

Abstract
A significant number of microorganisms from the human oral cavity remain uncultivated. This is a major impediment to the study of human health since some of the uncultivated species may be involved in a variety of systemic diseases. We used a range of innovations previously developed to cultivate microorganisms from the human oral cavity, focusing on anaerobic species. These innovations include (i) in vivo cultivation to specifically enrich for species actively growing in the oral cavity (the “minitrap” method), (ii) single-cell long-term cultivation to minimize the effect of fast-growing microorganisms, and (iii) modifications of conventional enrichment techniques, using media that did not contain sugar, including glucose. To enable cultivation of obligate anaerobes, we maintained strict anaerobic conditions in most of our cultivation experiments. We report that, on a per cell basis, the most successful recovery was achieved using minitrap enrichment (11%), followed by single-cell cultivation (3%) and conventional plating (1%). Taxonomically, the richest collection was obtained using the single-cell cultivation method, followed by minitrap and conventional enrichment, comprising representatives of 13, 9, and 4 genera, respectively. Interestingly, no single species was isolated by all three methods, indicating method complementarity. An important result is the isolation and maintenance in pure culture of 10 strains previously only known by their molecular signatures, as well as representatives of what are likely to be three new microbial genera. We conclude that the ensemble of new methods we introduced will likely help close the gap between cultivated and uncultivated species from the human oral cavity.
INTRODUCTION
According to 16S rRNA surveys, the typical oral community comprises over 700 bacterial species (1, 5, 31, 58), of which approximately 280 have been isolated in culture and formally named (37). It was estimated that less than half of bacterial species-level taxa from the oral cavity can be cultivated under anaerobic conditions (8, 16, 36, 37, 48). This is in general agreement with the estimate provided by the recently launched Human Oral Microbiome Database (HOMD; www.homd.org). Based on a 98.5% rRNA gene sequence similarity cutoff, this database lists 619 microbial phylotypes referred to as “oral taxa,” with one-third of them remaining uncultivated (19).
According to the HOMD, human oral microbiota comprises 13 phyla (Actinobacteria, Bacteroidetes, Chlamydiae, Chloroflexi, Euryarchaeota, Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes, SR1, Synergistes, Tenericutes, and TM7). An overwhelming number of species-level phylotypes (96%) fall into six phyla (Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Spirochaetes, and Fusobacteria). Three phyla (TM7, SR1, and Chloroflexi) are still not represented by a single cultivated oral species (19). Even though the proportion of uncultivated species is lower in the oral cavity (30 to 50%) (33, 56, 57) than in the environment (>99%) (27, 40), the “missing” oral species are a significant impediment to the study of human health. This is because there are indications that some of the uncultivated species may be involved in a variety of systemic diseases (4, 20, 44) and likely play an important role in the function of the oral microbial community. We have shown that some previously uncultivable microorganisms can be isolated by mimicking natural growth conditions, using in vivo incubation devices (25, 30, 34) or via enrichments (17, 46, 47), and also developed a method for single-cell long-term incubations (S. Buerger et al., submitted for publication). The main objective of this study was to apply these different approaches to the cultivation of oral microorganisms, assess their relative merits, and isolate new species from the list of presently uncultivated taxa.
MATERIALS AND METHODS
Samples and subject identification.
Samples of subgingival plaque for single-cell long-term cultivation and enrichment experiments were collected from nine individuals (subjects 1 through 9), who did not use antibiotics for 6 months before sampling. The overall concept was to screen subjects regardless of oral health status for high levels of microbial richness. Therefore, we did not perform clinical assessment of the subjects, but they were likely to be systemically and dentally healthy. Subjects refrained from oral hygiene (e.g., brushing and flossing). Subgingival samples were obtained with a sterile Gracey curette or toothpick at the Forsyth Institute and Northeastern University (Boston, MA). Informed consent was obtained from all enrolled individuals. The study protocol and informed consent were approved by the Institutional Review Board of Northeastern University and the Forsyth Institute.
The HOMIM.
The human microbial identification microarray (HOMIM) (37) allows the simultaneous detection of about 300 of the most prevalent oral bacterial species, including those that have not yet been cultivated. HOMIMs were used to screen six individuals for the presence and richness of uncultivated bacteria. Briefly, the 16S rRNA-based, reverse-capture oligonucleotide probes (typically 18 to 20 bases) were printed on aldehyde-coated glass slides (http://mim.forsyth.org/protocol.html). The 16S rRNA genes were PCR amplified from DNA extracts using 16S rRNA gene universal primers and labeled via incorporation of Cy3-dCTP in a second nested PCR. The labeled 16S amplicons were hybridized overnight to probes on the slides. After washing, the microarray slides were scanned using an Axon 4000B scanner, and crude data were extracted using GenePix Pro software. Data were normalized by comparing individual signal intensities to the average of signals from universal probes (38).
Media.
The following media were used in this study. Unless otherwise indicated, all individual components were purchased from Becton-Dickinson and Company (Sparks, MD) or Sigma-Aldrich (St. Louis, MO).
(i) BM.
Basic anaerobic medium (BM) contained the following components: yeast extract, 0.5 g/liter; Casamino Acids, 0.5 g/liter; NaHCO3, 0.5 g/liter; MgCl2 · 6H2O, 0.1 g/liter; NH4Cl, 0.4 g/liter; CaCl2 · 2H2O, 0.05 g/liter; FeCl2 · 4H2O, 0.05 g/liter; l-cysteine HCl, 0.5 g/liter; and resazurin, 0.0025 g/liter. In some cases, instead of Casamino Acids, we added either starch (1.0 g/liter) or xylan (1.0 g/liter), or BM was supplemented with 4 ml/liter of saliva collected from healthy individuals and sterilized by filtration or supplemented with 4 ml/liter defibrinated sheep blood (Quad Five, Ryegate, MT).
(ii) TY.
Trypticase-yeast extract (TY) contained the following components: Trypticase, 30.0 g/liter; yeast extract, 20.0 g/liter; hemin, 0.005 g/liter; MgCl2 · 6H2O, 0.1 g/liter; NH4Cl, 0.4 g/liter; CaCl2 · 2H2O, 0.05 g/liter; FeCl2 · 4H2O, 0.05 g/liter; l-cysteine HCl, 0.5 g/liter; and resazurin, 0.0025 g/liter.
(iii) TGY.
Trypticase-glucose-yeast extract (TGY) had the same composition as TY but was supplemented with 5.0 g/liter glucose.
For all media, l-cysteine HCl and minerals (except for NaHCO3) were prepared as a 100× concentrated stock solution, flushed with N2, and autoclaved. All sterile and reduced ingredients were combined in serum bottles inside an anaerobic glove box, sealed, and crimped.
For pour plating and isolation of single colonies, 15 g/liter of Bacto agar (BD) was added to liquid BM, while the yeast extract and Casamino Acids concentrations were increased to 5 g/liter.
Minitrap in vivo cultivation.
The trap method was described in detail elsewhere (22, 25). In short, a trap is a chamber or a series of minichambers containing sterile agar and separated from the outside environment by membranes. For this application, we used a miniature trap custom built by Hi-Tech Manufacturing LLC (Schiller Park, IL) (Fig. 1). The minitrap consisted of three surgical steel metal plates, each with 72 through holes 400 μm in diameter, with the through holes registered among the plates. The central plate (8.5 mm by 4 mm by 0.5 mm) was dipped into 1% (wt/vol) molten agar supplemented with BM. Once agar solidified and formed ≈0.1-μl plugs in the through holes, precut 1.0-μm-pore-size polycarbonate membranes (GE Water & Process Technologies, Burlington, MA) were applied from each side, pressed against the plate by two side plates 12 by 5.5 by 1 mm in size, and tightened with 2-mm screws. The minitrap was assembled aseptically, using only sterilized parts and assembly tools.
Fig 1.

Minitrap used for in vivo cultivation of oral microorganisms. (Left) Basic design (explanations in the text). (Right) General view of the subjects' dental appliance, with the minitrap glued to a window cut in the appliance.
At the Forsyth Institute, the minitrap was inserted into a window precut in a palatal appliance molded from the upper maxilla of subject 3. The minitrap was positioned on the left quadrant of the upper lingual side adjacent to the gum line for the premolar, 1st molar, and 2nd molar teeth. The minitrap was affixed with Superglue. The subject was allowed to eat, drink, and perform normal oral hygiene, except for flossing. After a 48-h-long in vivo incubation, the appliance was removed and placed into an anaerobic glove box. Using aseptic techniques, the minitrap was separated from the appliance and placed inside a sterile Balch test tube, closed with a serum stopper, sealed, and transported to Northeastern University within 1 h. The minitrap was aseptically disassembled under anoxic conditions (2% H2, 1% CO2, 97% N2), and the central plate with agar plugs was placed into a serum bottle with 50 ml of BM. The sealed bottle was agitated at 180 rpm for 30 min to wash out microbial cells trapped in the through holes. The resulting cell suspension (1 to 10 ml) was subsampled for cell enumeration by direct microscopy. Cells were collected on 0.22-μm-pore-size black polycarbonate membranes (GE Water & Process Technologies, Burlington, MA), dried at room temperature, stained with 1.0 g/liter of filter-sterilized acridine orange solution (3,6-bis[dimethylamino]acridine; Sigma-Aldrich), and visualized under a Leica DMLB microscope equipped with a Mercury short ARC photo optic lamp and K3 filter set (illumination path, 470 to 490 nm; observation path LP, 515 nm). Cells were counted on two replicate filters for each serial dilution, with 20 microscopic fields counted per filter, and the average ± standard deviation (SD) was calculated. We noted the presence of cell aggregates and evaluated the number of cells per aggregate. We were not able to disrupt these aggregates by further shaking or vortexing. Enumeration showed that, due to clumping, (180 ± 9) × 106 cells/sample comprised (60 ± 2) × 106 potential CFU per sample. Following enumeration, cells were serially diluted into molten BM agar at 46 to 48°C and poured into 16 petri dishes per dilution. Half of the plates were then incubated in pack-rectangular 2.5-liter jars (Mitsubishi Gas Chemical Co., Inc., Japan) under anaerobic conditions (2% H2, 1% CO2, 97% N2) and the other half were incubated aerobically, both at 37°C. The total number of CFU was determined after 4 and 10 days of incubation. Four anaerobic plates were used for isolation of pure cultures and the rest of the plates were used for microbial identification by sampling colony material, extracting DNA, and sequencing PCR-amplified 16S rRNA genes (see below). Sixty-nine single colonies grown in anaerobic plates were picked with a sterile syringe needle and subcultured into serum bottles with liquid BM or BM supplemented with saliva or sheep blood. All bottles that exhibited growth were subsampled and examined for culture purity microscopically. Mixed cultures were purified via single colony isolation on solid agar TY medium. Pure cultures of isolated anaerobic bacteria were identified by 16S rRNA gene sequencing (see below). Twenty-one colonies grown in anaerobic plates as well as 24 colonies grown in aerobic plates were also picked for direct identification without subculturing. Colony identification was performed from picked material after DNA extraction and 16S rRNA gene sequence analysis. Isolated pure cultures were maintained in liquid TY or TGY medium.
Single-cell, long-term cultivation.
Two experiments were conducted. In both cases, a sample of subgingival plaque from subject 3 was immediately inoculated into a sealed 130-ml serum bottle (Wheaton, Mays Landing, NJ) with 50 ml of BM. After inoculation, the serum bottle was placed on ice, and 1 ml was withdrawn for cell counting as described above. Small aggregates (2 to 10 cells/aggregate) were present in both experiments. In one experiment, we performed a total cell count as described above, diluted the cell suspension in BM, and aliquoted the mixture into 10 96-well microtiter plates such that a single well received on average one cell. In the second experiment, we counted only potential CFU, regardless of whether or not such a unit was a single cell or a cell aggregate, diluted the cell suspension in TY, and aliquoted the mixture into 10 96-well microtiter plates such that a single well received on average 1 potential CFU per well. Note that in the first experiment, the number of wells receiving no cell was larger than expected from a Poisson distribution, whereas in the second, each well received on average more than one cell. In either case, one row in the middle of each plate was filled with sterile medium to control for contamination. Plates were sealed with Breathe-Easy sealing membranes (Rpi Corp., Mount Prospect, IL) and incubated at 37°C for 20 to 30 days under anaerobic conditions in pack-rectangular 2.5-liter jars. Plates were periodically examined for visual microbial growth. Wells that exhibited turbidity were noted and subsampled. Subsamples were transferred into serum bottles with fresh medium, incubated at 37°C with agitation at 180 rpm, and examined for purity microscopically and by 16S rRNA gene sequencing (see below). Mixed cultures were purified via single-colony isolation on solid agar-TY medium. Pure cultures were identified by sequencing their 16S rRNA gene (see below).
Direct plating.
A subgingival sample was obtained from subject 1 as described above. The sample was immediately inoculated into a sealed 130-ml serum bottle with 50 ml of BM, agitated at 180 rpm for 30 min, and subsampled for cell counting under epifluorescence as before. Cell aggregates were noted, and the number of cells per aggregate was estimated. Cell suspensions were serially diluted in serum bottles filled with 50 ml of molten TY agar at 46 to 48°C and poured into plates. Sixteen plates were prepared, with one-half incubated aerobically and the other half anaerobically, both at 37°C. After 4 to 6 days of incubation, colonies were picked up with a sterile syringe needle. Grown material was used for identification via sequencing the 16S rRNA gene (see below).
Cultivation via enrichment.
Subgingival samples from subjects 1, 3, 4, and 7 to 9 were obtained as described above. To obtain primary enrichments, samples from four subjects were inoculated into BM. Samples from two other subjects were inoculated into media in which Casamino Acids were replaced with starch or xylan. The inoculated serum bottles were incubated at 37°C with agitation at 180 rpm for 3 to 5 days, followed by 2 or 3 more rounds of similar cultivation. After these rounds of enrichment, the contents were serially diluted in serum bottles filled with 50 ml of molten BM and TY agar at 46 to 48°C and poured into four to six petri dishes per enrichment. After solidifying, the petri dishes were incubated at 37°C in pack-rectangular 2.5-liter jars for 3 to 7 days. Per enrichment, 12 to 20 single colonies were picked with a sterile syringe needle and inoculated into liquid BM and TY medium for subculturing. All bottles that exhibited growth were subsampled and examined microscopically for culture purity. Mixed cultures were purified via single-colony isolation on solid agar TY medium. All steps were conducted in an anaerobic glove box. Pure cultures were identified by 16S rRNA gene sequencing (see below).
Microbial identification and molecular phylogeny.
Genomic DNA was extracted from microbial biomass with the GenElute genomic DNA kit (Sigma, St. Louis, MO) according to the supplier's instructions. PCR amplification of the 16S rRNA gene and sequencing were performed with Hot Star Taq DNA polymerase (Qiagen, Germantown, MD) and eubacterial universal primers 27F (5′-AGA GTT TGATCC TGG CTC AG-3′) (3) and 1492R (5′-TAC GGT TAC CTT GTT ACG ACT T-3′) (41) according to the supplier's instructions. Amplified PCR products were sequenced at Macrogen USA Corporation (Rockville, MD) with 907R (5′-CCG TCA ATT CCT TTR AGT TT-3′) (29) and 518F (5′-CCAGCAGCCGCGGTAATACG-3′) universal primers (45). Nucleotide sequences were aligned with sequences from GenBank using BioEdit v.7.0.5 (26) and ClustalX (54). Sequence identity was established using BLAST (2), HOMD (11, 19), and EzTaxon (12). Phylogenetic trees were reconstructed using the ME algorithm (43) via the MEGA4 program package (51). Phylogenetic trees were assembled using a bootstrap test with 1,000 replicates to evaluate robustness.
Strains ACC2, ACB1, and ACB7 have been deposited in DSMZ and BEI Resources under deposition no. DSM 24645, DSM 24637, and DSM 24638 and HMS-480, HMS-481, and HMS-482, respectively. Strains ACC19a, CM2, CM5, ICM47, ICM39, ICM58, FOBRC14, ICM7, MSTE9, AS15, OBRC8, OBRC7, ACB8, MSX73, FOBRC9, FOBRC6, BS35b, AS14, ACP1, CM50, CM52, CM59, CM382, and OBRC5-5 have been deposited in BEI Resources under deposition numbers HMS-483 to HMS-485, HMS-759 to HMS-778, and HM-780.
Nucleotide sequence accession numbers.
Sequences generated in this study have been deposited in GenBank under accession no. HM120209 to HM120217, HQ593875 to HQ593876, HQ616351 to HQ616401, HQ610180 to HQ610199, and JN091082 to JN091085.
RESULTS
HOMIM analyses.
Using HOMIMs, we analyzed 12 subgingival plaque samples, two each from subjects 1 to 6. The HOMIM profiles were substantially different among the subjects, with samples from subject 3 revealing the largest number of positive microarray reactions containing signatures of 58 oral taxa as defined by HOMD (Fig. 2). Subsequently, subject 3 was chosen as a source of subgingival plaque microorganisms for single-cell cultivation; subject 3 also volunteered for the minitrap experiment. For direct plating, we used samples from subject 1. For microbial isolation via enrichment, we used samples from subjects 1, 3, 4, and 7 to 9.
Fig 2.

HOMIM-enabled microbial composition of two subgingival plaque samples from subject 3. Dark green and light green correspond to the samples from the right and left sides of the mouth, respectively. The sizes of the bars reflect the relative band intensities of hybridization with target bacterial species.
Minitrap in vivo cultivation.
After the 48-h in vivo incubation of a single minitrap (Fig. 1), the number of cells that colonized the inner space was (180 ± 9) × 106. Some of the cells were in small aggregates (2 to 5 cells/aggregate), with the number of potential CFU totaling (60 ± 2) × 106. Plating counts on BM agar revealed that 11 and 7.6% of cells formed colonies under anaerobic and aerobic conditions, respectively. These are conservative estimates since some CFU were aggregates of cells with potential cogrowth of two or more cells.
From the 69 collected colonies, we were able to stably subculture 40 (in liquid medium only) and isolate and maintain 31 in pure culture. These 31 strains represented the microbial phyla Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes (see Fig. S1 in the supplemental material). Eleven more strains were identified from the colony material by 16S rRNA gene sequencing (Table 1). Among the subcultured isolates, four deserve specific mentioning. Two of them, ICM47 and ICM34, are the first cultured representatives of the “uncultured” taxon 172 as defined by HOMD (Table 1); both share 98.9% 16S rRNA gene sequence identity to Actinomyces odontolyticus. Another two isolates, ICM7 and ICM62, also closely related to each other, are only distantly related to the closest validly described species (91% 16S rRNA gene identity with Clostridium aerotolerans) (13) and therefore may represent a novel genus. We note, however, that these two isolates also showed 98.6 to 98.8% 16S rRNA gene identity to the Lachnospiraceae [G-1] oral strain F0167 from the HOMD taxon 107 (19). Here and elsewhere, we discriminate between identity with an established species vis a vis that with the closest reported isolate. The rationale is that, for a proper taxonomic description, the latter may be uninformative since its identity is often known from sequencing DNA from colony material without isolating, maintaining, and archiving the microorganism, whose availability is therefore uncertain.
Table 1.
Phylogenetic identity of microorganisms isolated by three different cultivation approaches applied to oral microorganisms from subject 3
| Isolate no. | Minitrap |
Single cell |
Enrichment |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | Closest relative in HOMD | Taxon | Strain | Closest relative in HOMD | Taxon | Strain | Closest relative in HOMD | Taxon | |
| 1 | ICM47 | Actinomyces sp. | 172 | CM83 | Actinomyces sp. | 171 | CM2 | Peptostreptococcaceae [XI][G-7] sp. | 081 |
| 2 | ICM34 | Actinomyces sp. | 172 | CM84 | Actinomyces naeslundii | 176 | CM5 | Peptostreptococcaceae [XI][G-7] sp. | 081 |
| 3 | ICM39 | Actinomyces odontolyticus | 701 | CM37 | Campylobacter showae | 763 | CM1 | Fusobacterium nucleatumsubsp. animalis | 420 |
| 4 | ICM41 | Actinomyces odontolyticus | 701 | CM51 | Capnocytophaga gingivalis | 337 | CM21 | Fusobacterium naviforme | 200 |
| 5 | ICM54 | Actinomyces odontolyticus | 701 | CM59 | Capnocytophaga granulosa | 325 | CM22 | Fusobacterium nucleatumsubsp. polymorphum | 202 |
| 6 | ICM58 | Atopobium parvulum | 723 | CM100 | Catonella morbi | 165 | CM3 | Fusobacterium nucleatum subsp. polymorphum | 202 |
| 7 | ICM42b | Atopobium parvulum | 723 | CM55 | Fusobacterium nucleatum subsp. polymorphum | 202 | CM12 | Oribacterium sinus | 457 |
| 8 | ICM57 | Atopobium parvulum | 723 | CM66 | Fusobacterium nucleatum subsp. polymorphum | 202 | CM7 | Streptococcus anginosus | 071 |
| 9 | NICM20a | Campylobacter concisus | 575 | CM92 | Granulicatella adiacens | 534 | CM6 | Streptococcus sp. | 543 |
| 10 | NICM6a | Gemella sanguinis | 757 | CM50 | Mogibacterium timidum | 042 | |||
| 11 | ICM7 | Lachnospiraceae [G-1] sp. | 107 | CM96 | Mogibacterium diversum | 593 | |||
| 12 | ICM62 | Lachnospiraceae [G-1] sp. | 107 | CM88 | Peptostreptococcus stomatis | 112 | |||
| 13 | ICM51 | Oribacterium sinus | 457 | CM38 | Prevotella sp. | 317 | |||
| 14 | ICM1 | Prevotella melaninogenica | 469 | CM75 | Prevotella denticola | 291 | |||
| 15 | ICM33 | Prevotella melaninogenica | 469 | CM103 | Selenomonas infelix | 639 | |||
| 16 | ICM65 | Prevotella melaninogenica | 469 | CM52 | Selenomonas sputigena | 151 | |||
| 17 | ICM55 | Prevotella salivae | 307 | CM382 | Slackia exigua | 602 | |||
| 18 | NICM28a | Prevotella histicola | 298 | CM36 | Streptococcus anginosus | 543 | |||
| 19 | ICM60 | Streptococcus sp. | 070 | CM86 | Streptococcus oralis | 707 | |||
| 20 | ICM16 | Streptococcussp. | 070 | CM60 | Veillonella parvula | 161 | |||
| 21 | ICM2 | Streptococcus sp. | 070 | CM79 | Veillonella dispar | 160 | |||
| 22 | NICM25a | Streptococcus sp. | 070 | ||||||
| 23 | ICM12 | Streptococcus sp. | 071 | ||||||
| 24 | ICM24 | Streptococcus sp. | 071 | ||||||
| 25 | AICM23b | Streptococcus sp. | 071 | ||||||
| 26 | ICM59 | Streptococcus sp. | 071 | ||||||
| 27 | NICM2a | Streptococcus sp. | 071 | ||||||
| 28 | ICM64 | Streptococcus sp. | 071 | ||||||
| 29 | AICM5b | Streptococcus parasanguinis | 411 | ||||||
| 30 | ICM46 | Streptococcus mitis | 677 | ||||||
| 31 | ICM10 | Streptococcus mitis | 677 | ||||||
| 32 | ICM45 | Streptococcus pneumoniae | 734 | ||||||
| 33 | NICM17a | Streptococcus salivarius | 755 | ||||||
| 34 | ICM4 | Streptococcus salivarius | 755 | ||||||
| 35 | AICM24b | Streptococcus salivarius | 755 | ||||||
| 36 | AICM14b | Streptococcus salivarius | 755 | ||||||
| 37 | NICM22a | Veillonella dispar | 160 | ||||||
| 38 | ICM51a | Veillonella atypica | 524 | ||||||
| 39 | ICM53 | Veillonella atypica | 524 | ||||||
Identified from an anaerobically grown colony without isolation into pure culture.
Identified from an aerobically grown colony without isolation into pure culture.
The remaining isolates represent new strains within 17 established oral taxa in eight genera: Actinomyces, Atopobium, Campylobacter, Gemella, Oribacterium, Prevotella, Streptococcus, and Veillonella (Table 1). Only two Streptococcus taxa, 071 and 755, were isolated both aerobically and anaerobically. Representatives of Campylobacter sp., Gemella sp., Prevotella sp., and Veillonella sp. (oral taxa 575, 757, 298, and 160, respectively) were isolated exclusively from anaerobic plates. One strain, a Streptococcus sp., oral taxon 411, was observed only on aerobic plates.
Single-cell long-term cultivation.
The two experiments conducted in this part of the project differed primarily by how we distributed individual cells and cell aggregates across the wells of microtiter plates. In experiment 1, the 840 noncontrol wells received 840 cells. Figure 3A shows that the number of new growth events, registered as visible turbidity in the well's contents, grew steadily over the first week of incubation, and stopped thereafter. At the end of the experiment, the total number of turbid wells was 27, translating into 3.2% recovery of inoculated cells. As in the case of the minitrap-based recovery, this is a conservative estimate given the possibility of two or more cells cogrowing in some wells. No growth occurred in the control (cell-free) wells.
Fig 3.

Accumulation of growth events in long-term cultivation experiments presented as a fraction of wells positive (PW) for growth. (A) Experiment 1; (B) experiment 2.
In experiment 2, the 840 noncontrol wells received 840 potential CFU, with some of them containing more than one cell. Expectedly, many more wells showed growth, but unlike the first experiment, this does not reflect the probability of a single cell forming growth (Fig. 3B). However, it provided ample material for microbial isolation. Collectively, the two growth experiments resulted in 21 pure cultures representing 13 different genera and 20 oral taxa (Table 1). Three isolates may represent novel species as they share less than 99% 16S rRNA gene sequence identity with previously cultivated species: one related to Prevotella loescheii and Prevotella sp. strain B31FD (97.8% and 99.3%, respectively), one related to Catonella morbi (98.9%), and one related to Capnocytophaga granulosa (99.1%). The remaining 18 isolates fell into 17 HOMD taxa and represent new strains within known species in the genera Actinomyces, Campylobacter, Capnocytophaga, Fusobacterium, Granulicatella, Mogibacterium, Peptostreptococcus, Prevotella, Selenomonas, Streptococcus, Slackia, and Veillonella.
Direct plating.
The subgingival sample used for direct plating contained (450 ± 98) × 106 cells. The cell suspension was essentially aggregate free, with the total number of potential CFU being statistically the same: (382 ± 102) × 106. Plate counts revealed that 1.2 and 0.8% of cells formed colonies under anaerobic and aerobic conditions, respectively. Two Streptococcus species from oral taxa 578 and 622 were detected under both aerobic and anaerobic conditions. Representatives of six HOMD taxa (four genera) were unique to anaerobic petri dishes; representatives of nine HOMD taxa (five genera) were specific to aerobic dishes. Only one anaerobically grown colony, NAC11, appeared to represent a novel species, with 98.1% 16S rRNA gene sequence identity to Streptococcus sp. Two aerobically grown colonies, AAC25 and AAC5, shared 98.8% and 98.9% 16S sequence identity with Kingella sp. and Propionibacterium sp., respectively, and may therefore represent novel species.
Cultivation via enrichment.
While the above experiments served two purposes—to compare the merits of different methods and provide cultures of novel strains—we also conducted a number of isolation experiments in a less quantitative fashion, aiming purely at microbial discovery. These were based on first cultivating mixed samples under strict anaerobic conditions in one or another reduced-nutrient liquid medium, followed by their anaerobic isolation on solid media. In total, we obtained six anaerobic enrichments. Microorganisms from four samples were enriched on BM with yeast extract and Casamino Acids, and two others were enriched on media with plant polymers (starch and xylan). The majority of enrichments became black after 2 to 3 days of incubation, most likely as a result of ferrous sulfide formation in the presence of cysteine desulfhydrase. Almost half of all colonies grown were black. In total, we isolated into pure culture 70 strains from five different phyla (Firmicutes, Fusobacteria, Proteobacteria, Actinobacteria, and Bacteroidetes) representative of 31 HOMD taxa (Fig. 4). (Note that isolates obtained exclusively from subject 3 are also listed in Table 1; their phylogeny is shown in Fig. S1 in the supplemental material.)
Fig 4.

Minimum evolution phylogenetic tree of 16S rRNA gene sequences of anaerobic bacteria isolated via anaerobic enrichment. Shown in boldface are strains sharing less than 99% gene sequence homology with the closest named species.
Microorganisms isolated via enrichments may be divided into three groups: (i) strains of significant taxonomic novelty sharing less than 95% 16S rRNA gene identity with validly described species, which may represent novel genera; (ii) strains likely representative of novel species within established genera (e.g., sharing over 95% but less than 99% of 16S rRNA gene identity with validly described species); and (iii) new strains within established species.
The first group is represented by seven strains, with three (ACC2, OBRC5-5, MSX33) from the family Lachnospiraceae and four (ACC19a, CM2, CM5, and OBRC8) from the family Eubacteriaceae. The closest relative of strain ACC2 is Moryella indoligenes isolated from clinical specimens (94.36% 16S rRNA gene sequence similarity) (10). Isolates OBRC5-5 and MSX33 showed 91.1 and 91.2% 16S rRNA gene identity with Dorea formicigenerans (53) and Clostridium aerotolerans (55), the closest validly described species. They also showed 98.7 to 98.9% 16S rRNA gene sequence similarity to oral strain F0167 (HOMD taxon 107) and unpublished Eubacterium sp. “Smarlab BioMol-2301166” AY230774 isolated from human tissues. Isolates ACC19a, CM2, CM5, and OBRC8 exhibited 93.9 to 94.3% 16S rRNA gene identity to Eubacterium yurii subsp. margaretiae (32) but also had 98.8 to 100% identity to Eubacteriaceae bacterium P4P_50 P4 (18) and 98.6 to 99.6% identity to the uncultivated HOMD taxon 081 Peptostreptococcaceae [XI][G-7] sp.
The second group comprises four novel microorganisms in the genera Streptococcus and Oribacterium (strains BS35a and CM6 and ACB1 and ACB7, respectively) and two in Selenomonas and Campylobacter (strains FOBRC9 and FOBRC14). The third group consists of the remaining 57 strains, which belong to the following species: Streptococcus spp., Lactobacillus sp., Selenomonas spp., Veillonella spp., Eubacterium spp., Oribacterium spp., Shuttleworthia sp., Clostridium sp., Fusobacterium spp., Olsenella sp., Klebsiella spp., Campylobacter spp., Atopobium spp., Bifidobacterium spp., and Prevotella sp.
DISCUSSION
The majority of microbial species in the biosphere resist cultivation in the laboratory (27, 40). This is often referred to as the phenomenon of microbial “uncultivability,” also termed the “great plate count anomaly” (50). This phenomenon has been known for over a century (59, 60) and is continuously referred to as one of the principal challenges in microbiology (28), but even today the underlying reasons remain by and large unresolved (14, 21).
Uncultivated species from the human microbiome are likely a reflection of this general phenomenon. The microbiota of the human gut consists mostly of as yet uncultivated species (23). Skin microorganisms appear to be easier to cultivate, with rRNA gene surveys reporting >90% of clones represent cultivable species (for examples, see reference 24). The cultivability of oral microorganisms occupies the middle ground in this spectrum and is currently estimated to be between about one-half and two-thirds of all species present (19, 33, 36, 37, 56, 57). Therefore, there are at least several hundred presently uncultivated species in the human mouth, and many aspects of microbial dynamics in the oral cavity in health and disease cannot be understood without accessing these “missing” species.
The main thrust of this paper is to explore various alternatives to conventional cultivation in order to isolate previously uncultivated species from the human microbiome. We first developed and used these methods for environmental applications (7, 22, 25, 30, 34, 35). From these applications, we learned two principal lessons. The first is that we can significantly increase the probability that a new microorganism will domesticate (i.e., grow in vitro), if we first grow it in vivo (6, 35). The latter could be achieved by incubating target microorganisms inside diffusion chambers placed in the organisms' natural environment (25, 30, 34). While the exact reasons for this domestication remain unclear (21, 22), the method gives a practical tool to improve the rate of microbial discovery. The second is an observation that when a cell's growth is unimpeded by neighbors, such as in our single-cell format in microtiter plates, the overall recovery increases (Buerger et al., submitted). This observation has been made repeatedly in the past, and dilution to extinction has already led to the cultivation of spectacular environmental species (for examples, see references 9 and 39). In addition to the above results, and when designing our cultivation strategy, we took into account earlier findings that strict anaerobic incubation often increases microbial recovery (42, 48, 49, 52). Finally, we hypothesized that conventional cultivation with nonselective media rich in sugars, such as Trypticase-glucose-yeast extract (TGY), brain heart infusion (BHI), Lactobacillus MRS, Wilkins-Chalgren, and many others may select for fast-growing species, thus masking growth of other, rarely cultivated or uncultivated strains (15, 56). Here we use a multifaceted ensemble of cultivation methods by applying the above methodological developments, together with more conventional direct plating and cultivation via enrichment, employing strictly anaerobic conditions in all but comparative experiments, and utilizing sugar-free media.
The principal result of this work is the cultivation of 10 different strains previously known only from their molecular signatures and likely representing 10 new species (their taxonomic description is in progress). Additionally, we isolated and maintained 20 novel species in pure culture, including those likely representing novel genera that are new to the human oral cavity. In collaboration with the Broad Institute (Cambridge, MA) and J. Craig Venter Institute (Rockville, MD), we are sequencing the genomes of 27 strains, and 5 of them have already been released (http://www.broadinstitute.org/scientific-community/data). We view the number of novel species isolated as an illustration of the success of our cultivation strategy, an important element of which was the maintenance of strictly anaerobic conditions starting from the sampling acquisition and throughout experimentation (unless incubation on air was done for comparative purposes). Since our strategy was also multifaceted, this begs the question—which approach was the most efficacious for microbial discovery? In spite of the question's apparent simplicity, the answer is not straightforward. The reason is that one obvious measure of the method's efficacy, the degree of recovery estimated as a percentage of plaque cells forming growth in vitro, is hard to apply. While we did calculate values of such recovery, which proved to be at least 7.6 to 11% in minitrap cultivation and 3.2% using the single-cell approach versus 0.8 to 1.2% for standard plating, a direct comparison of these figures may be misleading. For example, while the last two methods measure the recovery of plaque cells directly, the minitrap approach does not because it involves an enrichment (in vivo) step. Perhaps a more informative comparison is that among the species lists obtained by the methods used. Remarkably, these lists proved unique, sharing no single species (Fig. 5). Trivial undersampling is an unlikely explanation considering that culture collections obtained by a single approach but using sharply contrasting oxygen regimes do have species in common. More likely, the differences between the culture collections are due to the respective biases of the cultivation techniques used. Indeed, the single-cell long-term approach, in contrast to petri dish cultivation, likely enriches for species that grow slower and/or less competitively on nutrient agar and selects against microorganisms that require the presence of other species. The minitrap method selects for species active in the mouth at the time of incubation as only actively growing species would be expected to colonize the space within the minitrap, but may exclude larger and nonmotile cells. Therefore, even if one cultivation method is more efficacious than the other in discovering new species, the resulting culture collections would not necessarily be inclusive of each other. This nonredundancy of the cultivation methods is fully in line with our earlier experiences with environmental application of these methods (21, 22). A general—and in retrospect predictable—conclusion we draw from these observations is that an ensemble of novel and traditional cultivation techniques is a promising tool to close the gap between microorganisms available in culture and those present in the human oral cavity.
Fig 5.
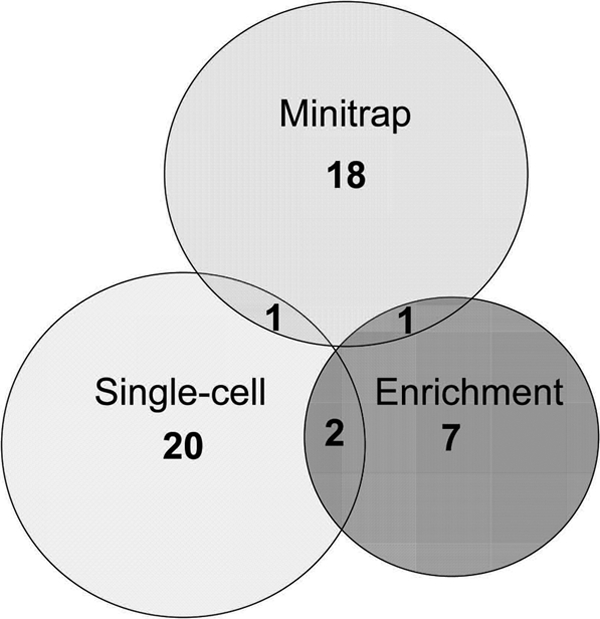
Overlap between culture collections obtained by three different cultivation approaches for a single subject (subject 3). Values in the center of each circle represent the total number of isolated oral taxa by method; values in the overlapping areas represent the numbers of coisolated oral taxa.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants 1RC1DE020707-01 and 3 R21 DE018026-02S1 to S.S.E.
We thank S. Reynolds for technical assistance.
Footnotes
Published ahead of print 4 November 2011
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. 2005. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 43:5721–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amann R, Snaidr J, Wagner M, Ludwig W, Schleifer KH. 1996. In situ visualization of high genetic diversity in a natural microbial community. J. Bacteriol. 178:3496–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S. 1996. Periodontal disease and cardiovascular disease. J. Periodontol. 67:1123–1137 [DOI] [PubMed] [Google Scholar]
- 5. Bik EM, et al. 2010. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 4:962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bollmann A, Lewis K, Epstein SS. 2007. Incubation of environmental samples in a diffusion chamber increases the diversity of recovered isolates. Appl. Environ. Microbiol. 73:6386–6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bollmann A, Palumbo AV, Lewis K, Epstein SS. 2010. Isolation and physiology of bacteria from contaminated subsurface sediments. Appl. Environ. Microbiol. 76:7413–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brook I, Frazier EH, Gher ME. 1991. Aerobic and anaerobic microbiology of periapical abscess. Oral Microbiol. Immunol. 6:123–125 [DOI] [PubMed] [Google Scholar]
- 9. Button DK, Schut F, Quang P, Martin R, Robertson BR. 1993. Viability and isolation of marine bacteria by dilution culture: theory, procedures, and initial results. Appl. Environ. Microbiol. 59:881–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carlier JP, K'Ouas G, Han XY. 2007. Moryella indoligenes gen. nov., sp. nov., an anaerobic bacterium isolated from clinical specimens. Int. J. Syst. Evol. Microbiol. 57:725–729 [DOI] [PubMed] [Google Scholar]
- 11. Chen T, et al. 2010. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010:baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chun J, et al. 2007. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 57:2259–2261 [DOI] [PubMed] [Google Scholar]
- 13. Collins MD, et al. 1994. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 44:812–826 [DOI] [PubMed] [Google Scholar]
- 14. Colwell RK, Gotelli NJ. 2001. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 4:379–391 [Google Scholar]
- 15. Connon SA, Giovannoni SJ. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 68:3878–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Daniluk T, et al. 2006. Aerobic and anaerobic bacteria in subgingival and supragingival plaques of adult patients with periodontal disease. Adv. Med. Sci. 51(Suppl. 1:81–85 [PubMed] [Google Scholar]
- 17. Dedysh SN, et al. 1998. Isolation of acidophilic methane-oxidizing bacteria from northern peat wetlands. Science 282:281–284 [DOI] [PubMed] [Google Scholar]
- 18. de Lillo A, et al. 2006. Novel subgingival bacterial phylotypes detected using multiple universal polymerase chain reaction primer sets. Oral Microbiol. Immunol. 21:61–68 [DOI] [PubMed] [Google Scholar]
- 19. Dewhirst FE, et al. 2010. The human oral microbiome. J. Bacteriol. 192:5002–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dodman T, Robson J, Pincus D. 2000. Kingella kingae infections in children. J. Paediatr. Child Health 36:87–90 [DOI] [PubMed] [Google Scholar]
- 21. Epstein SS. 2009. Microbial awakenings. Nature 457:1083. [DOI] [PubMed] [Google Scholar]
- 22. Epstein SS, Lewis K, Nichols D, Gavrish E. 2010. New approaches to microbial isolation, p 3–12. In Baltz RH, Davies JE, Demain A. (ed), Manual of industrial microbiology and biotechnology, vol 3. ASM, Washington, DC [Google Scholar]
- 23. Frank DN, Pace NR. 2008. Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 24:4–10 [DOI] [PubMed] [Google Scholar]
- 24. Gao Z, Tseng CH, Pei Z, Blaser MJ. 2007. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. U. S. A. 104:2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gavrish E, Bollmann A, Epstein S, Lewis K. 2008. A trap for in situ cultivation of filamentous actinobacteria. J. Microbiol. Methods 72:257–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall TA. 1999. Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 27. Handelsman J. 2004. Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68:669–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hurst CJ. 2005. Divining the future of microbiology. ASM News 71:262–263 [Google Scholar]
- 29. Ishii K, Fukui M. 2001. Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl. Environ. Microbiol. 67:3753–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaeberlein T, Epstein SS, Lewis K. 2002. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127–1129 [DOI] [PubMed] [Google Scholar]
- 31. Kroes I, Lepp PW, Relman DA. 1999. Bacterial diversity within the human subgingival crevice. Proc. Natl. Acad. Sci. U. S. A. 96:14547–14552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Margaret BS, Krywolap GN. 1986. Eubacterium yurii subsp. yurii sp. nov. and Eubacterium yurii subsp. margaretiae subsp. nov.: test tube brush bacteria from subgingival dental plaque. Int. J. Syst. Bacteriol. 36:145–149 [Google Scholar]
- 33. Marsh PD, Moter A, Devine DA. 2011. Dental plaque biofilms: communities, conflict and control. Periodontol. 2000. 55:16–35 [DOI] [PubMed] [Google Scholar]
- 34. Nichols D, et al. 2010. Use of Ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 76:2445–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nichols D, et al. 2008. Short peptide induces an “uncultivable” microorganism to grow in vitro. Appl. Environ. Microbiol. 74:4889–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paster BJ, et al. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183:3770–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paster BJ, Olsen I, Aas JA, Dewhirst FE. 2006. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol. 2000. 42:80–87 [DOI] [PubMed] [Google Scholar]
- 38. Preza D, et al. 2009. Microarray analysis of the microflora of root caries in elderly. Eur. J. Clin. Microbiol. Infect. Dis. 28:509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rappe MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630–633 [DOI] [PubMed] [Google Scholar]
- 40. Rappe MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57:369–394 [DOI] [PubMed] [Google Scholar]
- 41. Rickard AH, et al. 2005. Adhaeribacter aquaticus gen. nov., sp nov., a Gram-negative isolate from a potable water biofilm. Int. J. Syst. Evol. Microbiol. 55:821–829 [DOI] [PubMed] [Google Scholar]
- 42. Rosebury T, Reynolds JB. 1964. Continuous anaerobiosis for cultivation of spirochetes. Proc. Soc. Exp. Biol. Med. 117:813–815 [DOI] [PubMed] [Google Scholar]
- 43. Rzhetsky A, Nei M. 1992. Statistical properties of the ordinary least-squares, generalized least-squares, and minimum-evolution methods of phylogenetic inference. J. Mol. Evol. 35:367–375 [DOI] [PubMed] [Google Scholar]
- 44. Scannapieco FA. 1999. Role of oral bacteria in respiratory infection. J. Periodontol. 70:793–802 [DOI] [PubMed] [Google Scholar]
- 45. Seo SH, Lee SD. 2010. Altererythrobacter marensis sp nov., isolated from seawater. Int. J. Syst. Evol. Microbiol. 60:307–311 [DOI] [PubMed] [Google Scholar]
- 46. Sizova MV, Izquierdo JA, Panikov NS, Lynd LR. 2011. Cellulose- and xylan-degrading thermophilic anaerobic bacteria from biocompost. Appl. Environ. Microbiol. 77:2282–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sizova MV, Panikov NS, Tourova TP, Flanagan PW. 2003. Isolation and characterization of oligotrophic acido-tolerant methanogenic consortia from a Sphagnum peat bog. FEMS Microbiol. Ecol. 45:301–315 [DOI] [PubMed] [Google Scholar]
- 48. Slots J. 1977. Microflora in the healthy gingival sulcus in man. Scand. J. Dent. Res. 85:247–254 [DOI] [PubMed] [Google Scholar]
- 49. Socransky S, Macdonald JB, Sawyer S. 1959. The cultivation of Treponema microdentium as surface colonies. Arch. Oral Biol. 1:171–172 [DOI] [PubMed] [Google Scholar]
- 50. Staley JT, Konopka A. 1985. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 39:321–346 [DOI] [PubMed] [Google Scholar]
- 51. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 52. Tanner AC, et al. 2011. Cultivable anaerobic microbiota of severe early childhood caries. J. Clin. Microbiol. 49:1464–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taras D, Simmering R, Collins MD, Lawson PA, Blaut M. 2002. Reclassification of Eubacterium formicigenerans Holdeman and Moore 1974 as Dorea formicigenerans gen. nov., comb. nov., and description of Dorea longicatena sp nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 52:423–428 [DOI] [PubMed] [Google Scholar]
- 54. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vangylswyk NO, Vandertoorn JJTK. 1987. Clostridium aerotolerans sp. nov., a xylanolytic bacterium from corn stover and from the rumina of sheep fed corn stover. Int. J. Syst. Bacteriol. 37:102–105 [Google Scholar]
- 56. Vartoukian SR, Palmer RM, Wade WG. 2010. Strategies for culture of ‘unculturable’ bacteria. FEMS Microbiol. Lett. 309:1–7 [DOI] [PubMed] [Google Scholar]
- 57. Wade W. 2002. Unculturable bacteria—the uncharacterized organisms that cause oral infections. J. R. Soc. Med. 95:81–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wade WG, Munson MA, de Lillo A, Weightman AJ. 2005. Specificity of the oral microflora in dentinal caries, endodontic infections and periodontitis. Int. Congr. Ser. 1284:150–157 [Google Scholar]
- 59. Winslow C-EA, Willcomb GE. 1905. Tests of a method for the direct microscopic enumeration of bacteria. J. Infect. Dis. Suppl. 1:273–283 [Google Scholar]
- 60. Winterberg H. 1898. Zur Methodik der Bakterienzahlung. Zentralbl. Hyg. 29:75–93 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.