown in Fig. 12 . Then the background noise in each image was computed and extracted from the fluorescence signal. The integrated fluorescence signal was then calculated for each tissue specimen and normalized to specimen size. As observed, a clear difference has been obtained between the integrated fluorescence signals of the three pathologies.
Fig. 12.
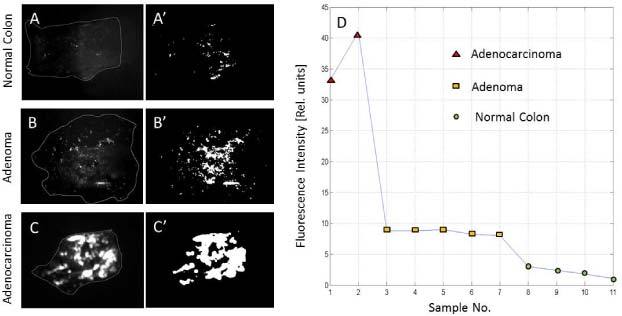
(A, B, C) Representative fluorescence images for normal colon, colon adenoma, and colon adenocarcinoma. (A′, B′, C′) Processed images with removed background noise. (D) Integrated fluorescence signal for 11 specimens that were assigned to the three pathologies.
These results suggest that enhanced contrast fluorescence imaging can be used to indicate cancer-suspect areas, while OCT imaging can be performed to further investigate these areas and detect alterations of the colon wall morphology. Quantitative OCT criteria can be developed as well to differentiate between various pathologies and more objectively stage cancer.
3.3 In-vivo OCT imaging study
In vivo OCT imaging was performed in several mice, as discussed above. Two representative examples are presented below.
I. Normal colon: The representative OCT-histology of the normal colon is shown in Fig. 13 . The OCT image shows the mucosa (M), submucosa (SM), muscularis propria (MP) layer, and the subserosa (SS)). The SS is a layer of tissue between the muscularis and serosa, which sometimes is admixed with some fat (F).Surprisingly, SS was not well differentiated in the animals investigated in the ex vivo OCT study. The SM was measured to be very thin in histology and not very well resolved in some of the OCT images. However, most of tissue boundaries remained clear and distinct at all depths, rotations, and time points. A highly scattering layer is observed on the top of the image; this is the OCT catheter protective Teflon tube.
Fig. 13.
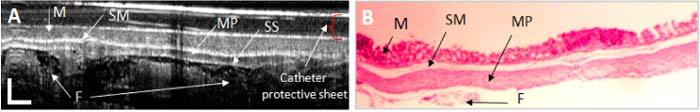
OCT image (A) and histology (B) of normal colon. The OCT image displays the normal mucosa (M), the thin submucosal (SM) layer, the muscularis propria (MP), and the subserosa (SS) boundary with some underlying fat (F) or connective tissue layers. OCT scale bar: 250 μm; Histology magnification: 5x.
II. Colon adenoma: The OCT images of the ApcMin mice that developed colonic adenomas showed substantial changes over the imaging time points. They consisted of mucosal thickening over time, or even growth of large polyps, and the disappearance of the mucosa/submucosa boundary. These changes in the mucosal-submucosal layers were more evident in the 12 weeks old ApcMin mice. All mice with the age of 12 weeks or higher have shown these changes. A representative example is shown in Fig. 14 .
Fig. 14.
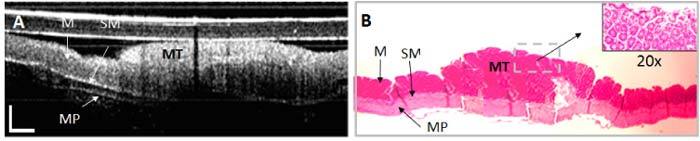
OCT (A) and histopathology (B) of adenoma in mouse upper colon. The adenoma almost triples the thickness of the mucosal layer in the inflamed area (mucosal thickening-MT). The adjacent normal mucosa (M), submucosa (SM), and muscularis propria (MP) are labeled. OCT scale bar: 250 μm; Histology magnification: 5x and 20x, respectively.
These examples demonstrate the suitability of OCT for in vivo use to differentiate between various colon pathologies in small animals. However, OCT imaging through the instrument channel of a 2-3 mm endoscopic probe, suitable for small animal anatomy, will require further miniaturization of the fiber catheter.
4. Discussion and Conclusions
Among gastrointestinal (GI) cancers, colorectal cancer is the most prevalent one in the Western world, while gastric cancer is somewhat more prevalent in the Eastern world [29]. Prognosis is variable and depends almost entirely on the specific type of cancer. However, in general prognosis is very good when cancer is detected at a very early stage and confined within the mucosa of the organ (stages I and II). Early stage cancer is usually curable with surgery [30]. If cancer is detected in later stages when it has already spread to regional lymph nodes (stage III), the cure rate degrades significantly [31]. Unfortunately, despite major advances in imaging, current endoscopic or radiologic imaging approaches may not detect these cancers in their early stage.
Various radiologic and optical imaging approaches have been investigated previously to increase GI cancer diagnostic sensitivity and specificity. Among optical approaches, OCT has shown high promise in diagnosing GI cancers [17,18,32]. OCT imaging was also successfully combined with fluorescence spectroscopy, fluorescence molecular imaging, or fluorescence lifetime imaging to improve cancer detection sensitivity and specificity in the GI tract or in other organs [33–37]. However, considering the fact that OCT and either of these complementary optical modalities have a relatively small field of view (FOV), it becomes impractical to use them for screening large organs without proper guidance. OCT use alone, or combined with any other small FOW optical spectroscopy or imaging method might become more appealing to the real clinical practice if a better guidance approach than current endoscopy becomes available. Therefore, the goal of this paper is to show that enhanced-contrast fluorescence imaging might be an appropriate tool for OCT guidance, and thus to limit its use to areas suspicious to adenoma or adenocarcinoma, as indicated by the fluorescence signal. Enhanced contrast fluorescence imaging might be possible by using a modified endoscope with and added fluorescence channel and either topical or i.v. administration of a cancer targeting agent loaded with a fluorophore. Previously, various contrast agents have been developed and tested in animal models to determine their suitability for GI cancer targeting [24,27,37]. Here we report on preliminary testing of a contrast agent based on micron-size MPs functionalized with an RGD peptide and loaded with a NIR dye, which has shown before promise in efficient targeting of various human cancer cell lines [22].
In this paper we summarize the results of an ex vivo fluorescence-guided OCT imaging study on BALB/c wild-type and ApcMin mice, where RGD functionalized PCLMPs were used as contrast agent to target activated colon cancer receptors. Although in vivo imaging would have been desirable, a combined fluorescence/OCT imaging probe suitable for small animal imaging was not available at the time this study took place. Such an endoscopic probe should have a maximum diameter of 3 mm and include an instrument channel for OCT imaging. OCT imaging was performed in both fluorescence-labeled and non-labeled areas of the colon and correlation with histological appearance of the colon wall was performed by a histopathologist blinded to OCT findings. Strong correlations between histology, fluorescently-labeled areas of the colon, and morphologic changes within the colon wall, as indicated by cross-sectional OCT imaging, were found. The OCT images of the fluorescently-labeled areas showed increased thickness of the mucosal layer of the colon, which is specific to adenomas, or uncontrolled cell growth (neoplasia), originating from glandular epithelium of the colorectal mucosa invading the submucosa or even muscularis propria, which is specific to adenocarcinoma. When quantifying the intensity of the fluorescence signal, a clear difference has been found between the three pathologies: weak background on the normal tissue, moderate signals from areas with adenomas, and strong signals from adenocarcinomas.
In conclusion, a novel multimodal approach for diagnosis of early stage epithelial cancers has been investigated. It consisted of topical administration of RGD functionalized PCL microparticles and fluorescence-guided OCT imaging. Preliminary studies on cell lines and a mouse model of colon cancer showed that it is possible to selectively target cancer cells and highlight colon cancer locations in a fluorescence image. Although the contrast agent stains cancer very effectively, and thus biopsy can be performed under fluorescence guidance only, OCT imaging remains an essential tool in the diagnostic process because it supplements biopsy findings, showing the depth and lateral extent of the lesion. It is known that biopsy provides the sub-cellular resolution needed to confirm or rule out cancer presence, but since it does not preserve well the morphology of the tissue, cancer depth spreading based on biopsy results is not accurate. Thus, the combination of OCT with biopsy can provide a more comprehensive set of data that will help clinicians to not only confirm cancer presence, but also determine its depth and lateral spreading.
The proposed cancer screening approach needs to be further investigated in vivo in a larger pool of animals and in a larger diversity of tissue pathologies. Therefore, further in vivo testing of this approach is planned in an orthotopic mouse model of colon cancer. If successful, this screening approach could be translated to the clinical practice for improving the efficacy of current endoscopy procedures by better differentiation between normal and early stage cancerous lesions of the colon, especially in flat dysplasia.
Acknowledgments
This work was funded by the National Institutes of Health (Grant 5R41 CA 132256). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Ethical conduct of research: BALB/c wild-type mice, purchased from Charles River Laboratories (Wilmington, MA), and ApcMin mice purchased from Jackson Laboratory, Barr Harbor, ME, were housed in the Division of Laboratory Animal Medicine (DLAM, room 21 Mugar Building) of Northeastern University. DLAM is an AAALAC-accredited facility for housing and care of laboratory animals. The facility is fully staffed with professional animal care personnel and a veterinarian. The protocol for the animal experiments has been approved by the Northeastern University and Physical Sciences Inc. Institutional Animal Care and Use Committees.
References and links
- 1.Tajiri H., Ohtsu A., Boku N., Muto M., Chin K., Matsumoto S., Yoshida S., “Routine endoscopy using electronic endoscopes for gastric cancer diagnosis: retrospective study of inconsistencies between endoscopic and biopsy diagnoses,” Cancer Detect. Prev. 25(2), 166–173 (2001). [PubMed] [Google Scholar]
- 2.Jeffery G., Hickey B., Hilder P. N., “Follow up strategies for patients treated for non-metastatic colorectal cancer (Review),” Cochrane Database Syst. Rev. 1, 1–27 (2008), http://generalsurgery.utoronto.ca/Assets/GenSurgery+Digital+Assets/Hospitals/cochranereview.pdf [DOI] [PubMed] [Google Scholar]
- 3.Hosokawa O., Watanabe K., Hatorri M., Douden K., Hayashi H., Kaizaki Y., “Detection of gastric cancer by repeat endoscopy within a short time after negative examination,” Endoscopy 33(4), 301–305 (2001). 10.1055/s-2001-13685 [DOI] [PubMed] [Google Scholar]
- 4.Lambert R., Rey J. F., “Endoscopy and early neoplasia: better but not the best,” Endoscopy 33(4), 348–352 (2001). 10.1055/s-2001-13690 [DOI] [PubMed] [Google Scholar]
- 5.Ott J. J., Ullrich A., Miller A. B., “The importance of early symptom recognition in the context of early detection and cancer survival,” Eur. J. Cancer 45(16), 2743–2748 (2009). 10.1016/j.ejca.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 6.Westcott E. D. A., Windsor A. C. J., “Can we improve the outcome of colorectal cancer by early diagnosis?” Postgrad. Med. J. 78(919), 255–256 (2002). 10.1136/pmj.78.919.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber W. A., Czernin J., Phelps M. E., Herschman H. R., “Technology Insight: novel imaging of molecular targets is an emerging area crucial to the development of targeted drugs,” Nat. Clin. Pract. Oncol. 5(1), 44–54 (2008). 10.1038/ncponc0982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massoud T. F., Gambhir S. S., “Integrating noninvasive molecular imaging into molecular medicine: an evolving paradigm,” Trends Mol. Med. 13(5), 183–191 (2007). 10.1016/j.molmed.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 9.Tafreshi N. K., Kumar V., Morse D. L., Gatenby R. A., “Molecular and functional imaging of breast cancer,” Cancer Contr. 17(3), 143–155 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Brindle K., “New approaches for imaging tumour responses to treatment,” Nat. Rev. Cancer 8(2), 94–107 (2008). 10.1038/nrc2289 [DOI] [PubMed] [Google Scholar]
- 11.Rosen M. A., Schnall M. D., “Dynamic contrast-enhanced magnetic resonance imaging for assessing tumor vascularity and vascular effects of targeted therapies in renal cell carcinoma,” Clin. Cancer Res. 13(2), 770s–776s (2007). 10.1158/1078-0432.CCR-06-1921 [DOI] [PubMed] [Google Scholar]
- 12.Pierce M. C., Javier D. J., Richards-Kortum R., “Optical contrast agents and imaging systems for detection and diagnosis of cancer,” Int. J. Cancer 123(9), 1979–1990 (2008). 10.1002/ijc.23858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivak M. V., Jr, Kobayashi K., Izatt J. A., Rollins A. M., Ung-Runyawee R., Chak A., Wong R. C., Isenberg G. A., Willis J., “High-resolution endoscopic imaging of the GI tract using optical coherence tomography,” Gastrointest. Endosc. 51(4), 474–479 (2000). 10.1016/S0016-5107(00)70450-0 [DOI] [PubMed] [Google Scholar]
- 14.Drezek R. A., Collier T., Brookner C. K., Malpica A., Lotan R., Richards-Kortum R. R., Follen M., “Laser scanning confocal microscopy of cervical tissue before and after application of acetic acid,” Am. J. Obstet. Gynecol. 182(5), 1135–1139 (2000). 10.1067/mob.2000.104844 [DOI] [PubMed] [Google Scholar]
- 15.Hariri L. P., Bonnema G. T., Schmidt K., Winkler A. M., Korde V., Hatch K. D., Davis J. R., Brewer M. A., Barton J. K., “Laparoscopic optical coherence tomography imaging of human ovarian cancer,” Gynecol. Oncol. 114(2), 188–194 (2009). 10.1016/j.ygyno.2009.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kraft M., Glanz H., von Gerlach S., Wisweh H., Lubatschowski H., Arens C., “Clinical value of optical coherence tomography in laryngology,” Head Neck 30(12), 1628–1635 (2008). 10.1002/hed.20914 [DOI] [PubMed] [Google Scholar]
- 17.Evans J. A., Bouma B. E., Bressner J., Shishkov M., Lauwers G. Y., Mino-Kenudson M., Nishioka N. S., Tearney G. J., “Identifying intestinal metaplasia at the squamocolumnar junction by using optical coherence tomography,” Gastrointest. Endosc. 65(1), 50–56 (2007). 10.1016/j.gie.2006.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouma B. E., Tearney G. J., Compton C. C., Nishioka N. S., “High-resolution imaging of the human esophagus and stomach in vivo using optical coherence tomography,” Gastrointest. Endosc. 51(4), 467–474 (2000). 10.1016/S0016-5107(00)70449-4 [DOI] [PubMed] [Google Scholar]
- 19.Testoni P. A., Mariani A., Mangiavillano B., Arcidiacono P. G., Di Pietro S., Masci E., “Intraductal optical coherence tomography for investigating main pancreatic duct strictures,” Am. J. Gastroenterol. 102(2), 269–274 (2007). 10.1111/j.1572-0241.2006.00940.x [DOI] [PubMed] [Google Scholar]
- 20.Iftimia N., Cizginer S., Deshpande V., Pitman M., Tatli S., Iftimia N. A., Hammer D. X., Mujat M., Ustun T., Ferguson R. D., Brugge W. R., “Differentiation of pancreatic cysts with optical coherence tomography (OCT) imaging: an ex vivo pilot study,” Biomed. Opt. Express 2(8), 2372–2382 (2011). 10.1364/BOE.2.002372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ai J., Biazar E., Jafarpour M., Montazeri M., Majdi A., Aminifard S., Zafari M., Akbari H. R., Rad H. G., “Nanotoxicology and nanoparticle safety in biomedical designs,” Int. J. Nanomedicine 6, 1117–1127 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lue N., Ganta S., Hammer D. X., Mujat M., Stevens A. E., Harrison L., Ferguson R. D., Rosen D., Amiji M., Iftimia N., “Preliminary evaluation of a nanotechnology-based approach for the more effective diagnosis of colon cancers,” Nanomedicine (Lond.) 5(9), 1467–1479 (2010). 10.2217/nnm.10.93 [DOI] [PubMed] [Google Scholar]
- 23.Yang G.-Y., Xu K.-S., Pan Z.-Q., Zhang Z.-Y., Mi Y.-T., Wang J.-S., Chen R., Niu J., “Integrin alphavbeta6 mediates the potential for colon cancer cells to colonize in and metastasize to the liver,” Cancer Sci. 99(5), 879–887 (2008). 10.1111/j.1349-7006.2008.00762.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Buono R., Pignatelli M., Bodmer W. F., Wright N. A., “The role of the arginine-glycine-aspartic acid-directed cellular binding to type I collagen and rat mesenchymal cells in colorectal tumour differentiation,” Differentiation 46(2), 97–103 (1991). 10.1111/j.1432-0436.1991.tb00870.x [DOI] [PubMed] [Google Scholar]
- 25.Meyer D., Nickols G., Pegg J., Westlin W., “An oral alpha-v-beta-3 antagonist, S-247, induces tumor regression and inhibits hypercalcemia of malignancy in a syngeneic mouse colon model,” Proc. Am. Assoc. Cancer Res. 42, 825 (2001). [Google Scholar]
- 26.Griggs D., Shannon K., Settle S., Duffin T., Nickols M., Schroeter S., Gickols N., Westlin W., “Anti-metastatic efficacy mediated by peptidomimetic alpha-v-beta-3 integrin antagonists in orthotopic and experimental models,” Proc. Am. Assoc. Cancer Res. 42, 263 (2001). [Google Scholar]
- 27.Ustun T. E., Iftimia N. V., Ferguson R. D., Hammer D. X., “Real-time processing for Fourier domain optical coherence tomography using a field programmable gate array,” Rev. Sci. Instrum. 79(11), 114301 (2008). 10.1063/1.3005996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kettunen H. L., Kettunen A. S. L., Rautonen N. E., “Intestinal immune responses in wild-type and Apcmin/+ mouse, a model for colon cancer,” Cancer Res. 63(16), 5136–5142 (2003). [PubMed] [Google Scholar]
- 29.N. Howlader, A. M. Noone, M. Krapcho, N. Neyman, R. Aminou, W. Waldron, S. F. Altekruse, C. L. Kosary, J. Ruhl, Z. Tatalovich, H. Cho, A. Mariotto, M. P. Eisner, D. R. Lewis, H. S. Chen, E. J. Feuer, K. A. Cronin, and B. K. Edwards, eds., Surveillance Epidemiology and End Results, Cancer Statistics Review, 1975–2008, National Cancer Institute, Bethesda MD, http://seer.cancer.gov/csr/1975-2008
- 30.Cappell M. S., “Pathophysiology, clinical presentation, and management of colon cancer,” Gastroenterol. Clin. North Am. 37(1), 1–24, v (2008). 10.1016/j.gtc.2007.12.002 [DOI] [PubMed] [Google Scholar]
- 31.Desch C. E., Benson A. B., 3rd, Somerfield M. R., Flynn P. J., Krause C., Loprinzi C. L., Minsky B. D., Pfister D. G., Virgo K. S., Petrelli N. J., American Society of Clinical Oncology , “Colorectal cancer surveillance: 2005 update of an American Society of Clinical Oncology practice guideline,” J. Clin. Oncol. 23(33), 8512–8519 (2005). 10.1200/JCO.2005.04.0063 [DOI] [PubMed] [Google Scholar]
- 32.Pfau P. R., Sivak M. V., Jr, Chak A., Kinnard M., Wong R. C., Isenberg G. A., Izatt J. A., Rollins A., Westphal V., “Criteria for the diagnosis of dysplasia by endoscopic optical coherence tomography,” Gastrointest. Endosc. 58(2), 196–202 (2003). 10.1067/mge.2003.344 [DOI] [PubMed] [Google Scholar]
- 33.Tumlinson A. R., Hariri L. P., Utzinger U., Barton J. K., “Miniature endoscope for simultaneous optical coherence tomography and laser-induced fluorescence measurement,” Appl. Opt. 43(1), 113–121 (2004). 10.1364/AO.43.000113 [DOI] [PubMed] [Google Scholar]
- 34.Ryu S. Y., Choi H. Y., Na J., Choi E. S., Lee B. H., “Combined system of optical coherence tomography and fluorescence spectroscopy based on double-cladding fiber,” Opt. Lett. 33(20), 2347–2349 (2008). 10.1364/OL.33.002347 [DOI] [PubMed] [Google Scholar]
- 35.Park J., Jo J. A., Shrestha S., Pande P., Wan Q., Applegate B. E., “A dual-modality optical coherence tomography and fluorescence lifetime imaging microscopy system for simultaneous morphological and biochemical tissue characterization,” Biomed. Opt. Express 1(1), 186–200 (2010). 10.1364/BOE.1.000186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan S., Roney C. A., Wierwille J., Chen C. W., Xu B., Griffiths G., Jiang J., Ma H., Cable A., Summers R. M., Chen Y., “Co-registered optical coherence tomography and fluorescence molecular imaging for simultaneous morphological and molecular imaging,” Phys. Med. Biol. 55(1), 191–206 (2010). 10.1088/0031-9155/55/1/011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roney C. A., Xu B., Xie J., Yuan S., Wierwille J., Chen C. W., Chen Y., Griffiths G. L., Summers R. M., “Rh-I-UEA-1 polymerized liposomes target and image adenomatous polyps in the APC(Min/+) mouse using optical colonography,” Mol. Imaging 10(4), 305–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]