

Abstract
The indigenous bacterial microbiome of the stomach, including lactobacilli, is vital in promoting colonization resistance against Candida albicans. However, there are gaps in our understanding about C. albicans gastric colonization versus disease, especially during the postantibiotic recovery phase. This study compared the gastric responses to C. albicans strains CHN1 and SC5314 in microbiome-disturbed and germfree mice to elucidate the contribution of the indigenous microbiota in C. albicans colonization versus disease and yeast-bacterium antagonism during the post-cefoperazone recolonization period. C. albicans can prevent the regrowth of Lactobacillus spp. in the stomach after cefoperazone and promote increased colonization by Enterococcus spp. Using a culture-independent analysis, the effects of oral cefoperazone on the gastric bacterial microbiota were observed to last at least 3 weeks after the cessation of the antibiotic. Disturbance of the gastric bacterial community by cefoperazone alone was not sufficient to cause gastritis, C. albicans colonization was also needed. Gastritis was not evident until after day 7 in cefoperazone-treated infected mice. In contrast, in germfree mice which lack a gastric microbiota, C. albicans induced gastric inflammation within 1 week of inoculation. Therefore, the gastric bacterial community in cefoperazone-treated mice during the first week of postantibiotic recolonization was sufficient to prevent the development of gastritis, despite being ineffective at conferring colonization resistance against C. albicans. Altogether, these data implicate a dichotomy between C. albicans colonization and gastric disease that is bacterial microbiome dependent.
INTRODUCTION
There are gaps in our understanding about Candida albicans gastric colonization during the postantibiotic recovery phase and the factors that lead to C. albicans-induced gastritis during this period. C. albicans is both a normal member of the gastrointestinal (GI) tract of healthy humans and an opportunistic pathogen. The indigenous microbiota of the GI tract is effective at preventing invading fungi, such as C. albicans, from long-term colonization and disease (17, 20–23, 41–43), and germfree mice, lacking the indigenous microbiota, are highly susceptible to Candida colonization (28). Mouse models of candidiasis demonstrate that disturbance of the microbiota or immunosuppression are necessary to promote Candida colonization. During gastric colonization, Candida spp. can induce inflammation or exist as noninflammatory commensal organisms (16, 37, 38). Thus, colonization and gastritis are two related but separable events, and the role of the bacterial microbiota in promoting or preventing gastritis is not well defined.
The bacterial microbiota of the stomach is vital in promoting gastric colonization resistance against the opportunistic pathogen C. albicans. The stomach is a preferential niche for Candida colonization such that gastrectomized rodents are unable to support Candida colonization (2). Within the stomach, Lactobacillus, an important contributor to host health, can prevent colonization of Candida through displacement of yeast from the epithelial layer of the stomach (47). Lactobacilli can also inhibit hyphal invasion and systemic infection (37, 47). Previous studies demonstrated that penicillin treatment reduces Lactobacillus populations and promotes yeast colonization of the gastric epithelium (37). Furthermore, in vitro assays in our laboratory have found that Lactobacillus spp. can significantly inhibit C. albicans germ tube formation (31). Although many studies have investigated the ability of bacteria to alter fungi, there few studies have investigated the ability of C. albicans to influence the GI bacteria.
The rapid identification of genes from bacterial populations sampled directly from their environment, such as the mucosa, has revolutionized microbial ecology. It is now possible to analyze microbial community composition in a given environment, without culturing microorganisms, by sequencing PCR amplicons generated using oligonucleotide primers that target phylogenetically conserved genetic sequences. Of particular interest is the 16S rRNA gene, which is highly conserved in all bacteria but displays sequence differences between taxons that can be exploited for culture-independent identification. DNA fingerprinting techniques such as terminal restriction fragment length polymorphism (T-RFLP) (25, 27) are economical, culture-independent, high-throughput techniques for analyzing the composition of the dominant phylotypes in a single complex microbial community. The fingerprint generated from T-RFLP analysis provides a measurement of bacterial community structure and can be used as a molecular surrogate of relative similarities or differences between complex microbial communities.
We have previously reported that pretreatment of mice with cefoperazone facilitates C. albicans CHN1 colonization (30–32). In the present study, we compared the gastric responses to this strain, as well as the commonly studied strain SC5314, in microbiome-disturbed and germfree states to further elucidate the contribution of the indigenous microbiome and C. albicans strain differences in colonization versus disease and in Candida modulation of the gastric bacterial microbiota during the postantibiotic recolonization period.
MATERIALS AND METHODS
Animals and housing.
Female C57BL/6 mice were purchased from Jackson Laboratories (Indianapolis, IN) and were housed under specific-pathogen-free conditions in enclosed filter-top cages. Food and sterile water were given ad libitum. Food remained constant throughout the experiment to minimize the effect of diet on the microbiota. The mice were maintained on grates to prevent coprophagy by the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan (Ann Arbor, MI) and the protocols were approved by an animal institutional review board. Germfree C57BL/6 mice were raised and housed in the ULAM germfree barrier facility at the University of Michigan. C. albicans-infected germfree mice were maintained in the barrier facility.
Antibiotic treatment.
Cefoperazone (0.5 mg/ml; Sigma-Aldrich, St. Louis, MO) was administered orally to mice ad libitum in drinking water. Antibiotic treatment was continued for 7 days prior to C. albicans colonization. After 7 days, antibiotic containing drinking water was replaced with sterile water.
C. albicans gastric inoculation.
C. albicans strain CHN1 (a human pulmonary clinical isolate) and C. albicans strain SC5314 (ATCC MYA-2876) were grown in Sabouraud dextrose broth (Difco, Detroit, MI) to stationary phase in a shaking flask at 37°C. For gavage, the cultures were washed in sterile nonpyrogenic saline, counted using a hemacytometer, and diluted to 2 × 108 CFU/ml in sterile nonpyrogenic saline. Mice were inoculated with C. albicans (107 CFU in 50 μl) by oral administration using a 24-gauge feeding needle attached to a 1-ml syringe. The syringe containing C. albicans was mounted on a Stepper repetitive pipette (Tridak, Brookfield, CT) to deliver an equivalent amount of inoculum to each mouse. The inoculums were serially diluted and grown on Sabouraud dextrose agar (SDA) to verify the number of CFU delivered.
Necropsy and microbiological culture.
Mice were euthanized by CO2 asphyxiation. Mouse stomachs were removed, opened along the greater curvature, and washed in phosphate-buffered saline to remove contents. Sections for bacterial 16S analysis were flash frozen in liquid nitrogen and stored at −80°C. Histological sections from the stomach were fixed with 4% buffered formalin and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (H&E) for the detection of inflammatory infiltrates. The remaining stomachs were homogenized in sterile water, serially diluted, and cultured on differential agars (SDA, violet bile agar (Difco), Trypticase soy agar supplemented with 5% sheep blood (BD Biosciences, Franklin Lakes, NJ), and de Man, Rogosa, and Sharpe (MRS) agar supplemented with 0.02% sodium azide (Difco) to determine culturable bacterial counts.
Colonies that grew on MRS plus azide agar were identified through a colony PCR method with previously published bacterial universal primers (33). Briefly, a sterile toothpick was used to sample each colony and individually added to a 50-μl reaction mixture for PCR amplification (20 pmol of each primer and PCR Mastermix (Roche Diagnostics Corp., Indianapolis, IN) with the following conditions: 95°C for 45 s, annealing at 60°C for 45 s, and elongation for 1.5 min with an additional 5 s for each cycle. A total of 30 cycles were performed, which was followed by a final elongation step at 72°C for 10 min. Purified DNA from the PCR was sequenced at the University of Michigan Sequencing Core.
Yeast numbers were quantified in mucosal samples through culturing on SDA agar supplemented with cefoperazone (0.1 mg/ml). Identity of the yeast was confirmed with wet mounts and replica plating on HardyChrom Candida indicator plates (Hardy Diagnostics, Santa Maria, CA).
DNA extraction.
Genomic DNA was extracted from stomach sections stored at −80°C using a modified commercial kit (DNeasy tissue kit; Qiagen, Germantown, MD). Samples were subjected to bead beating for 1 min in DNA isolation bead tubes (MoBio Laboratories, Carlsbad, CA) prior to kit use. DNeasy tissue protocol was modified to use 40 μl of proteinase K instead of the recommended 20 μl, and the samples were eluted with 100 μl of buffer AE instead of the suggested 200 μl.
T-RFLP.
The culture-independent T-RFLP assay analyzes bacterial community structure by amplifying the 16S rRNA gene from T-RFLP was performed as described previously (25). Briefly, full-length bacterial 16S rRNA genes were amplified from each sample by PCR amplification. The primers used in the amplification were a fluorescently labeled FAM-8F forward primer and an unlabeled 1525R reverse primer. Each 25-μl PCR mixture contained 20 pmol of each primer, 200 μM concentrations of each deoxynucleoside triphosphate, and 1.5 U of Taq DNA polymerase in a final concentration of 10 mM Tris-HCl, 50 mM KCl, and 1.5 mM MgCl2 (Ready-to-Go PCR beads; Amersham Pharmacia Biotech, Piscataway, NJ). PCR was performed under the following cycle conditions: an initial denaturation step at 94°C for 2 min and 30 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 45 s, and extension at 72°C for 90 s. A final extension step at 72°C for 5 min was performed. The PCR product was purified using a QIAquick PCR purification kit (Qiagen). A 200-ng portion of purified PCR amplicon was cut individually with the restriction enzyme MspI (New England Biolabs, Beverly, MA) for 2 h at 37°C. The DNA fragments were separated on an ABI 3730XL (Applied Biosystems Instruments, Foster City, CA) at the University of Michigan Sequencing Facility. The 5′-terminal restriction fragments (TRFs) were detected by excitation of the 6-FAM molecule attached to the forward primer.
T-RFLP analysis.
Raw T-RFLP chromatograms were analyzed by using Peakscanner (Applied Biosystems) to call the fragment sizes and to build a list of peaks (a peak file). This process is carried out for every sample, after which all of the peak files are exported as one bulk peak file. Further analysis was carried out using K9, an in-house designed program for T-RFLP data analysis (freely available at http://www-personal.umich.edu/∼jre/Microbiome_Core/K9.html). K9 separates the bulk peak file into all of the individual peak files, and the meta-tags are removed. Next, corrected peak files are generated by binning peak fragments to the corresponding whole number of fragment lengths. This binning allows uniform comparison of samples from different analyses and also allows for simple background subtraction to be performed where appropriate.
Rank abundance graphs .
Individual TRFs were used to create rank abundance curves for each experimental group. Briefly, for each experimental treatment and time point, TRFs are presented with base pair length plotted on the x axis. The peak height was normalized by determining the percentage of total TRFs for each individual sample. Within each treatment group, individual mice TRFs were combined, and the standard error of the mean is represented by the error bars. Experiments were performed at least twice with three to five mice per group per experiment.
Statistics.
All values reported in rank abundance curves are standard errors of the mean, where mean values are pooled from independent experiments and are noted for each experiment. Bacterial and fungal colonization levels were compared by two-way analysis of variance with a Bonferroni correction (GraphPad Prism 5; *, P < 0.05; **, P < 0.01).
RESULTS
C. albicans CHN1 gastric colonization and histopathology.
Our first objective was to determine whether antibiotic-mediated disruption of colonization resistance was necessary for C. albicans CHN1 to colonize the stomach of conventional C57BL/6 mice. Conventional female mice were treated with the broad-spectrum antibiotic cefoperazone in their water for 1 week (days −7 to 0), followed by a single oral gavage of C. albicans CHN1 at day 0 (antibiotic-free water was provided from days 0 to 21) (30–32). At days 7 and 21 after oral gavage, we analyzed the stomach for culturable CHN1 (Fig. 1). Neither untreated nor cefoperazone-treated conventional mice had culturable Candida albicans species present in the stomach at day 7 or 21. Other Candida species were infrequently observed (<10% of mice) at day 7 in cefoperazone-treated conventional mice and never at day 21 or any other treatment group (data not shown). Less than 40% of the mice given a single oral gavage of CHN1 without prior cefoperazone treatment had a low level of colonization at day 7, and this decreased to <10% of the mice at day 21 that had detectable C. albicans CHN1. However, 100% of the mice at day 7 that were treated with both cefoperazone and given a single oral gavage of C. albicans CHN1 had significant C. albicans colonization. Colonization above detectable levels persisted in >80% of the mice through day 21. These results demonstrate that, without broad-spectrum antibiotic treatment, C. albicans CHN1 cannot establish gastric colonization in conventional C57BL/6 mice.
FIG 1.
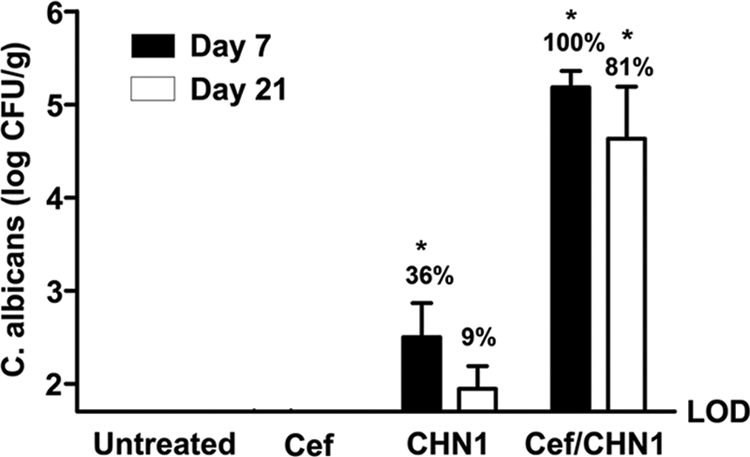
The presence of C. albicans CHN1 during antibiotic recolonization results in elevated fungal colonization of the stomach. The stomach was removed at day 7 and day 21 postantibiotic and differentially cultured to determine C. albicans CHN1 colonization in conventional untreated, antibiotic-treated (Cef), CHN1-colonized alone (CHN1), or cefoperazone-treated CHN1-colonized (Cef/CHN1) mice. Untreated and cefoperazone-only mice had no detectable C. albicans CHN1 colonization. At day 7 postantibiotic, 36% of the CHN1 mice and 100% of the Cef/CHN1 mice had detectable C. albicans CHN1. At day 21 postantibiotic, 9% of the CHN1 mice and 81% of the Cef/CHN1 mice had detectable C. albicans CHN1 in the stomach. Error bars represent the standard errors of the mean. LOD, limit of detection (*, P < 0.05 versus untreated).
We next examined these mice for histological evidence of gastric inflammation and/or changes in the mucosa. None of the mice that received only cefoperazone (not shown) or only C. albicans CHN1 (Fig. 2) developed histologically evident gastric inflammation at days 7 or 21. In contrast, cefoperazone-treated, C. albicans CHN1-colonized mice began to develop low-grade inflammation at the limiting ridge of the stomach at day 7, which progressed into significant mucosal erosion, neutrophil accumulation, and submucosal edema by day 21 (Fig. 2). Using periodic acid-Schiff (PAS) staining to detect hyphae, there was also significant Candida hyphal growth at the eroded mucosa and the underlying tissue at the limiting ridge at day 21 in cefoperazone-treated, C. albicans CHN1-colonized mice, which was not evident in the other groups (Fig. 3). Thus, C. albicans CHN1 gastric colonization following cefoperazone treatment results in limiting ridge inflammation and mucosal erosion that is coincident with Candida hyphal colonization.
FIG 2.

C. albicans CHN1 colonization during bacterial recolonization results in long-term gastric erosions at the murine limiting ridge. Histological sections of the murine stomach were stained with H&E to look for evidence of inflammation during cefoperazone-induced microbiota disruption. Untreated mice had no evidence of gastric erosions at any time point. CHN1-colonized mice at day 7 had a low incidence of gastric erosions and, by day 21, no erosions were seen. Cefoperazone-treated CHN1-colonized mice at day 7 had low-grade inflammation that progressed into erosions by day 21.
FIG 3.

Hyphal growth at the murine limiting ridge following bacterial dysbiosis. Histological sections of the murine stomach were stained with PAS to detect fungi during antibiotic recolonization. Untreated mice had no fungi present at any time point. Cef/CHN1 mice at day 7 had some detectable hyphal growth, but by day 21 postantibiotic, hyphal growth and invasion were detected at the limiting ridge (magnification, ×100). At higher magnifications, substantial hyphal growth was detected at the area of inflammation at the limiting ridge.
Cefoperazone effects in shaping the postantibiotic gastric bacterial community.
Our next objective was to analyze the postantibiotic disruption of the indigenous gastric microbial community caused by oral cefoperazone treatment. We used T-RFLP analysis to generate a global overview of changes in the gastric bacterial microbiome during recovery from antibiotic treatment. At day 7, and even day 21, post-cefoperazone treatment, the community structure of the gastric bacterial microbiota had not recovered to its preantibiotic state (Fig. 4). The disappearance of some TRFs and the appearance of new TRFs after the cessation of antibiotic indicated changes in the bacterial community structure of the stomach as a result of the earlier disruption by cefoperazone treatment. Furthermore, disturbance of the gastric bacterial community by cefoperazone alone is not sufficient to cause inflammation of the limiting ridge.
FIG 4.

Cefoperazone (Cef) treatment results in long-term alterations in the indigenous bacterial populations of the murine stomach in the presence or absence of C. albicans CHN1. The stomach was removed and analyzed using T-RFLP. Rank abundance plots were constructed from TRFs in each of the experimental groups at day 7 and day 21 postantibiotic. Error bars represent the standard errors of the mean, where the mean is pooled TRFs from individual mice within each experimental group.
C. albicans CHN1 modulation of the lactic acid bacterial community during postantibiotic recolonization.
We had previously demonstrated that Lactobacillus can antagonize C. albicans CHN1 hyphal transformation (31, 32). Therefore, we sought to determine whether the hyphal transformation of C. albicans CHN1 at day 21 in the stomachs of cefoperazone-treated mice might be associated with a change in indigenous Lactobacillus levels. Cefoperazone treatment resulted in a 100-fold-lower lactic acid bacterium levels in the stomach at both days 7 (Fig. 5A) and 21 (Fig. 5B). In untreated mice, the predominant lactic acid bacterium in the stomach was Lactobacillus, while at day 7 it had changed to Enterococcus (Fig. 5 and Table S1 in the supplemental material). However, between days 7 and 21, when the ratio between Lactobacillus and Enterococcus was returning to its preantibiotic state, the presence of C. albicans CHN1 antagonized the recolonization of Lactobacillus in the stomach. Enterococcus remained the numerically dominant lactic acid bacterium at day 21 in cefoperazone-treated CHN1-colonized mice, while Lactobacillus became predominant in mice treated with only cefoperazone (Fig. 5B).
FIG 5.

C. albicans CHN1 interacts with the indigenous lactic acid bacteria of the murine stomach. The stomach was removed at day 7 (A) and day 21 (B) postantibiotic and differentially cultured to determine total lactic acid bacterium (LAB) colonization levels in conventional untreated, antibiotic-treated (Cef), and Cef/CHN1 mice (graphs). Lactic acid bacterial colonies that grew on MRS plus azide agar were further identified and expressed as a fraction of the total LAB population in that group, using colony PCR as described in the methods (pie charts). Error bars represent the standard errors of the mean (*, P < 0.05 compared to untreated mice).
C. albicans SC5314 gastric colonization, histopathology, and lactic acid bacterium communities.
C. albicans SC5314 is a highly studied strain of C. albicans, and its genome has been fully sequenced. For comparison to CHN1, we also studied gastric pathogenesis by C. albicans SC5314, using both the same microbiota disruption protocol described above and germfree mice. Similar community changes in the gastric bacterial microbiota were observed by T-RFLP analysis (Fig. 6). Furthermore, the presence of C. albicans SC5314 was also able to antagonize the regrowth of lactobacilli in the stomachs of cefoperazone-treated C57BL/6 mice (Fig. 7). Colonization levels in the stomach were comparable between the two strains of C. albicans (not significant, P > 0.05), although the levels of strain SC5314 (Fig. 8A) were slightly lower than those observed for strain CHN1 (Fig. 1) despite identical inoculum doses. SC5314 also induced low-grade gastric inflammation by day 21 in cefoperazone-treated mice (Fig. 8).
FIG 6.

Cefoperazone (Cef) treatment results in long-term alterations in the indigenous bacterial populations of the murine stomach in the presence or absence of C. albicans SC5314.The stomach was removed and analyzed with T-RFLP. Rank abundance plots were constructed from TRFs in each of the experimental groups at day 7 and day 21 postantibiotic. Error bars represent the standard errors of the mean, where the mean is pooled TRFs from individual mice within each experimental group.
FIG 7.

C. albicans SC5314 interacts with the indigenous lactic acid bacteria of the murine stomach. The stomach was removed at day 7 (A) and day 21 (B) postantibiotic and differentially cultured to determine total lactic acid bacterium (LAB) colonization levels in conventional untreated, antibiotic-treated (Cef), and Cef/SC5314 mice (graphs). Lactic acid bacterial colonies that grew on MRS plus azide agar were further identified and expressed as a fraction of the total LAB population in that group, using colony PCR as described in the methods (pie charts). Error bars represent the standard errors of the mean (*, P < 0.05 compared to untreated mice).
FIG 8.

The presence of C. albicans SC5314 during cefoperazone recovery results in gastric colonization and erosions of the limiting ridge. The stomach was removed at day 21 postantibiotic and differentially cultured to determine C. albicans SC5314 colonization in conventional untreated, antibiotic-treated (Cef), SC5314-colonized alone (SC5314), or Cef/SC5314 mice (A). Histological sections were stained with H&E to look for evidence of inflammation during cefoperazone-induced microbiota disruption (B). At day 7 postantibiotic, 17% of the SC5314 mice and 83% of the Cef/SC5314 mice had detectable C. albicans CHN1. At day 21 postantibiotic, 20% of the SC5314 mice and 67% of the Cef/SC5314 mice had detectable C. albicans SC5314 in the stomach. LOD, limit of detection (*, P < 0.05 versus untreated).
Dissociation of C. albicans-mediated gastritis from colonization resistance.
Our final objective was to determine whether it is possible to separate resistance against Candida albicans colonization from protection against gastritis. We had demonstrated that both strains CHN1 and SC5314 could colonize by day 7 without inducing gastritis, while at day 21 colonization was coincident with gastritis (Fig. 1, 2, and 8). Using these same two strains, we also introduced them into microbiota-deficient (germfree) mice by gavage. Similar to cefoperazone-treated mice, equivalent levels of gastric colonization and inflammatory involvement of the limiting ridge were observed in germfree mice at day 21 (Fig. 9). However, at day 7, germfree mice colonized with C. albicans had robust gastritis (Fig. 9) compared to conventional mice treated with cefoperazone and colonized with C. albicans, which had a low level of histologically evident inflammation at this time point (Fig. 2). These data demonstrate that, in the absence of a gastric microbiota, C. albicans can colonize and induce gastric inflammation within 1 week of oral inoculation. Altogether, this implicates a dichotomy between C. albicans colonization and gastric disease that is bacterial microbiome dependent.
FIG 9.

C. albicans CHN1 and SC5314 effectively colonize germfree mice and result in gastric erosions. The stomach was removed at day 7 and day 21 postantibiotic and differentially cultured to determine C. albicans colonization in germfree mice. (A) All germfree colonized mice had detectable C. albicans at all time points. (B) Histological sections were stained with H&E to look for evidence of inflammation during C. albicans colonization. Germfree mice had no evidence of inflammation or erosions. Germfree mice colonized with CHN1 or SC5314 at day 7 and day 21 all had evidence of gastric inflammation. Error bars represent the standard errors of the mean (*, P < 0.05 versus uninfected).
DISCUSSION
This study focuses on C. albicans gastric colonization of mice during the postantibiotic recovery phase. Using culture-independent and culture-dependent approaches, we demonstrated here that cefoperazone causes long-term disturbances to the gastric bacterial microbiome, including a significant reduction in the numbers of lactobacilli and the outgrowth of enterococci. The introduction of C. albicans into this disrupted community antagonizes the regrowth of Lactobacillus (directly or indirectly) and promotes increased Enterococcus levels, implicating that C. albicans can antagonize Lactobacillus in vivo. Most importantly, these findings begin to address the factors involved in C. albicans switching from a commensal to a pathogen and identify that the indigenous microbiome plays a critical role in this process, both through colonization resistance and through an unknown mechanism that inhibits gastric inflammation.
This is one of the first reports to use culture-independent analysis to analyze antibiotic-mediated disruption of the murine gastric microbiome. Our group has previously demonstrated that disturbances of the cecal microbiome by cefoperazone can be detected 6 weeks after the cessation of the antibiotic (1). Culture-based studies of the murine gastric microbiota have revealed a diverse, but limited community (just over 20 culturable genera), including lactobacilli (10). Cefoperazone is a poorly absorbed broad-spectrum cephalosporin with excellent activity against anaerobic organisms (18). It can promote C. albicans overgrowth in mice and humans, suggesting that its spectrum of activity encompasses bacteria that are critical for colonization resistance against C. albicans outgrowth or invasion (24, 36, 44). Although the exact mechanisms still remain to be determined, it has been suggested that lactobacilli are critical (22, 23). The results from in vitro studies implicate the bacterial microbiome in blocking yeast adhesion to the epithelium and producing inhibitor substances (such as volatile fatty acids and secondary bile acids) that can reduce C. albicans adhesion, hyphal transformation, and invasion (22, 23, 31, 32, 47, 48). It is well documented that mice lacking a bacterial microbiome are readily colonized by C. albicans, whereas conventional mice are highly resistant (3, 16). We have demonstrated that the effects of oral cefoperazone on the gastric bacterial microbiota can last at least 3 weeks after the cessation of the antibiotic, allow colonization by C. albicans, and promote C. albicans-induced gastritis.
One unexpected result from these studies was that the well-documented interaction between Lactobacillus and C. albicans, whereby Lactobacillus antagonizes the growth, adhesion, and hyphal transformation of C. albicans, can be a bidirectional process. Previous studies have demonstrated the ability of Lactobacillus to displace Candida from the epithelial layer of the stomach (37), inhibit hyphal invasion (37, 47), and prevent germ tube formation (31, 32), but the novel observation from these studies is that Lactobacillus-Candida antagonism can be a two-way process whereby the presence of Candida can prevent the regrowth of Lactobacillus after antibiotics. This could be via direct microbe-microbe interactions or indirectly through the induction of mucosal inflammation. Dietary modulation can also create a temporary Lactobacillus-deficient state, which has been shown to predispose a host to C. albicans overgrowth (48). Feeding mice lactobacilli can reduce the numbers of C. albicans in the stomachs of colonized mice (45–47). Thus, it is likely that a major contributing factor underlying C. albicans colonization and the ensuing gastritis is a reduction in lactobacilli in the stomachs of cefoperazone-treated mice.
In addition to changes in Lactobacillus numbers, the presence of C. albicans during antibiotic recolonization promoted the persistence of another lactic acid bacterium, Enterococcus. This bacterium, especially E. faecalis (the predominant species isolated from our treated mice), is a major concern in critical care settings, both due to its pathogenicity and to concern regarding antibiotic resistance (6, 7, 35, 40). Enterococcus is well adapted to survival along the mucosa: it can adhere to different epithelial and extracellular matrix proteins (13) and survive in a broad range of pH environments (29). Little is known about lactic acid bacterium niche competition on the human mucosa (e.g., Lactobacillus-Enterococcus antagonism) or C. albicans-Enterococcus antagonism. However, since both C. albicans and Enterococcus are concerns in critical care settings, our studies suggest that dissecting their potential symbiosis may provide new insights for treatments.
T-RFLP does not provide exact identities of the bacteria in a complex, undefined community, such as found in the murine stomach; rather, it generates a TRF profile or “fingerprint” that represents the community, and changes in that fingerprint serve as a surrogate measurement for changes in bacterial community structure. T-RFLP cannot differentiate between abundance changes within a community and additions/loss of new membership. If the bacterial community is a defined community, i.e., the identities of all of the members are known, then T-RFLP is often used to approximate changes in this known membership and specific TRFs assigned to specific bacterial species. This assignment of TRFs to a specific species of bacteria relies upon knowledge of the 16S rRNA gene sequence of that bacteria (which can usually be obtained through a database such as the RDP). In our studies, it is very unlikely that the number of enterococci is sufficient to generate an observable TRF. However, the lactobacilli are likely numerous enough to generate a T-RFLP signal; however, all of them would need to contain the exact same site for the T-RFLP restriction enzyme to generate the same TRF. There are some candidate peaks that may indeed be lactobacilli, but we cannot say with certainty that these are indeed the TRFs. Thus, we have decided not to overinterpret our T-RFLP results and simply use them as a culture-independent methodology to identify that changes in the bacterial community structure of the stomach microbiota occur after oral cefoperazone therapy.
In the studies presented here, we presented separate sets of experiments demonstrating that both strain CHN1 and SC5314 were able to induce gastric inflammation at the limiting ridge. No histologically evident inflammation was observed in the duodenum, jejunum, ileum, cecum, or colon (data not shown). This is consistent with previous studies, which have demonstrated, using other C. albicans strains, that the limiting ridge is a primary site of hyphal invasion and inflammation in mice with an altered microbiota following oral inoculation of C. albicans (4, 16, 34, 37, 38). While the function of the limiting ridge is unknown, it is the physical junction between the keratinized epithelium of the murine forestomach and the glandular body. The limiting ridge is comparable to the junction between the esophagus (squamous epithelium) and the stomach in humans. This anatomical resemblance of the murine limiting ridge and the human esophageal-gastric junction, combined with the evidence that C. albicans has a predilection to colonize and cause inflammation of the ridge, finds human parallels in the medical literature, where there are reports of esophageal ulcers related to Candida infections (8, 15).
In humans, gastric ulcers associated with C. albicans colonization is a well-documented condition, although generally unappreciated in terms of etiologic agents of gastric ulceration. In one study of 293 patients aged 20 to 80 years, >50% patients with gastric ulcers and >10% with chronic gastritis had fungal colonization of the stomach, with C. albicans being the most frequently isolated fungus (50). In three separate studies of 188, 66, and 42 adult patients with benign gastric ulcers, C. albicans infiltration into the gastric lesions were identified in 7, 9, and 36% of the patients, respectively (12, 14, 26). The general conclusion of these studies was that the yeast were a secondary infection of the ulcer site, although causation versus secondary colonization were never actually examined in these studies. Finally, in another study of >150 patients, Candida spp. were found in the gastric mucosa of 17% of patients, and two-thirds of those samples were cocolonized with both H. pylori and Candida (19). Additional analysis identified a link between coexistence of H. pylori with Candida and gastric ulcers, suggesting synergism of these microbes in the development of gastric pathology. In subjects with gastric colonization by C. albicans, but no H. pylori, colonization levels on the gastric mucosa were low (<103 CFU/ml). Thus, while the pathogenesis of C. albicans in the mouse versus human gastric mucosa may be different, this yeast exerts a tropism for this tissue site that is very likely influenced by the microbiota.
In support of this general concept, we observed a dichotomy between C. albicans colonization and gastric disease, which was bacterial microbiome dependent. Gastric colonization is known to be independent of the T cell status of the host, while gastritis involves the generation of a Th1 response, neutrophil infiltrates, and local production of indoleamine 2,3-dioxygenase (5, 9, 11, 16). Germfree mice C57BL/6 mice can respond to colonization and gastric candidiasis by increasing the expression of defensins and innate inflammatory cytokines (39). Some Candida species, such as C. pintolopesii, can exist in the murine microbiome without inducing inflammation (38). However, studies of C. albicans in mice have largely focused on its pathogenic potential, rather than on its ability to exist as a commensal or mutualist in the microbiome. Thus, little is known about the microbiome-derived interactions that control this switch. Our results predict that the bacterial community in cefoperazone-treated mice changes between days 7 and 21, thereby allowing the already established colonization by C. albicans to become an inflammatory stimulus for the gastric mucosa, similar to that in mice that completely lack a microbiota. Future kinetic studies, using culture-independent techniques such as pyrosequencing of 16S amplicons or high-throughput sequencing of metagenomic transcripts will provide some insights into this process. The role of the microbiome in regulating inflammatory responses to members of the indigenous microbiome is an area of current interest for a number of diseases. The potential ramification of understanding the process of C. albicans colonization is illustrated by research from our lab and others that has demonstrated that gastrointestinal colonization by C. albicans in mice can promote sensitization against intranasally and orally delivered antigens, such as food (30–32, 49). Further, our data provide new insights into the development and potential management of gastric ulcers and C. albicans-induced gastritis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kate Eaton, Rod McDonald, and Kelly Mason for technical support.
This study was supported by grants RO1-AI064479 (G.B.H.), R01-DK087708 (J.Y.K.), R21-AI087869 (G.B.H.), R21-AI083473 (G.B.H.), R21-AI087869 (G.B.H.), Frederick G. Novy Fellowship (K.L.M.), KO8 DK0669907-01 (J.Y.K.), and P30-DK034933 (G.B.H., J.Y.K., and V.B.Y.).
Footnotes
Published ahead of print 10 October 2011
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Antonopoulos DA, et al. 2009. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect. Immun. 77: 2367–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Artwohl J, McClain A, Cera L. 1988. Population changes of indigenous murine Candida pintolopesii under various experimental conditions and routes of inoculation. Appl. Environ. Microbiol. 54: 2371–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balish E, Balish MJ, Salkowski CA, Lee KW, Bartizal KF. 1984. Colonization of congenitally athymic, gnotobiotic mice by Candida albicans. Appl. Environ. Microbiol. 47: 647–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balish E, Jensen J, Warner T, Brekke J, Leonard B. 1993. Mucosal and disseminated candidiasis in gnotobiotic SCID mice. J. Med. Vet. Mycol. 31: 143–154 [DOI] [PubMed] [Google Scholar]
- 5. Bistoni F, et al. 1993. Mucosal and systemic T helper cell function after intragastric colonization of adult mice with Candida albicans. J. Infect. Dis. 168: 1449–1457 [DOI] [PubMed] [Google Scholar]
- 6. Bonten MJ, Gaillard CA, van Tiel FH, van der Geest S, Stobberingh EE. 1995. Colonization and infection with Enterococcus faecalis in intensive care units: the role of antimicrobial agents. Antimicrob. Agents Chemother. 39: 2783–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonten MJ, et al. 1996. Epidemiology of colonization of patients and environment with vancomycin-resistant enterococci. Lancet 348: 1615–1619 [DOI] [PubMed] [Google Scholar]
- 8. Borges MC, Colares JK, Lima DM, Fonseca BA. 2009. Advantages and pitfalls of the polymerase chain reaction in the diagnosis of esophageal ulcers in AIDS patients. Dig Dis. Sci. 54: 1933–1939 [DOI] [PubMed] [Google Scholar]
- 9. Bozza S, et al. 2005. A crucial role for tryptophan catabolism at the host/Candida albicans interface. J. Immunol. 174: 2910–2918 [DOI] [PubMed] [Google Scholar]
- 10. Brown JF, Balish E. 1978. Gastrointestinal microecology of BALB/c nude mice. Appl. Environ. Microbiol. 36: 144–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cenci E, et al. 1995. T helper cell type 1 (Th1)- and Th2-like responses are present in mice with gastric candidiasis but protective immunity is associated with Th1 development. J. Infect. Dis. 171: 1279–1288 [DOI] [PubMed] [Google Scholar]
- 12. DiFebo G, Carnevale G, Sterrantino SF. 1985. Treatment of a case of advanced periodontitis: clinical procedures utilizing the “combined preparation” technique. Int. J. Periodontics Restorative Dent. 5: 52–62 [PubMed] [Google Scholar]
- 13. Franz CM, Holzapfel WH, Stiles ME. 1999. Enterococci at the crossroads of food safety? Int. J. Food Microbiol. 47: 1–24 [DOI] [PubMed] [Google Scholar]
- 14. Gotlieb-Jensen K, Andersen J. 1983. Occurrence of Candida in gastric ulcers: significance for the healing process. Gastroenterology 85: 535–537 [PubMed] [Google Scholar]
- 15. Hasosah MY, Showail M, Al-Sahafi A, Satti M, Jacobson K. 2009. Esophageal candidiasis in an immunocompetent girl. World J. Pediatr. 5: 152–154 [DOI] [PubMed] [Google Scholar]
- 16. Helstrom PB, Balish E. 1979. Effect of oral tetracycline, the microbial flora, and the athymic state on gastrointestinal colonization and infection of BALB/c mice with Candida albicans. Infect. Immun. 23: 764–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hummel RP, Oestreicher EJ, Maley MP, Macmillan BG. 1973. Inhibition of Candida albicans by Escherichia coli in vitro and in the germfree mouse. J. Surg. Res. 15: 53–58 [DOI] [PubMed] [Google Scholar]
- 18. Jacobus NV, Tally FP, Barza M, Gorbach SL. 1980. Susceptibility of anaerobic bacteria to cefoperazone and other beta-lactam antibiotics. Clin. Ther. 3: 34–38 [PubMed] [Google Scholar]
- 19. Karczewska E, et al. 2009. Assessment of coexistence of Helicobacter pylori and Candida fungi in diseases of the upper gastrointestinal tract. J. Physiol. Pharmacol. 60(Suppl 6): 33–39 [PubMed] [Google Scholar]
- 20. Kennedy MJ. 1981. Inhibition of Candida albicans by the anaerobic oral flora of mice in vitro. Sabouraudia 19: 205–208 [PubMed] [Google Scholar]
- 21. Kennedy MJ, Volz PA. 1983. Dissemination of yeasts after gastrointestinal inoculation in antibiotic-treated mice. Sabouraudia 21: 27–33 [DOI] [PubMed] [Google Scholar]
- 22. Kennedy MJ, Volz PA. 1985. Ecology of Candida albicans gut colonization: inhibition of Candida adhesion, colonization, and dissemination from the gastrointestinal tract by bacterial antagonism. Infect. Immun. 49: 654–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kennedy MJ, Volz PA. 1985. Effect of various antibiotics on gastrointestinal colonization and dissemination by Candida albicans. Sabouraudia 23: 265–273 [DOI] [PubMed] [Google Scholar]
- 24. Kinsman OS, Pitblado K. 1989. Candida albicans gastrointestinal colonization and invasion in the mouse: effect of antibacterial dosing, antifungal therapy and immunosuppression. Mycoses 32: 664–674 [DOI] [PubMed] [Google Scholar]
- 25. Kuehl CJ, Wood HD, Marsh TL, Schmidt TM, Young VB. 2005. Colonization of the cecal mucosa by Helicobacter hepaticus impacts the diversity of the indigenous microbiota. Infect. Immun. 73: 6952–6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minoli G, et al. 1984. A prospective study of relationships between benign gastric ulcer, Candida, and medical treatment. Am. J. Gastroenterol. 79: 95–97 [PubMed] [Google Scholar]
- 27. Moeseneder MM, Arrieta JM, Muyzer G, Winter C, Herndl GJ. 1999. Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 65: 3518–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naglik JR, Fidel PR, Jr, Odds FC. 2008. Animals models of mucosal Candida infection. FEMS Microbiol. Lett. 283: 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakajo K, et al. 2006. Resistance to acidic and alkaline environments in the endodontic pathogen Enterococcus faecalis. Oral Microbiol. Immunol. 21: 283–288 [DOI] [PubMed] [Google Scholar]
- 30. Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. 2005. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect. Immun. 73: 30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noverr MC, Huffnagle GB. 2004. Regulation of Candida albicans morphogenesis by fatty acid metabolites. Infect. Immun. 72: 6206–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noverr MC, Noggle RM, Toews GB, Huffnagle GB. 2004. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect. Immun. 72: 4996–5003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paster BJ, et al. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183: 3770–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phillips AW, Balish E. 1966. Growth and invasiveness of Candida albicans in the germ-free and conventional mouse after oral challenge. Appl. Microbiol. 14: 737–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sabria-Leal M, et al. 1994. Molecular epidemiology of gastric colonization by Enterococcus faecalis in a surgical intensive care unit. Diagn. Microbiol. Infect. Dis. 19: 197–202 [DOI] [PubMed] [Google Scholar]
- 36. Samonis G, Anaissie EJ, Bodey GP. 1990. Effects of broad-spectrum antimicrobial agents on yeast colonization of the gastrointestinal tracts of mice. Antimicrob. Agents Chemother. 34: 2420–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Savage DC. 1969. Microbial interference between indigenous yeast and lactobacilli in the rodent stomach. J. Bacteriol. 98: 1278–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Savage DC, Dubos RJ. 1967. Localization of indigenous yeast in the murine stomach. J. Bacteriol. 94: 1811–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schofield DA, Westwater C, Balish E. 2005. Divergent chemokine, cytokine and beta-defensin responses to gastric candidiasis in immunocompetent C57BL/6 and BALB/c mice. J. Med. Microbiol. 54: 87–92 [DOI] [PubMed] [Google Scholar]
- 40. Shimizu K, et al. 2011. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis. Sci. 56: 1171–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van der Waaij D. 1987. Colonization resistance of the digestive tract: mechanism and clinical consequences. Nahrung 31: 507–517 [DOI] [PubMed] [Google Scholar]
- 42. van der Waaij D, Berghuis JM. 1974. Determination of the colonization resistance of the digestive tract of individual mice. J. Hyg. (Lond.) 72: 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van der Waaij D, Van der Waaij BD. 1990. The colonization resistance of the digestive tract in different animal species and in man; a comparative study. Epidemiol. Infect. 105: 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Ogtrop ML, Guiot HF, Mattie H, van Furth R. 1991. Modulation of the intestinal flora of mice by parenteral treatment with broad-spectrum cephalosporins. Antimicrob. Agents Chemother. 35: 976–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wagner RD, Dohnalek M, Hilty M, Vezquez-Torres A, Balish E. 2000. Effects of probiotic bacteria on humoral immunity to Candida albicans in immunodeficient bg/bg-nu/nu and bg/bg-nul/+ mice. Rev. Iberoam. Micol. 17: 55–59 [PubMed] [Google Scholar]
- 46. Wagner RD, et al. 1997. Biotherapeutic effects of probiotic bacteria on candidiasis in immunodeficient mice. Infect. Immun. 65: 4165–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wagner RD, et al. 2000. Probiotic effects of feeding heat-killed Lactobacillus acidophilus and Lactobacillus casei to Candida albicans-colonized immunodeficient mice. J. Food Prot. 63: 638–644 [DOI] [PubMed] [Google Scholar]
- 48. Yamaguchi N, et al. 2005. Gastric colonization of Candida albicans differs in mice fed commercial and purified diets. J. Nutr. 135: 109–115 [DOI] [PubMed] [Google Scholar]
- 49. Yamaguchi N, et al. 2006. Gastrointestinal Candida colonization promotes sensitization against food antigens by affecting the mucosal barrier in mice. Gut 55: 954–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zwolinska-Wcislo M, Budak A, Trojanowska D, Bogdal J, Stachura J. 1998. Fungal colonization of the stomach and its clinical relevance. Mycoses 41: 327–334 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.