

Abstract
Various regulatory authorities such as the International Conference on Harmonization (ICH), the United States Food and Drug administration (FDA), and the Canadian Drug and Health Agency (CDHA) are emphasizing on the purity requirements and the identification of impurities in Active Pharmaceutical Ingredients (APIs). The various sources of impurity in pharmaceutical products are — reagents, heavy metals, ligands, catalysts, other materials like filter aids, charcoal, and the like, degraded end products obtained during \ after manufacturing of bulk drugs from hydrolysis, photolytic cleavage, oxidative degradation, decarboxylation, enantiomeric impurity, and so on. The different pharmacopoeias such as the British Pharmacopoeia, United State Pharmacopoeia, and Indian Pharmacopoeia are slowly incorporating limits to allowable levels of impurities present in APIs or formulations. Various methods are used to isolate and characterize impurities in pharmaceuticals, such as, capillary electrophoresis, electron paramagnetic resonance, gas–liquid chromatography, gravimetric analysis, high performance liquid chromatography, solid-phase extraction methods, liquid–liquid extraction method, Ultraviolet Spectrometry, infrared spectroscopy, supercritical fluid extraction column chromatography, mass spectrometry, Nuclear magnetic resonance (NMR) spectroscopy, and RAMAN spectroscopy. Among all hyphenated techniques, the most exploited techniques for impurity profiling of drugs are Liquid Chromatography (LC)-Mass Spectroscopy (MS), LC-NMR, LC-NMR-MS, GC-MS, and LC-MS. This reveals the need and scope of impurity profiling of drugs in pharmaceutical research.
Keywords: Characterization, chromatography, identification, impurities, NMR, mass spectrometry
INTRODUCTION
The impurities in drug products can be attributed not only to the drug substance or inert ingredients used for formulating a drug product; but they can also be brought into the drug product through the formulation process or by contact with packaging of the various impurities that can be found in drug products.
“Any component of the drug product that is not the chemical entity defined as the drug substance or an excipient in the drug product.” (ICH Q6A: Specifications).[1] It is important to give greater consideration to these detrimental impurities. In general, most of these impurities are small molecules. This is especially true in solid dosage forms where the limited mobility restricts the reactivity of larger molecules. For most drugs, the reactive species consist of water (which can hydrolyze some drugs or effect the dosage form performance), small electrophiles (e.g., aldehyde and carboxylic acid derivatives), peroxides (which can oxide some drugs), and metals (which can catalyze oxidation and other drug degradation pathways). Additionally, some impurites can cause toxicological problems. The presence of these unwanted chemicals, even in small amounts, may influence the efficacy and safety of the pharmaceutical products. Impurity profiling (i.e., the identity as well as the quantity of impurity in the pharmaceuticals), is now receiving critical attention from regulatory authorities. The different pharmacopoeias such as BP (British pharmacopoeias), USP (United States pharmacopoeias), IP (Indian pharmacopoeias), and so on, are slowly incorporating limits to the allowable levels of impurities present in active pharmaceutical ingredients (APIs) or formulations. The large number of compounds under investigation in drug discovery presents a significant analytical challenge for the detection, quantitation, and characterization of the compounds alone.[2] Here, in Figure 1, we have summarized all classes of impurities.
Figure 1.
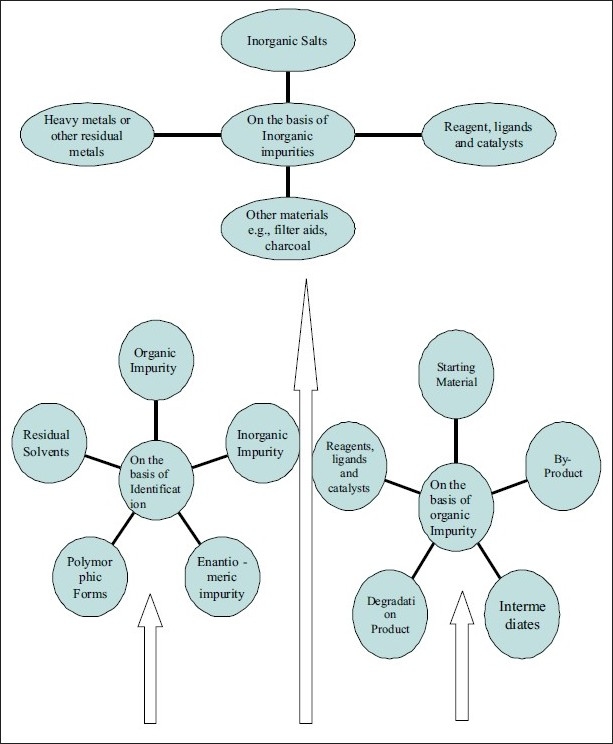
Flow chart depicting various kinds of impurities
SOURCES OF IMPURITY IN MEDICINES
According to the International Conference on Harmonization (ICH) guidelines, impurities associated with APIs are classified in different ways.
Organic Impurities
Organic impurities are the most common impurities found in every API unless proper care is taken in every step involved, throughout the multi-step synthesis. Although the end products are always washed with solvents, there is always a chance that the residual unreacted starting materials remain, unless the manufacturers are very careful about the impurities. In a paracetamol bulk, there is a limit test for p-aminophenol, which could be a starting material for one manufacturer or be an intermediate for others [Figure 2].
Figure 2.
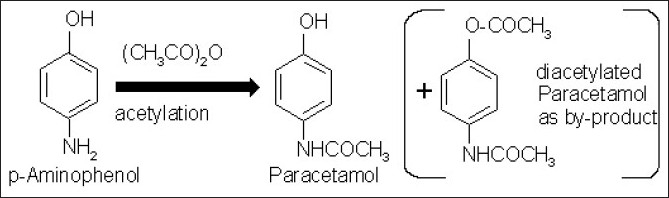
Production of paracetamol from intermediate, p-Amin-ophenol
Oxidative degradation
Hydrocortisone, methotrexate, adinazolam, hydroxyl group directly bonded to an aromatic ring (e.g., phenol derivatives such as catecholamines and morphine), conjugated dienes, heterocyclic aromatic rings, nitroso and nitrite derivatives, and aldehydes (e.g., flavones) are all susceptible to oxidative degradation.
Decarboxylation
Some dissolved carboxylic acids, such as p-aminosalicylic acid, lose carbon dioxide from the carboxyl group when heated, in the case of photoreaction of rufloxacin.[3]
Hydrolysis
Hydrolysis is a common phenomenon for the ester type of drugs, especially in liquid dosage forms. Examples include benzyl penicillin, barbitol, chloramphenicol, chlordiazepoxide, lincomycin, ethyl paraben,[4] and cefpodoxime proxetil.[5] Moreover, the hydrolysis scheme of benzocain has been depicted in Figure 3.
Figure 3.

Ester hydrolysis of benzocaine
PHOTOLYTIC CLEAVAGE
Pharmaceutical products are exposed to light while being manufactured as a solid or solution, and then they are packaged. Most compounds will degrade as solutions when exposed to high energy UV exposure (Ergometrine,[6] Nifedipine,[7] riboflavin, and phenothiazines are very labile to photo-oxidation.). Fluoroquinolones antibiotics are also found to be susceptible to photolytic cleavage.[8] In ciprofloxacin eye drops, the photocleavage reaction produces the ethylenediamine analog of ciprofloxacin.[9]
Enantiomeric Impurities
The single enantiomeric form of a chiral drug is now considered as an improved chemical entity that may offer a better pharmacological profile and an increased therapeutic index, with a more favorable adverse reaction profile. However, the pharmacokinetic profiles of levofloxacin (S-isomeric form) and ofloxacin (R-isomeric form) are comparable, suggesting the lack of advantages of a single isomer in this regard. For the manufacturers of a single enantiomeric drug (eutomer), the undesirable stereoisomers in drug control are considered in the same manner as other organic impurities.[10]
Inorganic Impurities
Inorganic impurities may also be derived from the manufacturing processes used for bulk drugs. They are normally known and identified, and include the following:
Reagents, ligands, and catalysts
The chances of having these impurities are rare: however, in some processes, these could create a problem unless the manufacturers take proper care during production.
Heavy metals
The main sources of heavy metals are the water used in the processes and the reactors (if stainless steel reactors are used), where acidification or acid hydrolysis takes place. These impurities of heavy metals can easily be avoided using demineralized water and glass-lined reactors.
Other materials (e.g., filter aids, charcoal etc.)
The filters or filtering aids such as centrifuge bags are routinely used in the bulk drugs manufacturing plants and in many cases, activated carbon is also used. The regular monitoring of fibers and black particles in the bulk drugs is essential to avoid these contaminations.
In-Process Production Impurities
Crystallization related impurities
Impurity can be any substance other than the material being crystallized. Therefore, even the solvent from which the crystals are grown can be considered as an impurity. When impurities are added specifically to produce a desired morphological effect they are referred to as additives. The presence of impurities or additives in a crystallization system can have a radical effect on crystal growth, nucleation, and agglomeration, asn also on the uptake of foreign ions in the crystal structure.[11]
Stereochemistry related impurities
It is of paramount importance to look for stereochemistry related compounds; that is, those compounds that have a similar chemical structure, but different spatial orientation. These compounds can be considered as impurities in the APIs. The single enantiomeric form of a chiral drug is now considered as an improved chemical entity that may offer a better pharmacological profile and an increased therapeutic index, with a more favorable adverse reaction profile, for example, the pharmacokinetic profile of levofloxacin (S-isomeric form) and ofloxacin (R-isomeric form) are comparable, other examples are levofloxacin (S-ofloxacin), esomeprazole (S-omeprazole), and lavalbuterol (R-albuterol).[10]
Solvents remain after processing
Residual solvents are organic volatile chemicals used during the manufacturing process or generated during the production. Some solvents that are known to cause toxicity should be avoided in the production of bulk drugs.[12] Depending on the possible risk to human health, residual solvents are divided into three classes [Table 1].
Table 1.
Classification of solvents on the basis of their limit in parts per million (ppm)

Synthetic intermediates and by-products
Impurities in pharmaceutical compounds or a new chemical entity (NCE) can originate during the synthetic process, from raw materials, intermediates, and / or by-products. Impurity profiling of tablets by GC-MS and MDMA (3, 4-Methylene dioxy methamphetamine) samples produced impurities in the intermediates via the reductive amination route.[13]
Impurities generated during storage
A number of impurities can originate during storage or shipment of drug products. It is essential to carry out stability studies to predict, evaluate, and ensure drug product safety.[14]
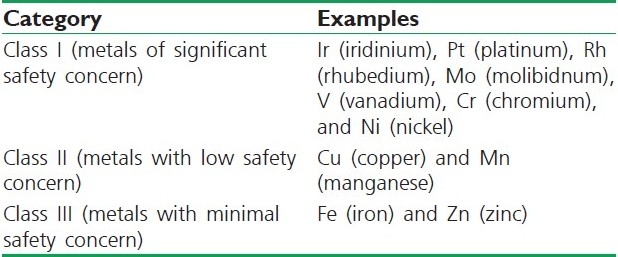
Metal impurities
Metal acts as an impurity in the APIs and excepients. Metals can be divided into three classes, as mentioned in Table 2.[15]
Table 2.
Classification of metals on the basis of their safety concern
Leachables / Extractables
Regulatory, safety, and scientific considerations in evaluating extractables and leachables is important, along with strategy studies, for analytical identification, quantification, and monitoring.[16]
ICH Guidelines
We have summarized various classes based on impurities according to the ICH guideline in Table 3.
Table 3.
Classification of Q guideline on the basis of impurities

ICH Limits for Impurities
According to the ICH guidelines on impurities in new drug products, identification of impurities below 0.1% level is not considered to be necessary, unless potential impurities are expected to be unusually potent or toxic. According to the ICH, the maximum daily dose qualification threshold to be considered is as follows; <2 g / day, 0.1 % or 1 mg per day intake (whichever is lower) >2 g / day, 0.05%.
-
i)
Organic Impurities
Each specific identified impurity
-
-Each specific unidentified impurity at or above 0.1%
-
-Any unspecific impurity, with a limit of not more than 0.1%
-
-Total impurities
-
-
-
ii)
Residual solvents
-
iii)
Inorganic impurities[17]
Isolation Methods
It is often necessary to isolate impurities. However, if instrumental methods are used, isolation of impurities is avoided, as it directly characterizes the impurities. Generally, chromatographic and non-chromatographic techniques are used for the isolation of impurities prior to its characterization. The term ‘chromatographic reactor’ refers to the use of an analytical-scale column, serving both as a flow-through reactor and a separation medium for the reactant(s) and product(s). High-performance liquid chromatography (HPLC) and the chromatographic reactor approach, with solution-phase hydrolysis kinetics can be used for an aprepitant (EmendTM) prodrug and fosaprepitant dimeglumine.[18] In loratidine, the impurity found was ofloratidine;[19] other examples include celecoxib[20] and amikacin.[21]
The structure of impurities — unknown degradation products in drug substances and drug products — must, according to the current FDA and EMEA guidelines, elucidate if they exceed a level of greater than 0.1%. Analytical Services provide the latest analytical techniques for structure elucidation (e.g., high-field NMR, LC-MSMS, GC-MS, and MALDI-TOF) as well as for preparative isolation of unknown impurities (e.g., semi and fully preparative HPLC). Software tools for the prediction of spectra support the study of our experts.
Solid-Phase Extraction Methods
Solid phase extraction (SPE) is an increasingly useful sample preparation technique. With SPE, many of the problems associated with liquid – liquid extraction can be prevented, such as incomplete phase separation, less-than-quantitative recoveries, use of expensive, breakable specialty glassware, and disposal of large quantities of organic solvents. SPE is more efficient than liquid – liquid extraction, yields quantitative extractions that are easy to perform, is rapid, and can be automated. Solvent use and laboratory time are reduced. SPE is used very often to prepare liquid samples and extract semi-volatile or nonvolatile analytes, and can also be used with solids that are pre-extracted into solvents. SPE products are excellent for sample extraction, concentration, and cleanup. They are available in a wide variety of chemistries, adsorbents, and sizes. Selecting the most suitable product for each application and sample is important.
Liquid – Liquid Extraction Methods
Liquid – liquid extraction, also known as solvent extraction and partitioning, is a method to separate compounds based on their relative solubilities in two different immiscible liquids, usually water and an organic solvent. It is an extraction of a substance from one liquid phase into another liquid phase. Liquid – liquid extraction is a basic technique in chemical laboratories, where it is performed using a separating funnel. This type of process is commonly performed after a chemical reaction as part of the workup.
Accelerated Solvent Extraction Methods
Accelerated Solvent Extraction (ASE) is a better technique for the extraction of solid and semisolid sample matrices, using common solvents, at elevated temperatures and pressures. ASE systems are available in the entry level ASE 150 system and the fully automated ASE 350. Extractions that normally take hours can be done in minutes using ASE with pH hardened pathways, using Dionium™ components. Compared to techniques such as Soxhlet and sonication, ASE generates results in a fraction of the time. The many steps involved in sample preparation can now be automated with the ASE flow-through technology. Filtration and clean up of solid samples can be achieved as part of the solvent extraction process in a single step. ASE offers a lower cost per sample than other techniques, reducing solvent consumption by up to 90%.
Supercritical Fluid Extraction
Supercritical Fluid Extraction (SFE) is the process of separating one component (the extractant) from another (the matrix), using supercritical fluids as the extracting solvent. Extraction is usually from a solid matrix, but can also be from liquids. SFE can be used as a sample preparation step for analytical purposes, or on a larger scale to either strip unwanted material from a product (e.g., decaffeination) or collect a desired product (e.g., essential oils). Carbon dioxide (CO2) is the most used supercritical fluid, sometimes modified by co-solvents such as ethanol or methanol. Extraction conditions for supercritical CO2 are above the critical temperature of 31°C and critical pressure of 72 bar. Addition of modifiers may slightly alter this.
Column Chromatography
Column chromatography in chemistry is a method used to purify individual chemical compounds from mixtures of compounds. It is often used for preparative applications on scales from micrograms to kilograms. The classical preparative chromatography column is a glass tube with a diameter of 50 mm and a height of 50 cm to 1 m with a tap at the bottom. Two methods are generally used to prepare a column; the dry method and the wet method. The individual components are retained by the stationary phase differently and separate from each other while they are running at different speeds through the column with the eluent. At the end of the column they elute one at a time. During the entire chromatography process the eluent is collected in a series of fractions. The composition of the eluent flow can be monitored and each fraction is analyzed for dissolved compounds, for example, by analytical chromatography, UV absorption or fluorescence. Colored compounds (or fluorescent compounds, with the aid of an UV lamp) can be seen through the glass wall as moving bands.
Flash Chromatography
Distillation, re-crystallization, and extraction are all important techniques for the purification of organic compounds. However, the technique used most commonly in modern organic research is ‘flash’ chromatography. In traditional column chromatography the sample to be purified is placed on top of a column containing some solid support, often silica gel. The rest of the column is then filled with a solvent (or a mixture of solvents), which then runs through the solid support under the force of gravity. The various components to be separated travel through the column at different rates and are then collected separately as they emerge from the bottom of the column. Unfortunately, the rate at which the solvent percolates through the column is slow. In flash chromatography, however, air pressure is used to speed up the flow of the solvent, dramatically decreasing the time needed to purify the sample.[22]
Thin Layer Chromatography
Thin layer chromatography (TLC) is a chromatography technique used to separate mixtures. Thin layer chromatography is performed on a sheet of glass, plastic or aluminum foil, which is coated with a thin layer of adsorbent material, usually silica gel, aluminium oxide or cellulose. This layer of adsorbent is known as the stationary phase.
After the sample has been applied on the plate, a solvent or solvent mixture (known as the mobile phase) is drawn up the plate via capillary action. As different analytes ascend the TLC plate at different rates, separation is achieved.
Thin layer chromatography finds many applications to determine the components that are contained in plants. It is also used for monitoring organic reactions and analyzing ceramides and fatty acids; for the detection of pesticides or insecticides in food and water; for analyzing the dye composition of fibers in forensics and identifying compounds present in a given substance, and for assaying the radiochemical purity of radiopharmaceuticals [Figure 4]. A number of enhancements can be made to the original method, to automate the different steps, to increase the resolution achieved with TLC, and to allow more accurate quantization. This method is referred to as HPTLC or ‘high performance TLC’.
Figure 4.

Separation of different chemical constituents by TLC
Gas Chromatography
Gas–liquid chromatography (GLC) or simply gas chromatography (GC), is a common type of chromatography used in analytical chemistry for separating and analyzing compounds that can be vaporized without decomposition. Typical uses of GC include testing the purity of a particular substance or separating the different components of a mixture (the relative amounts of such components can also be determined). In some situations, GC may help in identifying a compound. In preparative chromatography, GC can be used to prepare pure compounds from a mixture.[23]
High Performance Liquid Chromatography
High performance liquid chromatography (or high pressure liquid chromatography, HPLC) is a form of column chromatography used frequently in biochemistry and analytical chemistry, to separate, identify, and quantify compounds, based on their idiosyncratic polarities and interactions with the column's stationary phase. HPLC utilizes different types of stationary phases (typically, hydrophobic saturated carbon chains), a pump that moves the mobile phase(s) and analyte through the column, and a detector that provides a characteristic retention time for the analyte. The detector may also provide other characteristic information (i.e., UV / Vis spectroscopic data for the analyte if so equipped). Analyte retention time varies depending on the strength of its interactions with the stationary phase, the ratio / composition of the solvent(s) used, and the flow rate of the mobile phase.[24]
Supercritical Fluid Chromatography
Supercritical Fluid Chromatography (SFC) is a form of normal phase chromatography that is used for the analysis and purification of low-to-moderate molecular weight, thermally labile molecules. It can also be used for the separation of chiral compounds. Its principles are similar to those of HPLC, however SFC typically utilizes carbon dioxide as the mobile phase; therefore, the entire chromatographic flow path must be pressurized.
Capillary Electrophoresis
Capillary electrophoresis (CE), also known as capillary zone electrophoresis (CZE), can be used to separate ionic species by their charge and frictional forces and mass. In traditional electrophoresis, electrically charged analytes move in a conductive liquid medium under the influence of an electric field. Introduced in the 1960s, the technique of capillary electrophoresis (CE) was designed to separate species based on their size, to charge ratio in the interior of a small capillary filled with an electrolyte.
Electron Paramagnetic Resonance
Electron paramagnetic resonance (EPR) or electron spin resonance (ESR) spectroscopy is a technique for studying the chemical species that have one or more unpaired electrons, such as organic and inorganic free radicals or inorganic complexes possessing a transition metal ion. The basic physical concepts of EPR are analogous to those of NMR, but it is electron spins that are excited here instead of spins of the atomic nuclei. As the most stable molecules have all their electrons paired, the EPR technique is less widely used than NMR. However, this limitation to the paramagnetic species also means that the EPR technique is one of great specificity, as ordinary chemical solvents and matrices do not give rise to EPR spectra.
Gas Chromatography – Mass Spectroscopy
Gas chromatography – mass spectrometry (GC – MS) is a method that combines the features of gas – liquid chromatography and mass spectrometry, to identify different substances within a test sample. Applications of GC – MS include drug detection, fire investigation, environmental analysis, explosives investigation, and identification of unknown samples. Additionally, it can identify trace elements in materials that were previously thought to have disintegrated beyond identification. The GC – MS has been widely heralded as a ‘gold standard’ for forensic substance identification because it is used to perform a specific test. A specific test positively identifies the actual presence of a particular substance in a given sample. A non-specific test merely indicates that a substance falls into a category of substances. Although a non-specific test could statistically suggest the identity of the substance, this could lead to false positive identification.
Gravimetric Analysis
Gravimetric analysis describes a set of methods in analytical chemistry for the quantitative determination of an analyte based on the mass of a solid. A simple example is the measurement of solids suspended in a water sample: A known volume of water is filtered, and the collected solids are weighed. In most cases, the analyte must first be converted to a solid by precipitation, with an appropriate reagent. The precipitate can then be collected by filtration, washed, dried to remove traces of moisture from the solution, and weighed. The amount of analyte in the original sample can then be calculated from the mass of the precipitate and its chemical composition.
UV Spectrometry
Ultraviolet (UV) spectroscopy is a physical technique of the optical spectroscopy that uses light in the visible, ultraviolet, and near infrared ranges. The Beer-Lambert law states that the absorbance of a solution is directly proportional to the concentration of the absorbing species in the solution and the path length. Thus, for a fixed path length, UV / VIS spectroscopy can be used to determine the concentration of the absorber in a solution. It is necessary to know how rapidly the absorbance changes with concentration.
Infrared Spectroscopy
Infrared spectroscopy is the subset of spectroscopy that deals with the infrared region of the electromagnetic spectrum. It covers a range of techniques, the most common being a form of absorption spectroscopy. As with all spectroscopic techniques, it can be used to identify compounds and investigate sample compositions. A common laboratory instrument that uses this technique is an infrared spectrophotometer. The infrared portion of the electromagnetic spectrum is usually divided into three regions; the near-, mid- and far-infrared, named according to their relation to the visible spectrum. The far-infrared, approximately 400 – 10 cm-1 (1000 – 30 μm), lying adjacent to the microwave region, has low energy and may be used for rotational spectroscopy. The mid-infrared, approximately 4000 – 400 cm-1 (30 – 2.5 μm), may be used to study the fundamental vibrations and associated rotational-vibrational structure.
Fluorescence Spectroscopy
Fluorescence spectroscopy is called as fluorometry or spectrofluorometry. It is a type of electromagnetic spectroscopy, which analyzes the fluorescence from a sample. It involves using a beam of light, usually ultraviolet light, which excites the electrons in the molecules of certain compounds and causes them to emit light of a lower energy, typically, but not necessarily, visible light. A complementary technique is absorption spectroscopy. Devices that measure fluorescence are called fluorometers or fluorimeters.
Characterization Method
Highly sophisticated instrumentation, such as MS attached to a GC or HPLC, are inevitable tools in the identification of minor components (drugs, impurities, degradation products, metabolites) in various matrices. For characterization of impurities, different techniques are used; which are as follows;
Mass spectrometry
Mass spectrometers are used in the industry and academia for both routine and research purposes. The following list is just a brief summary of the major mass spectrometric applications:
Biotechnology: Is the analysis of proteins, peptides, and oligonucleotides.
Pharmaceuticals: Deal with drug discovery, combinatorial chemistry, pharmacokinetics, and drug metabolism.
Clinical: Deals with neonatal screening, hemoglobin analysis, and drug testing.
Environmental: Deals with polycyclic aromatic hydrocarbons (PAHs), Polychlorinated biphenyls (PCBs), water quality, and food contamination.
Geological: Deals with the oil composition.
The instruments include: Ionization source, for example, electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI); Analyzer mass to charge (m/z), for example, quadruple and magnet; FT-ICR detector, for example, the photomultiplier micro-channel plate electron multiplier.
Ionization methods include the following:
Atmospheric Pressure Chemical Ionization (APCI), Chemical Ionization (CI), Electron Impact (EI), Electrospray ionization (ESI), Fast atom bombardment (FAB), Field desorption / Field ionization (FD / FI), Matrix-assisted laser desorption ionization (MALDI), and Thermospray ionization (TSP).
NMR spectroscopy
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful and theoretically complex analytical tool. In NMR, the chemical environment of the specific nuclei is deduced from the information obtained about the nuclei.
Nuclear magnetic resonance (NMR) is a property that magnetic nuclei have in a magnetic field and the applied electromagnetic (EM) pulse or pulses, which cause the nuclei to absorb energy from the EM pulse and radiate this energy back out. The energy radiated back out is at a specific resonance frequency, which depends on the strength of the magnetic field and other factors. This allows for the observation of specific quantum mechanical magnetic properties of an atomic nucleus. Many scientific techniques exploit NMR phenomena, to study molecular physics, crystals, and non-crystalline materials, through NMR spectroscopy. NMR is also routinely used in advanced medical imaging techniques, such as in magnetic resonance imaging (MRI).
All stable nuclides that contain an odd number of protons and / or neutrons (see Isotope) have an intrinsic magnetic moment and angular momentum, in other words a spin > 0, while all nuclides with even numbers of both have spin 0. The most commonly studied nuclei are 1H (the most NMR-sensitive isotope after the radioactive 3H) and 13C, although nuclei from isotopes of many other elements (e.g., 2H, 10B, 11B, 14N, 15N, 17O, 19F, 23Na, 29Si, 31P, 35Cl, 113Cd, and 195Pt) are studied by high-field NMR spectroscopy as well.
A key feature of NMR is that the resonance frequency of a particular substance is directly proportional to the strength of the applied magnetic field. It is this feature that is exploited in imaging techniques. If a sample is placed in a nonuniform magnetic field, then the resonance frequencies of the sample's nuclei depend on where in the field they are located. As the resolution of the imaging techniques depends on how big the gradient of the field is, many efforts are made to develop more powerful magnets, often using superconductors. The effectiveness of NMR can also be improved using hyper polarization, and / or two-dimensional, three-dimensional, and higher dimension multi-frequency techniques.
Nuclear Magnetic Resonance phenomena are also utilized in low-field NMR, NMR spectroscopy, and MRI in the Earth's magnetic field (referred to as Earth's field NMR).[25]
Raman spectroscopy
Raman spectroscopy is a spectroscopic technique used to study vibrational, rotational, and other low-frequency modes in a system. It relies on the inelastic scattering or the Raman scattering of the monochromatic light, usually from a laser, in the visible, near infrared, or near ultraviolet range. The laser light interacts with the photons or other excitations in the system, resulting in the energy of the laser photons being shifted up or down. The shift in energy gives information about the phonon modes in the system. Infrared spectroscopy yields similar, but complementary, information. Typically, a sample is illuminated with a laser beam. Light from the illuminated spot is collected with a lens and sent through a monochromator. Wavelengths close to the laser line, due to elastic Rayleigh scattering, are filtered out, while the rest of the collected light is dispersed onto a detector.
Spontaneous Raman scattering is typically very weak, and as a result the main difficulty of Raman spectroscopy is separating the weak inelastically scattered light from the intense Rayleigh scattered laser light. Modern instrumentation almost universally employs notch or edge filters for laser rejection and spectrographs (either axial transmissive (AT) or Czerny-Turner (CT) monochromator) or FT (Fourier transform spectroscopy), and CCD detectors.
There are a number of advanced types of Raman spectroscopy, including surface-enhanced Raman, tip-enhanced Raman, polarized Raman, stimulated Raman (analogous to stimulated emission), transmission Raman, spatially-offset Raman, and hyper Raman. Heavily B-doped polycrystalline diamond films ([B]≥1019 cm-3) are studied by Raman spectroscopy and electron spin resonance. The formation of an impurity band is accompanied by a Fano-type interference for the one-photon scattering. Bands at 1200 and 500 cm-1 are observed in Raman spectroscopy for concentrations above 1020 cm-3. They are related to the maxima in the photon density of states, and are ascribed to disordered regions or crystalline regions of very small size. The concentration of defects associated with the paramagnetic signal observed around g = 2.0030 increases drastically above 1021B cm-3. The Mott insulator-metal transition is accompanied by the presence of a new paramagnetic signal (g = 2.0007 for 2 × 1020B cm-3, g = 1.9990 for 1021B cm-3) ascribed to free holes in the impurity band.[26]
APPLICATIONS OF ISOLATION AND CHARACTERIZATION OF IMPURITIES
Numerous applications have been sought in the areas of drug designing and monitoring. Quality, stability, and safety of pharmaceutical compounds, whether produced synthetically, extracted from natural products or produced by recombinant methods. The applications include alkaloids, amines, amino acids, analgesics, antibacterials, anticonvulsants, antidepressants, tranquilizers, antineoplastic agents, local anesthetics, macromolecules, steroids, and so on.[27]
CONCLUSION
Isolation and characterization of impurities is mandatory for acquiring and evaluating data that establishes biological safety, which reveals the need and scope for impurity profiling of drugs in pharmaceutical research. To isolate and quantify the impurities, various instrumental analytical techniques have been used routinely. Moreover the recognition and regulatory contemplation of organic impurities is an extremely complex problem owing to numerous sources ranging from microbial contamination to degradation products of APIs apart from traces of intermediates. Although, ICH has an out lighted course of action with regard to impurities, but still much more needs to be done. Hence, there is a strapping need to have unified specifications / standards with regard to impurities.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Keitel S. Impurity Profiles in Active Pharmaceutical Ingredients.EU / Swissmedic GMP Workshop Beijing University. 2006 Sep [Google Scholar]
- 2.Josephs JL, Sanders M, Shipkova P. Detection and Characterization of Pharmaceutical Metabolites, Degradants and Impurities by the Application of MS / MS Software Algorithms; Technical Program. 2007 Feb 25; [Google Scholar]
- 3.Condorelli G, De Guidi G, Giulfrido S. Molecular mechanisms of photosensitization induced by drugs XII. Photochemistry and photosensitization of rufloxacin: An unusual photodegradation path for the antibacterials containing a fluoroquinolone like chromophore. Photochem Photobiol. 1999;70:280–6. [PubMed] [Google Scholar]
- 4.Connor KA, Amidon GL, Stella VJ. 2nd ed. New York: John Wiley and Sons; 1986. Chemical Stability of Pharmaceuticals- A Handbook for Pharmacists; pp. 182–4. [Google Scholar]
- 5.Hoerle SL, Evans KD, Snider BG. New Jersey: Eastern Analytical Symposium; 1992. Nov 16-20, HPLC Determination of Impurities in a 3rd Generation Cephalosporin. [Google Scholar]
- 6.Roy J, Bhuiyan K, Faruque A. Injectable ergometrine: Stability and packaging for developing countries. Indian Drugs. 1997;34:634–6. [Google Scholar]
- 7.Kumar V, Sunder N, Potdar A. Critical factors in developing pharmnaceutical formulations- An overview.Part 2. Pharm Technol. 1992;16:86–8. [Google Scholar]
- 8.Smith A, Pennefather PM, Kaye SB. Fluoroquinolones - place in ocular therapy. Drugs. 2001;61:747–61. doi: 10.2165/00003495-200161060-00004. [DOI] [PubMed] [Google Scholar]
- 9.Roy J, Das SC. The effect of sunlight on ciprofloxacin eye drops. 2010 [In press] [Google Scholar]
- 10.Riley TN. Steric aspects of drug action. Pharmacist. 1998;23:40–51. [Google Scholar]
- 11.Hatakka H, Alatalo H, Palosaari S. Effect of Impurities and additives on crystal Growth. [Last accessed on 2010]. Available from: http://www2.lut.fi/~hhatakka/docit/impure.html .
- 12.Jacobs P, Dewe W, Flament A. A new validation approach applied to the GC determination of impurities in organic solvents. J Pharm Biomed Anal. 2005;40:294–304. doi: 10.1016/j.jpba.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 13.Gimeno P, Besacier F, Bottex M, Dujourdy L, Chaudron-Thozet H. A study of impurities in intermediates and 3, 4 methylenedioxymethamphetamine (MDMA) samples produced via reductive amination routes. Forensic Sci Int. 2005;155:141–57. doi: 10.1016/j.forsciint.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Ahuja S. New York: Marcel Dekker; 1998. Impurities Evaluation of Pharmaceuticals; p. 142. [Google Scholar]
- 15.Humfrey C. London: R and D, SCI; 2007. Jun 07, Keeping afloat in a sea of impurities, Global Safety Assessment AstraZeneca. [Google Scholar]
- 16.Markovic I. Evaluation of safety and quality impact of extractable and leachable substances in therapeutic biologic protein products: A risk-based perspective. Informa. 2007;6:487–91. doi: 10.1517/14740338.6.5.487. [DOI] [PubMed] [Google Scholar]
- 17.Bari SB, Kadam BR, Jaiswal YS. Impurity profile: Significance in Active Pharmaceutical Ingredient. Eurasian J Anal Chem. 2007;2:1. [Google Scholar]
- 18.Peter JS, Ahmed A, Yan W. An HPLC chromatographic reactor approach for investigating the hydrolytic stability of a pharmaceutical compound. J Pharm Biomed Anal. 2006;41:883–90. doi: 10.1016/j.jpba.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Radhakrishna T, Satynarayana J, Satynarayana A. Determination of Loratidine and its Related Impurities by HPLC. Indian Drugs. 2002;39:342. [Google Scholar]
- 20.Radhakrishna T, Satynarayana J, Satynarayana A. HPLC method for the Degradation of Celecoxib and its Related Impurities. Indian Drugs. 2002;40:166. [Google Scholar]
- 21.Zawilla NH, Li B, Hoogmartens J. Improved RP-LC method combined with pulsed electrochemical detection for the analysis of amikacin. J Pharm Biomed Anal. 2006;42:114. doi: 10.1016/j.jpba.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 22.Still WC, Kahn M, Mitra A. Flash chromatography. J Org Chem. 1978;43:2923–5. [Google Scholar]
- 23.Donald LP, Gary ML. Vol. 4. Thomson Brooks; 2006. Introduction to Organic Laboratory Techniques; pp. 797–817. [Google Scholar]
- 24.Look DC, Jones RL, Cantwell G. Characterization of homoepitaxial p-type ZnO grown by molecular beam epitaxy. Appl Phys Lett. 2002;81:1830. [Google Scholar]
- 25.Buckau G, Duschner H, Psarros N. Characterization of humic and fulvic acids from Gorleben groundwater. Fresenius J Anal Chem. 1990;338:245–52. [Google Scholar]
- 26.Gonon P, Gheeraert E, Deneuville A. Characterization of heavily B-doped polycrystalline diamond films using Raman spectroscopy and electron spin resonance. J Appl Phys. 1995;78:7059–2. [Google Scholar]
- 27.Lohr LL, Sharp TR, Alsante KM. Isolation and Identification of Process Related Impurities and Degradation Products from Pharmaceutical Drug Candidates.Part 2: The Roles of NMR and Mass Spectrometry. [Last accessed on 2010];Am Pharm Rev. 2001 14:232–9. Available from: http: // www.americanpharmaceuticalreview.com / past_articles_f.htm . [Google Scholar]