

Abstract
We have previously reported that mice deficient in the beta-glucan receptor Dectin-1 displayed increased susceptibility to Aspergillus fumigatus lung infection in the presence of lower interleukin 23 (IL-23) and IL-17A production in the lungs and have reported a role for IL-17A in lung defense. As IL-23 is also thought to control the production of IL-22, we examined the role of Dectin-1 in IL-22 production, as well as the role of IL-22 in innate host defense against A. fumigatus. Here, we show that Dectin-1-deficient mice demonstrated significantly reduced levels of IL-22 in the lungs early after A. fumigatus challenge. Culturing cells from enzymatic lung digests ex vivo further demonstrated Dectin-1-dependent IL-22 production. IL-22 production was additionally found to be independent of IL-1β, IL-6, or IL-18 but required IL-23. The addition of recombinant IL-23 augmented IL-22 production in wild-type (WT) lung cells and rescued IL-22 production by lung cells from Dectin-1-deficient mice. In vivo neutralization of IL-22 in the lungs of WT mice resulted in impaired A. fumigatus lung clearance. Moreover, mice deficient in IL-22 also demonstrated a higher lung fungal burden after A. fumigatus challenge in the presence of impaired IL-1α, tumor necrosis factor alpha (TNF-α), CCL3/MIP-1α, and CCL4/MIP-1β production and lower neutrophil recruitment, yet intact IL-17A production. We further show that lung lavage fluid collected from both A. fumigatus-challenged Dectin-1-deficient and IL-22-deficient mice had compromised anti-fungal activity against A. fumigatus in vitro. Although lipocalin 2 production was observed to be Dectin-1 and IL-22 dependent, lipocalin 2-deficient mice did not demonstrate impaired A. fumigatus clearance. Moreover, lung S100a8, S100a9, and Reg3g mRNA expression was not lower in either Dectin-1-deficient or IL-22-deficient mice. Collectively, our results indicate that early innate lung defense against A. fumigatus is mediated by Dectin-1-dependent IL-22 production.
INTRODUCTION
Aspergillus fumigatus, the etiological agent of invasive aspergillosis (IA), is a ubiquitous mold that causes severe, invasive, life-threatening infections in patients who are severely immunocompromised. Major risk factors include neutropenia, neutrophil dysfunction, and immunosuppressive therapies (19). Recent data from the Transplant Associated Infections Surveillance Network (TRANSNET), a network of 23 United States transplant centers, have shown that IA occurred in 43% of hematopoietic stem cell transplant (HSCT) recipients (20) and in 19% of solid organ transplant recipients (33) between March 2001 and March 2006. IA is also becoming more recognized in individuals with less severe levels of immunosuppression. This is increasingly observed in intensive-care unit (ICU) populations, often associated with such diseases as chronic obstructive pulmonary disorder (COPD), cirrhosis, alcoholism, and postinfluenza infection; various postsurgical settings; and adults presenting with heterozygous chronic granulomatous disease (reviewed in reference 1).
Our laboratory has had a longstanding interest in pulmonary innate immune mechanisms involved in controlling A. fumigatus. We have previously demonstrated a central role for the beta-glucan receptor Dectin-1 in innate lung immune responses against A. fumigatus (38). Mice deficient in Dectin-1 are highly susceptible to lung infection with A. fumigatus as a result of impaired inflammatory reactivity of alveolar macrophages and impaired recruitment of and defense by neutrophils (42). Among several cytokines we have reported to be induced in a Dectin-1-dependent manner during A. fumigatus lung infection, we have identified interleukin 17A (IL-17A) as a critical mediator in host defense (42). Dectin-1-deficient mice produced IL-17A at lower levels in the lungs after exposure to A. fumigatus, and neutralization of IL-17A in wild-type (WT) mice resulted in a compromised ability to clear A. fumigatus from the lungs, indicating a strong requirement for the mediator in pulmonary defense against A. fumigatus (42). In our most recent studies, we have identified neutrophils as a source of Dectin-1-dependent IL-17A production during A. fumigatus lung infection (43). IL-17A production by neutrophils required the presence of IL-23, which we have previously reported to be produced in a Dectin-1-dependent manner in the lungs (42) and, more recently, in a Dectin-1-dependent manner by dendritic cells (DCs) (43).
In addition to being critical for the maintenance of the Th17 lineage (23) and IL-17A-producing cells in general (27, 40), IL-23 is also a critical effector cytokine for the induction of IL-22 (37, 40), a class 2 α-helical cytokine of the IL-10 family of cytokines (34). Although often produced in concert with each other, growing data in several models have shown that IL-23, IL-17A, and IL-22 do not necessarily function equally. For example, mucosal defense against the gut pathogen Citrobacter rodentium is more dependent on IL-23 than IL-17A (26). In colitis, IL-23 deficiency is more effective in ameliorating disease than IL-17A deficiency (11, 22). In fact, IL-17A may be protective in intestinal inflammation (32). With respect to IL-22, infection models with the Gram-negative bacteria Klebsiella pneumoniae (2) and C. rodentium (50) showed a requisite role for IL-22 in protection. However, although produced by Th17/IL-17A-producing cells (29), IL-22 has been shown to act as an anti-inflammatory agent in hepatitis (47) and inflammatory bowel disease (IBD) (39). Paradoxically, IL-22 is thought to be a contributing factor in inflammation associated with psoriasis (49). Collectively, IL-23, IL-17A, and IL-22 often function in concordance with each other, but in certain models, the function of one may be more important than that of another. As we had previously identified a role for IL-17A in vivo during A. fumigatus lung infection (42), we questioned whether IL-22 was also required for innate immune-mediated defense against A. fumigatus.
MATERIALS AND METHODS
Mice.
C57BL/6NTac mice, 6 to 8 weeks of age, were purchased from Taconic Farms Incorporated (Germantown, NY). Dectin-1-deficient mice were generated on the 129/SvEv background as previously described (41), backcrossed for 10 generations to the C57BL/6 background, and bred at Taconic. IL-22-deficient mice (50) were provided by Wenjun Ouyang at Genentech and bred at the University of Alabama at Birmingham (UAB). Lipocalin 2-deficient mice (4) were provided by Yvonne Chan at the University of Pittsburgh. Mice were maintained in a specific-pathogen-free environment in microisolator cages within an American Association for Laboratory Animal Science-certified animal facility in the Lyons Harrison Research Building at the University of Alabama at Birmingham. Animal studies were reviewed and approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC).
Preparation of A. fumigatus, in vivo challenge, and lung fungal burden assessment.
A. fumigatus isolate 13073 (ATCC, Manassas, VA) was maintained on potato dextrose agar for 5 to 7 days at 37°C. Conidia were harvested by washing the culture flask with 50 ml of sterile phosphate-buffered saline supplemented with 0.1% Tween 20. The conidia were then passed through a sterile 40-μm nylon membrane to remove hyphal fragments and enumerated on a hemacytometer. Mice were lightly anesthetized with isoflurane and administered 5 × 107 to 7 × 107 A. fumigatus conidia in a volume of 50 μl intratracheally. For lung fungal burden analysis, the left lungs were collected and homogenized in 1 ml of phosphate-buffered saline (PBS). Total RNA was extracted from 0.1 ml of unclarified lung homogenate using the MasterPure yeast RNA purification kit (Epicentre Biotechnologies, Madison, WI), which includes a DNase treatment step to eliminate genomic DNA, as previously reported (28). The lung A. fumigatus burden was analyzed with real-time PCR measurement of the A. fumigatus 18S rRNA (GenBank accession number AB008401) (5) and quantified using a standard curve for A. fumigatus conidia (101 to 109) as previously described (28). Specifically, total RNA was isolated, using the MasterPure kit, from serial 1:10 dilutions of A. fumigatus conidia, beginning with 109, and real-time PCR amplification of A. fumigatus 18S rRNA was performed on each dilution. As a validation of the real-time PCR method, heat-killed A. fumigatus did not yield a signal by real-time PCR and was unable to grow on potato dextrose agar plates. In addition, no amplification controls (i.e., no reverse transcriptase included in the cDNA reaction mixture) yielded a signal of <0.001% by real-time PCR, indicating that the DNase treatment step was efficient at eliminating contaminating A. fumigatus DNA.
IL-22 neutralization.
For in vivo IL-22 neutralization, WT mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia in 50 μl, and 6 h thereafter, the mice were administered 50 μg of goat anti-mouse IL-22 (R&D Systems) or goat IgG isotype control antibody. Twenty-four hours after challenge, the mice were sacrificed, and the left lungs were collected and homogenized in 1 ml of PBS. Total RNA was extracted from 0.1 ml of unclarified lung homogenate using the MasterPure yeast RNA purification kit, and the lung A. fumigatus burden was analyzed with real-time PCR measurement of the A. fumigatus 18S rRNA, as described previously (42).
Lung cell isolation, culture, cytokine neutralizations, and IL-23 stimulation.
Mice were anesthetized with intraperitoneal ketamine/xylazine and sacrificed by exsanguination 18 h postinfection. Both lungs were collected and minced in Iscove's modified Dulbecco's medium (IMDM) (Sigma, St. Louis, MO) supplemented with 1% penicillin-streptomycin-glutamine (Mediatech, Herndon, VA), 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), and 0.4 mg/ml polymyxin B (Thermo Fisher), followed by incubation for 60 min with tissue culture grade type IV collagenase (1 mg/ml; Sigma, St. Louis, MO) in a 37°C orbital shaker at 100 rpm. The cell suspension was filtered through sterile 70-μm and 40-μm nylon filters, and red blood cells were lysed with ACK buffer (Lonza, Walkersville, MD) to create lung cell preparations. For lung cell cultures, cells were enumerated on a hemacytometer and plated at 1 × 106 cells in a volume of 0.2 ml. The supernatants were collected after 24 h, clarified by centrifugation, and stored at −80°C. IL-22 levels were quantified by enzyme-linked immunosorbent assay (ELISA) (42). In specific experiments, neutralizing antibodies were added to lung cells to assess the effects of cytokine neutralization on IL-22 production. For this, anti-mouse IL-1β, IL-6, IL-18, and IL-23 (all from R&D Systems) were added to lung cell cultures at a final concentration of 2 to 5 μg/ml for 24 h. Rat (IL-1β, IL-6, and IL-18) or goat (IL-23) isotype antibodies were added to lung cell cultures as a control. The supernatants were collected after 24 h and clarified by centrifugation, and IL-22 levels were quantified by ELISA (R&D Systems). In specific experiments, recombinant murine IL-23 (R&D Systems) was added to lung digest cells at 1 or 10 ng/ml for 24 h. The supernatants were collected after 24 h and clarified by centrifugation, and IL-22 levels were quantified by ELISA (R&D Systems). For lung neutrophil analysis by flow cytometry, cells were washed, and Fc receptors were blocked with Mouse BD Fc Block (BD Biosciences, San Diego, CA) at 4°C for 20 min. Thereafter, the cells were stained with a single-color LIVE/DEAD Fixable Dead Cell Stain (Invitrogen), followed by labeling with CD11b+ and Ly6G+ (1A8 clone; antibodies from BD Biosciences) (43).
Analysis of lung lavage fluid antifungal activity.
Wild-type, Dectin-1-deficient, and IL-22-deficient mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia. Twenty-four hours postinfection, a bronchoalveolar lavage was performed as previously described (31, 38). The lavage fluid was centrifuged to remove inflammatory cells and live A. fumigatus. Fifty microliters of clarified lavage fluid from each strain was incubated with 1 × 105 A. fumigatus conidia (in 150 μl of RPMI supplemented with 10% FBS and 1% penicillin-streptomycin) for 4 h at 37°C. Thereafter, the contents of the well was subjected to total RNA extraction using the MasterPure yeast RNA purification kit and analyzed for A. fumigatus viability as described above. RNA was also extracted from the lavage fluid to assess the presence of A. fumigatus after centrifugation, which demonstrated negligible levels (4 to 5 log units below that quantified in 50 μl lavage fluid plus 1 × 105 A. fumigatus conidia).
Lipocalin 2, S100a8, S100a9, and Reg3g analysis.
C57BL/6 WT, Dectin-1-deficient, and IL-22-deficient mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 18 h after exposure, lungs were collected and homogenized in TRIzol reagent (Invitrogen), and total RNA was isolated according to the manufacturer's instructions. Thereafter, RNA was transcribed to cDNA (iScript cDNA synthesis kit; Bio-Rad), and real-time PCR for S100a8 (Mm00496696_g1; Applied Biosystems), S100a9 (Mm00656925_m1; Applied Biosystems), and Reg3g (Mm00441127_m1; Applied Biosystems) was performed (iQ Supermix; Bio-Rad). mRNA levels were normalized to Gapdh mRNA levels (primers/probe from Applied Biosystems) using the 2−ΔΔCT method. For lipocalin 2 quantification, C57BL/6 WT, Dectin-1-deficient, and IL-22-deficient mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 24 h after exposure, the left lungs were homogenized in PBS supplemented with Complete Mini protease inhibitor tablets (Roche), clarified by centrifugation, and stored at −80°C. The supernatants from lung homogenates were analyzed for lipocalin 2 levels by ELISA (R&D Systems).
Statistics.
Data were analyzed using GraphPad Prism Version 5.0 statistical software. Comparisons between groups when data were normally distributed were made with Student's t test. Significance was accepted at a P value of <0.05.
RESULTS
IL-22 production after A. fumigatus challenge is dependent on Dectin-1.
As we have previously reported that Dectin-1-dependent IL-17A was a critical component of lung defense against A. fumigatus (42), we sought to determine whether IL-22 was also dependent on beta-glucan recognition via Dectin-1 and whether it was required for A. fumigatus host defense. The results in Fig. 1A show that IL-22 was robustly induced in the lungs after A. fumigatus challenge (naïve lungs have undetectable IL-22 [data not shown]) and required Dectin-1-mediated recognition of A. fumigatus, as mice deficient in Dectin-1 had severely compromised production of IL-22 in the lungs. We next collected lungs from C57BL/6 (WT) and Dectin-1-deficient (knockout [KO]) mice 18 h after A. fumigatus challenge and subjected them to enzymatic digestion to determine whether single-cell suspensions could replicate the differences in IL-22 levels observed in whole-lung homogenates, as we have recently reported for IL-17A (43). Upon ex vivo culturing of lung cells overnight, cells from Dectin-1-deficient mice had a >8-fold reduction in IL-22 production compared to WT lung digest cells (Fig. 1B). Thus, beta-glucan recognition via Dectin-1 mediates lung IL-22 production after A. fumigatus challenge.
Fig 1.

IL-22 production after A. fumigatus challenge is dependent on Dectin-1. (A) C57BL/6 WT and Dectin-1-deficient (KO) mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 48 h after exposure, IL-22 levels were quantified in lung homogenates by ELISA. The data are expressed as mean pg/ml plus standard error of the mean (SEM). Shown are cumulative data from three independent studies (n = 5 mice/group for each study). *** indicates a P value of <0.001 (unpaired two-tailed Student's t test). (B) C57BL/6 WT and Dectin-1-deficient (KO) mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 18 h after exposure, lungs were collected and enzymatically digested. Single-cell suspensions were isolated, and 1 × 106 cells were cultured for 24 h in a volume of 0.2 ml. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Shown are cumulative data from four independent studies. The data are expressed as mean pg/ml plus SEM. *** indicates a P value of <0.001 (unpaired two-tailed Student's t test).
IL-22 is required for early A. fumigatus lung clearance.
IL-17A is acknowledged to stimulate the antimicrobial immune effector functions of multiple cell types, including neutrophils, macrophages, and epithelial cells (12). We have previously reported that neutralization of IL-17A compromised lung clearance of A. fumigatus (42). However, as IL-22 appears to primarily activate epithelial cells (37, 45), we questioned whether neutralization of IL-22, based on its limited cellular targeting, would have a significant effect on innate immune clearance of A. fumigatus. The results in Fig. 2 show that neutralization of IL-22 in the lungs of C57BL/6 mice (Fig. 2A) resulted in a >5-fold increase in the A. fumigatus lung burden by 24 h postinfection (Fig. 2B). We confirmed this finding in mice deficient in IL-22, which demonstrated a >8-fold increase in the A. fumigatus lung burden by 24 h postinfection (Fig. 3A). Despite having higher A. fumigatus lung burdens, IL-22-deficient mice demonstrated significantly lower levels of multiple cytokines and chemokines previously implicated in lung host defense against A. fumigatus, including IL-1α, tumor necrosis factor alpha (TNF-α), CCL3/MIP-1α, and CCL/4MIP-1β (13, 30) (Fig. 3B). In turn, the lack of these proinflammatory cytokines and chemokines resulted in a >7-fold reduction in CD11b+ Ly-6G+ neutrophils in the lungs of IL-22-deficient mice (Fig. 3C). We also observed reductions in IL-12p40 and IL-12p70 (Fig. 3D), although gamma interferon (IFN-γ) levels were unaffected (data not shown). IL-22 has also been shown to induce CXCL9/Mig production in the lungs during bacterial pneumonia (2), and CXCL9/Mig, as well as CXCL10/IP-10, has been reported to have direct antimicrobial activity (9), suggesting the possibility that IL-22-induced CXCL9/Mig or CXCL10/IP-10 could function as an innate effector molecule against A. fumigatus. However, IL-22-deficient mice exposed to A. fumigatus did not demonstrate a reduction in CXCL9/Mig or CXCL10/IP-10 (Fig. 3E), diminishing the likelihood of a role for these molecules in IL-22-mediated defense against A. fumigatus. Intriguingly, IL-17A levels were significantly increased in the lungs of A. fumigatus-challenged IL-22-deficient mice (Fig. 3F), indicating that IL-17A production in the lungs is not dependent on IL-22. Thus, IL-22 is required for optimal clearance of A. fumigatus from the lungs.
Fig 2.

Neutralization of IL-22 compromises early A. fumigatus lung clearance. (A) C57BL/6 WT mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia. Six hours after challenge, the mice were administered 50 μg of goat anti-mouse IL-22 or goat IgG antibodies intratracheally. IL-22 levels were quantified in lung homogenates 24 h after challenge by ELISA. Shown are cumulative data from two independent studies (n = 5 mice per group per time point). The data are expressed as mean pg/ml plus SEM. ** indicates a P value of <0.01 (unpaired two-tailed Student's t test). (B) Real-time PCR analysis of A. fumigatus 18S rRNA levels in the lungs of WT mice administered anti-IL-22 or isotype control antibodies. Shown are cumulative data from two independent studies (n = 5 mice per group per time point). The data are expressed as mean A. fumigatus 18S rRNA plus SEM. ** indicates a P value of <0.01 (unpaired two-tailed Student's t test).
Fig 3.

IL-22-deficient mice have impaired A. fumigatus lung clearance. (A) C57BL/6 WT and IL-22-deficient (IL-22 KO) mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 24 h after exposure, the lung fungal burden was assessed by real-time PCR analysis of A. fumigatus 18S rRNA levels. Shown are cumulative data from three independent studies (n = 5 mice per group). The data are expressed as mean A. fumigatus 18S rRNA plus SEM. ** indicates a P value of <0.01 (unpaired two-tailed Student's t test). (B) Levels of IL-1α, TNF-α, CCL3, and CCL4 were quantified in lung homogenates collected 24 h postinfection by Bio-Plex. Shown are cumulative data from three independent studies (n = 5 mice per group per time point). The data are expressed as mean pg/ml plus SEM. * and *** indicate P values of <0.05 and <0.001, respectively (unpaired two-tailed Student's t test). (C) Lung cells were isolated via bronchoalveolar lavage, Fc blocked, stained with a LIVE/DEAD staining kit, and then stained with fluorochrome-conjugated CD11b and Ly-6G. Shown are representative data from one of two independent studies. The data are expressed as the absolute number of live cells in lung lavage fluid. * indicates a P value of <0.05 (unpaired two-tailed Student's t test). (D) IL-12p40 and IL-12p70 were quantified in lung homogenates collected 24 h postinfection by Bio-Plex. Shown are cumulative data from three independent studies (n = 5 mice per group per time point). The data are expressed as mean pg/ml plus SEM. * and ** indicate P values of <0.05 and 0.01, respectively (unpaired two-tailed Student's t test). (E and F) Levels of CXCL9 and CXCL10 (E) and IL-17A (F) were quantified in lung homogenates collected 24 h postinfection by ELISA. Shown are cumulative data from three independent studies (n = 5 mice per group per time point). The data are expressed as mean pg/ml plus SEM. ** indicates a P value of <0.01 (unpaired two-tailed Student's t test).
Impaired antifungal activity in lung lavage fluid from A. fumigatus-challenged Dectin-1-deficient and IL-22-deficient mice.
Mice deficient in the beta-glucan receptor Dectin-1 have impaired IL-17A (43) and IL-22 (Fig. 1) production in the lungs in response to A. fumigatus, and neutralization of IL-17A (42) or IL-22 (Fig. 2) compromises clearance of A. fumigatus from the lungs. As both IL-17A and IL-22 are efficient at eliciting soluble antimicrobial factors from epithelial cells (50), we hypothesized that defects in these factors would be reflected in the antifungal activity of lung lavage fluid. The results in Fig. 4A show that lung lavage fluid from Dectin-1-deficient mice demonstrated poor antifungal activity compared to lung lavage fluid from WT mice. Lung lavage fluid from IL-22-deficient mice also showed compromised antifungal activity, although it was not at the level of Dectin-1-deficient mice. We observed both a Dectin-1-dependent and an IL-22-dependent (Fig. 4B) induction of the siderophore binding protein lipocalin 2, which can be induced by IL-17A and IL-22 (2). As A. fumigatus requires iron for growth and encodes its own siderophores (35), we hypothesized that lipocalin 2 may act as an antifungal agent against A. fumigatus by limiting A. fumigatus iron acquisition. To our surprise, mice deficient in lipocalin 2 were not more susceptible to A. fumigatus lung infection (Fig. 4C). IL-22 has also been shown to induce other antimicrobial proteins (50). Real-time PCR analysis of Reg3g mRNA expression indicated low induction in response to A. fumigatus (1.5- to 2-fold) but intact expression in Dectin-1-deficient and IL-22-deficient mice (data not shown). Similarly, S100a8 and S100a9 mRNA expression was induced 15- to 25-fold in response to A. fumigatus but was not modulated in Dectin-1-deficient or IL-22-deficient mice (data not shown). Thus, one mechanism of susceptibility to A. fumigatus in the setting of Dectin-1 or IL-22 deficiency is a putative lack of or impairment in a soluble factor(s) with antifungal activity; however, this factor(s) does not appear to be lipocalin 2, S100A8, S100A9, or RegIIIγ.
Fig 4.
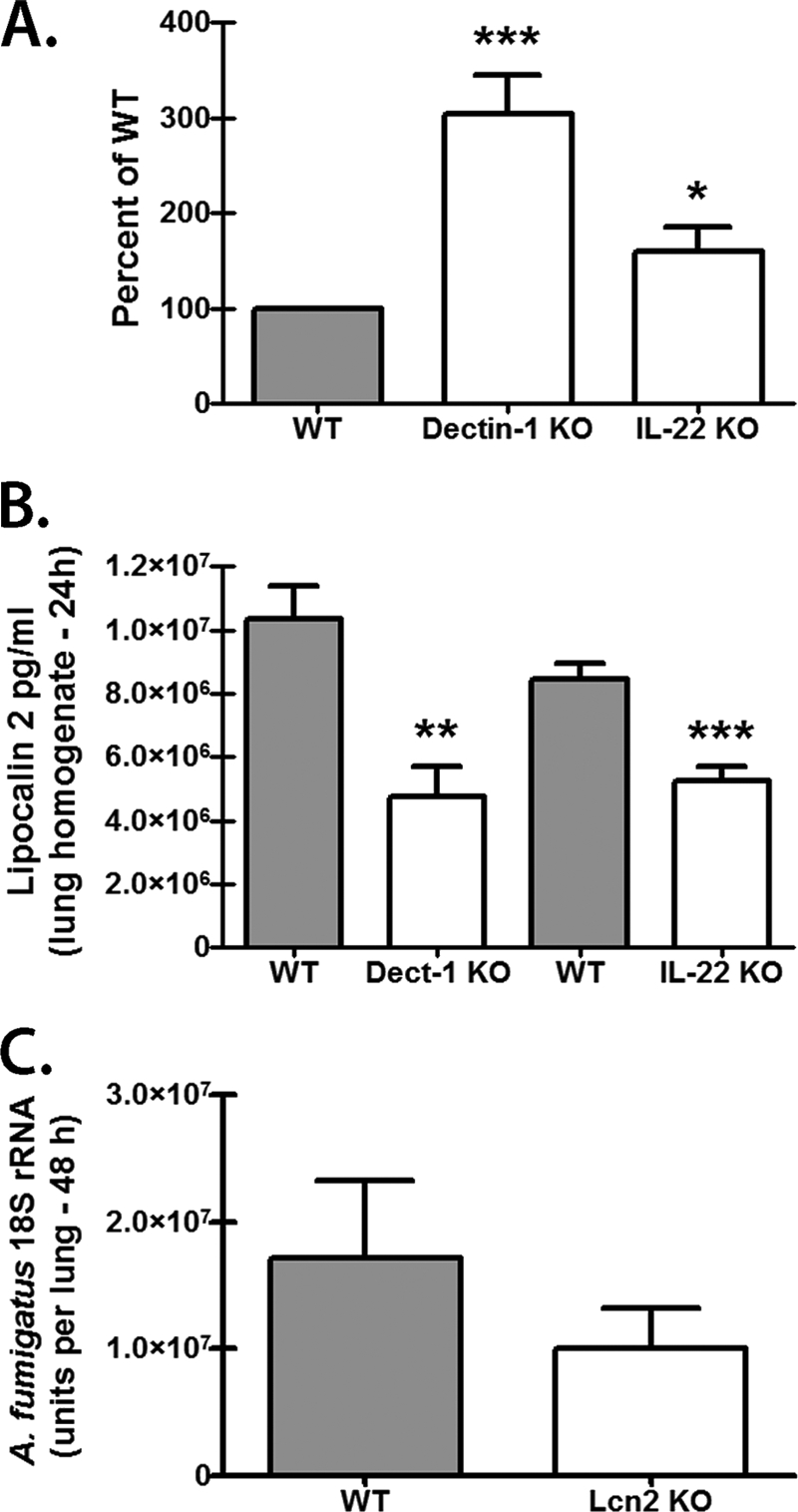
Impaired antifungal activity in lung lavage fluid from A. fumigatus-challenged Dectin-1-deficient and IL-22-deficient mice. (A) C57BL/6 WT, Dectin-1-deficient (Dectin-1 KO), and IL-22-deficient (IL-22 KO) mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 24 h after exposure, bronchoalveolar lavage was performed. The lung lavage fluid was processed to remove cells and A. fumigatus, and then, 50 μl of clarified lavage fluid from each strain was incubated with 1 × 105 A. fumigatus conidia (in 150 μl of RPMI supplemented with 10% FBS and 1% penicillin-streptomycin) for 4 h at 37°C. Thereafter, the contents of the well was subjected to total RNA extraction using the MasterPure yeast RNA purification kit and analyzed for A. fumigatus viability. For each experiment, the percent above WT was calculated by dividing the A. fumigatus 18S rRNA units in Dectin-1-deficient and IL-22-deficient cultures by the A. fumigatus 18S rRNA units in WT cultures. WT values were set at 100. Shown are cumulative data from eight independent studies. * and *** indicate P values of <0.05 and <0.001, respectively (paired two-tailed Student's t test). (B) C57BL/6 WT, Dectin-1-deficient, and IL-22-deficient mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 24 h after exposure, lungs were collected and homogenized and lipocalin 2 levels were quantified in the clarified homogenates by ELISA. Shown are cumulative data from two independent studies (n = 4 to 5 per group). ** and *** indicate P values of <0.01 and <0.001, respectively (unpaired two-tailed Student's t test). (C) C57BL/6 WT and lipocalin 2-deficient (Lcn2 KO) mice were challenged intratracheally with 5 × 107 to 7 × 107 A. fumigatus conidia, and 24 h after exposure, the lung fungal burden was assessed by real-time PCR analysis of A. fumigatus 18S rRNA levels. Shown are cumulative data from two independent studies (n = 5 mice per group). The data are expressed as mean A. fumigatus 18S rRNA plus SEM.
IL-22 production by lung cells in response to A. fumigatus is independent of IL-1β, IL-6, and IL-18 but requires IL-23.
We have previously employed the culture system in Fig. 1B to determine mechanisms associated with Dectin-1-dependent IL-17A production (43). IL-22 is recognized to be produced by IL-17A-producing CD4 T cells (Th17 cells), although other cellular sources have been described (27, 40). Along with IL-17A production, cytokines, such as IL-6, IL-23, and IL-1β, have also been shown to be important for IL-22 production by multiple cell types (21). In addition, IL-18 may synergize with IL-12 or IL-23 for IL-22 induction in NK cells (44, 48). We have recently shown that neutralization of IL-23 in lung cell cultures from A. fumigatus-challenged mice resulted in attenuated IL-17A production (43). Moreover, IL-23p19-deficient mice have reduced IL-22 production in a murine model of gastrointestinal candidiasis (10). Therefore, we questioned whether lung IL-22 production was similarly dependent on IL-1β, IL-6, IL-18, or IL-23 during A. fumigatus infection. We have previously reported that IL-6 and IL-1β were produced at lower levels by lung cells from Dectin-1-deficient mice (43), suggesting a possible role for these cytokines in lung cell IL-17A and IL-22 production during A. fumigatus infection. However, neutralization of IL-1β, IL-6, or IL-18 did not significantly reduce lung cell production of IL-22 (Fig. 5A). Although IL-1β neutralization appeared to lower IL-22 production, it did not reach statistical significance (P = 0.0685). Once again, however, IL-23 was a key factor in IL-22 induction, as neutralization of IL-23 resulted in a 75% decrease in IL-22 production by lung cells (Fig. 5A). The results in Fig. 5B show that supplementing IL-23 in lung cell cultures resulted in increased IL-22 production, even in lung cells from Dectin-1-deficient mice. Thus, IL-22 production by lung cells from A. fumigatus-challenged mice is partially dependent on IL-23, and IL-23 can restore IL-22 production in Dectin-1-deficient mice.
Fig 5.
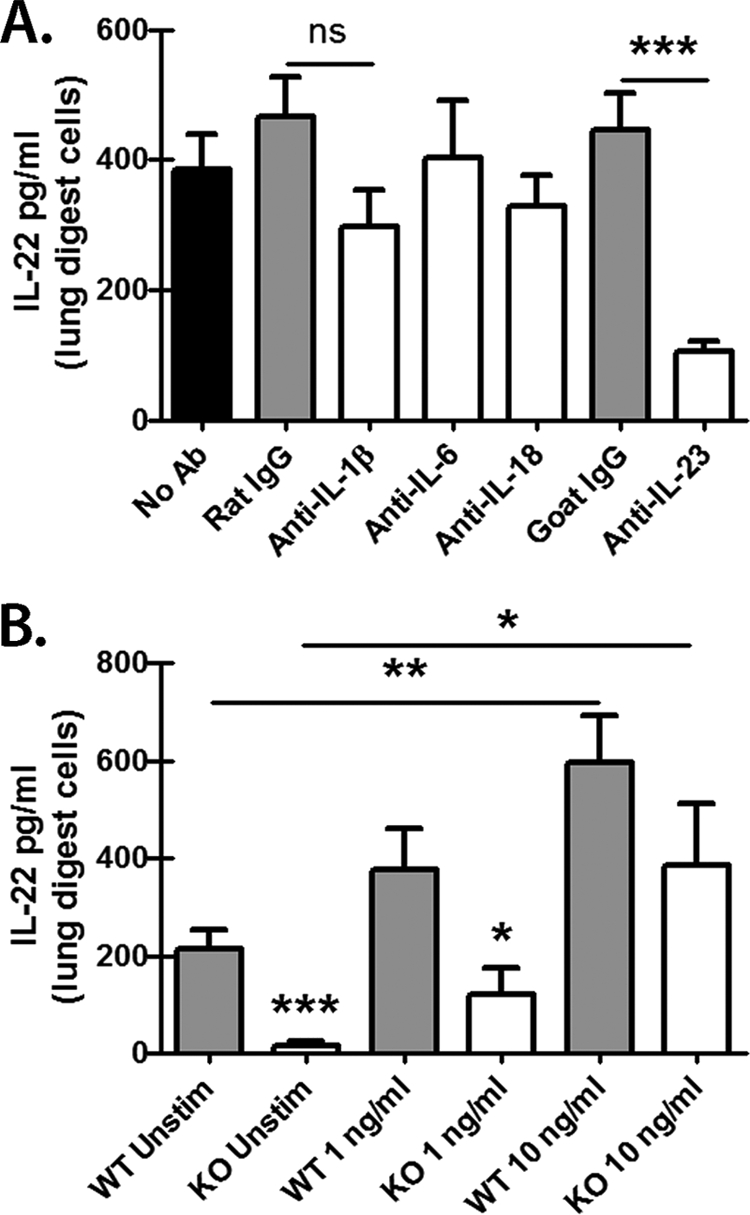
IL-22 production by lung cells in response to A. fumigatus is independent of IL-1β, IL-6, and IL-18 but requires IL-23. (A) Lung cells were isolated as described in Materials and Methods, and 1 × 106 cells were cultured for 24 h in a volume of 0.2 ml. Neutralizing antibodies against IL-1β, IL-6, IL-18, and IL-23 were added at a final concentration of 2 to 5 μg/ml at the beginning of the culture. Rat (IL-1β, IL-6, and IL-18) or goat (IL-23) isotype antibodies were included as a control. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Shown are cumulative data from two independent studies for each condition (isotype and neutralizing antibody) run in triplicate. The data are expressed as mean pg/ml plus SEM. *** indicates a P value of <0.001 (unpaired two-tailed Student's t test); ns, not significant. (B) Lung cells were isolated from WT and KO mice as described, and 1 × 106 cells were cultured for 24 h in a volume of 0.2 ml. Recombinant murine IL-23 was added at 1 and 10 ng/ml at the beginning of the culture. The controls included lung cells cultured in the absence of IL-23. IL-22 levels were quantified in clarified coculture supernatants by ELISA. Shown are cumulative data from three independent studies. The data are expressed as mean pg/ml plus SEM. *, **, and *** indicate P values of 0.05, 0.01, and 0.001, respectively (unpaired two-tailed Student's t test).
DISCUSSION
With the explosion of IL-17A-related research over the last 5 years, studies have discovered that CD4+ T cells producing IL-17A can also produce the cytokine IL-22, a member of the IL-10 family of cytokines (24). Analogous to the observations for IL-17A, additional cell types, such as lymphoid tissue inducer cells (40), NK cells (6), and γδ T cells (27), can also produce IL-22. Although IL-22 can act in both an anti-inflammatory (47) and a proinflammatory (49) manner, IL-22 has been reported to play a major role in stimulating epithelial antimicrobial activity and host defense against multiple mucosal pathogens (50), including the fungal organism Candida albicans (10). To date, only epithelial cells and keratinocytes have been identified as expressing the IL-22 receptor (45). We have previously identified a role for Dectin-1-dependent IL-17A in host defense against A. fumigatus (42, 43). As IL-17A may work in tandem with IL-22 (2, 26, 50) and IL-23 is reportedly required for IL-22 induction in several models (10, 37, 40), we extended our studies here to investigate the role of Dectin-1 in the induction of IL-22 and the role of IL-22 in A. fumigatus lung defense. In our initial studies, we were surprised at the magnitude of Dectin-1 dependency for IL-22 production in the lungs after A. fumigatus challenge. In both lung homogenates and lung cell cultures from Dectin-1-deficient mice, IL-22 was produced at less than 10% of that produced by WT mice. Although we hypothesize that some of this is due to compromised IL-23 production in Dectin-1-deficient mice (43), the dependency of IL-22 on Dectin-1 during A. fumigatus exposure is more striking than that observed in IL-23-deficient mice systemically exposed to C. albicans, which demonstrated IL-22 levels that were still a third of WT levels (10). Coupling this observation with our data indicating that IL-22 production by lung cells is reduced by three-fourths in the presence of IL-23 neutralization leads us to hypothesize that at sites of infection, additional mediators are likely involved in optimal IL-22 production (i.e., the remaining quarter to a third in both C. albicans and A. fumigatus infections). This does not appear to be IL-6, IL-1β, or IL-18, as we show that neutralization of these cytokines had no effect on IL-22 production by lung cells from A. fumigatus-exposed mice. As IL-23 signals through IL-12Rβ1 and IL-23Rα, it is thought to activate the STAT1, STAT4, STAT3, and STAT5 signaling pathways (14). With respect to Th17/IL-17A responses, STAT3 activation is clearly favored by IL-23 (23). Therefore, we can speculate that an additional mediator(s) may activate STAT3, and possibly other STATs as well, and synergize with IL-23 for optimal lung IL-22 production. Currently, studies are under way to identify additional cytokines that may be involved in IL-22 production by lung cells.
To thoroughly examine the role of IL-22 in lung host defense against A. fumigatus, we employed two independent experimental designs: (i) neutralization and (ii) genetic deficiency. Neutralization of IL-22, as well as IL-22 deficiency, led to significantly compromised clearance of A. fumigatus from the lungs. The level of impairment in fungal clearance was also more apparent with A. fumigatus than in a previous report with C. albicans, which demonstrated 2-fold changes in stomach (gastrointestinal infection) and kidney (systemic infection) burdens (3 days postchallenge) when IL-22 was genetically deficient (10). Neutralization of IL-22 in this model had little or no effect on the C. albicans stomach burden in C57BL/6 mice (8 days postchallenge) and increased the kidney burden by only a third in BALB/c mice (10). In contrast, our studies revealed that IL-22 neutralization resulted in a 5-fold increase in the lung A. fumigatus burden, whereas IL-22 genetic deficiency resulted in an 8-fold increase in the A. fumigatus burden. There are many possibilities as to why differences were observed in our study versus the C. albicans study. Clearly, these two pathogens are quite different in their tissue specificities and host defense requirements; thus, it is possible that host defense against one organism may require IL-22 more than the other. Moreover, our studies investigated the role of IL-22 in early/rapid host defense against A. fumigatus, i.e., 1 to 2 days postchallenge, in contrast to the 3- to 8-day time course of the C. albicans infections. In addition, it is also possible that the role of IL-22 may be more evident, perhaps even more important, in such tissues as the lung and gut, where the overwhelming majority of cells are epithelial cells and keratinocytes. Nevertheless, our studies point to an essential role for IL-22 at the earliest stages of A. fumigatus lung infection.
As mentioned previously, we have documented a role for IL-17A in A. fumigatus lung defense (42) and now extend this to IL-22. Dual roles for IL-17A and IL-22 have also been observed in lung infection with K. pneumoniae (17, 2) and gut infection with C. rodentium (26, 50). Surprisingly, however, IL-17A and IL-22 do not always play equal roles in host defense. Cutaneous infection with Staphylococcus aureus is worse in γδ T cell-deficient mice and is correlated with a lack of IL-17A, but not IL-22, production (7). In models of oral infection (8) and skin infection (18) with C. albicans, IL-17A, but not IL-22, was required for defense. Protective immunity to systemic infection with attenuated Salmonella enterica serovar Enteritidis is associated with IL-22, but not IL-17 (36), while infection with Borrelia burgdorferi induces a potent IL-22 response, yet IL-17A is completely absent (3). Finally, in Listeria monocytogenes infection, IL-17A is required for clearance (16), but not IL-22 (15), a finding also observed in Francisella tularensis infection (25). However, during A. fumigatus lung infection, our studies indicate that both IL-17A and IL-22 are simultaneously required for host defense. IL-17A levels are significantly increased in the lungs of IL-22-deficient mice challenged with A. fumigatus, yet lung clearance is impaired. In turn, we have reported that IL-17A neutralization leads to impaired A. fumigatus lung clearance (42), although IL-22 levels were not affected by IL-17A neutralization (1,276 ± 105 pg/ml, n = 10, versus 1,148 ± 98 pg/ml, n = 10, in lung homogenates for isotype- and anti-IL-17A-treated mice, respectively). Therefore, in a scenario where either IL-17A or IL-22 is absent, our data suggest that the remaining response is not sufficient to compensate.
A well-documented role of IL-22 in the context of host defense is in the induction of the epithelial antimicrobial response. Initial studies examining the function of IL-22 showed that stimulation of epithelial cells and keratinocytes with IL-22 led to the induction of antimicrobial defense factors, such as beta-defensins, S100 proteins, and RegIII proteins (50). IL-17A also has an acknowledged role in the induction of these factors (2), and IL-22 can often add to or synergize with IL-17A for the induction of the epithelial antimicrobial response. Recognizing that IL-22, along with IL-17A, can evoke this response in the lungs (2) led us to determine whether functional defects existed in the lungs of Dectin-1-deficient and IL-22-deficient mice exposed to A. fumigatus. To this end, we demonstrated that clarified lung lavage fluid (i.e., fluid that was free of live A. fumigatus and live host cells) from both Dectin-1-deficient and IL-22-deficient mice did not kill A. fumigatus as robustly as lung lavage fluid from WT mice. The defect in antifungal activity was more severe in lavage fluid from Dectin-1-deficient mice, which we hypothesize is a result of these mice having significant reductions in both IL-17A (42) and IL-22 (Fig. 1). Despite compromised S100A8 and S100A9 expression in IL-22-deficient mice intragastrically infected with C. albicans (10), we found that S100a8 and S100a9 mRNA expression was intact in the lungs of A. fumigatus-exposed Dectin-1-deficient and IL-22-deficient mice (data not shown). In addition, Reg3g levels were not found to be statistically lower in the lungs (data not shown). In contrast, we did observe a reduction in the lung levels of lipocalin 2, a siderophore binding protein induced by IL-22 (2), in both Dectin-1-deficient and IL-22-deficient mice, suggesting a possible role for lipocalin 2 in A. fumigatus lung defense. However, lipocalin 2-deficient mice did not demonstrate an impairment in A. fumigatus lung clearance, indicating that lipocalin 2 does not appear to play a major role in the susceptibility of Dectin-1-deficient and IL-22-deficient mice to A. fumigatus. Although we did not see an effect of lipocalin 2 deficiency on A. fumigatus lung clearance, we cannot exclude the possibility that other antimicrobial factors are compensating for the loss of lipocalin 2. For example, lactoferrin can mediate reactive oxygen species (ROS)-independent killing of A. fumigatus by neutrophils (46). Currently, studies are under way to identify the Dectin-1- and IL-22-dependent soluble antifungal factors induced in the lungs during A. fumigatus infection.
In summary, we have identified a role for IL-22 in early innate immune responsiveness to A. fumigatus lung infection. Induction of IL-22 was significantly dependent on A. fumigatus recognition by the beta-glucan receptor Dectin-1. Both neutralization of and genetic deficiency in IL-22 compromised early clearance of A. fumigatus from the lungs. IL-22 was critical for the induction of both lung inflammatory cytokines and chemokines, as well as the lung antifungal response. However, the Dectin-1- and IL-22-dependent lung antifungal response was independent of the known IL-17A- and IL-22-associated antimicrobial factor S100 proteins, RegIIIγ and lipocalin 2, suggesting a separate, yet-to-be-characterized Dectin-1- and IL-22-dependent antifungal mechanism. As with our recent report on IL-17A (43), our data further suggest that soluble mediators, in addition to IL-23, may also play a role in Dectin-1-dependent IL-22 production. However, as our data indicate that IL-17A and IL-22 are simultaneously needed for A. fumigatus lung clearance and that IL-23 is essential for the induction of both cytokines during A. fumigatus lung infection, IL-23 may be an effective immunotherapy for the treatment of IA in susceptible individuals. In conclusion, the current body of work adds depth to our understanding of the roles of Dectin-1 and the IL-23/IL-17A/IL-22 axis in innate lung defense against A. fumigatus.
ACKNOWLEDGMENTS
This work was supported was supported by Public Health Service grants AI068917 and HL096702.
Footnotes
Published ahead of print 28 October 2011
REFERENCES
- 1. Alangaden GJ. 2011. Nosocomial fungal infections: epidemiology, infection control, and prevention. Infect. Dis. Clin. North Am. 25: 201–225 [DOI] [PubMed] [Google Scholar]
- 2. Aujla SJ, et al. 2008. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14: 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bachmann M, et al. 2010. Early production of IL-22 but not IL-17 by peripheral blood mononuclear cells exposed to live Borrelia burgdorferi: the role of monocytes and interleukin-1. PLoS Pathog. 6: e1001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berger T, et al. 2006. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 103: 1834–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowman JC, et al. 2001. Quantitative PCR assay to measure Aspergillus fumigatus burden in a murine model of disseminated aspergillosis: demonstration of efficacy of caspofungin acetate. Antimicrob. Agents Chemother. 45: 3474–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cella M, et al. 2009. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457: 722–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho JS, et al. 2010. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 120: 1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conti HR, et al. 2009. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206: 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crawford MA, et al. 2010. Interferon-inducible CXC chemokines directly contribute to host defense against inhalational anthrax in a murine model of infection. PLoS Pathog. 6: e1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Luca A, et al. 2010. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 3: 361–373 [DOI] [PubMed] [Google Scholar]
- 11. Elson CO, et al. 2007. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132: 2359–2370 [DOI] [PubMed] [Google Scholar]
- 12. Gaffen SL. 2009. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9: 556–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao JL, et al. 1997. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J. Exp. Med. 185: 1959–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. 2009. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm. Allergy Drug Targets 8: 40–52 [DOI] [PubMed] [Google Scholar]
- 15. Graham AC, et al. 2011. IL-22 production is regulated by IL-23 during Listeria monocytogenes infection but is not required for bacterial clearance or tissue protection. PLoS One 6: e17171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamada S, et al. 2008. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against Listeria monocytogenes infection in the liver. J. Immunol. 181: 3456–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Happel KI, et al. 2005. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 202: 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. 2010. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J. Immunol. 185: 5453–5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kontoyiannis DP, Bodey GP. 2002. Invasive aspergillosis in 2002: an update. Eur. J. Clin. Microbiol. Infect. Dis. 21: 161–172 [DOI] [PubMed] [Google Scholar]
- 20. Kontoyiannis DP, et al. 2010. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin. Infect. Dis. 50: 1091–1100 [DOI] [PubMed] [Google Scholar]
- 21. Korn T, Bettelli E, Oukka M, Kuchroo VK. 2009. IL-17 and Th17 cells. Annu. Rev. Immunol. 27: 485–517 [DOI] [PubMed] [Google Scholar]
- 22. Kullberg MC, et al. 2006. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203: 2485–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langrish CL, et al. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201: 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liang SC, et al. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203: 2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin Y, et al. 2009. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity 31: 799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mangan PR, et al. 2006. Transforming growth factor-beta induces development of the TH17 lineage. Nature 441: 231–234 [DOI] [PubMed] [Google Scholar]
- 27. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. 2009. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31: 321–330 [DOI] [PubMed] [Google Scholar]
- 28. Mattila PE, Metz AE, Rapaka RR, Bauer LD, Steele C. 2008. Dectin-1 Fc targeting of Aspergillus fumigatus beta-glucans augments innate defense against invasive pulmonary aspergillosis. Antimicrob. Agents Chemother 52: 1171–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McGeachy MJ, et al. 2007. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 8: 1390–1397 [DOI] [PubMed] [Google Scholar]
- 30. Mehrad B, Strieter RM, Standiford TJ. 1999. Role of TNF-alpha in pulmonary host defense in murine invasive aspergillosis. J. Immunol. 162: 1633–1640 [PubMed] [Google Scholar]
- 31. Nelson MP, Metz AE, Li S, Lowell CA, Steele C. 2009. The absence of Hck, Fgr and Lyn tyrosine kinases augments lung innate immune responses to Pneumocystis murina. Infect. Immun. 77: 1790–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Connor W, Jr, et al. 2009. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 10: 603–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pappas PG, et al. 2010. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin. Infect. Dis. 50: 1101–1111 [DOI] [PubMed] [Google Scholar]
- 34. Pestka S, et al. 2004. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 22: 929–979 [DOI] [PubMed] [Google Scholar]
- 35. Schrettl M, et al. 2007. Distinct roles for intra- and extracellular siderophores during Aspergillus fumigatus infection. PLoS Pathog. 3: 1195–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schulz SM, et al. 2008. Protective immunity to systemic infection with attenuated Salmonella enterica serovar enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. J. Immunol. 181: 7891–7901 [DOI] [PubMed] [Google Scholar]
- 37. Sonnenberg GF, Fouser LA, Artis D. 2011. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12: 383–390 [DOI] [PubMed] [Google Scholar]
- 38. Steele C, et al. 2005. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 1: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugimoto K, et al. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118: 534–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takatori H, et al. 2009. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taylor PR, et al. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8: 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Werner J, et al. 2009. Requisite role for the Dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 182: 4938–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Werner JL, et al. 2011. Neutrophils produce IL-17A in a Dectin-1 and IL-23 dependent manner during invasive fungal infection. Infect. Immun. 79: 3966–3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wolk K, Kunz S, Asadullah K, Sabat R. 2002. Cutting edge: immune cells as sources and targets of the IL-10 family members? J. Immunol. 168: 5397–5402 [DOI] [PubMed] [Google Scholar]
- 45. Wolk K, et al. 2004. IL-22 increases the innate immunity of tissues. Immunity 21: 241–254 [DOI] [PubMed] [Google Scholar]
- 46. Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. 2007. Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J. Immunol. 178: 6367–6373 [DOI] [PubMed] [Google Scholar]
- 47. Zenewicz L, et al. 2007. Interleukin-22 but not Interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27: 647–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zenewicz LA, et al. 2008. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29: 947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng Y, et al. 2007. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445: 648–651 [DOI] [PubMed] [Google Scholar]
- 50. Zheng Y, et al. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14: 282–289 [DOI] [PubMed] [Google Scholar]