

Abstract
Living organisms are adept in forming inorganic materials (biominerals) with unique structures and properties that exceed the capabilities of engineered materials. Biomimetic materials syntheses are being developed that aim at replicating the advantageous properties of biominerals in vitro and endow them with additional functionalities. Recently, proof-of-concept was provided for an alternative approach that allows for the production of biomineral-based functional materials in vivo. In this approach, the cellular machinery for the biosynthesis of nano-/micropatterned SiO2 (silica) structures in diatoms was genetically engineered to incorporate a monomeric, cofactor-independent (“simple”) enzyme, HabB, into diatom silica. In the present work, it is demonstrated that this approach is also applicable for enzymes with “complex” activity requirements, including oligomerization, metal ions, organic redox cofactors, and posttranslational modifications. Functional expression of the enzymes β-glucuronidase, glucose oxidase, galactose oxidase, and horseradish peroxidase in the diatom Thalassiosira pseudonana was accomplished, and 66 to 78% of the expressed enzymes were stably incorporated into the biosilica. The in vivo incorporated enzymes represent approximately 0.1% (wt/wt) of the diatom biosilica and are stabilized against denaturation and proteolytic degradation. Furthermore, it is demonstrated that the gene construct for in vivo immobilization of glucose oxidase can be utilized as the first negative selection marker for diatom genetic engineering.
INTRODUCTION
Enzymes have become invaluable catalytic tools in many technological processes (4). They are superior to nonenzymatic catalysts regarding reaction rate, substrate specificity, and stereoselectivity (4). Furthermore, enzymes operate in aqueous solvents and usually under mild reaction conditions, which are highly desired for environmentally benign industrial processes (“green chemistry”) (4). However, enzymes also have disadvantages, because their production is often expensive and their lifetimes are usually shorter than for nonprotein catalysts (4). Therefore, important cost factors for enzyme-based production processes are stabilization and recovery of the enzyme after completion of the reaction (5).
Immobilization of enzymes on or in solid organic or inorganic materials solves the problem of reusability and, in some cases, can even enhance enzyme stability (5, 12, 13). Enzyme immobilization can be achieved through physisorption, covalent attachment, or in situ incorporation during formation of the solid material (organic polymerization or inorganic sol-gel formation) (5, 12, 13). Drawbacks of these approaches include risk of detachment and denaturation of the adsorbed enzyme, risk of enzyme inactivation during covalent attachment and incorporation into the solid material, and the use of caustic reagents (5). Furthermore, in each case the desired enzyme needs to be isolated (or at least enriched) from a suitable organism through labor-intensive procedures.
Recently, an entirely new concept for immobilization of enzymes in a SiO2 (silica) matrix was introduced (31), coined living diatom silica immobilization (LiDSI). LiDSI involves genetic engineering of diatoms, which are unicellular eukaryotic algae that produce silica-based cell walls. Diatom silica formation depends on specific proteins (silaffins, silacidins, cingulins) that remain permanently associated within the silica in vivo (23, 34, 41). It has previously been demonstrated that expression in the diatom Thalassiosira pseudonana of a fusion protein consisting of the silaffin tpSil3 and the bacterial enzyme hydroxylaminobenzene mutase (HabB) generates diatom strains that exhibit biosilica-associated HabB activity (31). The HabB did not interfere with the function of the silaffin domain, allowing for intracellular transport of the fusion protein into the silica-forming organelle, termed the silica deposition vesicle (SDV). Inside the SDV the silaffin fusion protein becomes incorporated into the forming biosilica, and thus becomes a component of the cell wall after exocytosis of the SDV contents (Fig. 1) (23).
Fig 1.

Hypothetical pathway for intracellular transport of silaffin-enzyme fusion proteins to the silica deposition vesicle (SDV) (23, 36). The fusion proteins are cotranslationally imported into the endoplasmic reticulum (ER), and the signal peptide (SP) for ER import is removed by signal peptidase. Further modifications of the fusion proteins may occur in the ER and after transport to and through the Golgi apparatus. The fusion proteins may reach the SDV via specific transport vesicles, and become incorporated into the silica-forming organic matrix in the SDV lumen. After completion of silica formation, the silaffin-enzyme fusion proteins become trapped in the deposited silica and, after exocytosis of the SDV contents, remain stably attached to the biosilica cell wall.
LiDSI is the first method to utilize the natural process of protein incorporation into a biologically produced mineral (biomineralization) for the immobilization of enzymes. There are multiple advantages of LiDSI compared to conventional enzyme immobilization methods:
(i) The enzyme to be immobilized does not need to be purified.
(ii) Immobilization proceeds under optimal (i.e., physiological) conditions for protein stability and is environmentally friendly.
(iii) Diatom silica is a desirable matrix for enzyme immobilization due to its hierarchical nano- to microporous structure, which exhibits exceptionally high mechanical stability (15), and is resistant to elevated temperatures (at least 100°C), high salt concentrations, and acidic conditions (pH 2 to 7).
(iv) Production of the immobilized enzyme is coupled to photosynthetic diatom growth and thus is a renewable and CO2-consuming process.
Due to these compelling advantages, studies were initiated to investigate the scope of LiDSI, identify the bottlenecks for its application, and develop strategies to overcome these limitations. Previously, LiDSI had been demonstrated only for enhanced green fluorescent protein (eGFP) and the enzyme HabB (31). HabB is a “simple” enzyme that consists of a single small polypeptide chain (164 amino acid residues) and does not require posttranslational modifications or cofactors for activity (9). In the present work, the applicability of LiDSI for “complex” enzymes that are active only as oligomers or require posttranslational modifications or cofactors for activity was investigated.
MATERIALS AND METHODS
Chemicals and commercial enzymes.
Flavin adenine dinucleotide (FAD), p-nitrophenyl-β-d-glucuronide, 2-amino-2-methylpropanediol, glucose oxidase from Aspergillus niger (≥100 U/mg, solid), and horseradish peroxidase (250 U/mg, solid) were purchased from Sigma-Aldrich. 5-Bromo-4-chloro-3-indolyl β-d-glucuronide was purchased from AppliChem. Glucose and o-dianisidine-dihydrogen chloride were purchased from Alfa Aesar. Galactose was purchased from JT Baker. Igepal-CA630 was purchased from Fluka.
Cloning of silaffin-enzyme fusion genes.
For construction of the silaffin-enzyme expression vector, a fragment of the T. pseudonana silaffin3 gene (28), denoted truncation 8 (T8) was amplified by PCR using oligonucleotides 5′-GCT AGA TAT CAT AAT CAT GAA GAC TTC TGC CAT TGT A-3′ and 5′-CGC CGA TAT CAA TGC TCT GCA GCT TCC CAC TCT TTC CCT TG-3′ (EcoRV sites in italics) and ligated into the EcoRV site of pTpNR/egfp (30). The unique PstI site (underlined) at the 3′ end of T8 and the unique NotI site at the 3′ end of the enhanced green fluorescent protein encoding gene, egfp, in the resulting vector pTpNR/T8-PstIegfpNotI allowed for the in-frame insertion of genes and deleted the egfp gene. The gene encoding β-glucuronidase, gusA, from Escherichia coli was amplified from pKmobGII (22) using the oligonucleotides 5′-GAA GCT GCA GAT GTT ACG TCC TGT AGA AAC C-3′ and 5′-GAA TGC GGC CGC TCA TTG TTT GCC TCC CTG-3′(PstI site underlined; NotI site in italics) and then ligated into the PstI and NotI sites of vector pTpNR/T8-PstIegfpNotI, resulting in the final plasmid pTpNR/T8-gusA.
The gene encoding glucose oxidase, gox, was PCR amplified from plasmid pEGLS (16) using oligonucleotides 5′-GAA GCT GCA GAT GGG AAT CGA AGC AAG TCT GC-3′ and 5′-GAA TGC GGC CGC TCA CTG CAT AGA AGC GTA ATC C-3′ (PstI site underlined; NotI site in italics). The gene encoding horseradish peroxidase, hrp, was PCR amplified from plasmid pBBG10 (37) using oligonucleotides 5′-CCT GCA GGA TG CAG TTA ACC CCT ACA TTC-3′ and 5′-GCG GCC GCT TAA GAG TTG CTG TTG ACC AC-3′ (SbfI site underlined; NotI site in italics). The gene encoding galactose oxidase, gaox, was PCR amplified from plasmid pR3 (24) using oligonucleotides 5′-GAG CTC GCC TCA GCA CCT ATC GGA A-3′ and 5′-GCG GCC GCT CAC TGA GTA ACG CGA ATC G-3′(SacI site underlined; NotI site in italics). The resulting PCR products were cloned into the PstI and NotI sites of plasmid pTpNR/T8-gusA, yielding pTpNR/T8-gox, pTpNR/T8-hrp, and pTpNR/T8-gaox, respectively. To construct a T8-GOx encoding fusion gene under the control of the fcp promoter and with the nourseothricin resistance gene, nat, the fcpNat(−KpnI) DNA fragment was cut out of plasmid pTpfcp/ nat(−Kpn) (34) using HindIII (blunt) and XbaI and ligated to the SmaI- and XbaI-digested vector pTpfcp/egfp (30), resulting in plasmid pTpfcp/egfp/Tpfcp/nat(−KpnI). The T8-gox gene was excised from the plasmid pTpNR/T8-gox with EcoRV and NotI and cloned into the EcoRV and NotI sites of plasmid pTpfcp/egfp/Tpfcp/nat(−KpnI), yielding pTpfcp/T8-gox/Tpfcp/nat(−KpnI).
All PCR products were verified by DNA sequencing. The plasmid constructs were introduced into T. pseudonana cells by microparticle bombardment using an established method (30).
Cultivation of T. pseudonana.
T. pseudonana cells were grown in NEPC medium under constant illumination as previously described (28). For T8-GAOx expression experiments, cells were also grown in NEPC medium supplemented with 1.6 μM CuCl2 · 2H2O. For FAD spiking experiments, select T8-GOx transformants were grown in the presence of 1.0 μM FAD for 48 h.
Enzyme assays. (i) β-Glucuronidase (GUS).
The activity of GUS was assayed by a histochemical method and a spectrophotometric method according to the method described by Jefferson (20). For the histochemical assay, cells were resuspended in 200 μl assay buffer [0.1 M Na-phosphate, pH 7.0, 10 mM EDTA, pH 8.0, 0.1% (vol/vol) Triton X-100, 1 mM K3Fe(CN)6, 2 mM 5-bromo-4-chloro-3-indolyl-beta-d-glucuronic acid] and incubated overnight at 37°C. Subsequently, cells were pelleted and inspected for the presence of a blue color. The spectrophotometric assay was used for quantification of GUS activity. T. pseudonana cells were resuspended in GUS assay buffer (1.0 ml; 0.1 M sodium phosphate, pH 7.0, 10 mM EDTA, pH 8.0, 0.1% [vol/vol] Triton X-100) supplemented with p-nitrophenyl-β-d-glucuronide and incubated for 30 to 60 min at 37°C. The reaction was stopped by the addition of 2-amino-2-methylpropanediol, and the absorbance of liberated 4-nitrophenol in the reaction mixture was measured at 415 nm.
(ii) Glucose oxidase (GOx) and galactose oxidase (GAOx).
To determine the activity of GOx (or GAOx), samples were incubated for 20 min at room temperature in 200 μl 50 mM sodium acetate, pH 5.2, containing 90 mM glucose (or galactose). Subsequently, the reaction mixture was centrifuged (5 min, 16,000 × g), and the supernatant were added to 55 μl ODS/HRP buffer (0.8 mM o-dianisidine-dihydrogen chloride, 2.5 U horseradish peroxidase, 0.1 M potassium phosphate buffer, pH 6.0). After 5 min at room temperature, the absorbance at 410 nm was measured using a plate reader (Biotek Synergy 2).
(iii) Horseradish peroxidase (HRP).
The activity of HRP in biosilica was quantified by incubation in 255 μl assay buffer (3.9 mM H2O2, 78 mM potassium phosphate, pH 6.0, 17 μM ODS) at room temperature for 20 min. Subsequently, the biosilica was pelleted by centrifugation (5 min, 16,000 × g), and the supernatant was used to measure absorbance at 410 nm. Background peroxide concentrations from cells were measured by incubating wild-type cells with H2O2 (3.9 mM) for 20 min at room temperature and by subsequent addition of ODS.
Isolation of diatom silica.
For T. pseudonana transformants expressing GUS, ∼1 ×106 cells were resuspended in lysis buffer (0.1 M sodium phosphate, pH 7.0, 1 mM phenylmethylsulfonyl fluoride) and lysed by vortexing for 1 min with glass beads (diameter, 0.25 to 0.30 mm). The insoluble material was washed three times with lysis buffer, extracted for 30 min at 37°C with 1% (vol/vol) Igepal-CA630 in lysis buffer, and finally subjected to three washes with lysis buffer to remove the detergent. Diatom silica was isolated from T. pseudonana wild-type cells according to the same method to serve as a negative control for GUS activity.
For all other T. pseudonana transformants, cells were resuspended in lysis buffer 2 (50 mM sodium acetate, pH 5.2, 1× protease inhibitor cocktail) and then lysed by vortexing (3 times, 30 s) with glass beads (diameter, 0.25 to 0.30 mm). The lysate was centrifuged (5 min, 16,000 × g, 4°C), and the crude biosilica was washed three times with lysis buffer 2. The organic material was extracted three times for 5 min at room temperature with 1% (vol/vol) Igepal-CA630 (in lysis buffer 2), and the biosilica was washed five times with 50 mM sodium acetate, pH 5.2.
For negative controls, diatom silica was isolated from wild-type T. pseudonana according to the methods described above.
Determination of GOx mass immobilized in diatom biosilica.
FAD was extracted from 14 mg biosilica (isolated from GX1) by incubation in 8 M urea (in 50 mM sodium acetate, pH 5.2) for 48 h at room temperature. After centrifugation (5 min, 16,000 × g) the supernatant was subjected to FAD fluorescence intensity measurements (excitation, 365 nm; emission, 525 nm; slit width, 5 nm) using a spectrofluorophotometer (Shimadzu).
Adsorption of GOx-PA to diatom silica in vitro.
T. pseudonana biosilica was isolated by extraction with a hot solution of SDS as described previously (28). A fixed amount of biosilica (10 μmol SiO2) was incubated with various amounts of glucose oxidase cross-linked to protamine (GOx-PA) (14) for 20 h at room temperature. Subsequently, the biosilica was pelleted by centrifugation (5 min, 16,000 × g), and the pellet and supernatant were collected (fraction Sup1). The pelleted biosilica was washed three times with 50 mM sodium acetate, pH 5.2, and the supernatants were combined with fraction Sup1. The GOx activities in Sup1 (“unbound” fraction) and in the final pellet (“bound” fraction) were determined as described above. The amount of GOx-PA required to saturate the biosilica surface was calculated from the GOx activity in the “bound” fraction of those samples, which exhibited GOx activity also in the “unbound” fraction.
GOx stability assays.
Biosilica-immobilized GOx and free GOx in solution were incubated under the following conditions: (i) 50 mM sodium acetate, pH 5.2, at 60°C for 2 h, (ii) 50 mM sodium phosphate, pH 6.0, containing 1 mg ml−1 pronase for 24 h at room temperature, and (iii) 60 days at room temperature or 4°C either in 50 mM sodium acetate, pH 5.2, or freeze-dried. GOx activity was determined after each treatment as described above.
To determine the reusability of biosilica immobilized GOx, the biosilica was resuspended in 50 mM sodium acetate, pH 5.2, 90 mM glucose, incubated for 20 min, and then pelleted by centrifugation (16,000 × g, 5 min). This procedure was repeated three times, and GOx activity was measured after each resuspension.
Cytotoxicity assays.
Cell suspensions (5 × 105 cells ml−1) of T. pseudonana wild-type and T8-GOx-expressing transformants were grown in NEPC medium supplemented with 50 mM glucose. At the times indicated, aliquots were removed from each sample to determine cell number and H2O2 concentration using the HRP assay described above.
Determination of SiO2 content in biosilica.
Biosilica isolated from diatoms was dissolved by treatment with 1 M NaOH for 30 min at 95°C. The solution was centrifuged (16,000 × g, 5 min) and the Si content of the supernatant was determined using the β-silicomolybdate method (19).
RESULTS
β-Glucuronidase.
The enzyme β-glucuronidase (GUS) from Escherichia coli (encoded by the gusA gene) is cofactor independent and is active only as a homotetramer (molecular mass, 272 kDa). It is widely used as a reporter enzyme in molecular genetic analysis of eukaryotic organisms, especially plants (18). To test whether this multimeric enzyme is susceptible to LiDSI, a fusion gene was constructed that consisted of the gusA gene fused to a silaffin encoding gene. It was hypothesized that assembly of the GUS subunits may be impeded if they are expressed as a fusion protein with the full-length polypeptide chain of silaffin tpSil3 (231 amino acid residues). Systematic truncation analysis with tpSil3-eGFP fusion proteins expressed in the diatom Thalassiosira pseudonana (N. Poulsen and N. Kröger, unpublished results) has revealed that 37-amino-acid-residue-bearing peptide truncation T8 (residues 164 to 200 of tpSil3), in combination with the N-terminal signal peptide (SP) for cotranslational endoplasmic reticulum (ER) import (amino acid residues 1 to 17 of tpSil3), is sufficient for targeting eGFP to the T. pseudonana cell wall (see Fig. S1 in the supplemental material). Therefore, an SP-T8-gusA fusion gene was constructed (Fig. 2A) and incorporated into plasmid pTpNR, which contains the Pnr2/Tnr2 expression cassette that allows for the control of the fusion protein based on the nitrogen source in the medium. In the presence of nitrate protein, expression is switched on and is switched off in the presence of ammonium (30). The recombinant plasmid was introduced into T. pseudonana cells along with a nourseothricin resistance gene using a previously established protocol (30), and clones were selected on agar plates containing nitrate and nourseothricin. A total of 39 nourseothricin-resistant clones were analyzed for GUS activity using a histochemical assay (20), and 11 clones were positive in this screen. As expected, GUS activity was detectable only when cells were grown on nitrate and was absent when ammonium was the sole nitrogen source (Fig. 2B). The variation in GUS activity among the transformants is likely due to random integration of the gusA gene into the diatom genome, resulting in transformant strains that carry different copy numbers of the transgene within genome regions of different transcriptional activities (30).
Fig 2.
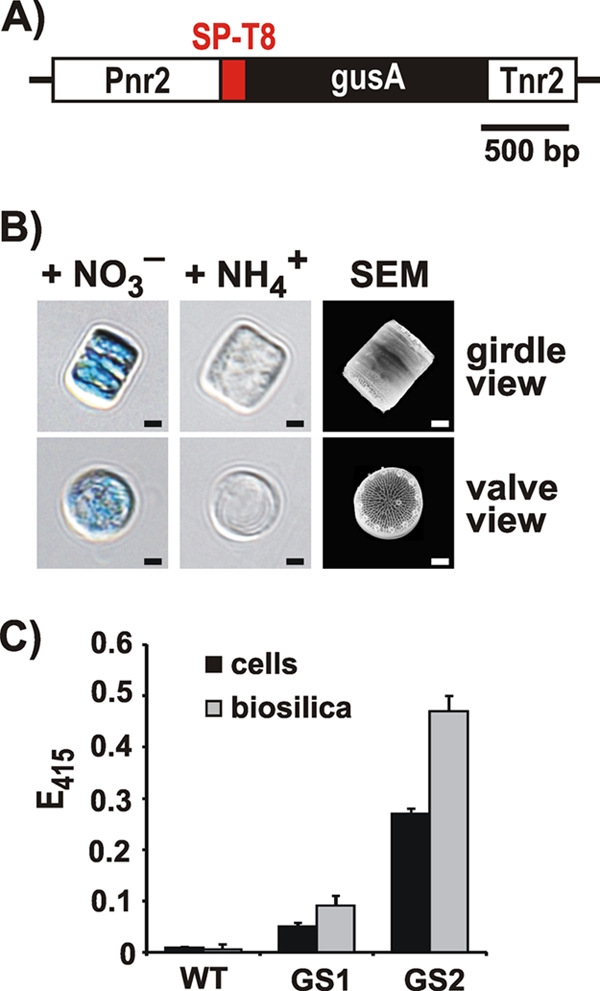
Expression of the SP-T8-GUS fusion protein in T. pseudonana. (A) The recombinant fusion gene encoding SP-T8-GUS was placed under the control of the inducible expression cassette Pnr2/Tnr2 that was derived from the T. pseudonana nitrate reductase gene (30). (B) Histochemical assay for GUS activity (20) with cells from T. pseudonana transformant clones that contain the T8-GUS fusion protein. The cells were grown with ammonium (NH4+) or nitrate (NO3−) as the sole nitrogen source in the medium. The scanning electron micrograph (SEM) shows intact biosilica cell walls from two diatom cells, one in girdle view (top) and the other in valve view (bottom). Scale bars: 1 μm. (C) Photometric assay for GUS activity (20) with intact cells and isolated biosilica from wild-type (WT) T. pseudonana and two independent transformant clones (GS1, GS2). Equal numbers of cells from each strain were used for biosilica isolation and live cell activity assays. The height of each bar represents the average value from the analyses of three independent experiments.
To test whether T8-GUS was stably attached to diatom silica, cells from two independent transformant clones were extracted with the nonionic detergent Igepal, which allows for isolation of cell-free biosilica (31). Interestingly, GUS activity was substantially higher in isolated biosilica compared to the intact cells. This effect was not caused by contaminating activity from other cellular hydrolases in the biosilica preparation, because biosilica from wild-type cells did not exhibit GUS activity (Fig. 2C). It is possible that cell wall-associated polysaccharides, glycoproteins, or other biomolecules that are present in intact diatom cells and absent in isolated diatom biosilica (17, 23) may inhibit GUS activity (2, 10).
The result described above demonstrated that silaffin fragment T8 (i) is able to target GUS for incorporation into diatom silica in vivo, (ii) does not interfere with homotetrameric assembly and activity of GUS, and (iii) provides a stable anchor for immobilizing GUS in biosilica.
Glucose oxidase.
The enzyme glucose oxidase (GOx) from Aspergillus niger is a homodimer of 128 kDa, and it requires the noncovalently bound redox cofactor FAD for activity (1 molecule FAD per GOx monomer). It is substantially glycosylated, containing predominantly mannose but also hexosamine and glucose (3). GOx catalyzes the oxidation of d-glucose by O2 to d-glucono-δ-lactone and H2O2. Immobilization of GOx is of interest for a wide variety of applications, including use in biosensors and biofuel cells (3). We hypothesized that GOx may be compatible with LiDSI, since immobilization of multimeric enzymes in diatom silica is feasible using the truncated silaffin domain T8 (see previous paragraph), and FAD is present inside the lumen of the ER (8) through which silaffin fusion proteins pass before they become incorporated into diatom silica (36).
To attempt LiDSI of GOx, a fusion gene encoding SP-T8-GOx was constructed, incorporated into diatom expression plasmid pTpNR, and introduced into T. pseudonana by biolistic particle delivery. The cells from 49% of the nourseothricin-resistant transformant clones exhibited GOx activity, whereas, as expected, no GOx activity was detected with wild-type cells (Fig. 3A). The T8-GOx expressing clones exhibited a wide range of GOx activity per cell (Fig. 3A), which is consistent with previous results from diatom transformation experiments using other genes (29–31) The differences in GOx activities in different clones may be caused by the following effects.
(i) Integration of the heterologous gene into the diatom genome is random; thus the transformant clones may contain different copy numbers of the heterologous gene, which may have also inserted into regions with different transcriptional activities.
(ii) The primary transformant clone may not be uniclonal, thus containing cells with different GOx activity levels.
(iii) Intracellular FAD availability may be limited, and thus the efficiency of loading apo-GOx (i.e., the cofactor-free enzyme) with cellular FAD may not be equally efficient in each clone; this may result in the incorporation of various amounts of inactive apo-GOx into the diatom silica.
Fig 3.

Activity and stability of T8-GOx expressed in T. pseudonana. (A) GOx activity in live cells of wild-type (WT) T. pseudonana and 10 independent transformant clones (GX1-10) that express the gene encoding fusion protein SP-T8-GOx under the control of the Pnr2/Tnr2 expression cassette. The activities were normalized to 109 cells. One unit corresponds to production of 1.0 μmol H2O2 per minute. (B) Biosilica was isolated from the cells of two independent T8-GOx containing transformant clones (GX1, GX2). Equal numbers of cells from each strain were used for biosilica isolation and live cell activity assays. The relative GOx activities of live cells of each transformant cell line were set to 100%. (C) Stability toward heat treatment (2 h at 60°C), and incubation with pronase (Pron.) of free GOx and biosilica-immobilized T8-GOx from clone GX1. The activity levels of free GOx and biosilica-immobilized T8-GOx before treatment were set to 100% (Ctr.).
Analysis of possibility i was beyond the scope of the present work, but possibility ii could be investigated by subcloning. A total of 2.0 × 103 cells of each T8-GOx-expressing clone GX1, GX3, GX6, and GX8 were plated on nourseothricin-containing agar plates. From each plate 10 subclones were cultivated in liquid medium and analyzed for GOx activity. Some of the subclones exhibited significantly higher activities than their parent clones (see Table S1 in the supplemental material), indicating that the parent clone was not uniclonal but rather consisted of a mixture of clones with different GOx activity levels. However, out of 40 subclones tested, none exhibited significantly higher activity than clone GX1. Therefore, it was concluded that clone GX1 contains the highest possible GOx activity that can be achieved with LiDSI using the fusion gene that encodes SP-T8-GOx.
To investigate whether FAD availability may play a role in the observed clonal variation of GOx activity (possibility iii), four T. pseudonana transformants (GX1, GX3, GX6, GX8) representing different levels of GOx activity were grown for 5 days in the presence of FAD. It was assumed that FAD addition may supplement the intracellular FAD pool and enable activation of cell wall-associated apo-GOx. FAD addition did not result in an increase in GOx activity for any of the transformant clones (see Fig. S2 in the supplemental material), suggesting that FAD availability was not a limiting factor for GOx activity.
As T8 is a rather short fragment (37 amino acids) compared to the mature tpSil3 protein (205 amino acid residues), it was not clear whether the T8-GOx fusion protein would remain attached to the diatom silica during biosilica isolation. To investigate this question, the biosilica from cells of transformant clones GX1 and GX2 was isolated by detergent extraction. The isolated biosilica exhibited 73.6% ± 0.8% (GX1) and 75.9% ± 1.1% (GX2) of the GOx activity that was present in the respective live cells (Fig. 3B). This demonstrated that silaffin fragment T8 was functional both in targeting of GOx to the SDV and serving as an anchor for tight association with the diatom silica.
It is conceivable that fusion of GOx to the T8 fragment and incorporation into biosilica, which contains a complex mixture of highly charged biopolymers (glycoproteins and polyamines) (23), may exert an inhibitory effect on the enzyme's activity. This effect would become noticeable in a reduction of the specific activity (i.e., catalytic activity per mol of enzyme). To determine the specific activity of diatom silica-immobilized T8-GOx, the amount of biosilica-associated fusion protein was quantified via the fluorescence of the cofactor FAD, which can be extracted from GOx by treatment with urea (1). The urea extract from biosilica of clone GX1 yielded 0.53 ± 0.02 μmol of FAD per mol of silica in fluorescence spectroscopy measurements (see Fig. S3 in the supplemental material). The catalytic activity of T8-GOx in GX1 biosilica was 2,980 ± 104 U per mol silica, which—considering the 1:1 stoichiometry of FAD:GOx—translates to a specific activity of 5.62 ×109 U per mol of GOx. This value represents 81.6 ± 3.6% of the specific activity of A. niger GOx in solution (6.97 ×109 ± 2.53 ×108 U per mol GOx), thus demonstrating that fusion to the truncated silaffin domain T8 and immobilization inside biosilica exert only a mildly inhibitory effect on the catalytic activity of GOx.
Immobilization in a solid support can lead to stabilization of an enzyme's activity (5, 12, 13). To investigate whether incorporation into diatom silica affects the stability of GOx activity, the biosilica isolated from transformant GX1 was subjected to various treatments that are known to cause inactivation of GOx in solution. When incubated at 60°C for 2 h, diatom silica-immobilized GOx retained 66.7% ± 1.3% of its activity, whereas the activity of GOx in solution decreased to 26.1% ± 0.9% (Fig. 3C). Biosilica-immobilized GOx was also substantially stabilized against treatment with pronase (i.e., a mixture of unspecific proteases) (27) compared to free GOx in solution (Fig. 3C). The stabilization of biosilica-immobilized GOx toward heat treatment and proteolytic attack has previously been observed also for enzymes that were immobilized in synthetic silica matrices in vitro (7, 14, 21). It has been hypothesized that the immobilized enzyme molecules reside within nanocavities inside the silica that provide a barrier against unfolding of the enzyme during heating and make the enzyme less accessible to proteases. Such cavities are not tightly sealed but rather are connected to the surrounding medium through a network of nanoscale channels that enable diffusion-mediated exchange of material with the surroundings (21).
Due to the increased conformational stability of biosilica-immobilized GOx, the shelf life of the enzyme was significantly increased both in aqueous solution as well as in the dry state compared to free GOx (Table 1). Furthermore, biosilica-immobilized GOx could easily be recovered by centrifugation after completion of the reaction with hardly any loss in activity (retention of 97.2% ± 0.6% activity after 3 cycles of reaction and recovery).
Table 1.
Stability of GOx during long-term storage (60 days)a
| Storage type and temp | Amta |
|
|---|---|---|
| Free GOx | LiDSI GOx | |
| In aqueous buffer | ||
| RTb | 43.0 ± 1.1 | 71.2 ± 0.8 |
| 4°C | 70.9 ± 2.1 | 81.2 ± 1.3 |
| Freeze-dried | ||
| RT | 66.3 ± 2.3 | 78.8 ± 1.6 |
| 4°C | 81.1 ± 1.3 | 87.2 ± 1.5 |
Numbers represent % activity relative to day 0.
RT, room temperature.
The above data demonstrated the successful production of silica-immobilized GOx that is readily reusable and exhibits enhanced stability and shelf life. To further improve the method, it would be desirable to enhance the loading of enzyme within the biosilica, which was only 0.77 ± 0.03 mg GOx per g of silica (∼0.1%) in the case of GX1, the transformant with the highest GOx activity. In this clone, expression of the T8-GOx fusion protein is driven by the nitrate reductase promoter (see above). It is possible that enzyme loading of diatom silica could be improved by utilizing a stronger promoter for T8-GOx expression. To test this possibility, the GOx fusion gene was placed into the Pfcp2/Tfcp2 expression cassette, because Pfcp2 (derived from gene LHCF9) appears to be the strongest promoter in T. pseudonana as suggested by the predominance of LHCF9 mRNA in the transcriptome (30). After introduction of the modified expression plasmid into T. pseudonana, nourseothricin-resistant transformant clones were analyzed for GOx activity. Out of 10 clones with GOx activity (GX*1 to GX*10), none exhibited a significantly higher GOx activity than clone GX1 (see Fig. S4 in the supplemental material). This result suggests that factors other than promoter strength are limiting the amount of enzyme that can be incorporated into diatom silica through LiDSI.
To obtain a reference point for high enzyme loading in diatom silica, GOx-PA, a silica binding derivative of A. niger GOx (14), was attached to biosilica isolated from T. pseudonana wild-type cells. GOx-PA has the same specific activity as GOx (14). GOx-PA-saturated T. pseudonana biosilica exhibited an enzyme activity level of 1.38 × 104 ± 6.74 × 102 U per mol silica (see Fig. S5 in the supplemental material), which was 4.6 times the GOx activity level of the biosilica obtained from the T8-GOx expressing transformant clone GX1. These data indicate that enzyme loading of diatom silica through LiDSI is not limited by the available surface area.
The GOx-catalyzed reaction produces H2O2 which is a strongly cytotoxic agent (11). Therefore, the rate of H2O2 production in T8-GOx containing cells and the ability to induce cell death in the presence of glucose were investigated. For this experiment, three T8-GOx expressing clones were chosen (GX1, GX6, GX8) that were representative of the different levels of cellular GOx activity of the T. pseudonana transformants. After addition of glucose to the medium, cells were kept under optimal growth conditions, and the number of live cells (Fig. 4A) and H2O2 concentration in the medium (Fig. 4B) were monitored at 24-h intervals. In the presence of glucose, wild-type growth was unperturbed compared to that in glucose-free medium. Cell numbers continuously increased until day 8 and in subsequent days remained stationary (Fig. 4A). In the culture of clone GX1, which expressed the highest GOx activity, the number of live cells started to decline after day 2, and after 8 days virtually all cells in the cultures of this clone were dead (Fig. 4A). In contrast, in the culture of clone GX8, which expressed the lowest level of GOx activity, cell numbers developed as in the wild-type culture up to day 5 before cell lethality became noticeable (Fig. 4A). The lethality rate of cells in the culture of clone GX6, which expressed a medium level of GOx activity, was intermediate between those of clones GX1 and GX8 (Fig. 4A). The differences in cell lethality rates in the glucose-containing cultures of the T8-GOx-expressing clones were in perfect agreement with the differences in the rates of H2O2 production (Fig. 4B), demonstrating that H2O2 produced from GOx-mediated glucose oxidation caused diatom cell death.
Fig 4.
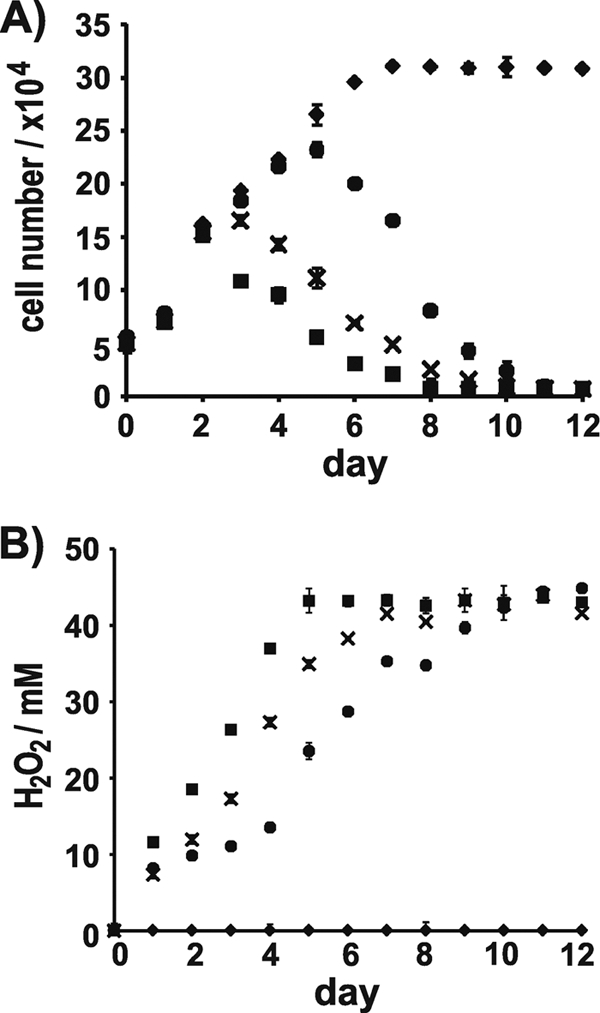
Cytotoxicity of T. pseudonana strains grown in the presence of glucose. (A) Development of cell number and (B) H2O2 content in cell cultures of WT (♦) and of T8-GOx containing transformants GX1 (■), GX6 (×), and GX8 (•). The height of each bar represents the average value from the analyses of three independent experiments.
The results from the cell lethality assays demonstrate that T8-GOx has the properties of a “suicide gene” in the presence of glucose. To our knowledge, this is the first negative selection marker described for diatoms to date. Such markers are useful tools for “knocking out” genes by homologous recombination (6), and thus T8-GOx could be utilized for establishing this important genetic technique in diatoms.
Horseradish peroxidase.
The enzyme horseradish peroxidase (HRP) from Amoracia rusticana is a 44-kDa monomer that requires one heme group and two Ca2+ ions for activity (40). It catalyzes the reaction of H2O2 with a wide variety of reducing substrates, including aromatic phenols, indoles, and amines (40). HRP is a widely used enzyme in immunoassays, for organic synthesis, in treatment of wastewaters, and for biosensors (33). Insertion of Ca2+ ions into T8-HRP expressed in diatoms is expected to occur in the ER or Golgi apparatus as both compartments are intracellular Ca2+ storage compartments in eukaryotic cells (26). However, hardly anything is known about the mechanism and intracellular site for heme group insertion into apo-hemo-proteins (35). Therefore, it was unclear whether LiDSI of HRP could be successful.
To investigate this question, an SP-T8-HRP encoding fusion gene was constructed, inserted into the expression plasmid under the control of the Pnr2/Tnr2 expression cassette, and introduced into T. pseudonana as described above. Of 37 nourseothricin-resistant clones 25 were positive in HRP assays, whereas wild-type cells were negative (Fig. 5A). After extraction of clones HP1, HP2, and HP3 with detergent, 72 to 78% of the HRP activities of the corresponding live cells were retained in the isolated biosilica (Fig. 5A). This result demonstrated that Ca2+ and heme are available for incorporation into apo-proteins on the intracellular route from the ER to the SDV in diatoms (Fig. 1).
Fig 5.
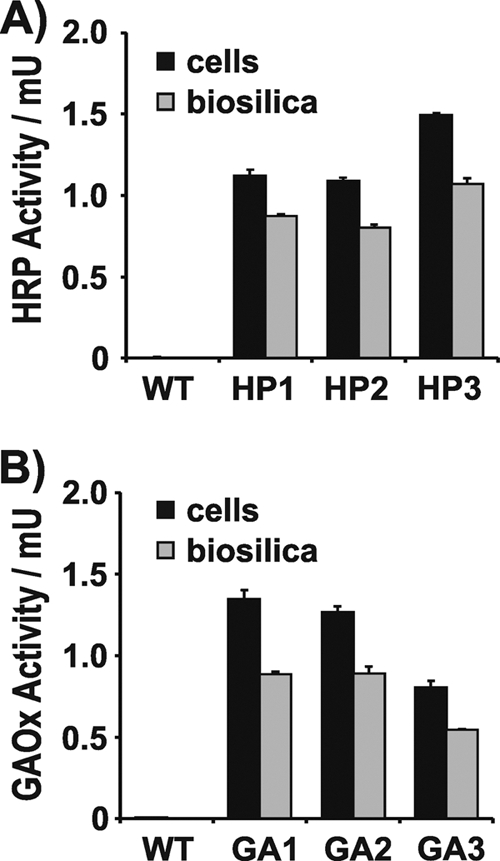
Expression in T. pseudonana of SP-T8-HRP (A) and SP-T8-GAOx (B). The activities of intact cells and isolated biosilica from wild-type T. pseudonana and three independent T. pseudonana transformant clones are shown. Equal numbers of cells from each strain were used for biosilica isolation and live cell activity assays. For HRP, 1 unit corresponds to 1.0 μmol H2O2 consumed per minute. For GAOx, 1 unit corresponds to 1.0 μmol H2O2 produced per minute. The height of each bar represents the average value from the analyses of three independent experiments.
Galactose oxidase.
Lastly, it was investigated whether LiDSI is compatible with Cu-dependent redox enzymes. Galactose oxidase (GAOx) from the fungus Fusarium sp. was chosen as a model enzyme for these studies. GAOx is a 68-kDa monomeric enzyme containing a mononuclear Cu center and an unusual posttranslational modification (tyrosine-cysteine cross-link) in the active site, which is important for redox activity of the enzyme (42).
A fusion gene encoding SP-T8-GAOx under the control of the Pnr2/Tnr2 expression cassette was constructed and introduced into the T. pseudonana genome. Fifteen nourseothricin resistant clones were obtained, and none of them exhibited GAOx activity. It was speculated that the availability of Cu ions in the culture medium may have been limited, thereby not allowing for functional expression of an additional Cu-dependent enzyme. Therefore, the experiment was repeated with the addition of CuCl2 to the culture medium. This time only 10 nourseothricin-resistant clones were obtained, but 6 of them exhibited GAOx activity. The biosilica isolated from the cells of three clones with GAOx activity retained 66 to 71% of the catalytic activity (Fig. 5B), thus demonstrating that most of the T8-GAOx fusion protein was stably associated with diatom silica.
The successful demonstration of LiDSI for GAOx supports the hypothesis that incorporation of the Cu ion and formation of the functionally essential tyrosine-cysteine cross-link happen spontaneously during GAOx maturation and do not require protein cofactors (42).
DISCUSSION
In the present study has been demonstrated the applicability of LiDSI for oligomeric enzymes and for redox enzymes that require FAD, heme, and Cu ions as cofactors. The success of LiDSI for cofactor-dependent enzymes is limited to cofactors that are available within the intracellular compartments through which the silaffin fusion proteins pass on their route to the cell wall (ER, Golgi apparatus, SDV) (Fig. 1). In cases in which specific chaperones are required for cofactor incorporation or subunit assembly, such chaperones would need to be coexpressed with the silaffin-enzyme fusion protein to generate a functional enzyme complex. LiDSI should be suitable also for disulfide-bond containing enzymes and receptor proteins (e.g., immunoglobulins), because the ER lumen naturally contains the redox milieu and chaperones for disulfide bond formation (32). It seems unlikely that LiDSI would be applicable for proteins carrying multiple transmembrane spanning domains, because the diatom cell wall is generally believed to be composed of silica, (glyco-)proteins, and polysaccharides and not to be surrounded by a lipid bilayer (23). However, recent analyses using various spectroscopic techniques have challenged this view, as significant amounts of lipids were identified as biosilica-associated components in the diatoms T. pseudonana and Phaeodactylum tricornutum (38, 39).
LiDSI is not only an environmentally friendly (“green”) method for enzyme immobilization, it provides at the same time a desirable immobilization matrix. The hierarchical nano-to-microscale porosity of diatom silica endows this material with high mechanical stability, and advantageous mass transport characteristics for applications in macromolecular separations, biochemical sensors, and flowthrough catalysis (15, 25, 43). Furthermore, LiDSI does not require that the enzyme be isolated, and the immobilization proceeds under physiological conditions. This will be advantageous when isolation of the enzyme is tedious and expensive, and/or when the isolated enzyme is rather unstable (4, 5). However, a disadvantage of LiDSI is that currently only ∼0.1% (wt/wt) enzyme loading of the silica can be achieved. In comparison, in vitro silica immobilization methods yield typically ∼10 times higher enzyme loadings (12). It was demonstrated here that up to 0.43% (wt/wt) enzyme could be attached just to the T. pseudonana silica surface in vitro. This loading is likely not even the maximum that can potentially be achieved with diatom silica using LiDSI, as LiDSI-immobilized enzymes are both exposed on the surface and embedded inside the diatom silica (reference 31 and this work). It is hypothesized that a major bottleneck for enzyme loading by LiDSI is the capacity for intracellular transport of silaffin fusion genes to the SDV. Being able to widen this potential bottleneck requires knowledge about the peptide sequence(s) and the corresponding receptor protein(s) that mediate silaffin targeting to the SDV. Such knowledge should allow for the design of improved peptide sequences for SDV targeting and for genetic engineering of T. pseudonana strains that contain substantially enhanced levels of targeting receptors. The discovery that silaffin truncation peptide T8 contains sufficient targeting information for delivery to the SDV, which is described in the present work, is a first step toward identifying the molecular mechanism of the SDV targeting process.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported through grants from the Department of Energy–BES (Biomolecular Materials grant ER46592), the National Science Foundation (DMR grant 0845939; identification of targeting peptide T8) to N.K., and a Postdoctoral Fellowship from the Deutsche Forschungsgemeinschaft to A.S. (SCHE1637/1-1; immobilization of GUS).
We thank the following colleagues for providing enzyme encoding genes: Dirk Schüler (LMU München) for sharing plasmid pKmobGII (gusA gene), Oded Shoseyov (The Hebrew University of Jerusalem) for plasmid pEGLS (gox gene), and Frances Arnold (California Institute of Technology) for plasmids pBBG10 (hrp gene) and pR3 (gaox gene).
Footnotes
Published ahead of print 4 November 2011
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Akhtar MS, Ahmad A, Bhakuni V. 2002. Guanidium chloride- and urea-induced unfolding of the dimeric enzyme glucose oxidase. Biochemistry 41:3819–3827 [DOI] [PubMed] [Google Scholar]
- 2. Bahieldin A, et al. 2005. Evidence for non-proteinaceous inhibitor(s) of β-glucuronidase in wheat (Triticum aestivum L.) leaf and root tissues. Plant Cell Tissue Organ Cult. 82:11–17 [Google Scholar]
- 3. Bankar SB, Bule MV, Singhal RS, Ananthanarayan L. 2009. Glucose oxidase—an overview. Biotechnol. Adv. 27:489–501 [DOI] [PubMed] [Google Scholar]
- 4. Bommarius A, Riebel BR. 2004. Introduction to biocatalysis. Wiley-VCH, Weinheim, Germany [Google Scholar]
- 5. Brady D, Jordan J. 2009. Advances in enzyme immobilization. Biotechnol. Lett. 31:1639–1650 [DOI] [PubMed] [Google Scholar]
- 6. Capecchi MR. 1989. Altering the genome by homologous recombination. Science 244:1288–1292 [DOI] [PubMed] [Google Scholar]
- 7. Chen Q, Kenausis GL, Heller A. 1998. Stability of oxidases immobilized in silica gels. J. Am. Chem. Soc. 120:4582–4585 [Google Scholar]
- 8. Csala M, Bánhegyi G, Benedetti A. 2006. Endoplasmic reticulum: a metabolic compartment. FEBS Lett. 580:2160–2165 [DOI] [PubMed] [Google Scholar]
- 9. Davis JK, et al. 2000. Sequence analysis and initial characterization of two isozymes of hydroxylaminobenzene mutase from Pseudomonas pseudoalcaligenes JS45. Appl. Environ. Microbiol. 66:2965–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fior S, Gerola PD. 2009. Impact of ubiquitous inhibitor on the GUS gene reporter system: evidence from the model plants Arabidopsis, tobacco and rice and correction methods for quantitative assays of transgenic and endogenous GUS. Plant Methods 5:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fuglsang CC, Johansen C, Christgau S, Nissen JA. 1995. Antimicrobial enzymes: applications and future potential in the food industry. Trends Food Sci. Technol. 61:390–396 [Google Scholar]
- 12. Gill I, Ballesteros A. 2000. Bioencapsulation within synthetic polymers (part 1): sol-gel encapsulated biologicals. Trends Biotechnol. 18:282–296 [DOI] [PubMed] [Google Scholar]
- 13. Gill I, Ballesteros A. 2000. Bioencapsulation within synthetic polymers (part 2): non-sol-gel protein-polymer biocomposites. Trends Biotechnol. 18:469–479 [DOI] [PubMed] [Google Scholar]
- 14. Haase NR, Shian S, Sandhage KH, Kröger N. 2011. Biocatalytic nanoscale coatings through biomimetic layer-by-layer mineralization. Adv. Funct. Mater. 21:4243–4251 [Google Scholar]
- 15. Hamm CE, et al. 2003. Architecture and material properties of diatom shells provide effective mechanical protection. Nature 421:841–843 [DOI] [PubMed] [Google Scholar]
- 16. Heyman A, Levy I, Altman A, Shoseyov O. 2007. SP1 as a novel scaffold building block for self-assembly nanofabrication of submicron enzymatic structures. Nano Lett. 7:1575–1579 [DOI] [PubMed] [Google Scholar]
- 17. Hoagland KD, Rosowski JR, Gretz MR, Roemer SC. 1993. Diatom extracellular polymeric substances—function, fine structure, chemistry, and physiology. J. Phycol. 29:537–566 [Google Scholar]
- 18. Huttly A. 2009. Reporter genes. Methods Mol. Biol. 478:39–69 [DOI] [PubMed] [Google Scholar]
- 19. Iler RK. 1979. The chemistry of silica, p 97 Wiley, New York, NY [Google Scholar]
- 20. Jefferson RA. 1987. Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol. Biol. Rep. 5:387–405 [Google Scholar]
- 21. Jin W, Brennan JD. 2002. Properties and applications of proteins encapsulated within sol-gel derived materials. Anal. Chim. Acta 461:1–36 [Google Scholar]
- 22. Katzen F, Becker A, Ielmini MV, Oddo CG, Ielpi L. 1999. New mobilizable vectors suitable for gene replacement in gram-negative bacteria and their use in mapping of the 3′ end of the Xanthomonas campestris pv. campestris gum operon. Appl. Environ. Microbiol. 65:278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kröger N, Poulsen N. 2008. Diatoms—from cell wall biogenesis to nanotechnology. Annu. Rev. Genet. 42:83–107 [DOI] [PubMed] [Google Scholar]
- 24. Lis M, Kuramitsu HK. 1997. Galactose oxidase-glucan binding domain fusion proteins as targeting inhibitors of dental plaque bacteria. Antimicrob. Agents Chemother. 41:999–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Losic D, Rosengarten G, Mitchell JG, Voelcker NH. 2006. Pore architecture of diatom frustules: potential nanostructured membranes for molecular particle separations. J. Nanosci. Nanotechnol. 6:982–989 [DOI] [PubMed] [Google Scholar]
- 26. Micaroni M, Mironov AA, Rizzuto R. 2010. The role of Ca2+ in the regulation of intracellular transport, p 143–160 In Mironov AA, Pavelka M. (ed), The Golgi apparatus. Springer, Vienna, Austria [Google Scholar]
- 27. Narahashi Y. 1970. Pronase. Methods Enzymol. 19:651–664 [Google Scholar]
- 28. Poulsen N, Kröger N. 2004. Silica morphogenesis by alternative processing of silaffins in the diatom Thalassiosira pseudonana. J. Biol. Chem. 279:42993–42999 [DOI] [PubMed] [Google Scholar]
- 29. Poulsen N, Kröger N. 2005. A new molecular tool for transgenic diatoms: control of mRNA and protein biosynthesis by an inducible promoter-terminator cassette. FEBS J. 272:3413–3423 [DOI] [PubMed] [Google Scholar]
- 30. Poulsen N, Chesley PM, Kröger N. 2006. Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J. Phycol. 42:1059–1065 [Google Scholar]
- 31. Poulsen N, Berne C, Spain J, Kröger N. 2007. Silica immobilization of an enzyme via genetic engineering of the diatom Thalassiosira pseudonana. Angew. Chem. Int. Ed. Engl. 46:1843–1846 [DOI] [PubMed] [Google Scholar]
- 32. Riemer J, Bulleid N, Herrmann JM. 2009. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science 324:1284–1287 [DOI] [PubMed] [Google Scholar]
- 33. Ryan BJ, Carolan N, O'Fágáin C. 2006. Horseradish and soybean peroxidases: comparable tools for alternative niches? Trends Biotechnol. 24:355–363 [DOI] [PubMed] [Google Scholar]
- 34. Scheffel A, Poulsen N, Shian S, Kröger N. 2011. Nanopatterned protein microrings from a diatom that direct silica morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 108:3175–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Severance S, Hamza I. 2009. Trafficking of heme and porphyrins in metazoa. Chem. Rev. 109:4596–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheppard VC, Poulsen N, Kröger N. 2010. Characterization of an endoplasmic reticulum-associated silaffin kinase from the diatom Thalassiosira pseudonana. J. Biol. Chem. 285:1166–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith AT, et al. 1990. Expression of a synthetic gene for horseradish peroxidase C in Escherichia coli and folding and activation of the recombinant enzyme with Ca2+ and heme. J. Biol. Chem. 265:13335–13343 [PubMed] [Google Scholar]
- 38. Tesson B, et al. 2008. Contribution of multi-nuclear solid state NMR to the characterization of the Thalassiosira pseudonana cell wall. Anal. Bioanal. Chem. 390:1889–1898 [DOI] [PubMed] [Google Scholar]
- 39. Tesson B, et al. 2009. Surface chemical composition of diatoms. Chembiochem 10:2011–2024 [DOI] [PubMed] [Google Scholar]
- 40. Veitch NC. 2004. Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 65:249–259 [DOI] [PubMed] [Google Scholar]
- 41. Wenzl S, Hett R, Richthammer P, Sumper M. 2008. Silacidins: highly acidic phosphopeptides from diatom shells assist in silica precipitation in vitro. Angew. Chem. Int. Ed. Engl. 47:1729–1732 [DOI] [PubMed] [Google Scholar]
- 42. Whittaker JW. 2003. Free radical catalysis by galactose oxidase. Chem. Rev. 103:2347–2363 [DOI] [PubMed] [Google Scholar]
- 43. Yang W, Lopez PJ, Rosengarten G. 2011. Diatoms: self asembled nanostructures, and templates for bio/chemical sensors and biomimetic membranes. Analyst 136:42–53 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.