

Abstract
Cefuroxime Axetil (Poorly water soluble drug), when prepared as solid dispersion showed improved solubility and dissolution. Therefore, the main purpose of this investigation was to increase the solubility and dissolution rate of Cefuroxime Axetil by the preparation of its solid dispersion with urea, using the solvent evaporation method. Physical mixtures and solid dispersions of Cefuroxime Axetil were prepared by using urea as a water-soluble carrier in various proportions (1:1, 1:2, 1:3, 1:4, 1:5, 1:6, and 1:7 by weight), by employing the solvent evaporation method. The drug release profile was studied and it was found that the dissolution rate and the dissolution parameters of the drug from the physical mixture as well as solid dispersion were higher than those of the intact drug. The Fourier Transform Infrared (FTIR) spectra revealed no chemical incompatibility between the drug and urea. Drug-polymer interactions were investigated using differential scanning calorimetry (DSC) and Powder X-Ray Diffraction (PXRD).
Keywords: Cefuroxime Axetil, solid dispersion, solvent evaporation method, urea
INTRODUCTION
Cefuroxime Axetil (1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 72-(Z)-(O-methyl-oxime), 1-acetate 3-carbamate. Its molecular formula is C20H22N4O10S, and it has a molecular weight of 510.48. Cefuroxime Axetil is used orally for the treatment of patients with mild-to-moderate infections, caused by susceptible strains of the designated microorganisms. One of the major problems of this drug is its very poor solubility in biological fluids, which results in poor bioavailability after oral administration. It shows erratic dissolution problems in the gastric and intestinal fluids, due to its poor water solubility. The rate of absorption and / or extent of bioavailability for such insoluble drugs is controlled by the rate of dissolution in the gastrointestinal fluids.[1] The peak plasma concentration (Cmax) and the time taken to reach Cmax (tmax) depend on the extent and rate of dissolution of the drug, respectively. The effort to improve the dissolution and solubility of a poorly water-soluble drug remains one of the most challenging tasks in drug development. Several methods have been introduced to overcome this problem, such as, solid dispersions, complexation, Zydis technology, and the use of hydrophilic carriers.
Solid dispersion, which was introduced in the early 1970s,[2] refers to a group of solid products consisting of at least two different components, generally a hydrophilic matrix and a hydrophobic drug. The matrix can be either crystalline or amorphous. The drug can be dispersed molecularly, in amorphous particles (clusters) or in crystalline particles.[2] The solid dispersion technique has been used for a wide variety of poorly soluble drugs such as nimesulide,[3] ketoprofen,[4] tenoxicam,[5] nifedipine,[6] nimodipine,[7] ursodeoxycholic acid,[8] and albendazole.[9] Various hydrophilic carriers, such as polyethylene glycols,[10] polyvinylpyrrolidone,[11] hydroxypropyl methylcellulose,[12] gums,[6] sugar,[13] mannitol,[14] and urea,[8] have been investigated for improvement of dissolution characteristics and bioavailability of poorly aqueous soluble drugs [Figure 1].
Figure 1.
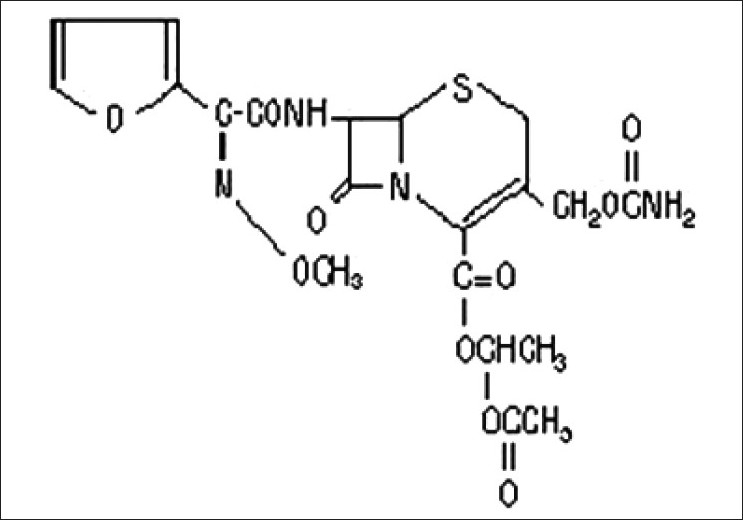
Chemical structure of Cefuroxime Axetil
Solid dispersion can be prepared by various methods such as the solvent evaporation and melting methods. The solid dispersion technique has been extensively used to increase the solubility of a poorly water-soluble drug. According to this method, a drug is thoroughly dispersed in a water-soluble carrier by a suitable method of preparation. The mechanism by which the solubility and the dissolution rate of the drug are increased includes: reduction of the particle size of the drug to a submicron size or to a molecular size in a case where solid solution is obtained. The particle size reduction generally increases the rate of dissolution; secondly, the drug is changed from an amorphous to a crystalline form, the high energetic state that is highly soluble; finally, the wettability of the drug particle is improved by the hydrophilic carrier. Cefuroxime Axetil-urea systems, prepared by the solvent evaporation method, show an improvement in dissolution rates of the drug from the solid dispersions, as compared with the pure drug and physical mixtures. This study presents the formulation of solid dispersions of Cefuroxime Axetil, with urea as the hydrophilic carrier.
MATERIALS AND METHODS
Materials
Cefuroxime Axetil was obtained as a generous gift from FDC Limited, Mumbai. Urea (Analytical grade) was purchased from Qualikems Fine Chemicals Pvt. Ltd., New Delhi. All other chemical reagents were of analytical grade.
Methods
Preparation of the physical mixture
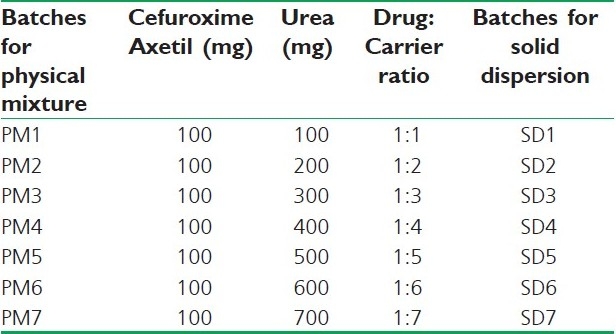
An accurately weighed amount of Cefuroxime Axetil and urea (carrier) in various drug-to-carrier weight ratios were thoroughly blended in a glass mortar for five minutes. The composition of various batches is shown in Table 1. The products were kept in a desiccator for further study.
Table 1.
Composition of batches containing Cefuroxime Axetil and urea
Preparation of solid dispersion
The solid dispersions of Cefuroxime Axetil and urea (carrier) in various drug-to-carrier weight ratios were prepared by the solvent evaporation method. One hundred milligrams of Cefuroxime Axetil were dissolved in 20 ml of methanol in a beaker and the carrier was added and mixed to dissolve at 40°C on a hot plate, to get a clear solution. Then the solvent was allowed to evaporate until a constant weight was obtained. The Solid Dispersions prepared were crushed, pulverized, and sifted through mesh number 80 and stored in desiccators.
Estimation of Cefuroxime Axetil
Cefuroxime Axetil was estimated at 278 nm by using a UV spectrophotometer[15] (Systronics Double Beam Spectrophotometer 2202). The standard curve for the estimation was prepared in 0.1 M HCl in a concentration range of 2 – 30 μg/ml. In this concentration range good linearity was observed with a correlation coefficient of (R2) 0.9981. The graph obeyed the Beer-Lambert's law in the selected concentration range.
Characterization of Samples
Fourier transform infrared spectroscopy
All the prepared solid dispersions were subjected to FTIR spectroscopic studies, to determine drug-carrier interaction. The FTIR spectra were recorded on samples prepared in potassium bromide (KBr) disks, using a Fourier Transform IR spectrophotometer (Perkin Elmer, RXI FTIR System). The samples were prepared in KBr disks by means of a hydrostatic press. The scanning range was 400 to 4000 cm-1 and the resolution was 2 cm-1.
Differential scanning calorimetric studies
Differential scanning calorimetry (DSC) measurements were carried out on a scanning calorimeter (DSC Q10 V9.0 Build 275, Universal V4.1D TA Instruments). The instrument was calibrated using indium as the standard. Samples (5 – 10 mg) were placed in sealed aluminium pans and heated from 70°C to 150°C at a rate of 10°C/min under nitrogen atmosphere (60 ml/min), with an empty pan as a reference.
X-ray diffraction studies
The powder X-ray diffraction (XRD) was performed by X’pert Pro with Spinner PW3064 using Ni-filtered, CuKα radiation, a voltage of 45 kV, and a current of 40 mA with a scintillation counter. The instrument was operated with a continuous scanning speed of 4°/min over a range of 5°C to 40°C.
Drug Content
Solid dispersions equivalent to 100 mg of Cefuroxime Axetil were weighed accurately and dissolved in a suitable quantity of 0.1M HCl. The solutions were filtered and the drug content was determined at 278 nm by a UV spectrophotometer (Systronics Double Beam Spectrophotometer 2202) after suitable dilution. The percentage yield of each formulation was also calculated.
Saturation Solubility
To evaluate the increase in the solubility of Cefuroxime Axetil, physical mixture and solid dispersions and saturation solubility measurements were conducted. The known excess (approximately 50 mg) of Cefuroxime Axetil was added to 100 mL of 0.1M HCl. The samples were rotated at 20 rpm in a water bath (37.0±0.5°C) for 48 hours. The samples were then filtered, suitably diluted, and analyzed, with the help of a UV spectrophotometer at 278 nm.
Dissolution Studies
The dissolution studies were performed using a US Pharmacopeia XXIV type II dissolution test apparatus. The samples equivalent to 100 mg Cefuroxime Axetil were placed in a dissolution vessel containing 900 mL of 0.1M HCl maintained at 37.0±0.5°C and stirred at 100 rpm. The samples were collected periodically and replaced with a fresh dissolution medium. After filtration, the concentration of Cefuroxime Axetil was determined spectrophotometrically at 278 nm.
RESULT AND DISCUSSION
Fourier Transform Infrared Spectroscopy
Fourier Transform Infrared Spectroscopy (FT-IR) studies were done to detect the possible interactions between Cefuroxime Axetil and urea. The characteristic peaks of Cefuroxime Axetil, urea, and physical mixtures are presented in Table 2. It was revealed that there were no differences in the positions of the absorption bands, thus, providing evidence for the absence of interactions in the solid state between Cefuroxime Axetil and urea.
Table 2.
FT-IR peaks of pure Cefuroxime Axetil, urea, and physical mixture of Cefuroxime Axetil and urea

Differential Scanning Calorimetric Studies
Differential scanning calorimetry shows a sharp endothermic fusion peak at 134.2°C, which corresponds to the melting point of Cefuroxime Axetil [Figure 2].
Figure 2.
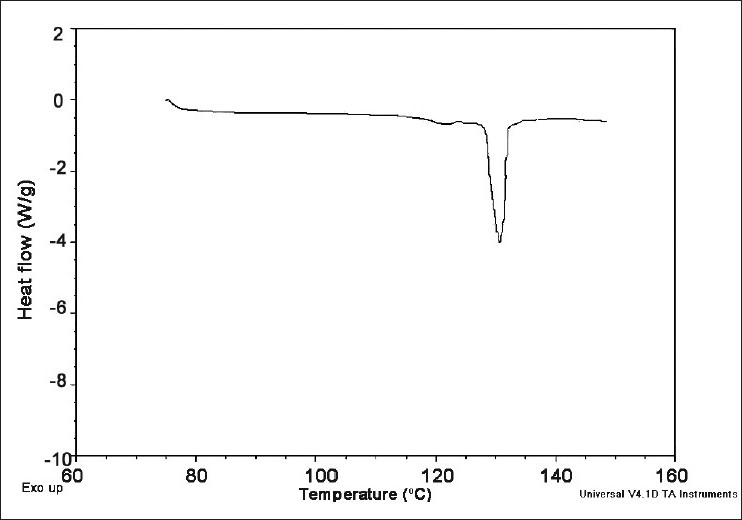
Differential scanning calorimetry of Cefuroxime Axetil
X-Ray Diffraction Studies
The diffraction spectra of Cefuroxime Axetil and urea show numerous distinct peaks indicating that both are present in a highly crystalline state. The XRD pattern of the solid dispersion of sample SD5 exhibits all the characteristic diffraction peaks of urea and crystalline Cefuroxime Axetil, although of lower intensity. This study reveals that some Cefuroxime Axetil still exists in the crystalline state in the solid dispersion.
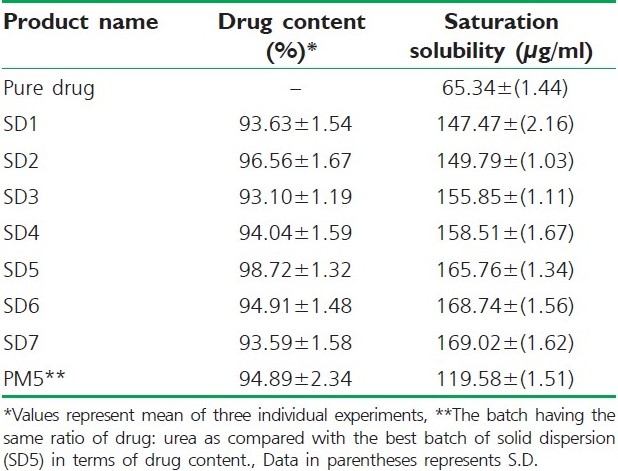
Drug content and saturation solubility
The drug content and saturation solubility were determined and the results are presented in Table 3.
Table 3.
Drug content and saturation solubility of different formulations
Dissolution Studies
The dissolution rate of pure Cefuroxime Axetil was very poor, and during 120 minutes, a maximum of about 30.23% of the drug was released. The reason for poor dissolution of the pure drug could be poor wettability and / or agglomeration or particles size. It was found that the dissolution rate of the drug increased according to the increasing amount of hydrophilic carrier (urea) in the physical mixture batches. This was due to the increase in solubility of the drug by the presence of hydrophilic carrier surrounding the drug particles. Figure 3 shows the comparative release profile of various solid dispersions of Cefuroxime Axetil with urea, physical mixture containing 1 : 5 ratio of drug : urea and pure drug. From the release profile it can be seen that the dissolution of Cefuroxime Axetil in solid dispersions increases with the increase in urea up to 1 : 5 ratio of drug : urea. This increase in the dissolution rate may due to an increase in drug wettability and solubilization of the drug by carriers. After this particular ratio, with further increase in the amount of urea, the dissolution rate was decreased. The decrease in dissolution may be due to the higher amounts of carrier that takes time to dissolve. It is found that drug release from the physical mixture is faster than that of the pure drug and slower than that of solid dispersions. From the results, it can be concluded that the dissolution rate of Cefuroxime Axetil increases by preparing solid dispersion with urea.
Figure 3.
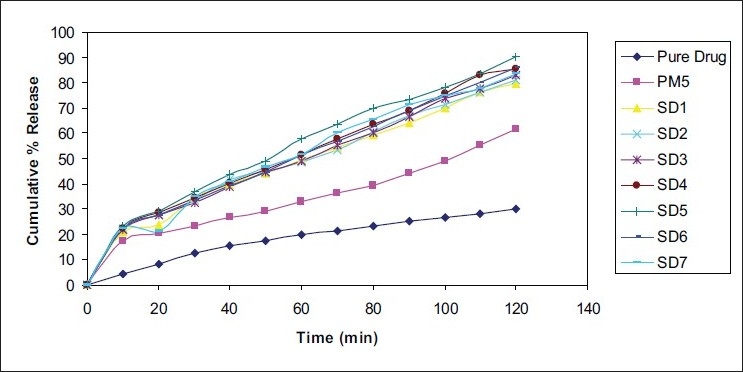
Comparative in vitro release profiles of Cefuroxime Axetil from solid dispersions and physical mixture containing urea
ACKNOWLEDGMENT
Authors would like to thank FDC Limited, Mumbai for the generous gift of Cefuroxime Axetil.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Reynolds JE, editor. 29th ed. London: The Royal Pharmaceutical Society of Great Britain; 1993. Martindale; The Extra Pharmacopoeia; p. 295. [Google Scholar]
- 2.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 3.Babu GV, Kumar NR, Himasankar K, Seshasayana A, Murthy KV. Nimesulide-modified gum karaya solid mixtures: Preparation, characterization and formulation development. Drug Dev Ind Pharm. 2003;29:855–64. doi: 10.1081/ddc-120024181. [DOI] [PubMed] [Google Scholar]
- 4.Rogers JA, Anderson AJ. Physical characteristics and dissolution profiles of ketoprofen-urea solid dispersions. Pharm Acta Helv. 1982;57:276–81. [Google Scholar]
- 5.El-Gazayerly ON. Characterization and evaluation of tenoxicam coprecipitates. Drug Dev Ind Pharm. 2000;26:925–30. doi: 10.1081/ddc-100101319. [DOI] [PubMed] [Google Scholar]
- 6.Vippagunta SR, Maul KA, Tallavajhala S, Grant DJ. Solid-state characterization of nifedipine solid dispersions. Int J Pharm. 2002;236:111–23. doi: 10.1016/s0378-5173(02)00019-4. [DOI] [PubMed] [Google Scholar]
- 7.Murali Mohan Babu GV, Prasad CH, Ramana Murthy KV. Evaluation of modified gum karaya as carrier for the dissolution enhancement of poorly water soluble drug nimodipine. Int J Pharm. 2002;234:1–17. doi: 10.1016/s0378-5173(01)00925-5. [DOI] [PubMed] [Google Scholar]
- 8.Okonogi S, Yonemochi E, Oguchi T, Puttipipatkhachorn S, Yamamoto K. Enhanced dissolution of ursodeoxycholic acid from the solid dispersion. Drug Dev Ind Pharm. 1997;23:1115–21. [Google Scholar]
- 9.Torrado S, Torrado S, Torrado JJ, Cadorniga R. Preparation, dissolution and characterization of albendazole solid dispersions. Int J Pharm. 1996;140:247–50. [Google Scholar]
- 10.Margarit MV, Rodrýguez IC, Cerezo A. Physical characteristics and dissolution kinetics of solid dispersions of ketoprofen and polyethylene glycol 6000. Int J Pharm. 1994;108:101–7. [Google Scholar]
- 11.Yagi N, Terashima Y, Kenmotsu H, Sekikawa H, Takada M. Dissolution behavior of probucol from solid dispersion systems of probucol-polyvinylpyrrolidone. Chem Pharm Bull (Tokyo) 1996;44:241–4. [Google Scholar]
- 12.Kushida I, Ichikawa M, Asakawa N. Improvement of dissolution and oral absorption of ER-34122, a poorly water soluble dual 5-lipoxygenase /cyclooxygenase inhibitor with anti-inflammatory activity by preparing solid dispersion. J Pharm Sci. 2002;91:258–66. doi: 10.1002/jps.10020. [DOI] [PubMed] [Google Scholar]
- 13.Danjo K, Nakata T, Otsuka A. Preparation and dissolution behavior of ethenzamide solid dispersions using various sugars as dispersion carriers. Chem Pharm Bull (Tokyo) 1997;45:1840–4. [Google Scholar]
- 14.Arias MJ, Ginés JM, Moyano JR, Pérez-Martínez JI, Rabasco AM. Influence of preparation method of solid dispersions on dissolution rate: Study of triammterene-D-mannitol system. Int J Pharm. 1995;123:25–31. [Google Scholar]
- 15.The Indian Pharmacopeia. Ministry of Health and Family Welfare. Government of India. 2007 [Google Scholar]