

Abstract
An unnatural base-pair architecture with base pairs 2.4 Å larger than the natural DNA-based genetic system (xDNA) is evaluated for its ability to function like DNA, encoding amino acids in the context of living cells. xDNA bases are structurally analogous to natural bases but with benzene added, increasing their sizes and resulting in a duplex that is wider than native B-DNA. Plasmids encoding green fluorescent protein were constructed to contain single and multiple xDNA bases (as many as eight) in both strands, and were transfected into E. coli. Although yielding fewer colonies than the natural control plasmid, in all cases in which a modified plasmid (containing one, two, three, or four consecutive size-expanded base pairs) the correct codon bases were substituted, yielding green colonies. All four xDNA bases (xA, xC, xG, xT) were found to encode the correct partners in the replicated plasmid DNA, both alone and in longer segments of xDNA. Controls with mutant cell lines having repair functions deleted were found to express the gene correctly, ruling out repair of xDNA and confirming polymerase reading of the unnatural bases. Preliminary experiments with polymerase deletion mutants suggested combined roles of replicative and lesion-bypass polymerases in inserting correct bases opposite xDNA bases and in bypassing the xDNA segments. These experiments demonstrate a biologically functioning synthetic genetic set with larger-than-natural architecture.
Introduction
Studies aimed at mimicking the functional properties of DNA with alternative genetic structures have lent basic insights into why the naturally evolved DNA has its specific structure, and have offered alternative structures that may retain biophysical and biochemical function. In addition, modified nucleic acids have been useful as basic tools for probing biochemical systems and technological tools for diagnosing disease. Although many studies exist of altered sugarphosphate backbones for DNA, far fewer studies have been carried out on biochemical and biological function of alternative structures for the DNA bases,1-3 which are the carriers and encoders of genetic information.
In an ongoing effort to mimic the functional properties of DNA with a new genetic architecture, we have previously explored the synthesis, self-assembly, structure, and enzyme substrate properties of size-expanded DNA (xDNA),4-8 in which base pairs are made larger than the natural ones by benzo-homologation. Results have shown that xDNA forms stable, righthanded, sequence-selective helices,4-8 and that individual xDNA bases in a template can encode successful formation of base pairs by DNA polymerase enzymes.9 However, correct function in vitro is far from active functioning in a living cell, where many enzymes must operate simultaneously. To date, no unnatural genetic architectures have been used with general success to encode amino acids of a protein in a living system.
A number of laboratories have worked on artificial bases and base pairs to augment the natural DNA genetic system.10-13 In a different approach, we have investigated whether all base pairs in the DNA framework could be replaced with pairs of larger geometry, comprising an alternative genetic scaffold (Figure 1). Other research groups have recently adopted an enlargedbase-pair strategy as well.14-16 In our approach, benzopyrimidines pair with purines and benzopurines17 with pyrimidines, yielding up to eight letters of information encoding capability.5,7 The study of this nonnatural genetic set gives basic insight into the chemical, structural, and biophysical properties that govern encoding and transmission of genetic information. Moreover, the unusual properties of xDNA, including high helix stability and inherent fluorescence,18 suggest applications in the biomedical sciences.
Figure 1.
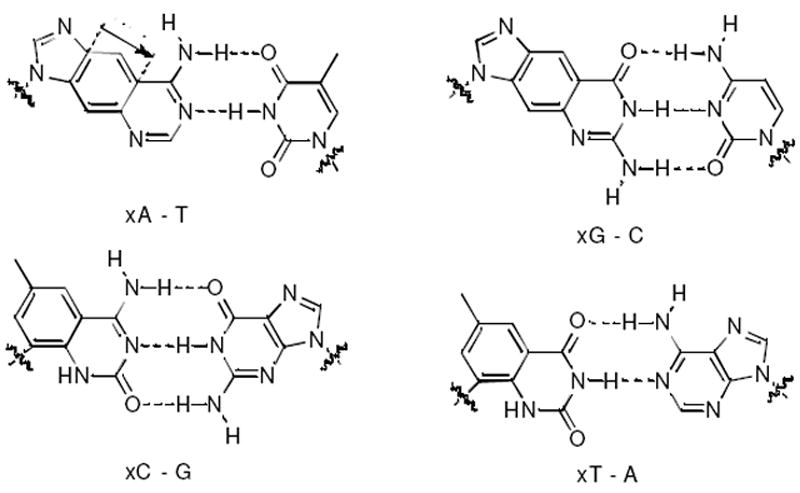
The size-expanded architecture of xDNA, in which xA, xC, xT, xG bases are paired opposite natural bases. To encode phenotype in a cell, the 2.4 Å-expanded bases must be read by cellular polymerases accurately and with adequate efficiency.
To date, studies on xDNA have been performed primarily in vitro, characterizing its pairing, structure and fluorescence properties. Ultimately, however, to have a fully functional genetic system, it is necessary to have machinery capable of efficiently recognizing and propagating the unnatural base pair architecture during replication. Given that the natural replication enzymes have evolved to function with normally-sized DNA, this is a challenging task. Nevertheless, we have begun to explore the ability of existing DNA machinery to tolerate xDNA’s size- expanded geometry. Since cells possess several classes of DNA polymerases to carry out replication bypassing a variety of lesions,19 it is possible that some enzymes may tolerate sterically enlarged xDNA bases with reasonable efficiency.
Recent in vitro studies with DNA polymerases suggested that single xDNA bases could be recognized moderately well to make one base pair. In addition, one xDNA base in single-stranded M13 phage was found to be bypassed by the E. coli replication machinery with good success, for two of the four bases.1 Encouraged by these results, we decided to evaluate this unnatural genetic set more thoroughly in the context of a biological system by asking whether xDNA could encode information as expressed by a phenotype, and whether multiple xDNA bases could be used in one vector. Here we report the use of multiple xDNA bases to encode biological information, as amino acids in a protein, green fluorescent protein (GFP), that confers a visible phenotype.
Experimental Section
Synthesis and purification of oligonucleotides
Synthesis of dxA, dxG, dxT, dxC phosphoramidites were carried out as previously reported.4,7,17 Oligodeoxynucleotides were synthesized on an Applied Biosystems 394 DNA/RNA synthesizer on a 1 μmole scale and possessed a 3’-OH, and a 5’-phosphate group (Chemical Phosphorylation II reagent from Glen Research). Coupling employed standard β-cyanoethyl phosphoramidite chemistry, but with extended coupling time (600 s) for nonnatural nucleotides. All oligomers were deprotected in concentrated ammonium hydroxide (55 °C, 16 h), purified by preparative 20% denaturing polyacrylamide gel electrophoresis, and isolated by excision and extraction from the gel, followed by dialysis against water, or desalting via PolyPak cartridges. The recovered material was quantified by absorbance at 260 nm with molar extinction coefficients determined by the nearest neighbor method. Molar extinction coefficients for unnatural oligomers were estimated by adding the measured value of the molar extinction coefficient of the unnatural nucleoside (at 260 nm) to the calculated value for the natural DNA fragments. Molar extinction coefficients for xDNA nucleosides used were as follows: dxA, ε260=19,800 M-1•cm-1; dxG, ε260=8,100 M-1•cm-1; dxT, ε260=1,200 M-1•cm-1; dxC, ε260=5,800 M-1•cm-1. Nonnatural oligomers were characterized by MALDI-TOF mass spectrometry. Their observed and expected masses are listed in Table S1.
Plasmid digestion
The pGFPuv vector ((Clontech, Cat No:632312), 3 μL, ~ 2 μg, see SI for details) was then digested by addition of 3 μL each of the restriction enzymes BsrGI and MluI (New England Biolabs), 5 μL of NEB Buffer 2, 5 μL of 10X BSA, and 31 μL of H2O (autoclaved) to an Eppendorf tube. The digest was allowed to proceed in a thermocycler at 37 °C for 6 h, followed by 65 °C for 20 min to denature the enzymes. After digestion, removal of the 5’-phosphate (to prevent self-ligation of the cut plasmid) was accomplished by addition (to the same Eppendorf tube) of 5 μL of antarctic phosphatase (or calf intestinal phosphatase (NEB) was used with the corresponding buffer) and 5 μL of 10X antarctic phosphatase buffer and then incubation for 1 h at 37 °C followed by denaturing at 65 °C for 20 min. The plasmid was purified on a 0.9% agarose (LMP) gel run at 20 volts for 12 h. The gel bands were visualized under a UV (365 nm) transilluminator, cut out (quickly, ~3 s), and the purified DNA was extracted with a plasmid gel extraction kit (Qiagen #28704). Concentration was determined by UV absorbance units at 260 nm.
Ligation of 46 bp xDNA-containing segments into digested pGFPuv vector
Ligations of xDNA-containing inserts were carried out at various vector:insert ratios (1:3, 1:5, 1:6, 1:8, 1:10). Solutions of xDNA inserts were prepared/annealed by adding 2.5 μL of each strand (100 ng/μL), 0.5 μL 1M MgCl2, and 94.5 μL H2O (autoclaved). This DNA solution was denatured in a thermocycler by heating to 72 °C for 3 min, then cooling slowly to room temperature. Appropriate volumes of the pGFPuv vector (~50 ng/μL), and annealed inserts (~2.5 ng/μL) were combined in an Eppendorf tube with 1 μL of 5X T4 DNA ligase buffer, 1 μL of T4 DNA ligase (New England Biolabs), and H2O (autoclaved) to reach a volume of 10 μL. The ligation reaction was carried out at 16 °C for 12 h in a thermocycler.
Transformation of modified vectors into E. coli
Transformation of the ligated inserts into BL21 (DE3) competent cells (Stratagene) was carried out per the transformation protocol described by Stratagene. For each transformation, ~1 to 10 ng of xDNA-containing insert were transformed into the cells, and after transformation, ~100 to 150 μL of the cells were plated on LB agar plates containing ampicillin and isopropyl thiogalactosidase (IPTG), and allowed to incubate at 37 °C for 20 h. Colonies were briefly visualized under long wavelength UV (365 nm), and the number of white and green colonies were documented. Transformation efficiencies were compared with that of a control ligation reaction under identical conditions using unmodified duplex DNA, which was otherwise identical in sequence to the xDNA-containing segment.
Colony picking and sequencing of plasmid DNA
Green colonies (as well as white colonies) from each transformation were picked, grown overnight at 37 °C in LB media, and the plasmid DNA was isolated using a mini prep kit (Qiagen #27104). The cloned region within the gene on the isolated DNA was sequenced (Sequetech, Mountain View, CA or Quintara Biosciences, Berkeley, CA) with the appropriate primers (GFPseq1.24).
Bacterial strains
The bacterial strains used in this study are shown in Table 2; they were purchased from the E. coli Genetic Stock Center at Yale University. Bacteria were routinely grown in LB media containing 50 μg/mL kanamycin.
Table 2.
Bacterial strain genotypes tested for ability to replicate xDNA segments.
| Strain | Deleted Gene | Function/Gene Product | Genotype |
|---|---|---|---|
| BW25113 | -- | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514 | |
| JW2703-2 | mutS | methyl-mediated mismatch repair | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, ΔmutS738∷kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW0704-1 | nei | endonuclease VII | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), Δnei-764∷kan, λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW2928-1 | mutY | adenine glycosylase: G-A repair | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, ΔmutY736∷kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW3610-2 | mutM (fpg) | formamidopyrimidine-DNA glycosylase | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, ΔmutM744∷kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW4019-2 | uvrA | excision nuclease: molecular matchmaker | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, rph-1, Δ(rhaD-rhaB)568, ΔuvrA753∷kan, hsdR514 |
| JW0221-1 | dinB | Pol IV | F-, Δ(araD-araB)567, ΔdinB749∷kan, ΔlacZ4787(∷rrnB-3), λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW1173-1 | umuC | Pol V | F-, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, ΔumuC773∷kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW0059-1 | polB (dinA) | Pol II | F-, ΔpolB770∷kan, Δ(araD-araB)567, ΔlacZ4787(∷rrnB-3), λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
| JW0205-1 | dnaQ (mutD) | Pol III ε subunit: 3’-> 5’ proofreading | F-, Δ(araD-araB)567, ΔdnaQ744∷kan, ΔlacZ4787(∷rrnB-3), λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514 |
Results and Discussion
A high copy plasmid vector (pGFPuv) containing a gene encoding green fluorescent protein was used as a biological template in the experiments (see SI for details). A 46 base pair segment within the GFP gene (encoding amino acids 93-107) was excised, and a synthetic insert of identical length containing single or multiple xDNA bases was ligated into the vector in place of the excised segment (Table 1). The synthetic insert contained silent mutations to distinguish it from unmodified vector, and contained an equal number of xDNA bases in both strands so that the replicative enzymes would need to address them regardless of the direction of DNA synthesis. Ultimately, if the cellular machinery were capable of faithfully reading the xDNA bases, the resulting bacterial colonies would fluoresce green under UV illumination.
Table 1.
Modified GFP inserts containing various size-expanded base pairs with a summary of sequencing results for green colonies.a
| Synthetic insert | Sequences | Seq. Results |
|---|---|---|
| thr ile ser phe lys asp asp gly | ||
| xGFP.0 (control) |
|
No change |
| xGFP.1 (one xG-C bp) |
|
xG replaced by G |
| xGFP.2 (one xA-T bp) |
|
xA replaced by A |
| xGFP.3 (one xC-G bp) |
|
xC replaced by C |
| xGFP.4 (one xT-A bp) |
|
xT replaced by T |
| xGFP.5 (two consec. xDNA bps) |
|
xCxA replaced by CA |
| xGFP.6 (three consec. xDNA bps) |
|
xCxAxA replaced by CAA |
| xGFP.7 (four consec. xDNA bps) |
|
xCxAxAxA replaced by CAAA |
size-expanded bases in red. Silent mutations in blue. Total insert size is 46 nt (Table S1).
The xDNA-containing vectors were transformed into E. coli. After incubation, green and white colonies were picked and the DNA sequenced to determine the fate of encoding by the xDNA bases. It should be noted that xDNAs were only present in the vector template and were not present as deoxynucleoside triphosphates; thus one round of plasmid replication produces natural DNA in the bacterial colonies.
Significantly, results showed that while efficiency of colony formation was generally reduced relative to the unmodified control (by ca. 10-fold), green colonies were observed in all cases where a single size-expanded base pair was present (xGFP.1-4), demonstrating that the xDNA bases were bypassed by the bacteria’s machinery, and encoded a correctly functioning amino acid (Figure 2). All green colonies that were sequenced contained the silent mutations (except for a few cases of self-ligated vector missing insert), thus confirming that there was no contamination from natural DNA in these experiments (this was true for all experiments with xDNA containing inserts). Furthermore, on sequencing the plasmid DNA of the green colonies, we found that in all the synthetic inserts the correct natural base was present opposite the xDNA base in the template (Table 1). Thus the data suggested that each of the four xDNA bases was read correctly by the replication machinery.
Figure 2.
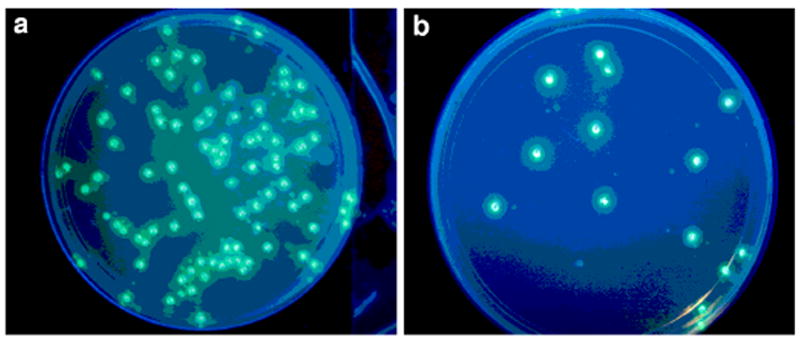
Example of GFP expression from xDNA-containing gene segment, as shown by green colonies on LB plates after 20h at 37°C. A) control (unmodified plasmid insert; (b) xGFP.7 insert containing 8 xDNA base pairs (four consecutive xDNA bases in each strand).
To test the limits of xDNA encoding, we then synthesized inserts containing 2-4 consecutive size-expanded DNA bases (xGFP.5, xGFP.6, and xGFP.7). Note that the insert actually contains double this number of xDNA pairs, 4, 6, and 8 pairs respectively, since both strands were modified. Extension of unnatural base pairs by polymerases has proved challenging in vitro, especially for consecutive substitutions, thus these inserts represent stringent cases for the replicative machinery. However, although transformation efficiencies remained similar to those with single modifications, green colonies were observed in all cases. Yet more significant was the ability for the information to be replicated faithfully; for example, the vector xGFP.7 (containing xCxAxAxA) was replaced by CAAA in all green colonies examined that contained the silent mutations (Table 1). This is striking, considering the presence of two 4-bp expanded DNA segments near one another.
A number of white colonies (not successfully expressing GFP, but expressing antibiotic resistance) were sequenced as well. Interestingly, in nearly all cases the xDNA base was replaced by the correct natural base, and the lack of GFP expression was a result of frameshifts (insertions/deletions) that occurred elsewhere in the gene (Table S6). All white colonies that contained the insert also contained the silent mutations. In only one case was the xDNA-encoded pair deleted (one of ten colonies sequenced from xGFP.3).
Further exploration of the coding limits of xDNA was undertaken by synthesizing more demanding sequences containing six and eight consecutive xDNA pairs, and sequences containing multiple isolated (nonconsecutive) size-expanded base pairs (see Tables S4, S5). However, experiments with these inserts not only yielded lower expression efficiencies, but none of the observed green colonies contained the silent mutation, even after multiple trials and varying vector:insert ratios. This could occur either from xDNA interference with enzymatic ligation in assembling the plasmid, or from failed read-through of these longest segments by polymerases. Overall, the limit in these experiments appeared to be reached with the xGFP.7 vector.
The finding of protein expression from xDNA-containing gene segments suggests that cellular polymerases are able to successfully read the expanded bases as genetic information. However, we considered another possible explanation: namely, that DNA repair enzymes were removing xDNA and filling in the missing information by reading the natural bases opposite. This could occur by one of three different mechanisms: base excision repair (BER), methyl-directed mismatch repair, or nucleotide excision repair (NER).
In base excision repair, glycosylases remove damaged bases, and then use a lyase mechanism to remove the abasic sugar prior to polymerase filling in the missing nucleotide. BER enzymes in E. coli include those encoded by nei, mutY, and mutM genes.20 In the case of xDNA, the large bases might conceivably be recognized by such enzymes as “damage”; however, the expanded pyrimidines are attached by stable C-glycosidic bonds and so cannot be excised by this mechanism. Thus full removal and repair of the xDNA is not possible for the inserts xGFP.3 through xGFP.7, all of which contain x-pyrimidines. However, it remained a possibility for inserts xGFP.1 and xGFP.2.
In the absence of data, it is also formally possible that the methyl-directed repair or NER pathways could repair xDNA segments. In these pathways, damage is recognized and excised in segments of DNA. A damaged strand is nicked upstream and downstream of the damage; ultimately the missing information is filled in by polymerases reading the undamaged strand opposite. While such repair would be difficult for the present inserts, where substitutions appear near one another in both strands (see below), it still remained a possibility.
To test the possibility of repair of xDNA segments, we obtained five bacterial strains having known BER, methyl-directed, and NER repair functions (mutM, mutY, nei, uvrA, mutS) deleted (see Table 2). If GFP expression from the xDNAs arose from a repair process, knocking out this gene should result in loss of expression. We transfected each of these strains with three examples of xDNA segments (inserts xGFP.2, xGFP.6 and xGFP.7) and measured colony counts and sequences as before, with multiple replicates of each case. Results showed no drop in colony counts, GFP expression or correct encoding for any of these knockout strains (Fig. 3 and SI). Considering the mechanisms of possible repair, it is perhaps not surprising that no evidence for repair of xDNA was seen in these experiments. Half of the xDNA nucleosides (the benzopyrimidines) contain unnatural C-C glycosidic bonds, rendering them inert to base excision. Second, if methyl-directed or NER repair were occurring for the xDNA segments, then one might expect equally efficient repair regardless of the length of the xDNA segments. However, our results show decreasing bypass efficiency as the segment is made longer, which is more consistent with polymerase activities rather than repair. Finally, in our segments, this repair would require the nicking to occur in the few nucleotides between the two closely-spaced segments of xDNA, which would seem unlikely. The more probable nicking outside the xDNA segments would remove one xDNA segment but still leave the other one in the opposite strand and require that it be read through by polymerases. Overall, the results establish that polymerasemediated reading of xDNA, rather than repair, is responsible for the GFP expression phenotype.
Figure 3.
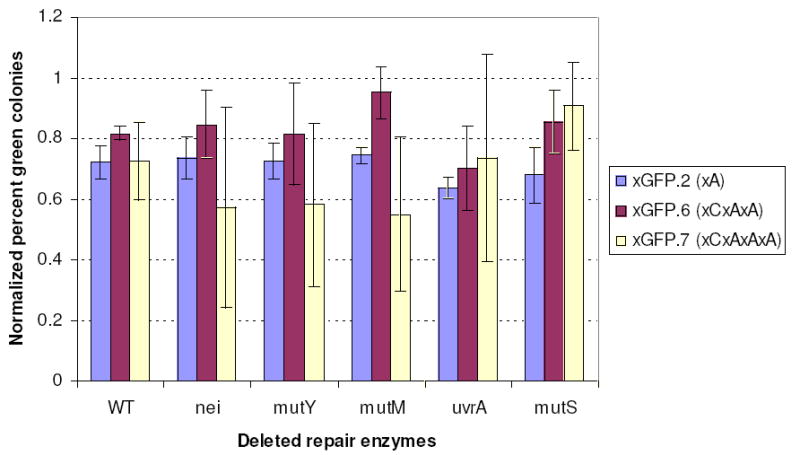
Effect of deleted repair functions on xDNA gene expression. Shown are percent green colonies for xDNA-containing inserts xGFP.2, xGFP.6 and xGFP.7 normalized to the unmodified control DNA (GFP.0) in five bacterial strains with known repair functions deleted.
The above data revealed that up to four consecutive xDNA pairs are read correctly by E. coli replication enzymes with surprisingly high fidelity. Previous experiments with bacterial polymerases have been carried out only in vitro. Data showed that DNA pol I (Klenow fragment) bypassed single xDNA pairs with low efficiency and fidelity, likely due to the enzyme’s steric preference for purine-pyrimidine pairs and its relative rigidity.1 Interestingly, experiments with Pol IV from Sulfolobus solfataricus showed considerably improved extension of xDNA pairs, demonstrating greater flexibility in this Y-family enzyme.9 This suggests that Y-family enzymes such as Pols II, IV, or V might be involved in the current cellular context. To begin to address this, we performed preliminary experiments with an additional set of bacterial strains having Y-family polymerases Pol II (polB), Pol IV (dinB), and Pol V (umuC) deleted (Table 2). Results measuring GFP expression from xDNA-containing genes showed (Fig. S1) that in each polymerase deletion mutant, most modified DNAs had little or no drop in expression. Interestingly, single xDNA substitutions showed some apparent reliance on these polymerases, while multiple substitutions did not, suggesting that the abrupt changes in size at xDNA/DNA junctions may benefit from such a repair enzyme for bypass. No one enzyme deletion yielded full loss of bypass activity, which suggests that either the different Y-family enzymes can substitute for one another in this bypass activity, and/or that replicative polymerases I and III also contribute to the replication of xDNA.
To further investigate the involvement of the Y-family polymerases in replication of the xDNA bases, the expression of GFP was examined in cells irradiated with UV light to induce the SOS response and upregulate the expression of Pol II, Pol IV, and Pol V. Induction of the SOS polymerases did not significantly affect the green colony counts and GFP expression (Table S8, Fig. S2), suggesting that, rather than repair polymerases alone, the replicative enzymes Pol I and Pol III play a significant role in the xDNA replication. Overall, the results are most consistent with a combination of enzymes processing the xDNA; for example, by replicative enzymes incorporating nucleotides opposite the large bases, followed by extension beyond them by a Y-family enzyme.9
An early study tested the ability of isolated xDNA bases in a single-stranded M13 vector to be bypassed in E. coli, and found some similarities and differences with the current experiments.1 In the earlier work, xA and xC were found to be replaced correctly by A and C after replication, consistent with the current results. However, in the previous work, xG and xT bases were incorrectly replaced (by A in both cases), indicating frequent mispairing with T, especially in the xG case. In contrast to this are our current findings that single xT and xG bases were correctly replicated in every clone sequenced. We hypothesize that the difference may be due either to the difference in sequence context, or to the differences in genome constructs. The current work was carried out within the context of a double-stranded plasmid, while the previous study utilized a single-stranded phage; as the machinery and mechanisms involved in replicating double stranded plasmid DNA versus a single stranded genome are not identical, different polymerase enzymes could possibly be involved in the initial nucleotide insertion step.
Taken together, the results show that native enzymes in E. coli are capable of accurately reading genetic information encoded in segments of xDNA, and expressing this information in a visible phenotype. This is the first example in which an intact biological system has been shown to be capable of reading all components of an unnatural genetic set. The only other examples of biological expression of modified DNA previously involved a modified backbone rather than bases,21,22 or a simple substitution on a natural base.2 The results are a precedent for the eventual use of unnatural genetic sets for expression of designed biological activities in vivo, a long-term goal of chemical synthetic biology.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM63587) for support.
Footnotes
Supporting Information Available. Experimental details, sequencing data, and characterization of synthetic DNAs/xDNAs.
References
- 1.Delaney JC, Gao J, Liu H, Shrivastav N, Essigmann JM, Kool ET. Angew Chem Int Ed. 2009;48:4524–4527. doi: 10.1002/anie.200805683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marlière P, Patrouix J, Döring V, Herdewijn P, Tricot S, Cruveiller S, Bouzon M, Mutzel R. Angew Chem Int Ed. 2011;50:7109–7114. doi: 10.1002/anie.201100535. [DOI] [PubMed] [Google Scholar]
- 3.Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc Natl Acad Sci USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H, Gao J, Lynch SR, Saito YD, Maynard L, Kool ET. Science. 2003;302:868–871. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]
- 5.Krueger A, Lu H, Lee A, Kool E. Acc Chem Res. 2007;40:141–50. doi: 10.1021/ar068200o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Gao J, Kool ET. J Am Chem Soc. 2005;127:1396–1402. doi: 10.1021/ja046305l. [DOI] [PubMed] [Google Scholar]
- 7.Gao J, Liu H, Kool ET. Angew Chem Int Ed. 2005;44:3118–3122. doi: 10.1002/anie.200500069. [DOI] [PubMed] [Google Scholar]
- 8.Lynch SR, Liu H, Gao J, Kool ET. J Am Chem Soc. 2006;128:14704–14711. doi: 10.1021/ja065606n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H, Krueger AT, Gao J, Liu H, Kool ET. Org Biomol Chem. 2010;8:2704–2710. doi: 10.1039/c002766a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henry AA, Romesberg FE. Curr Op Chem Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Krueger AT, Kool ET. Chem Biol. 2009;16:242–248. doi: 10.1016/j.chembiol.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirao I. Curr Op Chem Biol. 2006;10:622–627. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 13.Krueger AT, Kool ET. Curr Op Chem Biol. 2007;11:588–594. doi: 10.1016/j.cbpa.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hikishima S, Minakawa N, Kuramoto K, Fujisawa Y, Ogawa M, Matsuda A. Angew Chem Int Ed. 2005;44:596–598. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]
- 15.Battersby T, Albalos M, Friesenhahn M. Chem Biol. 2007;14:525. doi: 10.1016/j.chembiol.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Doi Y, Chiba J, Morikawa T, Inouye M. J Am Chem Soc. 2008;130:8762–8768. doi: 10.1021/ja801058h. [DOI] [PubMed] [Google Scholar]
- 17.Leonard NJ. Acc Chem Res. 1982;15:128–135. [Google Scholar]
- 18.Krueger AT, Kool ET. J Am Chem Soc. 2008;130:3989–3999. doi: 10.1021/ja0782347. [DOI] [PubMed] [Google Scholar]
- 19.Nohmi T. Ann Rev Microbiol. 2006;60:231–253. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 20.David SS, S’Shea VL, Kundu S. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pochet S, Kaminski PA, Van Aerschot A, Herdewijn P, Marliere P. C R Biool. 2003;326:1175–1184. doi: 10.1016/j.crvi.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 22.El-Sagheer AH, Sanzone AP, Gao R, Tassavoli A, Brown T. Proc Natl Acad Sci USA. 2011;108:11338–11343. doi: 10.1073/pnas.1101519108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.