

Abstract
Gastric cancer (GC) is the fourth most common cancer and the second most frequent cause of cancer-related deaths, accounting for 10.4% of cancer deaths worldwide. Despite the improvements, estimated cure rates for patients with advanced stages remain poor, and in the metastatic setting, chemotherapy is the mainstay of palliative therapy and results in objective response rates (ORRs) of only 20–40% and median overall survivals (OS) of 8–10 months. Therefore, many investigators believe that the potential for making significant progress lies in understanding and exploiting the molecular biology of these tumors to investigate new therapeutic strategies to combat GC, such as specific immunotherapy. In this paper, we analyze the different approaches used for immune-based (especially dendritic and T cells) therapies to gastric cancer treatment and discuss the results obtained in preclinical models as in clinical trials.
1. Introduction
Gastric cancer (GC) is the fourth most common cancer and the second most frequent cause of cancer-related deaths, accounting for 10.4% of cancer deaths worldwide [1], with high incidence in definite area (China, Eastern Europe, and Japan) [2]. Surgical tumor resection remains the primary curative treatment for GC but the overall 5-year survival rate remains poor, ranging between 20–25%. The addition of combined modality strategies (pre- or postoperative chemo/radiotherapy or perioperative chemotherapy) results in 5-year survival rates of only 30–35% [3–6]. preoperative chemo/radiotherapy produces pathologic complete responses (pCRs) in no more than 20–30% of patients [5], while preoperative chemotherapy alone is only rarely associated with pCRs [3, 7]. Worldwide, despite the improvements, estimated cure rates for patients with advanced stages remain poor and, in the metastatic setting, chemotherapy is the mainstay of palliative therapy and results in objective response rates (ORRs) of only 20–40% with a median overall survivals (OS) of 8–10 months [8].
Therefore, many investigators believe that the potential for making significant progress lies in exploiting the molecular biology of tumors to investigate new therapeutic strategies: such as epithelial growth factor receptor (EGFR) inhibitors [9], antiangiogenic agents [10], apoptosis promoters [11], and specific immunotherapy [12, 13].
Evidence from different investigations suggests a role for the immune system in the treatment of cancer: tumours are 100 times more likely to occur in people who take immunosuppressive medications than in people with normal immune function [14]. Patients who have undergone renal transplantation have an estimated 3 to 5 times higher overall incidence of malignancy in the long term than general population [15]. Conversely, heightened antitumor activity of the immune system has been suggested in many reports of spontaneous cancer regression [16]. A positive correlation between tumor infiltrating T lymphocytes and patients survival has been observed [17], and spontaneous tumor-specific T cell responses have been found in patients with different tumours [18]. Immune defence against cancer is mediated through antigen-specific and nonspecific immune mechanisms, that are provided by cells of the macrophage and NK cell lineage and/or by soluble factors such as inflammatory cytokines.
The functioning of the antigen-specific immune system is based on a division of tasks between T cells and B cells (Figure 1). Various reagents (vaccines, infusion of T cells or cytokines) can stimulate the immune system through one of several mechanisms: (1) stimulation of the antitumor response, either by increasing the number of effector cells or by producing soluble mediators (e.g., cytokines); (2) alteration of tumor cells to increase their immunogenicity and susceptibility to immunological defences; or (3) improvement of susceptibility to cytotoxic drugs or radiotherapy, such as stimulating bone marrow function with granulocyte colony stimulating factor (G-CSF). However, the cancer also developed a number of different strategies to escape immune surveillance such as loss of tumor antigen expression, MHC downregulation, expression of Fas-L that can induce apoptosis in activated T cells, secretion of cytokines such as IL-10 (Interleukin-10) and/or TGF-ß (Tumor grow factor-β) and generation of regulatory T cells (Treg) [19, 20]. The requirement for an immune-based strategy against cancer is the induction of an effective tumor-specific immunity in order to break immunological tolerance to the tumor and generate antitumor immunity. To achieve this goal, several strategies both in preclinical models and in clinical trials are currently being investigated.
Figure 1.
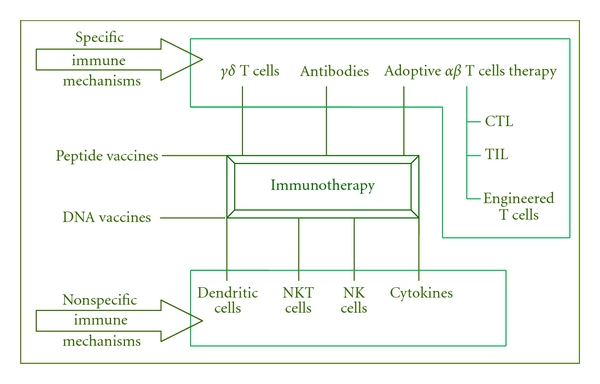
Different immunotherapeutic approaches.
In this paper, we examine the currently used different approaches for immune-based gastric cancer therapies and the results obtained in some clinical trials. These trials have had variable eligibility requirements and have grouped together combinations of patients with esophageal, gastro-esophageal (GE) junction, and/or gastric primary tumors. Histologies on these trials are squamous cell carcinomas (SCC), which mostly occur in the proximal third of the esophagus and adenocarcinomas, which arise in the distal two-thirds of the esophagus, GE junction, and stomach.
2. Innate and Adoptive Immunity Cells as Antitumoral Therapy in Gastric Cancer
2.1. Dendritic Cell-Based Vaccination
Dendritic cells (DCs) are the most powerful professional antigen-presenting cells (APC) at the interface between innate and adaptive immunity with the ability to activate many effector cells (NK, T, B, and NKT cells).
Studies in mice shown that ex vivo generated DCs can induce antigen-specific T cell immunity and are superior to other types of tools (e.g., vaccines) [21]. These studies are the basis to design DC-based anticancer vaccines in clinical trials (Figure 2).
Figure 2.
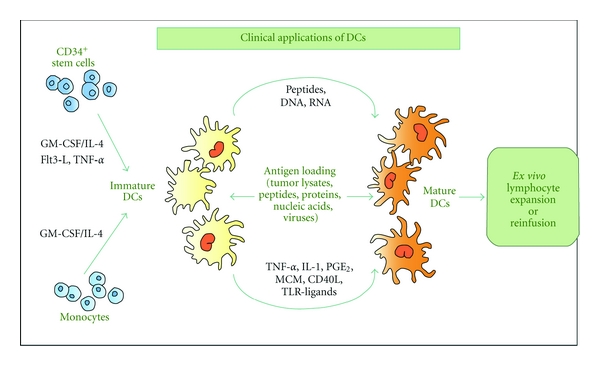
Circulating monocytes and hematopoietic (CD34+) stem cells are practicable sources of DCs for clinical applications. Following incubation with growth factors to induce cell differentiation and to increase immunogenicity, DCs are incubated with the antigen (peptides, proteins, nucleic acids, viral particles, or tumor lysates). Antigen-loaded DCs can then be reinjected into the patient or used for ex vivo expansion of antitumor lymphocytes which will then be infused.
Antigen presentation by dendritic cells is a central element in the induction of the cellular immune responses necessary for tumor immunotherapy [22, 23]. Since DCs have this intrinsic ability to prime immune responses, there has been a great deal of interest in the use of these cells for cancer therapy [24].
More than 150 DC-based clinical studies for the treatment of solid or haematological malignancies have been reported so far (http://www.mmri.mater.org.au/).
Melanoma is the most frequent type of cancer treated with DC-based immunizations, with nearly 40 published clinical studies, followed by 20 studies on prostate cancer, 16 of renal cell carcinoma, 12 of breast cancer, and 9 of multiple myeloma, leukemia, colorectal cancer, and gliomas.
About the gastric cancer, it was demonstrated that patients with many DCs infiltration had lower lymph node metastases and lymphatic invasion than patients with fewer DCs infiltration. The 5-year survival rates of patients with many DCs infiltration were 78%, better than that of patients with fewer DCs infiltration [25]. According to the function of DCs described above, we know that DCs are related to clinical stage, invasion, metastasis, and GC prognosis [26]. Therefore, it will be feasible that DC-based tumor vaccines become a new effective immune-adjuvant GC therapy, which can decrease the incidence and recurrence rates after operation for gastric cancer.
Galletto [27] gained DCs derived from adherent blood mononuclear cells of five GC patients, which were exposed to apoptotic autologous tumor (AAT) cells and cultured for 24 h with monocyte-conditioned medium to achieve full DCs maturation. Tumor-specific response was evaluated as single-cell cytokine release in an enzyme-linked immunospot (ELISPOT) and cytotoxicity in a cold target inhibition 51Cr-release assay. Data has shown that T cell memory against gastric cancer antigens could be triggered by tumor-loaded autologous DCs.
In addition, Kono [28] reported that tumor vaccination therapy with DCs pulsed with HER-2/neuro-peptides may be a potential candidate for the novel treatment of patients with gastric cancer. Nine GC patients with recurrent or unresectable cancer are enrolled in the clinical trial. The tumors were proved to overexpress HER-2/neu by immune-histochemistry. Vaccinations with DCs pulsed with HER-2 (p369) peptide were performed at 2-week intervals. DCs administered intradermally in a single site at a supra-clavicular location. In 3 of 9 patients, the tumor markers (CEA or CA19-9) were decreased after vaccination. Two had a tumor regression of more than 50%, and two presented a mixed response. The vaccines can be applied safely without significant side effects.
Due to a short lifespan of DCs, however, clinical application of current DC vaccines has been limited. One of factors threatening DC survival is antigen-specific CD8+ cytotoxic T lymphocytes (CTLs) that acquire cytolytic activities after activation by DCs presenting the relevant MHC-I: antigen peptide complexes [29, 30].
The early removal of DCs at each of priming and boosting stages would seriously limit the capacity of DCs to prime and expand CTL immunity, respectively [31–33].
In the past, activation of AKT/protein kinase B (PKB), a major effector of phosphatidylinositol 3-kinase (PI3K), has been reported as a critical factor in both activation and survival of DCs [34, 35]. Most recently, Kim [36] improved the potency of a DC vaccine with a small interfering RNA (siRNA) targeting phosphatase and tensin homologue (PTEN), which is known to be a central negative regulator of the PI3K/AKT signal transduction cascade [37]. Downregulation of PTEN in DCs resulted in AKT-dependent maturation, which in turn caused a significant upregulation of surface expression in costimulatory molecules and the chemokine receptor, CCR7, leading to an increase of in vitro T cell activation activity and in vivo migration to a draining lymph node, respectively. Moreover, these PTEN siRNA-transfected DCs (DC/siPTEN) acquired an increased survival from the apoptotic death caused by GM-CSF deprivation or antigen-specific CD8+ T cell killing. Most importantly, DC/siPTEN generated more tumor antigen-specific CD8+ T cells and stronger antitumor effects in vaccinated mice than did control DCs (DC/siGFP).
In conclusion, these results indicate that manipulation of the PI3K/AKT pathway via siRNA system could improve the efficacy of a DC-based tumor vaccine, for example, in gastric cancer therapy. Immunosuppressive cytokines such as IL-10 produced by DCs (autocrine) and other regulatory immune cells (paracrine) downregulate functional profiles of DCs through specific cell surface receptors such as IL-10R. In recent times, the same group demonstrated that by blocking the immunosuppressive axis with small interfering RNA targeting IL-10 receptor, it is possible to enhance dendritic cell-based vaccine potency providing the groundwork for future clinical translation of siRNA-mediated to enhance DC-based vaccine immunotherapy [38].
Most promising are also the data obtained by He and colleagues [39] about the possibility of enhancing antitumor immunity in vitro using granulocyte-macrophage colony stimulating factor (GM-CSF) gene-modified DCs. They demonstrated that after GM-CSF gene modification, DCs can produce high level of GM-CSF, which tend to be more maturated, and the capacity of activating the proliferation of allogenic T lymphocytes is enhanced greatly. GM-CSF gene modified DCs can induce specific CTL to target tumor cells in vitro.
2.2. Therapies with Natural Killer (NK) Cells
NK cells prevent the dissemination of metastatic tumors in mice and in humans [40], and tumor infiltration by NK cells may be associated with a better prognosis in squamous cell lung, gastric and colorectal carcinoma [41]. The important role of NK cells in host defence against cancer provides opportunities to contrast the disease by manipulating the NK cell “arms”; but, unfortunately, the achievement of NK cell-based therapeutic breakthroughs has been hampered by several factors: (a) poor and late learning of the mechanisms involved in activation of NK functional activity; (b) incomplete definition of the distinct NK cell subsets endowed with specific functions; (c) different mouse and human NK cell biology; (d) the small number of NK cells in peripheral blood; (e) the difficulties associated with large-scale production of cytolytic NK cells in compliance with good manufacturing practices (GMP); (f) the need to activate the NK cells in order to induce NK-cells-mediated killing; (g) the constraints imposed by autologous inhibitory receptor-ligand interactions [42].
In 1982, Grimm demonstrated that IL-2 generated lymphokine-activated killer (LAK) cells able to kill human tumor cell lines and that this phenomenon was mainly mediated by NK cells [43]. The first clinical study combining LAK cells and IL-2 was initiated by SA Rosenberg in 25 patients with advanced cancer. By the end of the 1990s, all published phase II and phase III randomized trials showed a clinical response rate of 15–20% [44]. High doses of recombinant IL-2 were administered systemically to ensure survival and sustained activation of infused LAK cells, but the high incidence of severe adverse effects precluded further development of these adoptive transfer-based therapies [45]. In addition, since the IL-2 era, different clinical trials have assessed the effects of systemic administration of other cytokines involved in NK cell differentiation and activation (Table 1).
Table 1.
Clinical trials with various strategies to modulate NK cell function.
| Treatment | Type of cancer | Trial phase (patient number) | NK cell functions | Clinical effects | Comment | References |
|---|---|---|---|---|---|---|
| IL-2 | AML | 3 (n = 320) | ND | 40% 3-years LFS (26% control) |
[46] | |
| IL-2 | M | 2, 3 (n = 270) | ND | 16% OR | High Toxicity | [47] |
| IL-2 | RCC | 1, 2, 3 (n = 1.712) | ND | 15% OR | [48] | |
| IL-12 | RCC M |
1 (n = 14) | ND | 1 PR, 3 SD | ↑ IFN-γ serum concentration correlate with clinical response | [49] |
| IL-12 | RCC M |
1 (n = 28) | ND | 1 PR | ↑ IFN-γ and CXCL10 serum concentration | [50] |
| IL-18 | RCC, M, Hd | 1 (n = 28) | ↑ FasL on NK | 2 PR | ↑ IFN-γ, GM-CSF, IL-18 binding protein in serum | [51] |
| IL-21 | M | 1 (n = 29) | ↑ Cytotoxicity | 1 CR | ↑ Perforn, granzyme B mRNA inPBMC | [52] |
| IFN-α | CML | 2 (n = 26) | ↑ Cytotoxicity and IFN-γ | 60–80% OR | ↑ NK cell activity correlated with remission | [53] |
| Flt3L | BC, Hd | 1 (n = 8) | ND | ↑ Immature circulating DC | [54] | |
| Rituximab | B-cell NHL | 1 (n = 43) | ↑ ADCC | 53% OR | ↑ NK cell expansion correlated with remission | [55] |
| Rituximab | Indolent NHL | 2 (n = 57) | ↑ ADCC | 8.8% OR | 28% of FcγR allotypes OR in resistant FcR allotypes | [56] |
| Rituximab | B-cell NHL | 1 (n = 10) | ↑ ADCC | 1 PR, 4 SD | [57] | |
| Trastuzumab | BC | 1 (n = 15) | ↑ ADCC ↑ IFN-γ |
1 CR, 2 SD | ↑ IFN-γ only in responding patients | [58] |
| Bisp-antibody CD16/CD30 | Hd | 1, 2 (n = 15) | ↑ Cytotoxicity | 25% OR | [59] | |
| Bisp-antibody CD16/CD30 | Hd | 1, 2 (n = 15) | ↑ ADCC | 29% OR | [60] |
AML/CML: Acute/Chronic myeloid leukemia; BC: Breast Cancer; RCC: Renal Cell Carcinoma; M: Melanoma; Hd: Hodgkin's Disease; LFS: Leukemia-Free Survival; NHL: Non-Hodgkin's Lymphoma; ND: Not Determined; OR: Overall Response; PR: Partial Response; CR: Complete Response; SD: Stable Disease; Bisp: Bispecific.
In regard to the gastric cancer, most recently Voskens confirmed that large quantities of cytotoxic NK cells can be expanded from PBMC in the presence of K562 cells (expressing membrane-bound IL-15 and 4-1BBLigand) from healthy individuals and interestingly from patients with different solid tumors [61]. Ex vivo expansion tended to alter the balance of NK cell receptor expression towards those that activate and mediate cytotoxicity. This activity resulted in cytotoxicity against various allogeneic tumor targets and more importantly, against autologous-derived gastric tumor targets. Blocking studies identified multiple activating receptor-ligand interactions that would be predicted to mediate NK cell cytotoxicity. Moreover, these activating receptor-ligand interactions were operative in ADCC (antibody-dependent cellular cytotoxicity) as in an autologous as in allogeneic setting.
Importantly, as a mean for prospect clinical translation, GMP compliant cytolytic NK cells could efficiently be expanded from lymphocyte-enriched cell fractions obtained from PBMC by counter current elutriation.
This study unequivocally demonstrated that human NK cells acquire cytolytic activity against autologous gastric tumor cells after ex vivo expansion and suggested a therapeutic potential for autologous expanded NK cells, both directly and in combination with monoclonal antibodies in future cell-based immunotherapy.
2.3. T-Cell-Based Antigastric Cancer Treatments
Evidence that T cells can help to control tumor growth has been provided by the analysis of tumor prevalence in immunodeficient mice and humans [14]. Furthermore, recent evidence indicates that tumor infiltration by human T cells is a powerful predictive biomarker of survival for ovarian and colorectal cancers [82].
Therapeutic cancer vaccines are entering the clinical medicine, but despite more than 60 years of research on this field, there are currently no FDA-approved adoptive T cell therapy protocols for cancer. However, the recent explosion of knowledge in the fields of T cell and cancer biology has enabled new approaches that might bring adoptive T cell transfer into daily clinical practice. Recent knowledge acquired from adoptive transfer in lymphodepleted hosts [83], the ability to overcome barriers raised by Tregs [84], and the use of improved culture systems [36] have not been tested yet in randomized clinical trials.
There are different types of T cell-based anticancer therapy approaches, using (a) cytotoxic T lymphocytes (CTL), (b) tumor-infiltrating lymphocytes (TIL), or (c) engineered T cells.
Improved CTLs cell culture technology has permitted the first clinical tests for adoptive transfer of CTLs, and this technique [85, 86] seems to result in substantial activity in patients with melanoma; CTLs were used to treat patients with metastatic melanoma, and 8 out of 20 patients had antitumor immune responses [87]. These results were confirmed in an independent trial in which engraftment of the CTLs, as measured by an elevated frequency of circulating T cells able to bind tetramers loaded with MART-1 peptides, was detectable up to two weeks after T cell transfer in all patients [88].
Recently, Kim [89] have shown the antitumor activity of ex vivo expanded T cells against the human gastric cancer. For this purpose, human peripheral blood mononuclear cells were cultured with medium (+ IL-2) in anti-CD3 antibody-coated flasks for 5 days, followed by incubation in medium with IL-2 for 9 days. The resulting populations were mostly CD3+ T cells (97%) and comprised 1% CD3− CD56+, 36% CD3+ CD56+, 11% CD4+, and 80% CD8+. This heterogeneous cell population was also called cytokine-induced killer (CIK) cells. CIK cells strongly produced IFN-γ, moderately TNF-α, but not IL-2 and IL-4. At an effector-target cell ratio of 30 : 1, CIK cells destroyed 58% of MKN74 human gastric cancer cells, as measured by the 51Cr-release assay. In addition, CIK cells at doses of 3 and 10 million cells per mouse inhibited 58% and 78% of MKN74 tumor growth in nude mouse xenograft assays, respectively. This study suggests that CIK cells may be used as an adoptive immunotherapy for gastric cancer patients.
The adoptive immunotherapy of gastric cancer with CIK cells has been also reported in preclinical and clinical studies [90, 91]. MHC-I restricted CTLs from GC patients recognize tumor-associated antigen and react specifically against self-tumor cells [91, 92]. One tumor-specific antigen, MG7-antigen, shows great potential for predicting early cancer as well as for inducing immune responses to gastric cancer [93, 94]. Using HLA-A-matched allogeneic GC cells to induce tumor-specific CTLs appears to be an alternative immunotherapy option for gastric cancer [95].
Also, CIK cells in combination with chemotherapy have shown benefits for patients with advanced gastric cancers [96, 97]. The serum levels of the tumor markers were significantly decreased, the host immune function was increased, and the short-term curative effect, as well as the quality of life, was improved in patients treated by chemotherapy plus CIK cells compared to those in patients treated by chemotherapy alone. CIK cells killed MGC-803 GC cells by inducing apoptosis in the early stage and by inducing necrosis in the late stage through downregulation of p53, c-myc, and bcl-2, and upregulation of bax [98].
In summary, despite the introduction of immune cell-based immunotherapy, the paucity of preclinical and clinical studies limited the broad application of immunotherapy for the treatment of patients with gastric cancer. Here, preclinical evidence proved that CIK cell immunotherapy can be used in GC patients.
Adoptive transfer therapy with TILs requires the isolation of T cells from neoplastic biopsies or surgical tissue and the selection of tumor-specific T cells ex vivo (Figure 3). The adoptive transfer of TILs has been promising in preclinical models [99], but clinical experiences were almost uniformly disappointing [100, 101].
Figure 3.
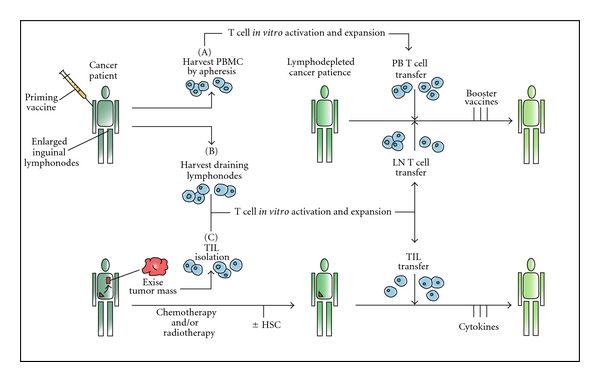
Different schemes for adoptive transfer of autologous, vaccine-primed, in-vitro-expanded T cells. Patients are primed with tumor vaccine followed by lymphocyte harvest. Autologous T cells are harvested from peripheral blood (I) or draining lymph nodes (II), undergo polyclonal in vitro activation and expansion, and are reinfused after lymphodepleting chemotherapy. Antigen-specific immune function is measured subsequent to the administration of booster vaccines. (III) TILs can be isolated from resected surgical samples and expanded in vitro for adoptive transfer after lymphodepleting chemotherapy. Most adoptive transfer therapy approaches using TILs have involved the use of IL-2 infusion following T cell transfer in order to select tumor-specific T cells.
Technical difficulties in producing tumor-specific T cells currently represent a barrier to randomized clinical trials. Only 30%–40% of the biopsies yield satisfactory T cell populations, and the whole process requires about 6 weeks before the T cells would be ready for infusion [102]. Furthermore, nearly all clinical experiences with TILs have been done in patients with melanoma, because of the easy surgical availability of the tumor tissue. However, should technical limitations of current tissue culture approaches be overcome, recent studies indicate that the presence of TILs positively correlate with patients survival in ovarian and colorectal cancer [103, 104], thus prompting the use of this protocol for other commonly encountered epithelial neoplasias.
Recently, Amedei [13] analyzed the functional properties of the T cell response to different GC-associated antigen peptides in patients with gastric adenocarcinoma. To this purpose, they have cloned and characterized TILs of gastric cancer. A T cell response specific to different tested peptides was documented in 17 out of 20 patients, selected for their HLA-A02 and/or -A24 alleles. Most of the cancer peptide-specific TILs expressed a T helper 1 (Th1)/T cytotoxic 1 (Tc1) cytokine profile and cytotoxic activity against target cells. Moreover, the effector functions of cancer peptide-specific T cells obtained from the peripheral blood of the same patients were also studied and the majority of peripheral blood peptide-specific T cells also expressed the Th1/Tc1 functional profile.
In conclusion, in most of patients with gastric adenocarcinoma, a specific type-1 T cell response to GC antigens was detectable and would have the potential of hampering tumor cell growth. However, in order to get tumor cell killing in vivo, the activity and the number of cancer peptide-specific Th1/Tc1 cells probably need to be enhanced by vaccination with the appropriate cancer antigenic peptides or by injection of the autologous tumor peptide-specific T cells expanded in vitro. These studies have laid the groundwork for a possible vaccination of GC patients with specific peptides of tumor-associated antigens able to raise an effective immune response to gastric cancer.
Genetic modification of T cells to improve antitumor effects is an attractive strategy in many settings [105]; although there is little clinical experience with engineered T cells for cancer therapy, it is notable that clinical trials to date using cells engineered to express suicide molecules indicated that the approach is safe.
Molmed and colleagues infused allogeneic-engineered donor lymphocytes (HSV-TK lymphocytes) [106] into 8 patients with hematologic malignancies who had suffered complications after receiving allogeneic bone marrow transplants [107]. The lymphocytes survived up to a year, and complete or partial tumor remission was achieved in five patients, but tumor regression coincided with the onset of GVHD.
Recently, investigators have developed suicide systems consisting of fusion proteins containing a human FAS or caspase death domain and a modified FKBP [108, 109]. These approaches have the advantage that the suicide switches are nonimmunogenic because they are based on endogenous proteins. T cells expressing these modified chimeric proteins are induced to undergo apoptosis when exposed to a drug that dimerizes the modified FKBP.
A principal limitation of adoptive T cell therapy in some tumors is their poor antigenicity; therefore, neither T cells with high avidity for tumor-specific antigens, nor T cells with the desired specificity remain in the patient following chemotherapy. One strategy to overcome this limitation is now being tested in clinical trials; this approach has been to endow T cells with chimeric receptors that have antibody-based external receptor structures and cytosolic domains that encode signal transduction modules of the T cell receptor [110].
Another purpose of engineered T cells is to enhance survival of CTLs, because they have short-term persistence in the host without antigen-specific T helper cells and/or cytokine infusions. The Greenberg's group had transduced human CTLs with chimeric GM-CSF–IL-2 receptors that deliver an IL-2 signal when cells bind GM-CSF [111]. Stimulation of the CTLs with antigen caused GM-CSF secretion and resulted in an autocrine growth loop such as that of proliferating CTL clones in the absence of exogenous cytokines. A related strategy to rejuvenate T cell function is to engineer T cells to ectopically express CD28 [112] or the catalytic subunit of telomerase [113].
Both these strategies could be used in the treatment of gastric cancer, but currently no clinical trials based on one of the two principles have been registered.
3. Monoclonal Antibodies (moAbs) Specific to Molecular Target of Gastric Cancer
3.1. Epidermal Growth Factor Receptor
The most extensively explored approach is the inhibition of EGFR. EGFR or ERBB1 is a member of the ERBB transmembrane growth factor receptor family, which initiates signal transduction by activation of a receptor-associated tyrosine kinase (TK). The ligands of the EGFR are epidermal growth factor (EGF) and TGF-β. Binding of a ligand to the EGFR causes the activation of the TK and of other effector signals, that are regulators of intracellular and intercellular processes (apoptosis, proliferation, angiogenesis, and metastasis) [114].
EGFR is constitutively expressed in a number of tissues, including the skin, gut, and kidney. This normal EGFR expression can explain acneform skin rash and diarrhea (and magnesium-wasting syndrome) which are the major toxicities associated with anti-EGFR therapy [115, 116].
In esophagogastric cancers (EGCs), EGFR overexpression occurs in 30–90% of tumors and correlate with increased invasion and a worse prognosis [62–64, 117]. In general, EGFR overexpression is more common with SCC than adenocarcinoma histology. In addition, it is now well established that the mutational status of K-ras, an oncogene downstream of the EGFR which is involved in an intricate array of signal transduction pathways, dictates responsiveness to anti-EGFR therapies in colorectal cancer (CRC) and patients whose tumors were found to have K-ras mutations derived no benefit from anti-EGFR antibodies treatment [65–67].
In EGCs, relatively little is known about the incidence of mutated K-ras status, much less its predictive value for anti-EGFR therapy. In recent studies, 0 of 3824 and 2 of 23 patients (8.7%) [118] were found to have mutated K-ras.
Current anti-EGFR therapies that have been evaluated in EGCs include oral TK inhibitors (TKIs: erlotinib, gefitinib) and monoclonal antibodies (moAbs: cetuximab, panitumumab, and matuzumab).
Cetuximab (C225, Erbitux, Imclone Systems) is a partially humanized murine IgG1 moAb that binds to the EGFR, which blocks binding of ligand to the EGFR and subsequent activation of the EGFR TK [119], as well it stimulates EGFR internalization [68] and can initiate immune-mediated mechanisms of cytotoxicity [antibody- or complement-dependent and complement-dependent cell-mediated cytotoxicity [69, 70]].
Cetuximab has been approved by the FDA (US Food and Drug Administration) for the treatment of irinotecan-refractory CRC, based on a phase III trial of cetuximab/irinotecan that demonstrated a superior ORR and time-to-progression (TTP) for the combination over irinotecan alone [71].
Recently, cetuximab has also been approved by the FDA in combination with concurrent radiotherapy for the treatment of advanced head and neck squamous cell cancers (HNSCCs) [72, 120]. Cetuximab has undergone more extensive evaluation in EGCs than any other targeted agent. In the locally advanced setting, six trials have been reported. Four of these trials have combined cetuximab with radiation and various chemotherapy regimens as preoperative therapy prior to planned surgical resection [73–76]; a fifth trial evaluated cetuximab alone with preoperative radiotherapy [77], while the final study in gastric and GE junction adenocarcinoma involved preoperative therapy with cetuximab/cisplatin/irinotecan, followed by surgery and adjuvant chemo/radiotherapy with cetuximab/5-FU/leucovorin [78].
With the exception of the trial to be described further below, all of these trials have been reported only in abstract form. Where reported, pCR rates have ranged from 13% to 40% for preoperative chemo/radiotherapy trials. Toxicities have generally been consistent with other combined modality trials, except for the phase II evaluation of cetuximab with cisplatin/irinotecan/radiation [79, 80, 121]
In a phase II trial, cetuximab was combined with carboplatin/paclitaxel and concurrent radiation [74]. The trial reported a clinical CR rate of 70% and a pCR rate of 27% in the 49 patients who underwent surgery. Survival data were not reported. Toxicities on this trial included grade 3/4 anaphylaxis in 5% of patients, grade 3/4 esophagitis in 16% of patients, and grade 3 skin toxicity in 25%. Final publication of these studies and careful analysis of toxicities and survival data are eagerly awaited.
In the first-line metastatic setting, seven trials have evaluated combinations of cetuximab with different chemotherapy regimens [81, 122–127] and all have reported encouraging ORRs of 40–69% and median OS of 9.5–17 months. Toxicities have generally been consistent with the additive toxicities of cetuximab and the respective chemotherapy regimens. Grade 3/4 toxicities included neutropenia in 6–46% of patients, diarrhea in 4–33% of patients, skin toxicity in 6–24% of patients, and anaphylaxis to cetuximab in <5% of patients.
The relationship between EGFR positivity and response to cetuximab on these trials remains unclear, while the trial with cetuximab and FOLFIRI [121] did not note any relationship between absolute EGFR positivity and benefit from cetuximab. In contrast, the trial of cetuximab and FOLFOX [67] noted that EGFR-positive tumors were associated with an ORR of 100%.
In addition, other four trials have evaluated cetuximab in the second-line setting and beyond. Preliminary data from these trials suggest that this is not a promising approach. The Southwest Oncology Group (SWOG) performed an evaluation of cetuximab as a second-line therapy in advanced esophageal and GE junction adenocarcinoma. Preliminary results for 55 evaluable patients revealed only 1 confirmed PR (partial response) and 2 unconfirmed PRs; median PFS was only 1.8 months [128]. Similarly disappointing results were reported in two studies that attempted to reverse chemotherapy resistance by incorporating cetuximab therapy [129, 130].
Finally, a study evaluated second-line cetuximab/irinotecan in patients with progression on platinum-based chemotherapy and reported an ORR of 6% and median PFS of 3.2 months [131].
Matuzumab (EMD72000, Merck) is another humanized IgG1 moAb against EGFR. In a phase I evaluation, 1 of 2 patients with esophageal SCC had a durable six-month PR [132].
Another phase I evaluation combined matuzumab with the ECX regimen (epirubicin/cisplatin/capecitabine) as a first-line therapy for patients with EGFR-positive gastric and GE junction adenocarcinoma [133]. Of 45 patients screened, 21 (47%) were found to have EGFR-positive tumors. The ORR in 20 evaluable patients was 65% and the median TTP was 5.2 months. Therapy was well tolerated and major toxicity on this trial was grade 3 fatigue.
Lastly, panitumumab (ABX-EGF, Vectibix, Amgen) is a fully humanized IgG2 moAb against EGFR that has been approved by the FDA for the treatment of chemorefractory EGFR-positive CRC, based on a phase III trial that demonstrated improvement in ORR and PFS over best supportive care [134].
A phase I evaluation of panitumumab in refractory solid tumors demonstrated SD for seven months in 1 of 3 patients with esophageal cancer [135].
Based on the promising studies described above, there are many ongoing confirmatory cooperative group studies. For example, based on the results of the phase II study by Safran [74], the phase III Radiation Therapy Oncology Group 04036 trial is currently comparing weekly cisplatin/paclitaxel/radiation ± cetuximab as definitive therapy in locally advanced esophageal cancer.
The REAL3 trial in the United Kingdom is randomizing patients with advanced EGCs to the EOX (epirubicin/oxaliplatin/capecitabine) regimen ± panitumumab, while MATRIX EG, an European phase II randomized trial of the ECX regimen ± matuzumab, has completed accrual.
Table 2 summarizes the results of trials involving anti-EGFR antibody therapies analyzed.
Table 2.
Major trials using anti-EGFR monoclonal antibodies.
| Treatment | Enrolled patients | Disease stage | Response rate | Time to progression | Overall survival | References |
|---|---|---|---|---|---|---|
| Cetuximab + Cis/CPT/RT | 5 EAC, 12 GEJAC | Locally advanced | 13% (2/15) | NS | NS | [62] |
| Cetuximab + Carbo/paclitaxel/RT | 45 EAC, 3 GAC, 12 SCC | Locally advanced | 27% (13/49) | NS | NS | [63] |
| Cetuximab + Cis/docetaxel/RT | 15 EAC/GEJAC, 13 SCC | Locally advanced | 32% (9/28) | NS | NS | [64] |
| Cetuximab + FOLFOX/RT | 9 AC, 18 SCC | Locally advanced | 40% (4/10) | NS | NS | [65] |
| Cetuximab/RT | 20 EAC 11 GEJAC, 9 SCC | Locally advanced | 36% (13/36) | NS | NS | [66] |
| Cetuximab/Cis/CPT + surgery + Cetuximab/5-FU/LV/RT | 20 GAC/GEJAC | Locally advanced | 0% (of 18) | NS | NS | [67] |
| Cetuximab + FOLFIRI | 4 GEJAC, 34 GAC | Metastatic (EGFR +ve) |
44% (of 34) | 8 months | 16 months | [68] |
| Cetuximab + FUFIRI | 15 GEJAC, 34 GAC | Metastatic | 42% (of 48) | 8.5 months | 16.6 months | [69] |
| Cetuximab + FUFOX | 25 GEJAC, 27 GAC | Metastatic | 65% (of 46) | 7.6 months | 9.5 months | [70] |
| Cetuximab + 5-FU/Cis verses 5-FU/Cis | 32 SCC 30 SCC |
Metastatic | 19% 13% |
5.7 months 3.6 months |
9.5 months 5.5 months |
[71] |
| Cetuximab + CI 5-FU/LV/Cis | 35 GAC | Metastatic | 69% | 11 months | 14.5 months | [72] |
| Cetuximab + Cis/docetaxel | 8 GEJAC, 40 GAC | Unresectable/meta static |
41% (of 42) | NS | NS | [73] |
| Cetuximab + oxaliplatin/CPT | 51 GAC | Metastatic | 63% (of 35) | 6.2 months | 9.5 months | [74] |
| Cetuximab | 55 EAC/GEJAC | Metastatic (2nd-line) |
2% | 1.8 months | 4 months | [75] |
| Cetuximab + Cis/CPT | 1 EAC, 7 GEJAC, 1 SCC | Metastatic (PD on Cis/CPT) |
11% | 1.3 months | NS | [76] |
| Cetuximab + docetaxel | 38 NS | Metastatic (PD on docetaxel) |
6% (of 35) | 2.1 months | 5.2 months | [77] |
| Cetuximab + CPT | 19 EAC/GEJAC, 8 GAC, 4 SCC | Metastatic (PD on platinum) |
6% | 3.2 months | NS | [78] |
| Matuzumab | 2 SCC | Metastatic (phase I) | 1 of 2 patients with 6-month partial response | [79] | ||
| Matuzumab + ECX | 5 EAC, 7 GEJAC, 9 GAC | Metastatic (phase I) | 65% (of 20) | 5.2 months | NS | [80] |
| Panitumumab | 3 NS | Metastatic (phase I) | 1 of 3 patients with 7-month stable disease | [81] | ||
5-FU: 5-fluorouracil; EAC: esophageal adenocarcinoma; Carbo: carboplatin; CI: continuous infusion; Cis: cisplatin; CPT: irinotecan; ECX: epirubicin/cisplatin/capecitabine; FOLFIRI: biweekly bolus 5-FU/leucovorin, irinotecan, infusional 5-FU; FUFIRI: weekly irinotecan/leucovorin/infusional; 5-FUFUFOX: weekly oxaliplatin/leucovorin/infusional 5-FU; LV: leucovorin; GAC: gastric adenocarcinoma; GEJAC: gastroesophageal junction adenocarcinoma; N/A: not applicable; NS: not stated; ORR: objective response rate; OS: overall survival; pCR: pathologic complete response; PD: progressive disease; RT: radiation therapy; SCC: squamous cell carcinoma; TTP: time-to-progression.
3.2. HER2/ErbB2
Her-2/neu (ERBB2) is another member of the ERBB TK receptor family. Peptide ligand binding to the extracellular domains of these receptors leads to homo- and heterodimerization of the receptors and subsequent tyrosine autophosphorylation. At least nine different homo- and heterodimers of the ERBB proteins exist, with their formation displaying a distinct hierarchy. In this network, Her-2/neu plays a major coordinating role since each receptor with a specific ligand appears to prefer Her-2/neu as its heterodimeric partner. This preference is further biased by overexpression of Her-2/neu, as seen in many types of human cancer cells, particularly breast cancer [136].
In EGCs, Her-2/neu overexpression has been variably demonstrated in esophageal SCC (mean 23%, range 0–52%) and GE junction adenocarcinoma (mean 22%, range 0–43%) [137, 138]. In esophageal SCC, Her-2/neu overexpression has been correlated with extramural invasion and poor response to neo-adjuvant chemotherapy [139]. In gastric and GE junction adenocarcinoma, some studies have demonstrated a correlation between Her-2 amplification with increasing of invasion, distant organ metastasis, and overall poor survival [140]
The anti-Her2/neu moAb therapy that have been evaluated in EGCs is Trastuzumab [Herceptin, Genentech], a humanized IgG1 moAb against the Her-2/neu receptor.
Trastuzumab can exert its effects by several mechanisms, including preventing Her-2 receptor dimerization, increasing destruction of the receptor, inhibiting shedding of the extracellular domain, and inducing antibody-dependent cytotoxicity [141]. Based on several trial studies, it has been approved by the FDA for use in combination with chemotherapy as adjuvant therapy for Her-2/neu- and node-positive breast cancer [142–146].
Trastuzumab is generally very well tolerated, with the exception of rare cardiac dysfunction, which is postulated to occur because Her-2 signaling is important for cardiac development [147, 148].
In the locally advanced setting, the use of trastuzumab produced encouraging results. In a phase I/II trial, increasing doses of trastuzumab were combined with cisplatin/paclitaxel and radiation for patients whose tumors were 2 + or 3 + Her-2/ [149].
Fourteen patients [74%] had either 3 + overexpression; of these, 8 (57%) achieved a clinical CR, 6 subsequently underwent surgery, and 3 were found to have achieved a pCR.
Therefore, the pCR rate was 16% for all patients enrolled and was 43% for patients who went to surgery.
The median OS was 24 months. Toxicities were comparable to other preoperative chemo/radiation trials and there was no cardiac toxicity.
In the metastatic setting, results of the phase III ToGA trial of 5-FU or capecitabine and cisplatin ± trastuzumab were recently reported [150]. This study enrolled patients with gastric or GE junction adenocarcinoma that were Her-2/neu-positive, and of tumors from 3807 patients, 22.1% were found to be Her-2/neu-positive.
Five hundred and ninety-four patients were randomized. There was a statistically significant increase in ORR (47.3% versus 34.5%), median PFS (6.7 versus 5.5 months) and median OS (13.8 versus 11.1 months) in favor of the trastuzumab-containing arm. There was no unexpected toxicity in the trastuzumab-containing arm, including symptomatic heart failure.
The ToGA trial represents the first positive phase III evaluation of a targeted therapy in EGCs. It indicates that trastuzumab is a new option for a subset of patients with gastric and GE junction adenocarcinoma and suggests that such patients should now be routinely tested for Her-2/neu expression.
3.3. Vascular Endothelial Growth Factor
Therapies directed against VEGF are the focus of major ongoing research in solid tumor malignancies. Of the identified angiogenic factors, VEGF appears the most potent and essential as regulator of angiogenesis (as normal as pathologic). VEGF exerts its angiogenic effects by binding to several high-affinity transmembrane receptors, most notably VEGF receptors (VEGFR) types 1 (flt-1) and 2 (KDR, flk-1) [151].
Increased VEGF expression has been measured in most human tumors [152] and in detail, in esophageal cancer, VEGF is overexpressed in 30–60% of patients and there is a correlation between high levels of VEGF expression, advanced stage and poor survival [153–157]. Studies in SCCs have indicated that expression of VEGF in tumors correlates with more advanced tumor stage, the presence of nodal and distant metastases, and a poorer survival outcome [155, 158].
Increased VEGF expression on tumors and increased serum VEGF levels have been correlated with worse prognoses also in gastric cancer [159, 160]
As anti-VEGF therapies, there are the multitarget TKIs, sunitinib and sorafenib, and most importantly the moAb bevacizumab (Avastin, Genentech), a humanized IgG1 monoclonal antibody against VEGF, investigated in different solid tumors.
The addition of bevacizumab to chemotherapy has been shown in several phase III clinical trials to improve the ORR and TTP in patients with CRC [161] NSCLC [162] and breast cancer [163]; on the basis of these studies, bevacizumab has been approved for the treatment of these cancers.
In addition to direct antiangiogenic effects, it has been postulated that bevacizumab leads to decreases in interstitial fluid pressures and increases chemotherapy drug delivery [164, 165].
About the side effects, in the phase III evaluation in advanced CRC, bevacizumab was noted to increase the incidence of grade 3 hypertension (11.0% versus 2.3%) compared to chemotherapy alone and colonic perforation noted in 1.5% of patients [161]. In the phase III evaluation in NSCLC, treatment with bevacizumab plus chemotherapy also led to an increased incidence of grade 3/4 proteinuria (3.1% versus 0%) and bleeding (4.4% versus 0.7%) compared to chemotherapy alone [162].
A considerable number of encouraging studies has been reported in the metastatic setting; for example, in a multicenter phase II evaluation led by MSKCC, bevacizumab and cisplatin/irinotecan were studied as first-line therapy in 47 patients with advanced gastric and GE junction adenocarcinoma [10]. The addition of bevacizumab significantly improved the TTP (8.3 months; 95% CI, 5.5 to 9.9 months) and OS (12.3 months; 95% CI, 11.3 to 17.2 months), compared to a historical TTP of 5 months. Toxicities that could have been related to bevacizumab included a 6% incidence of gastric perforation and a 2% incidence of myocardial infarction. Grade 3/4 thromboembolic events were observed in 25.5% of patients [similar to the 30% incidence observed using preoperative cisplatin/irinotecan therapy] [166].
Based on the promising results of this study, Kelsen completed a second evaluation of bevacizumab. In a study of 44 treatment-naïve patients with advanced esophagogastric adenocarcinoma, bevacizumab was combined with a modified regimen of docetaxel/cisplatin/fluorouracil (mDCF) [167]. Of 39 patients with measurable disease, the ORR was 67%. Median PFS was 12 months and median OS was 16.2 months. Therapy appeared to be more tolerable than the parent DCF regimen and included febrile neutropenia in 4% of patients perforation and bleeding in 1 patient each (2%) and grade 3/4 thromboembolism in 31% of patients.
Similarly high ORRs and toxicities were obtained in other evaluations of bevacizumab with different chemotherapy regimens [168, 169].
In the second-line setting, one evaluation of bevacizumab and docetaxel in 26 patients has been reported [170]. The ORR was 24% in 17 evaluable patients, which compares favorably to the ORR of 17% reported in two prior phase II evaluations of docetaxel in chemotherapy-naïve EGC patients [171, 172]. Grade 3/4 toxicities on this trial included gastrointestinal bleeding (12%) and arterial thromboses (8%).
Actually a confirmatory international phase III trial of capecitabine/cisplatin ± bevacizumab in advanced gastric cancer is underway. In the United Kingdom, the ongoing Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC)-B trial randomizes patients with resectable gastric and GE junction adenocarcinoma to perioperative ECX chemotherapy ± bevacizumab, building on the positive results for perioperative adjuvant chemotherapy observed in the original MAGIC trial [3].
4. Conclusion
Despite advances in clinical diagnostics, surgical techniques, and the improvement of chemo- and radiotherapy regimens, the prognosis of gastric cancer remains poor and the need for novel treatment options, such as immunotherapy, is very critical.
In this paper, we have reported the different immunotherapy strategies used against cancer, some of which are exploited in clinical trials with good results in terms of patient survival.
As new therapeutic strategies in the treatment of gastric cancer, we analyzed the immunotherapeutic approach based as on using of cellular component of the immune response (innate and acquired) as on usage of monoclonal antibody versus targets of gastric cancer.
In relation to cells of innate immunity, we observed that antitumor vaccination therapy with DCs pulsed with HER-2/neu-peptides may represent a potential candidate for the novel treatment of patients with gastric cancer. Accordingly in 33% of patients treated with this approach, the tumor markers were decreased after vaccination and 22% had a tumor regression of more than 50% with no significant side effects [28].
Moreover, the recent results about the enhancing of the antitumor immunity by (GM-CSF) gene-modified DCs [39] and blocking the immunosuppressive axis with small interfering RNA targeting IL-10 receptor [38] give hope for the immediate future in a more effective of GC immunotherapy DC-based.
Voskens [61] recently demonstrated that human NK cells acquire cytolytic activity against autologous gastric tumor cells after ex vivo expansion and suggested a therapeutic potential for autologous expanded NK cells, as directly as in combination with monoclonal antibodies in future cell-based immunotherapy.
However, using T-lymphocytes in GC immunotherapy lately, Kim [89] demonstrated the relevance of antitumor activity against the human GC of ex vivo expanded T cells, called cytokine-induced killer (CIK) cells. At an effector-target cell ratio of 30 : 1, CIK cells were able to destroy 58% of MKN74 human gastric cancer cells, suggesting that CIK cells might be used as an adoptive immunotherapy for GC patients.
Considering the use of moAbs specific for molecular targets of GC, the most extensively explored approach is the inhibition of EGFR. Current anti-EGFR therapies that have been evaluated in EGCs include the moAbs: cetuximab, panitumumab, and matuzumab, that have been used, either alone or in combination with chemotherapy in several clinical trials and the results obtained are quite good [74, 78, 123]. This type of therapy was well tolerated with little side effects.
Other moAbs that have been evaluated as therapy for EGCs are trastuzumab, a humanized IgG1 moAb against, the Her-2/neu receptor, and bevacizumab, a humanized IgG1 monoclonal antibody against VEGF. In the literature, there are several clinical trials (some in phase III) that use these two monoclonal antibodies in the EGC treatment either alone or with chemotherapy and good results were obtained in the survival of patients enrolled.
Currently, the major challenge in the field is to conduct randomized clinical trials demonstrating sufficient clinical benefits to justify the logistics and costs of customized cellular therapies. In many clinical trials, patients are enrolled at an advanced stage of gastric cancer, and this aspect could determine an unfavourable outcome; it would be interesting to plan clinical trials in the early stage of cancer because it would be possible that gastric cancer immunotherapeutic approaches confer a survival advantage when applied earlier during the course of the disease, such as in the adjuvant setting.
The big hurdle to make immunotherapy approach successful for gastric cancer remains the immune evasion strategies set up by the tumor resulting in avoidance of both innate and adaptive immunity.
Investigations during the past few years have provided novel insights into the cellular and molecular mechanisms involved in the bidirectional cross-talk between tumor cells and the immune system. Understanding this functional dialogue and the hierarchical status of different tumor-immune escape mechanisms at different stages of tumor progression will guide the design of novel therapeutic strategies aiming to destroy the “tumor fortress.”
Thus, it will be of particular interest to investigate the kinetics of the interactions between different inhibitory molecules and endogenous factors that influence the expansion and trafficking of Tregs and tolerogenic DCs within tumor-draining lymph nodes and the tumor surroundings.
On the basis of clinical and experimental evidence, it is reasonable to conclude that, successful therapy for gastric cancer must involve a combined approach, which should involve systemic chemotherapy and transplantation to reduce the burden or to eliminate immune suppressive cells, together with tailor-made immunotherapies customized to each single patient.
Acknowledgment
The authors thank Italian Ministry of University and Research and Ente Cassa di Risparmio di Firenze for supporting their studies.
References
- 1.Parkin DM. International variation. Oncogene. 2004;23(38):6329–6340. doi: 10.1038/sj.onc.1207726. [DOI] [PubMed] [Google Scholar]
- 2.Bruckner HW, Kufe DW, Pollock RE, Weichselbaum RR. Cancer Medicine. Ontario, Canda: 2003. [Google Scholar]
- 3.Cunningham D, Allum WH, Stenning SP, et al. MAGIC Trial Participants. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. New England Journal of Medicine. 2006;355:11–20. doi: 10.1056/NEJMoa055531. [DOI] [PubMed] [Google Scholar]
- 4.Tepper J, Krasna MJ, Niedzwiecki D, et al. Phase III trial of trimodality therapy with cisplatin, fluorouracil, radiotherapy, and surgery compared with surgery alone for esophageal cancer: CALGB 9781. Journal of Clinical Oncology. 2008;26(7):1086–1092. doi: 10.1200/JCO.2007.12.9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh TN, Noonan N, Hollywood D, Kelly A, Keeling N, Hennessy TP. A comparison of multimodal therapy and surgery for esophageal adenocarcinoma. New England Journal of Medicine. 1996;335(7):462–467. doi: 10.1056/NEJM199608153350702. [DOI] [PubMed] [Google Scholar]
- 6.Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. New England Journal of Medicine. 2001;345(10):725–730. doi: 10.1056/NEJMoa010187. [DOI] [PubMed] [Google Scholar]
- 7.Kelsen DP, Ginsberg R, Pajak TF, et al. Chemotherapy followed by surgery compared with surgery alone for localized esophageal cancer. New England Journal of Medicine. 1998;339(27):1979–1984. doi: 10.1056/NEJM199812313392704. [DOI] [PubMed] [Google Scholar]
- 8.Enzinger PC, Mayer RJ. Esophageal cancer. New England Journal of Medicine. 2003;349(23):2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 9.Vanhoefer U, Tewes M, Rojo F, et al. Phase I study of the humanized antiepidermal growth factor receptor monoclonal antibody EMD72000 in patients with advanced solid tumors that express the epidermal growth factor receptor. Journal of Clinical Oncology. 2004;22(1):175–184. doi: 10.1200/JCO.2004.05.114. [DOI] [PubMed] [Google Scholar]
- 10.Shah MA, Ramanathan RK, Ilson DH, et al. Multicenter phase II study of irinotecan, cisplatin, and bevacizumab in patients with metastatic gastric or gastroesophageal junction adenocarcinoma. Journal of Clinical Oncology. 2006;24(33):5201–5206. doi: 10.1200/JCO.2006.08.0887. [DOI] [PubMed] [Google Scholar]
- 11.Ocean AJ, Schnoll-Sussman F, Keresztes R. Phase II study of PS-341 (bortezomib) with or without irinotecan in patients (pts) with advanced gastric adenocarcinomas (AGA) Journal of Clinical Oncology. 2006;18:p. 14040. [Google Scholar]
- 12.Elkord E, Hawkins RE, Stern PL. Immunotherapy for gastrointestinal cancer: current status and strategies for improving efficacy. Expert Opinion on Biological Therapy. 2008;8(4):385–395. doi: 10.1517/14712598.8.4.385. [DOI] [PubMed] [Google Scholar]
- 13.Amedei A, Niccolai E, Della Bella C, et al. Characterization of tumor antigen peptide-specific T cells isolated from the neoplastic tissue of patients with gastric adenocarcinoma. Cancer Immunology, Immunotherapy. 2009;58(11):1819–1830. doi: 10.1007/s00262-009-0693-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goedert JJ. The epidemiology of acquired immunodeficiency syndrome malignancies. Seminars in Oncology. 2000;27(4):390–401. [PubMed] [Google Scholar]
- 15.Zeier M, Hartschuh W, Wiesel M, Lehnert T, Ritz E. Malignancy after renal transplantation. American Journal of Kidney Diseases. 2002;39(1):p. E5. doi: 10.1053/ajkd.2002.29926. [DOI] [PubMed] [Google Scholar]
- 16.Challis GB, Stam HJ. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncologica. 1990;29(5):545–550. doi: 10.3109/02841869009090048. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. New England Journal of Medicine. 2003;348(3):203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 18.Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clinical Cancer Research. 2003;9(12):4296–4303. [PubMed] [Google Scholar]
- 19.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9(6):669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 20.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nature Reviews Immunology. 2006;6(4):295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 21.Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD., Jr. Peptide-pulsed dendritic cells induce antigen-specific, CTL-mediated protective tumor immunity. Journal of Experimental Medicine. 1996;183(1):283–287. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caux C, Ait-Yahia S, Chemin K, et al. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Seminars in Immunopathology. 2000;22(4):345–369. doi: 10.1007/s002810000053. [DOI] [PubMed] [Google Scholar]
- 23.Figdor CG, Jolanda I, de Vries M, Joost Lesterhuis W, Melief CJM. Dendritic cell immunotherapy: mapping the way. Nature Medicine. 2004;10(5):475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 24.Gilboa E. DC-based cancer vaccines. Journal of Clinical Investigation. 2007;117(5):1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishigami S, Natsugoe S, Tokuda K, et al. Clinical impact of intratumoral natural killer cell and dendritic cell infiltration in gastric cancer. Cancer Letters. 2000;159(1):103–108. doi: 10.1016/s0304-3835(00)00542-5. [DOI] [PubMed] [Google Scholar]
- 26.Ishigami S, Natsugoe S, Uenosono Y, et al. Infiltration of antitumor immunocytes into the sentinel node in gastric cancer. Journal of Gastrointestinal Surgery. 2003;7(6):735–739. doi: 10.1016/s1091-255x(03)00076-3. [DOI] [PubMed] [Google Scholar]
- 27.Galetto A, Contarini M, Sapino A, et al. Ex vivo host response to gastrointestinal cancer cells presented by autologous dendritic cells. Journal of Surgical Research. 2001;100(1):32–38. doi: 10.1006/jsre.2001.6158. [DOI] [PubMed] [Google Scholar]
- 28.Kono K, Takahashi A, Sugai H, et al. Dendritic cells pulsed with HER-2/neu-derived peptides can induce specific T-cell responses in patients with gastric cancer. Clinical Cancer Research. 2002;8(11):3394–3400. [PubMed] [Google Scholar]
- 29.Kim TW, Hung CF, Ling M, et al. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. Journal of Clinical Investigation. 2003;112(1):109–117. doi: 10.1172/JCI17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes and Development. 2001;15(12):1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang TH, Lee JH, Noh KH, et al. Enhancing dendritic cell vaccine potency by combining a BAK/BAX siRNA-mediated antiapoptotic strategy to prolong dendritic cell life with an intracellular strategy to target antigen to lysosomal compartments. International Journal of Cancer. 2007;120(8):1696–1703. doi: 10.1002/ijc.22377. [DOI] [PubMed] [Google Scholar]
- 32.Kim TW, Hung CF, Boyd D, et al. Enhancing DNA vaccine potency by combining a strategy to prolong dendritic cell life with intracellular targeting strategies. Journal of Immunology. 2003;171(6):2970–2976. doi: 10.4049/jimmunol.171.6.2970. [DOI] [PubMed] [Google Scholar]
- 33.Kang TH, Lee JH, Noh KH, et al. Modification of professional antigen-presenting cells with small interfering RNA in vivo to enhance cancer vaccine potency. Cancer Research. 2005;65(1):309–316. [PubMed] [Google Scholar]
- 34.Park D, Lapteva N, Seethammagari M, Slawin KM, Spencer DM. An essential role for Akt1 in dendritic cell function and tumor immunotherapy. Nature Biotechnology. 2006;24(12):1581–1590. doi: 10.1038/nbt1262. [DOI] [PubMed] [Google Scholar]
- 35.Yu Q, Kovacs C, Yue FY, Ostrowski MA. The role of the p38 mitogen-activated protein kinase, extracellular signal-regulated kinase, and phosphoinositide-3-OH kinase signal transduction pathways in CD40 ligand-induced dendritic cell activation and expansion of virus-specific CD8+ T cell memory responses. Journal of Immunology. 2004;172(10):6047–6056. doi: 10.4049/jimmunol.172.10.6047. [DOI] [PubMed] [Google Scholar]
- 36.Kim JH, Kang TH, Noh KH, et al. Enhancement of DC vaccine potency by activating the PI3K/AKT pathway with a small interfering RNA targeting PTEN. Immunology Letters. 2010;134(1):47–54. doi: 10.1016/j.imlet.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27(41):5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- 38.Kim JH, Kang TH, Noh KH, et al. Blocking the immunosuppressive axis with small interfering RNA targeting interleukin (IL)-10 receptor enhances dendritic cell-based vaccine potency. Clinical and Experimental Immunology. 2011;165(2):180–189. doi: 10.1111/j.1365-2249.2011.04410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He SB, Sun K, Wang L, Li DC, Zhang YY. GM-CSF gene-modified dendritic cell vaccine enhances antitumor immunity in vitro. Zhonghua Zhong Liu Za Zhi. 2010;32(6):410–414. [PubMed] [Google Scholar]
- 40.Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(6):2731–2736. doi: 10.1073/pnas.050588297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coca S, Perez-Piqueras J, Martinez D, et al. The prognostic significance of intratumoral natural killer cells in patients with colerectal carcinoma. Cancer. 1997;79(12):2320–2328. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 42.Orange JS, Ballas ZK. Natural killer cells in human health and disease. Clinical Immunology. 2006;118(1):1–10. doi: 10.1016/j.clim.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 43.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. Journal of Experimental Medicine. 1982;155(6):1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Law TM, Motzer RJ, Mazumdar M, et al. Phase III randomized trial of interleukin-2 with or without lymphokine- activated killer cells in the treatment of patients with advanced renal cell carcinoma. Cancer. 1995;76(5):824–832. doi: 10.1002/1097-0142(19950901)76:5<824::aid-cncr2820760517>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 45.Kammula US, White DE, Rosenberg SA. Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer. 1998;83(4):797–805. [PubMed] [Google Scholar]
- 46.Brune M, Castaigne S, Catalano J, et al. Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial. Blood. 2006;108(1):88–96. doi: 10.1182/blood-2005-10-4073. [DOI] [PubMed] [Google Scholar]
- 47.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. Journal of Clinical Oncology. 1999;17(7):2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 48.Bukowski RM. Natural history and therapy of metastatic renal cell carcinoma: the role of interleukin-2. Cancer. 1997;80(7):1198–1220. doi: 10.1002/(sici)1097-0142(19971001)80:7<1198::aid-cncr3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 49.Gollob JA, Mier JW, Veenstra K, et al. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-γ induction is associated with clinical response. Clinical Cancer Research. 2000;6(5):1678–1692. [PubMed] [Google Scholar]
- 50.Gollob JA, Veenstra KG, Parker RA, et al. Phase I trial of concurrent twice-weekly recombinant human interleukin-12 plus low-dose IL-2 in patients with melanoma or renal cell carcinoma. Journal of Clinical Oncology. 2003;21(13):2564–2573. doi: 10.1200/JCO.2003.12.119. [DOI] [PubMed] [Google Scholar]
- 51.Robertson MJ, Mier JW, Logan T, et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clinical Cancer Research. 2006;12(14):4265–4273. doi: 10.1158/1078-0432.CCR-06-0121. [DOI] [PubMed] [Google Scholar]
- 52.Davis ID, Skrumsager BK, Cebon J, et al. An open-label, two-arm, phase I trial of recombinant human interleukin-21 in patients with metastatic melanoma. Clinical Cancer Research. 2007;13(12):3630–3636. doi: 10.1158/1078-0432.CCR-07-0410. [DOI] [PubMed] [Google Scholar]
- 53.de Castro FA, Bonini Palma PV, Morais FR, et al. Immunological effects of interferon-α on chronic myelogenous leukemia. Leukemia and Lymphoma. 2003;44(12):2061–2067. doi: 10.1080/1042819031000110973. [DOI] [PubMed] [Google Scholar]
- 54.Chen W, Chan ASH, Dawson AJ, Liang X, Blazar BR, Miller JS. FLT3 ligand administration after hematopoietic cell transplantation increases circulating dendritic cell precursors that can be activated by CpG oligodeoxynucleotides to enhance T-cell and natural killer cell function. Biology of Blood and Marrow Transplantation. 2005;11(1):23–34. doi: 10.1016/j.bbmt.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Gluck WL, Hurst D, Yuen A, et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin's lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clinical Cancer Research. 2004;10(7):2253–2264. doi: 10.1158/1078-0432.ccr-1087-3. [DOI] [PubMed] [Google Scholar]
- 56.Khan KD, Emmanouilides C, Benson DM, et al. A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin's lymphoma. Clinical Cancer Research. 2006;12(23):7046–7053. doi: 10.1158/1078-0432.CCR-06-1571. [DOI] [PubMed] [Google Scholar]
- 57.Berdeja JG, Hess A, Lucas DM, et al. Systemic interleukin-2 and adoptive transfer of lymphokine-activated killer cells improves antibody-dependent cellular cytotoxicity in patients with relapsed B-cell lymphoma treated with rituximab. Clinical Cancer Research. 2007;13(8):2392–2399. doi: 10.1158/1078-0432.CCR-06-1860. [DOI] [PubMed] [Google Scholar]
- 58.Parihar R, Nadella P, Lewis A, et al. A phase I study of interleukin 12 with trastuzumab in patients with human epidermal growth factor receptor-2-overexpressing malignancies: analysis of sustained interferon γ production in a subset of patients. Clinical Cancer Research. 2004;10(15):5027–5037. doi: 10.1158/1078-0432.CCR-04-0265. [DOI] [PubMed] [Google Scholar]
- 59.Renner C, Hartmann F, Jung W, Deisting C, Juwana M, Pfreundschuh M. Initiation of humoral and cellular immune responses in patients with refractory Hodgkin's disease by treatment with an anti-CD16/CD30 bispecific antibody. Cancer Immunology Immunotherapy. 2000;49(3):173–180. doi: 10.1007/s002620050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hartmann F, Renner C, Jung W, et al. Anti-CD16/CD30 bispecific antibody treatment for Hodgkin's disease: role of infusion schedule and costimulation with cytokines. Clinical Cancer Research. 2001;7(7):1873–1881. [PubMed] [Google Scholar]
- 61.Voskens CJ, Watanabe R, Rollins S, Campana D, Hasumi K, Mann DL. Ex-vivo expanded human NK cells express activating receptors that mediate cytotoxicity of allogeneic and autologous cancer cell lines by direct recognition and antibody directed cellular cytotoxicity. Journal of Experimental and Clinical Cancer Research. 2010;29(1, article 134) doi: 10.1186/1756-9966-29-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kitagawa Y, Ueda M, Ando N, Ozawa S, Shimizu N, Kitajima M. Further evidence for prognostic significance of epidermal growth factor receptor gene amplification in patients with esophageal squamous cell carcinoma. Clinical Cancer Research. 1996;2(5):909–914. [PubMed] [Google Scholar]
- 63.Gibault L, Metges JP, Conan-Charlet V, et al. Diffuse EGFR staining is associated with reduced overall survival in locally advanced oesophageal squamous cell cancer. British Journal of Cancer. 2005;93(1):107–115. doi: 10.1038/sj.bjc.6602625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilkinson NW, Black JD, Roukhadze E, et al. Epidermal growth factor receptor expression correlates with histologic grade in resected esophageal adenocarcinoma. Journal of Gastrointestinal Surgery. 2004;8(4):448–453. doi: 10.1016/j.gassur.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Allegra CJ, Jessup JM, Somerfield MR, et al. American society of clinical oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. Journal of Clinical Oncology. 2009;27(12):2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 66.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of Clinical Oncology. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 67.van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. New England Journal of Medicine. 2009;360(14):1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 68.Baselga J, Norton L, Masui H, et al. Antitumor effects of doxorubicin in combination with anti-epidermal growth factor receptor monoclonal antibodies. Journal of the National Cancer Institute. 1993;85(16):1327–1333. doi: 10.1093/jnci/85.16.1327. [DOI] [PubMed] [Google Scholar]
- 69.Kawaguchi Y, Kono K, Mimura K, Sugai H, Akaike H, Fujii H. Cetuximab induce antibody-dependent cellular cytotoxicity against EGFR-expressing esophageal squamous cell carcinoma. International Journal of Cancer. 2007;120(4):781–787. doi: 10.1002/ijc.22370. [DOI] [PubMed] [Google Scholar]
- 70.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nature Reviews Cancer. 2006;6(9):714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 71.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan- refractory metastatic colorectal cancer. New England Journal of Medicine. 2004;351(4):337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 72.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. New England Journal of Medicine. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 73.Enzinger P, Yock T, Suh W. Phase II cisplatin, irinotecan, cetuximab and concurrent radiation therapy followed by surgery for locally advanced esophageal cancer. Journal of Clinical Oncology. 2006;24:p. 4064. [Google Scholar]
- 74.Safran H, Suntharalingam M, Dipetrillo T, et al. Cetuximab With concurrent chemoradiation for esophagogastric cancer: assessment of toxicity. International Journal of Radiation Oncology Biology Physics. 2008;70(2):391–395. doi: 10.1016/j.ijrobp.2007.07.2325. [DOI] [PubMed] [Google Scholar]
- 75.Ruhstaller T, Pless M, Dietrich D, et al. Cetuximab in combination with chemoradiotherapy before surgery in patients with resectable, locally advanced esophageal carcinoma: a prospective, multicenter phase lb-II trial (SAKK 75/06) Journal of Clinical Oncology. 2011;29:626–631. doi: 10.1200/JCO.2010.31.9715. [DOI] [PubMed] [Google Scholar]
- 76.de Vita F, Orditura M, Martinelli E, et al. A multicenter phase II study of induction chemotherapy with FOLFOX-4 and cetuximab followed by radiation and cetuximab in locally advanced oesophageal cancer. British Journal of Cancer. 2011;104(3):427–432. doi: 10.1038/sj.bjc.6606093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Agarwala A, Hanna N, McCollum A. Preoperative cetuximab and radiation (XRT) for patients (pts) with surgically resectable esophageal and gastroesophageal junction (GEJ) carcinomas: a pilot study from the Hoosier Oncology Group and the University of Texas Southwestern. Journal of Clinical Oncology. 2009;27:p. 4557. [Google Scholar]
- 78.Ma H, Ryan T, Newman E. Neoadjuvant therapy of gastric cancer with irinotecan, cisplatin, and cetuximab followed by surgical resection and adjuvant chemoradiation. In: Proceedings of Graphics Interface American Society of Clinical Oncology (ASCO '09), vol. 58; 2009. [Google Scholar]
- 79.Ilson DH, Bains M, Kelsen DP, et al. Phase I trial of escalating-dose irinotecan given weekly with cisplatin and concurrent radiotherapy in locally advanced esophageal cancer. Journal of Clinical Oncology. 2003;21(15):2926–2932. doi: 10.1200/JCO.2003.02.147. [DOI] [PubMed] [Google Scholar]
- 80.Ku G, Bains M, Rizk N, et al. Phase II trial of pre-operative cisplatin/irinotecan and radiotherapy for locally advanced esophageal cancer: PET scan after induction therapy may identify early treatment failure. In: Proceedings of Graphics Interface American Society of Clinical Oncology (ASCO '07), vol. 9; 2007. [Google Scholar]
- 81.Moehler M, Mueller A, Trarbach T, et al. Cetuximab with irinotecan, folinic acid and 5-fluorouracil as first-line treatment in advanced gastroesophageal cancer: a prospective multi-center biomarker-oriented phase II study. Annals of Oncology. 2011;22(6):1358–1366. doi: 10.1093/annonc/mdq591. [DOI] [PubMed] [Google Scholar]
- 82.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 83.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Research. 2001;61(12):4766–4772. [PubMed] [Google Scholar]
- 85.June CH. Principles of adoptive T cell cancer therapy. Journal of Clinical Investigation. 2007;117(5):1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gutenberg-Universitat Mainz J, Eberts D, Fatho M, Lennerz V, et al. Melanoma-associated Mhc Class I associated oligopeptides and the uses thereof. WO/2007/025760, 2007.
- 87.Pharmaceuticals A. Cytotoxic T Lymphocyte-stimulation peptides for prevention, treatment, and diagnosis of melanoma. WO/2001/032193, 2001.
- 88.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim YJ, Lim J, Kang JS, et al. Adoptive immunotherapy of human gastric cancer with ex vivo expanded T cells. Cancer Letters. 2010;33(11):1789–1795. doi: 10.1007/s12272-010-1111-7. [DOI] [PubMed] [Google Scholar]
- 90.Sangiolo D. Cytokine induced killer cells as promising immunotherapy for solid tumors. Journal of Cancer. 2011;2:363–368. doi: 10.7150/jca.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoshino T, Seki N, Kikuchi M, et al. Hla class-I-restricted and tumor-specific CTL in tumor-infiltrating lymphocytes of patients with gastric cancer. International Journal of Cancer. 1997;70(6):631–638. doi: 10.1002/(sici)1097-0215(19970317)70:6<631::aid-ijc1>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 92.Kono K, Rongcun Y, Charo J, et al. Identification of HER2/neu-derived peptide epitopes recognized by gastric cancer-specific cytotoxic T lymphocytes. International Journal of Cancer. 1998;78(2):202–208. doi: 10.1002/(sici)1097-0215(19981005)78:2<202::aid-ijc14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 93.Guo DL, Dong M, Wang L, Sun LP, Yuan Y. Expression of gastric cancer-associated MG7 antigen in gastric cancer, precancerous lesions and H. pylori-associated gastric diseases. World Journal of Gastroenterology. 2002;8(6):1009–1013. doi: 10.3748/wjg.v8.i6.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu K, Nie Y, Guo C, Chen Y, Ding J, Fan D. Molecular basis of therapeutic approaches to gastric cancer. Journal of Gastroenterology and Hepatology. 2009;24(1):37–41. doi: 10.1111/j.1440-1746.2008.05753.x. [DOI] [PubMed] [Google Scholar]
- 95.Nie Y, Wu K, Yang J, et al. Induction of T lymphocytes specific to human gastric cancer using HLA-A matched allogeneic gastric tumor cells. Journal of Immunotherapy. 2003;26(5):403–411. doi: 10.1097/00002371-200309000-00003. [DOI] [PubMed] [Google Scholar]
- 96.Jiang J, Xu N, Wu C, et al. Treatment of advanced gastric cancer by chemotherapy combined with autologous cytokine-induced killer cells. Anticancer Research. 2006;26(3 B):2237–2242. [PubMed] [Google Scholar]
- 97.Wu C, Jiang J, Shi L, Xu N. Prospective study of chemotherapy in combination with cytokine-induced killer cells in patients suffering from advanced non-small cell lung cancer. Anticancer Research. 2008;28(6 B):3997–4002. [PubMed] [Google Scholar]
- 98.Sun S, Li XM, Li XD, Yang WS. Studies on inducing apoptosis effects and mechanism of CIK cells for MGC-803 gastric cancer cell lines. Cancer Biotherapy and Radiopharmaceuticals. 2005;20(2):173–180. doi: 10.1089/cbr.2005.20.173. [DOI] [PubMed] [Google Scholar]
- 99.Alexander RB, Rosenberg SA. Long term survival of adoptively transferred tumor-infiltrating lymphocytes in mice. Journal of Immunology. 1990;145(5):1615–1620. [PubMed] [Google Scholar]
- 100.Kono K, Takahashi A, Ichihara F, et al. Prognostic significance of adoptive immunotherapy with tumor-associated lymphocytes in patients with advanced gastric cancer: a randomized trial. Clinical Cancer Research. 2002;8(6):1767–1771. [PubMed] [Google Scholar]
- 101.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor- infiltrating lymphocytes and interleukin 2. Journal of the National Cancer Institute. 1994;86(15):1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 102.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. Journal of Immunotherapy. 2003;26(4):332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tomšová M, Melichar B, Sedláková I, Šteiner I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecologic Oncology. 2008;108(2):415–420. doi: 10.1016/j.ygyno.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 104.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 105.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3(5):431–437. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 106.Molmed SPA, Traversari C, Bordignon C. Antigen transduced T cells used as a delivery system for antigens. WO/2004/035768, 2004.
- 107.Bonini C, Ferrari G, Verzeletti S, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 108.Clackson T, Yang W, Rozamus LW, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Straathof KC, Spencer DM, Sutton RE, Rooney CM. Suicide genes as safety switches in T lymphocytes. Cytotherapy. 2003;5(3):227–230. doi: 10.1080/14653240310001497. [DOI] [PubMed] [Google Scholar]
- 110.Yeda Research and Development Co, LTD, Eshhar Z, Schindler D, Waks T, Gross G. Chimeric receptor genes and cells transformed therewith. WO/1993/019163, 1993.
- 111.Fred Hutchinson Cancer Research Center, Greenberg PD, Nelson BH. Chimeric cytokine receptors in lymphocytes. WO/1994/022914, 1994.
- 112.Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28− CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. Journal of Experimental Medicine. 2003;198(6):947–955. doi: 10.1084/jem.20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sierra Sciences Inc., Andrews WH, Foster CA, Fraser S, Mohammadpour H. Methods and compositions for modulating telomerase reverse transcriptase (Tert) expression. WO/2002/016658, 2002.
- 114.Yarden Y, Ullrich A. Growth factor receptor tyrosine kinases. Annual Review of Biochemistry. 1988;57:443–478. doi: 10.1146/annurev.bi.57.070188.002303. [DOI] [PubMed] [Google Scholar]
- 115.Lynch TJ, Jr., Kim ES, Eaby B, Garey J, West DP, Lacouture ME. Epidermal growth factor receptor inhibitor-associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist. 2007;12(5):610–621. doi: 10.1634/theoncologist.12-5-610. [DOI] [PubMed] [Google Scholar]
- 116.Schrag D, Chung KY, Flombaum C, Saltz L. Cetuximab therapy and symptomatic hypomagnesemia. Journal of the National Cancer Institute. 2005;97(16):1221–1224. doi: 10.1093/jnci/dji242. [DOI] [PubMed] [Google Scholar]
- 117.Itakura Y, Sasano H, Shiga C, et al. Epidermal growth factor receptor overexpression in esophageal carcinoma: an immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer. 1994;74(3):795–804. doi: 10.1002/1097-0142(19940801)74:3<795::aid-cncr2820740303>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 118.Janmaat ML, Gallegos-Ruiz MI, Rodriguez JA, et al. Predictive factors for outcome in a phase II study of gefitinib in second-line treatment of advanced esophageal cancer patients. Journal of Clinical Oncology. 2006;24(10):1612–1619. doi: 10.1200/JCO.2005.03.4900. [DOI] [PubMed] [Google Scholar]
- 119.Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clinical Cancer Research. 1995;1(11):1311–1318. [PubMed] [Google Scholar]
- 120.Vermorken JB, Trigo J, Hitt R, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. Journal of Clinical Oncology. 2007;25(16):2171–2177. doi: 10.1200/JCO.2006.06.7447. [DOI] [PubMed] [Google Scholar]
- 121.Pinto C, di Fabio F, Siena S, et al. Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study) Annals of Oncology. 2007;18(3):510–517. doi: 10.1093/annonc/mdl459. [DOI] [PubMed] [Google Scholar]
- 122.Lordick F, Luber B, Lorenzen S, et al. Cetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic gastric cancer: a phase II study of the Arbeitsgemeinschaft Internistische Onkologie (AIO) British Journal of Cancer. 2010;102(3):500–505. doi: 10.1038/sj.bjc.6605521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lorenzen S, Schuster T, Porschen R, et al. Cetuximab plus cisplatin-5-fluorouracil versus cisplatin-5-fluorouracil alone in first-line metastatic squamous cell carcinoma of the esophagus: a randomized phase II study of the Arbeitsgemeinschaft Internistische Onkologie. Annals of Oncology. 2009;20(10):1667–1673. doi: 10.1093/annonc/mdp069. [DOI] [PubMed] [Google Scholar]
- 124.Yeh K, Hsu C, Hsu C. Phase II study of cetuximab plus weekly cisplatin and 24-hour infusion of high-dose 5-fluorouracil and leucovorin for the first-line treatment of advanced gastric cancer. Journal of Clinical Oncology. 2009;27:p. 4567. [Google Scholar]
- 125.Zhang X, Xu J, Shen L. A phase II study of cetuximab with cisplatin and capecitabine as first-line treatment in advanced gastric cancer. In: Proceedings of Graphics Interface American Society of Clinical Oncology (ASCO '09), vol. 39; 2009. [Google Scholar]
- 126.Pinto C, di Fabio F, Barone C, et al. Phase II study of cetuximab in combination with cisplatin and docetaxel in patients with untreated advanced gastric or gastro-oesophageal junction adenocarcinoma (DOCETUX study) British Journal of Cancer. 2009;101(8):1261–1268. doi: 10.1038/sj.bjc.6605319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Woell E, Kühr T, Eisterer W, et al. Biweekly oxaliplatin and irinotecan chemotherapy in advanced gastric cancer. A first-line multicenter phase II trial of the arbeitsgemeinschaft medikamentoese tumortherapie (AGMT) Anticancer Research. 2008;28:2901–2905. [PubMed] [Google Scholar]
- 128.Gold P, Goldman B, Iqbal S, et al. Cetuximab as second-line therapy in patients with metastatic esophageal adenocarcinoma: a phase II southwest oncology group study (S0415) Journal of Thoracic Oncology. 2010;5(9):1472–1476. doi: 10.1097/JTO.0b013e3181e77a92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ku G, Shah M, Tang L. Cetuximab (C225) plus irinotecan/cisplatin (CPT/ CDDP) for CPT/CDDP-refractory esophageal cancer. In: Proceedings of the Graphics Interface American Society of Clinical Oncology (ASCO '08), vol. 54; 2008. [Google Scholar]
- 130.Tebbutt N, Cummins MM, Sourjina T, et al. Australasian Gastro-Intestinal Trials Group. Randomized, non-comparative phase II study of weekly docetaxel with cisplatin and 5-fluorouracilor with capecitabine in oesophagogastric cancer: the AGITG ATTAX trial. British Journal of Cancer. 2010;102:475–481. doi: 10.1038/sj.bjc.6605522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schønnemann K, Bjerregaard J, Jensen H. Biweekly cetuximab and irinotecan as second-line therapy to patients with platinium-resistant gastroesophageal cancer. In: Proceedings of the Graphics Interface American Society of Clinical Oncology (GI ASCO '09), vol. 73; 2009. [Google Scholar]
- 132.Vanhoefer U, Tewes M, Rojo F, et al. Phase I study of the humanized antiepidermal growth factor receptor monoclonal antibody EMD72000 in patients with advanced solid tumors that express the epidermal growth factor receptor. Journal of Clinical Oncology. 2004;22(1):175–184. doi: 10.1200/JCO.2004.05.114. [DOI] [PubMed] [Google Scholar]
- 133.Rao S, Starling N, Cunningham D, et al. Phase I study of epirubicin, cisplatin and capecitabine plus matuzumab in previously untreated patients with advanced oesophagogastric cancer. British Journal of Cancer. 2008;99(6):868–874. doi: 10.1038/sj.bjc.6604622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy- refractory metastatic colorectal cancer. Journal of Clinical Oncology. 2007;25(13):1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 135.Foon K, Yang XD, Weiner LM, et al. Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. International Journal of Radiation Oncology Biology Physics. 2004;58(3):984–990. doi: 10.1016/j.ijrobp.2003.09.098. [DOI] [PubMed] [Google Scholar]
- 136.Casalini P, Iorio MV, Galmozzi E, Ménard S. Role of HER receptors family in development and differentiation. Journal of Cellular Physiology. 2004;200(3):343–350. doi: 10.1002/jcp.20007. [DOI] [PubMed] [Google Scholar]
- 137.Al-Kasspooles M, Moore JH, Orringer MB, Beer DG. Amplification and over-expression of the EGFR and erbB-2 genes in human esophageal adenocarcinomas. International Journal of Cancer. 1993;54(2):213–219. doi: 10.1002/ijc.2910540209. [DOI] [PubMed] [Google Scholar]
- 138.Ross JS, McKenna BJ. The HER-2/neu oncogene in tumors of the gastrointestinal tract. Cancer Investigation. 2001;19(5):554–568. doi: 10.1081/cnv-100103852. [DOI] [PubMed] [Google Scholar]
- 139.Brien TP, Odze RD, Sheehan CE, McKenna BJ, Ross JS. Her-2/neu gene amplification by FISH predicts poor survival in Barrett's esophagus-associated adenocarcinoma. Human Pathology. 2000;31(1):35–39. doi: 10.1016/s0046-8177(00)80195-1. [DOI] [PubMed] [Google Scholar]
- 140.Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. New England Journal of Medicine. 2007;357(1):39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 141.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Herceptin Adjuvant (HERA) Trial Study Team Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. New England Journal of Medicine. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 142.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. New England Journal of Medicine. 2005;353(16):1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 143.Smith I, Procter M, Gelber RD, et al. HERA study team. 2-Year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomized controlled trial. Lancet. 2007;369:29–36. doi: 10.1016/S0140-6736(07)60028-2. [DOI] [PubMed] [Google Scholar]
- 144.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. Journal of Clinical Oncology. 1999;17(9):2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 145.Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. Journal of Clinical Oncology. 2002;20(3):719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 146.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against her2 for metastatic breast cancer that overexpresses HER2. New England Journal of Medicine. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 147.Crone SA, Zhao YY, Fan L, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nature Medicine. 2002;8(5):459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 148.Seidman A, Hudis C, Pierri MK, et al. Cardiac dysfunction in the trastuzumab clinical trials experience. Journal of Clinical Oncology. 2002;20(5):1215–1221. doi: 10.1200/JCO.2002.20.5.1215. [DOI] [PubMed] [Google Scholar]
- 149.Safran H, DiPetrillo T, Akerman P, et al. Phase I/II study of trastuzumab, paclitaxel, cisplatin and radiation for locally advanced, HER2 overexpressing, esophageal adenocarcinoma. International Journal of Radiation Oncology Biology Physics. 2007;67(2):405–409. doi: 10.1016/j.ijrobp.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 150.Bang JY, van Cutsem E, Feyereislova A, et al. ToGA Trial Investigators Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomized controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 151.Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. European Journal of Cancer. 2006;42(18):3127–3139. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 152.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocrine Reviews. 1997;18(1):4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 153.Inoue K, Ozeki Y, Suganuma T, Sugiura Y, Tanaka S. Vascular endothelial growth factor expression in primary esophageal squamous cell carcinoma. Association with angiogenesis and tumor progression. Cancer. 1997;79(2):206–213. doi: 10.1002/(sici)1097-0142(19970115)79:2<206::aid-cncr2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 154.Kitadai Y, Haruma K, Tokutomi T, et al. Significance of vessel count and vascular endothelial growth factor in human esophageal carcinomas. Clinical Cancer Research. 1998;4(9):2195–2200. [PubMed] [Google Scholar]
- 155.Kleespies A, Guba M, Jauch KW, Bruns CJ. Vascular endothelial growth factor in esophageal cancer. Journal of Surgical Oncology. 2004;87(2):95–104. doi: 10.1002/jso.20070. [DOI] [PubMed] [Google Scholar]
- 156.Shih CH, Ozawa S, Ando N, Ueda M, Kitajima M. Vascular endothelial growth factor expression predicts outcome and lymph node metastasis in squamous cell carcinoma of the esophagus. Clinical Cancer Research. 2000;6(3):1161–1168. [PubMed] [Google Scholar]
- 157.Imdahl A, Bognar G, Schulte-Mönting J, Schöffel U, Farthmann EH, Ihling C. Predictive factors for response to neoadjuvant therapy in patients with oesophageal cancer. European Journal of Cardio-thoracic Surgery. 2002;21(4):657–663. doi: 10.1016/s1010-7940(02)00044-1. [DOI] [PubMed] [Google Scholar]
- 158.Shimada H, Hoshino T, Okazumi S, et al. Expression of angiogenic factors predicts response to chemoradiotherapy and prognosis of oesophageal squamous cell carcinoma. British Journal of Cancer. 2002;86(4):552–557. doi: 10.1038/sj.bjc.6600129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Maeda K, Chung YS, Ogawa Y, et al. Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer. 1996;77(5):858–863. doi: 10.1002/(sici)1097-0142(19960301)77:5<858::aid-cncr8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 160.Yoshikawa T, Tsuburaya A, Kobayashi O, et al. Plasma concentrations of VEGF and bFGF in patients with gastric carcinoma. Cancer Letters. 2000;153(1-2):7–12. doi: 10.1016/s0304-3835(99)00426-7. [DOI] [PubMed] [Google Scholar]
- 161.Hurwitz H, Fehrenbacher L, Novotny W, et al. Kabbinavar bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New England Journal of Medicine. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 162.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. New England Journal of Medicine. 2006;355(24):2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 163.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. New England Journal of Medicine. 2007;357(26):2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 164.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nature Medicine. 2001;7(9):987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 165.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nature Medicine. 2004;10(2):145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shah MA, Ilson D, Kelsen DP. Thromboembolic events in gastric cancer: high incidence in patients receiving irinotecan- and bevacizumab-based therapy. Journal of Clinical Oncology. 2005;23(11):2574–2576. doi: 10.1200/JCO.2005.81.908. [DOI] [PubMed] [Google Scholar]
- 167.Shah MA, Jhawer M, Ilson DH, et al. Phase II study of modified docetaxel, cisplatin, and fluorouracil with bevacizumab in patients with metastatic gastroesophageal adenocarcinoma. Journal of Clinical Oncology. 2011;29(7):868–874. doi: 10.1200/JCO.2010.32.0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Enzinger P, Ryan D, Clark JW, et al. Weekly docetaxel, cisplatin, and irinotecan (TPC): results of a multicenter phase II trial in patients with metastatic esophagogastric cancer. Annals of Oncology. 2009;20(3):475–480. doi: 10.1093/annonc/mdn658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.El-Rayes B, Zalupski M, Bekai-Saab T, et al. A phase II study of bevacizumab, oxaliplatin, and docetaxel in locally advanced and metastatic gastric and gastroesophageal junction cancers. Annals of Oncology. 2010;21(10):1999–2004. doi: 10.1093/annonc/mdq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Enzinger P, Fidias P, Meyerhardt J. Phase II study of bevacizumab and docetaxel in metastatic esophageal and gastric cancer. In: Proceedings of the Graphics Interface American Society of Clinical Oncology (ASCO '06), vol. 68; 2006. [Google Scholar]
- 171.Einzig AI, Neuberg D, Remick SC, et al. Phase II trial of docetaxel (Taxotere) in patients with adenocarcinoma of the upper gastrointestinal tract previously untreated with cytotoxic chemotherapy: the Eastern Cooperative Oncology Group (ECOG) results of protocol E1293. Medical Oncology. 1996;13(2):87–93. doi: 10.1007/BF02993858. [DOI] [PubMed] [Google Scholar]
- 172.Heath EI, Urba S, Marshall J, Piantadosi S, Forastiere AA. Phase II trial of docetaxel chemotherapy in patients with incurable adenocarcinoma of the esophagus. Investigational New Drugs. 2002;20(1):95–99. doi: 10.1023/a:1014476602804. [DOI] [PubMed] [Google Scholar]