

Abstract
Pulmonary arterial remodeling is a pathological process seen in a number of clinical disease states, driven by inflammatory cells and mediators in the remodeled artery microenvironment. In murine models, Th2 cell-mediated immune responses to inhaled antigens, such as purified Aspergillus allergen, have been reported to induce remodeling of pulmonary arteries. We have previously shown that repeated intranasal exposure of healthy C57BL/6 mice to viable, resting Aspergillus fumigatus conidia leads to the development of chronic pulmonary inflammation and the coevolution of Th1, Th2, and Th17 responses in the lungs. Our objective was to determine whether repeated intranasal exposure to Aspergillus conidia would induce pulmonary arterial remodeling in this mixed Th inflammatory microenvironment. Using weekly intranasal conidial challenges, mice developed robust pulmonary arterial remodeling after eight exposures (but not after two or four). The process was partially mediated by CD4+ T cells and by interleukin-4 (IL-4) production, did not require eosinophils, and was independent of gamma interferon (IFN-γ) and IL-17. Furthermore, remodeling could occur even in the presence of strong Th1 and Th17 responses. Rather than serving an anti-inflammatory function, IL-10 was required for the development of the Th2 response to A. fumigatus conidia. However, in contrast to previous studies of pulmonary arterial remodeling driven by the A. fumigatus allergen, viable conidia also stimulated pulmonary arterial remodeling in the absence of CD4+ T cells. Remodeling was completely abrogated in IL-10−/− mice, suggesting that a second, CD4+ T cell-independent, IL-10-dependent pathway was also driving pulmonary arterial remodeling in response to repeated conidial exposure.
INTRODUCTION
Remodeling of pulmonary arteries is a pathological process seen in a number of clinical disease states. Progressive arterial remodeling combined with an aberrant milieu favoring vasoconstriction causes pulmonary hypertension that often leads to right-sided heart failure (6, 7, 22, 23). A study of asthma patients showed that bronchial arteries exhibited an increase in the intimal area, associated with smooth muscle proliferation and calcification of the elastica, and a corresponding decrease in the luminal area (16). In addition, pulmonary artery pathology has also been found in those with fatal asthma (39). While the pathophysiology of pulmonary arterial remodeling remains largely unclear, recent advances implicate a role for inflammation in the process that results in an imbalance between cellular proliferation and apoptosis, the presence of inflammatory cells in the remodeled artery microenvironment, and an association with increased levels of inflammatory mediators, including various chemokines and cytokines (12, 19). In murine models, Th2 cell-mediated immune responses to inhaled purified antigens have been reported to induce remodeling of pulmonary muscular arteries (9, 36, 37), although pulmonary inflammation and arterial remodeling can occur without the development of pulmonary hypertension (9). Overall, emerging data point to a possible mechanism whereby Th2 immune responses in the lungs may promote pulmonary arterial remodeling. However, much remains to be understood about the role of other types of cell-mediated inflammatory responses and cross-regulation between types of T-helper responses in pulmonary arterial remodeling.
Aspergillus fumigatus is a ubiquitous mold that releases significant numbers of airborne conidia (27), and many allergic and asthmatic patients are sensitive to A. fumigatus allergens (11). In addition to promoting the Th2 immune response that leads to asthma and allergic bronchopulmonary aspergillosis (11, 15, 25), A. fumigatus conidia are known also to stimulate innate Th1 and Th17 immune responses in the lung (3, 4, 21, 33, 41, 50). We have recently demonstrated that repeated inhalation of viable, resting A. fumigatus conidia by C57BL/6 mice results in a dynamic inflammatory response without fungal colonization or growth (30). The response was characterized by the engagement of several arms of the adaptive immune system, including Th1, Th2, and Th17 cells, as well as robust eosinophilia and remodeling of the lung architecture.
Pulmonary arterial remodeling can be stimulated by a highly polarized Th2 immune response to an Aspergillus antigen extract in immunized wild-type but not interleukin-4 (IL-4) knockout mice (9). Our objective was to determine whether repeated intranasal exposure to viable, resting Aspergillus conidia would induce pulmonary arterial remodeling in a mixed Th1 and Th2 inflammatory microenvironment. If so, then our objective was to determine whether CD4+ T cells, IL-4, IL-5, and/or gamma interferon (IFN-γ) was required for both remodeling and the pulmonary inflammatory response. In addition, we also examined the role of the regulatory cytokine IL-10 in modulating the polarization of T cell response, magnitude of pulmonary inflammation, and subsequent pulmonary arterial remodeling that develops upon repeated Aspergillus conidial exposure.
MATERIALS AND METHODS
Mice.
IL-4−/− (B6.129P2-Il4tm1Cgn/J), IL-10−/− (B6.129P2-Il10tm1Cgn/J), IFN-γ−/− (B6.129S7-Ifngtm1Ts/J), and wild-type (C57BL/6J) mice were obtained from Jackson Laboratories (Bar Harbor, ME). IL-5−/− and IL-17−/− mice on a C57BL/6 background were obtained from a breeding colony maintained at the University of Michigan. Breeders for the IL-17−/− mice were kindly provided by Yoichiro Iwakura (Tokyo University). Mice were housed under pathogen-free conditions in enclosed filter-topped cages. Clean food and water were given ad libitum. The mice were handled and maintained using microisolator techniques, with daily monitoring by veterinarian staff. All studies involving mice were undertaken with the approval of the University Committee on Use and Care of Animals at the University of Michigan Medical School.
Aspergillus fumigatus.
Strain ATCC 13073 was grown on Sabouraud dextrose agar (Difco) for 14 days. Conidia were harvested by washing plates with sterile phosphate-buffered saline (pH 7.4) with 0.1% Tween 80 (phosphate-buffered saline [PBS]-Tween), followed by filtration of the suspension through two layers of sterile gauze to remove hyphae. Conidia were washed in PBS-Tween, counted on a hemocytometer, diluted to 108 spores/ml in sterile PBS-Tween, and stored at 4°C for up to 4 months.
Respiratory exposure protocol.
For each exposure, 2 × 106 conidia/mouse were administered to the nostrils of mice in a volume of 20 μl. Repeat intranasal exposures were administered every 7 days. Prior to intranasal inoculation, mice were anesthetized by intraperitoneal injection of a ketamine-xylazine solution (2.5 mg of ketamine [Fort Dodge Animal Health, Fort Dodge, IA]/mouse plus 0.1 g of xylazine [Lloyd Laboratories, Shenandoah, IA]/mouse).
Lung histology.
Lungs were fixed by inflation with 10% neutral buffered formalin. After paraffin embedding, 5-μm sections were cut and stained with hematoxylin and eosin (H&E), periodic acid-Schiff stain (to detect mucus), Masson's trichrome (to detect collagen), Verhoeff-van Gieson stain (to detect elastin), and Gomori's methenamine-silver (to detect fungi).
Bronchoalveolar lavage (BAL) for cell recovery.
Airway contents were recovered by instillation and retrieval of 1 ml of sterile PBS through a tracheotomy tube. The first lavage fluid was centrifuged, and the cell-free fluid was immediately frozen down at −20°C. Cells collected with three total lavages were pooled. After erythrocyte lysis using NH4Cl buffer (0.83% NH4Cl, 0.1% KHCO3, 0.037% Na2 EDTA, pH 7.4), cells were washed, resuspended in complete medium (RPMI 1640, 10% fetal calf serum, 2 mmol/liter l-glutamine, 50 μmol/liter 2-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate), and enumerated under a light microscope in the presence of trypan blue using a hemocytometer.
Flow cytometry for cell surface molecules.
Cells were washed and resuspended at a concentration of 107 cells/ml in fluorescent antibody (FA) buffer (Difco) and 0.1% NaN3. Fc receptors were blocked by the addition of unlabeled anti-CD16/32 antibody (Fc block; BD Pharmingen, San Diego, CA). After Fc receptor blocking, 0.5 × 106 to 1 × 106 cells were stained in a final volume of 100 μl in 12- by 75-mm polystyrene tubes (Becton Dickinson, Franklin Lakes, NJ) for 30 min at 4°C. Fluorochrome-conjugated antibodies directed against the following antigens were obtained from BD Pharmingen and were used according to the manufacturer's instructions: CD45 (30-F11), CD3 (145-2C11), CD4 (RM4-5), CD8 (53-6.7), CD11b (M1/70), CD11c (HL3), CD19 (1D3), Gr1 (RB6-8C5), Siglec-F (E50-2440), IFN-γ (XMG 1.2), IL-4 (11B11), IL-10 (JES5-16E3), IL-17 (TC11-18H10), and CCR3 (83103). Cells were washed twice with FA buffer and resuspended in 100 μl, and an equal volume of 4% formalin was added to fix the cells. A minimum of 20,000 events were acquired on a FACSCalibur flow cytometer (BD Pharmingen) using Cell-Quest software (BD Pharmingen). The acquired data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Flow cytometry for intracellular molecules.
Prior to intracellular cytokine staining, cells were stimulated in vitro for 5 h with phorbol myristate acetate (PMA) (50 ng/ml) and ionomycin (1 μg/ml) in the presence of brefeldin A (BD Pharmingen) to promote the intracellular accumulation of cytokines. After stimulation, cells were washed twice prior to surface molecule staining. After surface molecule staining, intracellular molecules were stained using the BD Cytofix/Cytoperm kit according to the manufacturer's instructions (BD Pharmingen).
CD4+ T cell depletion.
Mice were treated with intraperitoneal injections of 300 μg of the GK1.5 anti-CD4 monoclonal antibody (MAb) on the day prior to the first exposure to conidia and boosted with 100 μg of MAb every 7 days. Antibody was prepared from ascites by dilution in PBS and filtering through a 0.45-μm syringe filter. The efficiency of T cell depletion was assessed by flow cytometric analysis using anti-CD4 Ab RM 4-5, which binds to a region of CD4 distinct from GK1.5. Depletion was >99% for CD4+ T cells in the lungs as well as in the spleen. Anti-CD4 antibody (GK1.5) prevents the development of an anti-rat Ig response, because an anti-rat Ig response requires CD4+ T cell help. Thus, PBS diluent was used for control mice instead of isotype-matched rat Ig that would lead to a mouse anti-rat Ig response, potentially resulting in Ab-antigen complexes and immune deviation or inflammation.
Immunohistochemistry.
Immunohistochemistry was performed as previously described (15). Lungs were inflated and fixed in 10% buffered formalin overnight, embedded in paraffin, and sectioned. Following deparaffinization and rehydration, sections were blocked and then stained with anti-α-smooth muscle actin (α-SMA) antibody (clone 1A4; Dako, Carpinteria, CA). Color development for α-SMA staining was achieved with DAB (Vector Labs, Burlingame, CA). Sections were then counterstained with hematoxylin and mounted.
Morphological leukocyte differential analysis.
Macrophages, neutrophils, lymphocytes, and eosinophils were visually counted by standard morphological criteria in Wright-Giemsa-stained samples of lung cell suspensions cytospun onto glass slides (Shandon Cytospin, Pittsburgh, PA). For Wright-Giemsa staining, the slides were fixed for 2 min with a one-step, methanol-based Wright-Giemsa stain (Harleco; EM Diagnostics, Gibbstown, NJ) followed by steps 2 and 3 of the Diff-Quik whole-blood stain kit (Diff-Quik, Baxter Scientific, Miami, FL). A total of 200 to 300 cells were counted from randomly chosen high-power microscope fields for each sample.
Morphometric analysis of pulmonary arteries.
Excised lungs were inflated via the trachea with 1 ml of 10% buffered formalin. The trachea was ligated, and the lungs were immersed in 10% buffered formalin for 24 h. The left lung from each mouse was removed, embedded in alginate, and sectioned into 3-mm slices along the long axis. A coin flip determined whether odd or even sections were chosen for analysis. After the chosen slices were embedded in paraffin, 5-μm sections were cut, fixed to glass slides, and stained with hematoxylin and eosin. Morphometry was performed using an Olympus BX40 microscope fitted with a DP71 digital camera (Olympus, Melville, NY) and an XYZ microscope stage (Prior Scientific, Rockland, MA) controlled by a Dell computer running Visopharm version 2.14 software (Visopharm, Denmark). Slides were coded and analyzed by the reader in a blinded fashion. Using a 4× low-power lens objective, the entire area of lung from each tissue section was outlined and included in the sampling area. Under ×40 magnification, a computer-assisted sampling protocol chose a random starting field from the selected area, and subsequent fields were determined by using a fixed step in the x and y positions. Thus, a random sampling of the entire tissue section was examined, allowing for objective measurements. All muscular pulmonary arteries appearing in the selected fields were assessed. For each artery, diameter (in both the long and short axes), total area, and luminal area were measured and recorded. A value of percent lumen was obtained by dividing the luminal area by the total area and multiplying by 100%. Vessels were excluded from analysis if the length/width measurements were >2.5 to exclude tangentially sliced arteries. Vessels were excluded from analysis if the total area was >20,000 μm2 to exclude large pulmonary arteries. Data for each artery measured were pooled within each experimental group from 2 to 4 separate sets of experiments.
Statistical analyses.
All values are reported as means ± standard errors of the means (SEM) unless otherwise noted. Mean values were pooled from 2 to 4 independent sets of experiments unless otherwise noted. Differences between two groups were evaluated with a two-tailed Student t test, and all differences between three or more groups were evaluated with a one-way analysis of variance (ANOVA) with Bonferroni's multiple comparison post hoc test. P values of ≤0.05 were considered statistically significant.
RESULTS
Assembly and validation of a flow cytometry-based differential stain for airway leukocytes.
Our first objective was to assemble and validate a multicolor flow cytometry panel of cell surface stains to analyze the leukocyte infiltrate in the airways of mice at intervals during repeated exposure to A. fumigatus conidia. Based upon combinations of staining strategies previously reported (13, 31, 43, 48), a panel of cell surface antibodies against CD45, CD11c, and Gr1 was used for differential flow cytometry staining of cells obtained by bronchoalveolar lavage (BAL). CD45+ BAL cells fell within one of four distinct populations on a Gr1/CD11c plot (Fig. 1A). These cells were sorted, subsequently stained with Wright-Giemsa, and identified by morphology. The Gr1− CD11c− population in the BAL fluid consisted of lymphocytes, Gr1+ CD11clow of neutrophils, Gr1mid CD11clow of eosinophils, and Gr1mid CD11c+ of alveolar macrophages (Fig. 1B). Additional cell surface markers, as well as forward-scatter and side-scatter properties, were used to confirm the identity of each cell population (Fig. 1C). Alveolar macrophages express high levels of CD11c and Siglec-F and low levels of CD11b, F4/80, CCR3, CD3, and CD19 (Fig. 1C), as well as exhibiting high autofluorescence (data not shown). These are all cell surface characteristics previously reported for alveolar macrophages (13, 43, 48). Dendritic cells, while expressing high levels of CD11c, do not express Siglec-F and do not have high autofluorescence (13, 31, 43, 48). Neutrophils are Gr1high and CD11bhigh, while eosinophils express high levels of CD11b, Siglec-F, and CCR3 (43). Finally, the lymphocyte population contained cells that were either CD3+ or CD19+. Thus, having validated this panel of antibodies, we used this flow cytometric approach for analysis of infiltrating leukocyte populations throughout the remainder of this study.
Fig 1.

Assembly and validation of a flow cytometric panel for leukocyte differential analysis. (A) After four A. fumigatus conidial exposures, airway leukocytes were recovered by BAL and analyzed by flow cytometry with isotype control antibody, anti-CD11c antibody, or anti-Gr1 antibody as labeled on the density plots. (B) After four A. fumigatus conidial exposures, BAL cells were analyzed and sorted by fluorescence-activated cell sorting (FACS). The sorted cells were then stained with modified Wright-Giemsa stain and examined under a light microscope. The forward-scatter (FSC) and side-scatter (SSC) properties and Gr1 and CD11c expression profiles are shown for the total cell population in the first and second density plots of the top row. Four distinct cell populations were identified on the basis of CD11c and Gr1 expression and gated as shown. The arrows point to the FSC and SSC properties of the gated populations and then to the representative photomicrographs of the cells sorted by this gating strategy. All photomicrographs are shown at the same scale (the black bar represents 20 μm in length). The purity of each sorted population was greater than 90% as determined by microscopic evaluation of the cells using standard morphological criteria (included photomicrographs and data not shown), and the populations are labeled as macrophages, neutrophils, lymphocytes, or eosinophils based on these analyses. (C) After several A. fumigatus exposures, BAL cells were gated according to the strategy described above for Fig. 1A and 1B, and the resulting populations were further characterized by the expression of certain cell surface molecules of interest. As depicted with the density plots, lymphocytes and eosinophils were enriched with primary gates based on FSC and SSC properties and subsequently gated from the enriched population of cells based on Gr1 and CD11c expression profiles, whereas macrophages and neutrophils were gated from the total population of cells based on Gr1 and CD11c expression profiles. The corresponding histograms show the cell surface staining with isotype control antibodies (shaded) or antibodies against F4/80, CD11b, Siglec-F, or CCR3 or an antibody pool against CD3 and CD19 (white).
Inflammation and arterial remodeling of the lung in response to repeated conidial exposure.
We determined leukocyte numbers and composition in the BAL fluid 24 h after two, four, and eight conidial challenges. At baseline (zero exposures), the BAL fluid consisted predominantly of alveolar macrophages. Two exposures resulted in only a small increase in the number of inflammatory cells in the airways, which was accounted for by an increase in neutrophils (Fig. 2). After four exposures, the total number of airway leukocytes increased significantly and was dominated by eosinophils and lymphocytes. After eight exposures, the number of BAL leukocytes was still significantly elevated, but the composition of the infiltrate had changed, with roughly equal proportions of all four leukocyte populations, indicating a shift in the ratio of eosinophils to macrophages, with increasing numbers of conidial exposures promoting an influx of macrophages into the airways (Fig. 2). These data on the kinetics of airway leukocyte influx are consistent with our previously published kinetic studies on the influx of leukocytes into the lung parenchyma following repeated A. fumigatus conidial exposure (30) and document an evolving inflammatory response during repeated exposure to conidia.
Fig 2.

Characterization of the inflammatory response to repeated A. fumigatus conidial exposure. (A) Airway leukocytes were recovered from each mouse by BAL and counted using a hemocytometer. Bar graphs depict the means ± SEM of data compiled from the means from two or more independent sets of experiments (n = 3 or 4 mice per group per experiment). *, P < 0.001 for 4 and 8 versus 0 and 2 exposures. (B) Flow cytometric leukocyte differential analyses were performed on BAL cells for each group based on FSC, SSC, Gr1, and CD11c, as detailed in the legend to Fig. 1, following zero, two, four, and eight conidial challenges.
In evaluating the mice repeatedly exposed to conidia, we noted the development of progressive remodeling of small- to medium-sized pulmonary arteries with concomitant luminal narrowing, especially at or below the level of the terminal bronchioles, where the inflammatory response was focused (Fig. 3A). After two exposures, inflammatory cells accumulated in the arterial lumen and in the periarterial space, but the arterial anatomy appeared normal (Fig. 3A to D). The inflammation was more severe after four exposures, where the lumen was often packed with inflammatory cells and fibrous, extracellular material appeared, but the arterial walls still appeared relatively normal (Fig. 3A to D). However, after eight exposures, the arterial walls themselves were extensively remodeled. While small numbers of resting, swollen, and germinating conidia could be identified in the lungs after eight exposures, they were not associated with the remodeled arteries, and hyphal masses were not evident in any of the sections (data not shown). After 8 exposures, the fully remodeled arteries exhibited extensive fibrosis of the wall (Fig. 3B), neointimal formation (Fig. 3C), and hyperplasia of the medial smooth muscle cell layer that is normally 1 cell layer thick (Fig. 3D). The pulmonary arterial remodeling was quite extensive, but it did not involve pulmonary veins or large pulmonary arteries (Fig. 3E). These experiments demonstrated that repeated intranasal exposure of mice to A. fumigatus conidia will result in the development of progressive pulmonary arterial remodeling that affects small- to medium-sized arteries with concomitant luminal narrowing.
Fig 3.

Histopathology of pulmonary inflammation and arterial remodeling after eight weekly intranasal exposures to A. fumigatus conidia. (A) Photomicrographs of pulmonary arteries adjacent to terminal bronchioles are depicted in H&E-stained sections (black bars = 50 μm). (B) Collagen deposition in remodeled arteries is demonstrated in trichrome-stained sections where collagen stains light blue (black bars = 50 μm). (C) Neointimal formation is depicted in Verhoeff-van Giesen-stained sections, where elastin stains darkly (black bars = 20 μm). Arrowheads point to the elastica interna, separated from the arterial lumen by the thickened intima. (D) Muscularization of the arterial walls is demonstrated in sections stained for β-smooth muscle actin by immunohistochemistry (black bars = 20 μm). Negative-control-stained sections are shown in the offset photos. (E) The extent of arterial remodeling is depicted in sections stained for β-smooth muscle actin by immunohistochemistry, where arteries are identified by arrowheads and veins are identified by asterisks. A negative-control-stained section is shown in the offset photo.
Our next objective was to evaluate the conidium-exposed mice for physiological alterations affecting the pulmonary arterial system. As a surrogate for pulmonary hypertension in our mice, we directly measured the right ventricular systolic pressure (RVSP) in anesthetized, ventilated mice. As a group, mice exposed eight times to Aspergillus conidia did not develop a significant increase in the RVSP (data not shown). However, a few of the mice did develop pressures that were higher than any of the unchallenged mice (data not shown). Pulmonary hypertension causes strain on the right ventricle that can lead to right ventricular hypertrophy and can be associated with chronic hypoxia, which induces a compensatory increase in the serum hemoglobin concentration. However, there were no significant differences in the right ventricular mass or in the serum hemoglobin concentration between the various groups after eight conidial exposures (data not shown). Thus, similar to a previous study by Daley et al. (9) in a sensitized mouse model of A. fumigatus allergen-driven Th2 pulmonary inflammation, we observed that pulmonary inflammation and arterial remodeling in mice could occur prior to or without the development of pulmonary hypertension.
Role of CD4 T cells in allergic inflammation and remodeling.
Our next objective was to determine whether CD4 T cells were required for the arterial remodeling process, similar to that reported in the allergen sensitization model described above (9). In intact mice, there was an influx of CD4+ T cells into the airways after 4 exposures, which coincided with the influx of eosinophils (Fig. 1B and 4A). We could also adoptively transfer the allergic phenotype into naïve mice using CD4+ T cells isolated from four A. fumigatus conidium-exposed mice, which then responded with a vigorous allergic response to conidial challenge, while naïve and nonimmune transfer recipients did not respond (data not shown). To deplete the mice of CD4+ T cells, we injected them with anti-CD4 MAb (GK1.5) prior to and throughout the period of eight exposures to A. fumigatus conidia. This resulted in depletion of >99% of CD4+ T cells in the BAL fluid cell populations, mediastinal lymph nodes, and spleen (data not shown). In the absence of CD4+ T cells, the innate host defense to A. fumigatus remained intact, as evidenced by the lack of fungal growth on Gomori methenamine silver (GMS)-stained lung sections after 4 weekly exposures, but the recruitment of eosinophils was ablated (Fig. 4B). Thus, the innate immune response remained intact for the clearance of conidia from the lung, whereas the allergic response to A. fumigatus following four conidial challenges was dependent on the presence of CD4+ T cells.
Fig 4.
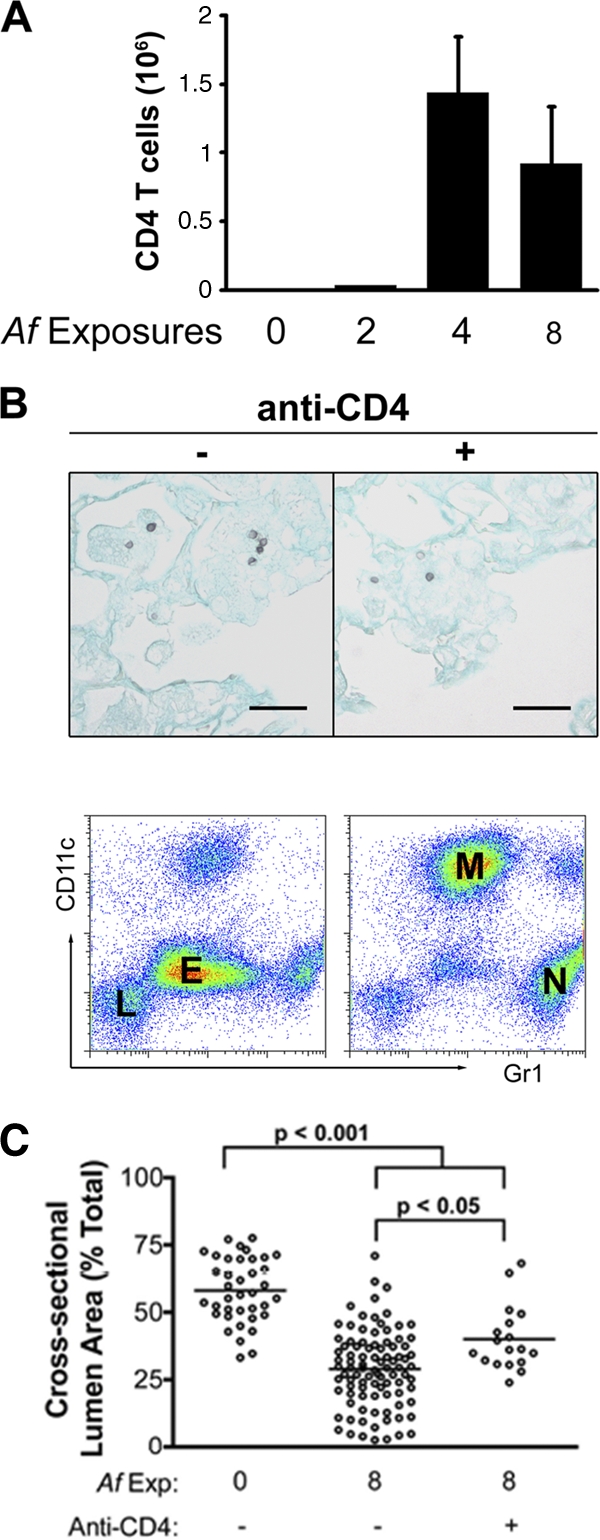
Quantification of pulmonary CD4+ T cell influx and the inflammatory response and arterial remodeling following CD4+ T cell depletion. (A) The number of CD4+ T cells recovered by BAL was determined as described in materials and methods. The graph depicts the means ± SEM of data compiled from the means from two independent sets of experiments (n = 3 or 4 mice per group per experiment). (B) A. fumigatus conidia in the lungs after four challenges are shown in representative photomicrographs of Gomori methenamine silver-stained sections for control and anti-CD4 antibody-treated mice. Black bars represent 20 μm in length. Flow cytometric differential analyses were also performed on BAL cells pooled from 3 or 4 mice per group. Labels on the Gr1 versus CD11c plot indicate the population densities for macrophages (M), neutrophils (N), lymphocytes (L), and eosinophils (E). (C) The cross-sectional lumen area was measured in randomly selected small- to medium-sized muscular pulmonary arteries in unchallenged mice, mice challenged eight times with A. fumigatus conidia, and mice challenged eight times with conidia while undergoing continuous CD4+ T cell depletion during this period. Five to 10 randomly selected vessels were analyzed per mouse. Each measured artery is shown on the scatter plot, and the mean for each group is indicated by the horizontal line (n = 6 to 8 mice/group).
We next compared the extent of arterial remodeling in CD4+ T cell-deficient, CD4+ T cell-sufficient, and unchallenged mice using morphometric analysis of randomly selected small- to medium-sized muscular pulmonary arteries. We measured the outer circumference and the inner, luminal circumference of randomly selected, cross-sectioned arteries in order to calculate the percent luminal area. Consistent with the severe histopathological changes, mice with an intact CD4+ T cell population showed a significant decrease in the luminal areas of their small- and medium-sized pulmonary arteries after eight conidial exposures compared to those of unexposed mice (Fig. 4C). In the absence of CD4+ T cells, there was significantly less arterial remodeling following repeated conidial exposure. However, there also remained a significant level of pulmonary arterial remodeling, even in the absence of CD4+ T cells (Fig. 4C). Thus, while CD4+ T cells play a major role in pulmonary arterial remodeling during repeated conidial exposure, there is also a CD4+ T cell-independent mechanism operating in parallel, which is different than that observed following sensitization with the A. fumigatus allergen alone (9).
Cytokine signaling in arterial remodeling and Th2-mediated allergic inflammation.
We next investigated the potential contribution of IFN-γ, IL-4, IL-5, and IL-10 to arterial remodeling. In our previous study, we noted similar numbers of IFN-γ-secreting CD4+ Th1 cells and IL-4-secreting CD4+ Th2 cells at the height of the allergic inflammatory response and that CD4+ Treg cells were recruited to the airways as well (30). IL-5 is a CD4+ Th2 cell effector cytokine that regulates eosinophil development and recruitment, but unlike IL-4, it is not required for the differentiation of CD4+ Th2 cells. In order to assess the role of eosinophils in arterial remodeling, we therefore examined IL-5-deficient mice as well.
We evaluated the development of pulmonary arterial remodeling in IFN-γ−/−, IL-4−/−, IL-5−/−, and IL-10−/− mice after eight conidial exposures. As in wild-type C57BL/6 mice and in CD4+ T cell-depleted mice, all cytokine-deficient mice were protected from fungal colonization or growth after inhalation of viable conidia (data not shown). Eosinophilia was absent in A. fumigatus-challenged IL-5-deficient mice (data not shown). Similar to wild-type mice, IFN-γ−/− and IL-5−/− mice developed pulmonary arterial remodeling with luminal narrowing after prolonged exposure, indicating that the Th1 response and the eosinophil component of the Th2 response are not required in this process (Fig. 5A and B). IL-17−/− mice also developed pulmonary arterial remodeling following eight conidial exposures (B. J. Murdock, N. R. Falkowski, A. A. Sadighi Akha, R. A. McDonald, E. S. White, G. B. Toews, and G. B. Huffnagle, submitted for publication). There was significantly less arterial remodeling in IL-4−/− mice than in other mice (Fig. 5A and B), but similar to CD4+ T cell-depleted mice (Fig. 4C and 5B), they were not completely protected. In contrast, IL-10−/− mice were completely protected from pulmonary arterial remodeling (Fig. 5A and B), suggesting a role for IL-10 in both the CD4+ T cell-dependent and -independent remodeling response. These data indicate that pulmonary arterial remodeling is independent of a Th1 or Th17 response and can be driven by IL-4 production, but does not require eosinophils, during a Th2 response to A. fumigatus conidia.
Fig 5.

Pulmonary arterial remodeling in cytokine knockout mice after eight intranasal exposures to A. fumigatus conidia. (A) Photomicrographs of pulmonary arteries adjacent to terminal bronchioles as depicted in H&E-stained sections after 8 exposures (black bars = 50 μm). (B) The cross-sectional lumen area was measured in randomly selected small- to medium-sized muscular pulmonary arteries. Five to 10 randomly selected vessels were analyzed per mouse. Each measured artery is shown on the scatter plot, and the mean for each group is indicated by the horizontal line (n = 6 to 8 mice/group) (significant P values are included). For reference, the gray bar illustrates the 25th quartile, mean (dashed line), and 75th quartile for untreated wild-type C57BL/6 mice (Fig. 3B). (C) For comparison with A. fumigatus-treated mice in panel A, a photomicrograph depicting an H&E-stained section of a pulmonary artery adjacent to a terminal bronchiole in an untreated C57BL/6 mouse is shown (black bar = 50 μm).
This observation prompted us to assess what role IL-4 and IL-10 play in driving the allergic response during repeated conidial exposure. Compared to C57BL/6 mice, there was not a significant change in total cell numbers or CD4+ T cells in IFN-γ−/−, IL-4−/−, and IL-10−/− mice, though there was a trend toward fewer total leukocytes in the absence of IL-4 and IL-10. Differences in total cell numbers were likely due to a significant reduction in the number of airway eosinophils recovered after four exposures in IL-4−/− and IL-10−/− mice (Fig. 6A). Eosinophil recruitment was also deficient in IL-5−/− mice (data not shown). The frequencies of IFN-γ-producing CD4+ T cells in IL-4−/− and IL-10−/− mice were not different from that found in wild-type mice. Similarly, the frequency of IL-4-producing CD4+ T cells in IFN-γ−/− mice was not significantly different from that in wild-type mice (Fig. 6B and C). In contrast, when IL-4 production by CD4+ T cells in wild-type and IL-10−/− mice was compared, we found a significant reduction in the frequency of IL-4-producing CD4+ T cells in the airways of IL-10−/− mice (Fig. 6B and C). Taken together, these data show that repeated conidial exposure stimulates IL-10-mediated responses that are required for both (i) the development of the Th2 response to A. fumigatus and (ii) the CD4+ T cell-independent pulmonary arterial remodeling that can occur even in the absence of the Th2 response.
Fig 6.

Pulmonary inflammation in cytokine knockout mice after eight intranasal exposures to A. fumigatus conidia. (A) After 4 weekly exposures, leukocytes were recovered from the airway, and total leukocytes, CD4+ T cells, and eosinophils were enumerated in IFN-γ−/−, IL-4−/−, and IL-10−/− mice. The bar graphs are normalized to compare all the experiments in one graph and depict the mean ± SEM of the ratios generated by comparing the cell numbers for a particular knockout mouse to the wild-type controls included with each experiment (n = 3 experiments/knockout, 3 to 5 mice/experiment). *, P < 0.05. (B) The bar graphs depict the means ± SEM of the ratios generated by normalizing to the wild-type controls included with each experiment for the percentage of CD4+ T cells that produced cytokine (n = 3 experiments/knockout, 3 to 5 mice/experiment). *, P < 0.05. (C) The production of cytokines by CD4+ T cells isolated from the BAL fluid after four conidial challenges was assessed in wild-type and IL-10−/− mice by intracellular flow cytometry. The density plots show the percentage of cytokine-producing cells within the gated CD4+ T cell population.
DISCUSSION
In this study, we have been able to demonstrate that long-term repeated exposure to A. fumigatus conidia results in pulmonary arterial remodeling. The process can be driven by CD4+ T cells and IL-4 production but does not require eosinophils, is independent of a Th1 or Th17 response, and can occur even in the presence of strong Th1 and Th17 responses (30). Rather than serving an anti-inflammatory function, IL-10 is required for the development of the Th2 response to A. fumigatus conidia, similar to our previously reported study demonstrating a role for IL-10 in the development of a Th2 response to another fungus, Cryptococcus neoformans (20). However, in contrast to the A. fumigatus allergen, viable conidia also stimulate pulmonary arterial remodeling in the absence of CD4+ T cells, albeit to a lower degree. Remodeling is completely abrogated in IL-10−/− mice, suggesting that a CD4+ T cell-independent, IL-10-dependent pathway exists for pulmonary arterial remodeling.
In humans, persistent superficial colonization of the bronchial mucosa by A. fumigatus can result in allergic bronchopulmonary aspergillosis (ABPA). It is characterized by an early allergic response and late-phase pulmonary injury that includes bronchopulmonary remodeling, inflammation with CD4 T cell activation, increased Th2 cytokine levels, and eosinophilia (17). Emerging evidence suggests that pulmonary inflammation associated with Th2-mediated inflammation could represent a novel pathway for the pathogenesis of pulmonary arterial remodeling. While the pathology seen in asthma is often described as limited to the airways, pulmonary artery pathology is also found in those with fatal asthma (39). A recent study of asthma patients showed that bronchial arteries exhibited an increase in intimal area, associated with smooth muscle proliferation and calcification of the elastica, and a corresponding decrease in luminal area (16). In murine models, Th2 cell-mediated immune responses to inhaled purified antigens have been reported to induce remodeling of pulmonary muscular arteries (36, 37), including a prolonged Th2 cell-mediated immune response to the soluble A. fumigatus antigen (9).
In our studies, we observed extensive remodeling of small- and medium-sized arteries within the lungs, including thickened arterial walls and narrowed arterial lumens, and obliteration of the luminal space in the most severely involved segments. There was smooth muscle cell hyperplasia, fibrosis, and neointimal formation. These features of fully remodeled arteries were preceded by the accumulation of cells in the arterial lumen, suggesting that cells recruited via the circulation likely participated. This process was similar to arterial remodeling in human disease, which involves all layers of the arterial wall: intima, media, and adventitia (6, 23). Surprisingly, but entirely consistent with a previous study by Daley et al. in a mouse model of allergen-driven pulmonary inflammation (9), we observed that pulmonary inflammation and arterial remodeling in mice could occur without the development of pulmonary hypertension. The studies by Swain et al. provide some potential insight into the dichotomy between these processes (44). The persistent pulmonary inflammation that develops when CD4 T cells in Pneumocystis-infected mice are temporarily depleted, and then allowed to return, drives the development of an IL-4 signaling-independent pulmonary hypertension that correlates with the return of CD4 T cells and perivascular fibrosis. Vascular adventitial remodeling reduces distensibility and compliance of the vessels, which can in turn contribute to increased vessel pressures (47). In our studies, 8 weeks of weekly exposure to viable Aspergillus conidia did produce pulmonary arterial remodeling but not a significant fibrotic response.
Our current and previous studies (30) have demonstrated that pulmonary arterial remodeling is preceded by a significant influx of macrophages into the Th1 and Th2 cytokine-rich environment of the lungs. This environment is likely to result in the differentiation of both M1 and M2 macrophages, similar to what we recently reported for the mixed Th1/Th2 environment during pulmonary Cryptococcus neoformans infections (1). IL-4 and IL-10 are potent drivers of M2 differentiation and M2-derived factors (such as matrix metalloproteinase-9 and factor XIIIA), as well as M1-derived molecules (such as inducible nitric oxide synthase [iNOS] and tumor necrosis factor alpha [TNF-α]), and are constituents of actively remodeling arterial environments (2, 14, 28, 45). Macrophages are well-documented central mediators of arterial remodeling in experimental atherosclerosis in apolipoprotein E knockout mice, and in these mice, there is an early influx of M2 followed by a late influx of M1 macrophages into atherosclerotic plaques (24). Thus, it is likely that macrophages, as well as T cells, play a key role in pulmonary arterial remodeling during repeated A. fumigatus conidial exposure.
Mice lacking IL-4-producing CD4+ Th2 cells, but not IFN-γ-producing CD4+ Th1 cells, were protected from arterial remodeling despite the presence of an otherwise vigorous inflammatory response to inhaled conidia. Daley et al. also demonstrated that arterial remodeling associated with experimental asthma was significantly abrogated in IL-4-deficient mice (9). We also observed that in IL-10-deficient mice, the development of IL-4-producing CD4+ Th2 cells and arterial remodeling was inhibited. These data are interesting in light of the notion that IL-10 is a pleiotropic cytokine that has immunomodulatory and anti-inflammatory properties (29). IL-10 is known to be produced by macrophages, dendritic cells (DC), neutrophils, B cells, and various sets of CD4+ and CD8+ T cells (29, 34). While we do not have any data with Aspergillus fumigatus, we have preliminary data with Cryptococcus neoformans, in which early IL-10 production is important for T cell polarization (20), that suggest that DC are a major source of that cytokine. IL-10 regulates T cell responses, but many of the effects of IL-10 on T cells are indirect, being mediated via a direct effect of IL-10 on antigen-presenting cells. In an experimental model of asthma, IL-10 gene transfer to the airway suppressed the development of allergic airway sensitization (40). In previous reports on the immune response to A. fumigatus, IL-10−/− mice developed an exaggerated Th1 response to viable conidia (10), and the allergic responses to antigen extracts were similar in IL-10−/− and wild-type mice (18). Similar to the present study, other reports described the attenuation of Th2 responses in IL-10-deficient mice. Those studies investigated a pulmonary infection with the fungus C. neoformans (20), an ovalbumin sensitization and challenge model of allergic airway disease (49), and a murine model of allergic dermatitis (26). Thus, the role of IL-10 in the manifestation of Th2 responses requires further investigation, and IL-10 should not simply be thought of as an inhibitor of allergic airway inflammation.
Our studies also demonstrate that the loss of the Th2 response in IL-10−/− mice was not due to trafficking of reactive T cells to the intestinal tract. Rivera et al. has recently reported that intestinal inflammation in IL-10−/− mice results in the altered trafficking of Aspergillus-specific Th1 cells from the lungs to the gastrointestinal (GI) tract (32). The presence and kinetics of intestinal inflammation in IL-10−/− mice are mouse colony dependent, due to differences in the microbiota of mice housed in different facilities (38, 42). We performed histological analysis of the IL-10−/− mice used in these studies and could not detect any early histological signs of colitis during their 3 months of housing in the University of Michigan animal facility (histology not shown). However, we also provided direct evidence that altered Th cell trafficking was not occurring during repeated exposure to A. fumigatus conidia, because the numbers of Th1 and Th2 cells in the lungs of A. fumigatus conidium-challenged IL-10−/− mice were identical to (or even greater than) those found in the lungs of similarly exposed wild-type mice, including Th1 cells (Fig. 6).
Our current studies with IL-10−/− mice, combined with our previous studies and reports from other labs, clearly demonstrate that there is a difference in the immunoregulatory function of IL-10 during the response to a single allergen versus to a complex, metabolically active particle, such as A. fumigatus conidia (5, 10, 18, 20, 26, 35, 40, 46, 49). Numerous studies have demonstrated that IL-10 can downregulate Th2 responses to purified antigens, such as ovalbumin or Aspergillus extract. The mechanism underlying this involves antigen-presenting cell-mediated upregulation of regulatory T cell responses. In contrast, our lab has now demonstrated that for pulmonary host responses to live C. neoformans yeast (20) and A. fumigatus conidia, IL-10 is centrally involved in the development of the Th2 response. This concept may be more broadly applicable to live fungi as an immunologic challenge, because systemic candidiasis and aspergillosis are also both exacerbated in IL-10 knockout mice, as is gastrointestinal infection by Candida albicans (5, 8, 10, 35, 46). The mechanism underlying this remains to be fully elucidated but may involve regulation of nitric oxide-mediated killing by macrophages, dendritic cell development, and innate inflammatory mediator production. Thus, in chronic fungal infections or scenarios of repeated exposure to live fungal organisms, IL-10 production contributes to lack of fungal clearance and/or immunopathology, such as pulmonary arterial remodeling, as shown in our study.
ACKNOWLEDGMENTS
We thank Roderick McDonald for expert technical assistance and John Erb-Downward for critical reading of the manuscript.
This work was supported by grants R01AI064479, R21AI087869, and R21AI083473 (to G.B.H.) from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.
Footnotes
Published ahead of print 7 November 2011
REFERENCES
- 1. Arora S, et al. 2011. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect. Immun. 79: 1915–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bakker EN, et al. 2008. Blood flow-dependent arterial remodelling is facilitated by inflammation but directed by vascular tone. Cardiovasc. Res. 78: 341–348 [DOI] [PubMed] [Google Scholar]
- 3. Bozza S, et al. 2002. Dendritic cells transport conidia and hyphae of Aspergillus fumigatus from the airways to the draining lymph nodes and initiate disparate Th responses to the fungus. J. Immunol. 168: 1362–1371 [DOI] [PubMed] [Google Scholar]
- 4. Brieland JK, et al. 2001. Cytokine networking in lungs of immunocompetent mice in response to inhaled Aspergillus fumigatus. Infect. Immun. 69: 1554–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cenci E, et al. 1993. Interleukin-4 and interleukin-10 inhibit nitric oxide-dependent macrophage killing of Candida albicans. Eur. J. Immunol. 23: 1034–1038 [DOI] [PubMed] [Google Scholar]
- 6. Chan SY, Loscalzo J. 2008. Pathogenic mechanisms of pulmonary arterial hypertension. J. Mol. Cell. Cardiol. 44: 14–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chin KM, Rubin LJ. 2008. Pulmonary arterial hypertension. J. Am. Coll. Cardiol. 51: 1527–1538 [DOI] [PubMed] [Google Scholar]
- 8. Clemons KV, et al. 2000. Role of IL-10 in invasive aspergillosis: increased resistance of IL-10 gene knockout mice to lethal systemic aspergillosis. Clin. Exp. Immunol. 122: 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Daley E, et al. 2008. Pulmonary arterial remodeling induced by a Th2 immune response. J. Exp. Med. 205: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Del Sero G, et al. 1999. Antifungal type 1 responses are upregulated in IL-10-deficient mice. Microbes Infect. 1: 1169–1180 [DOI] [PubMed] [Google Scholar]
- 11. Denning DW, O'Driscoll BR, Hogaboam CM, Bowyer P, Niven RM. 2006. The link between fungi and severe asthma: a summary of the evidence. Eur. Respir. J. 27: 615–626 [DOI] [PubMed] [Google Scholar]
- 12. Dorfmuller P, Perros F, Balabanian K, Humbert M. 2003. Inflammation in pulmonary arterial hypertension. Eur. Respir. J. 22: 358–363 [DOI] [PubMed] [Google Scholar]
- 13. Fulton SA, et al. 2004. Inhibition of major histocompatibility complex II expression and antigen processing in murine alveolar macrophages by Mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect. Immun. 72: 2101–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galis ZS, et al. 2002. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ. Res. 91: 852–859 [DOI] [PubMed] [Google Scholar]
- 15. Gibson PG. 2006. Allergic bronchopulmonary aspergillosis. Semin. Respir. Crit. Care Med. 27: 185–191 [DOI] [PubMed] [Google Scholar]
- 16. Green FH, Butt JC, James AL, Carroll NG. 2006. Abnormalities of the bronchial arteries in asthma. Chest 130: 1025–1033 [DOI] [PubMed] [Google Scholar]
- 17. Greenberger PA, Patterson R. 1987. Allergic bronchopulmonary aspergillosis. Model of bronchopulmonary disease with defined serologic, radiologic, pathologic and clinical findings from asthma to fatal destructive lung disease. Chest 91: 165S–171S [PubMed] [Google Scholar]
- 18. Grunig G, et al. 1997. Interleukin-10 is a natural suppressor of cytokine production and inflammation in a murine model of allergic bronchopulmonary aspergillosis. J. Exp. Med. 185: 1089–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hassoun PM, et al. 2009. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 54: S10–S19 [DOI] [PubMed] [Google Scholar]
- 20. Hernandez Y, et al. 2005. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J. Immunol. 174: 1027–1036 [DOI] [PubMed] [Google Scholar]
- 21. Hohl TM, et al. 2005. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 1: e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Humbert M, et al. 2004. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 43: 13S–24S [DOI] [PubMed] [Google Scholar]
- 23. Jeffery TK, Morrell NW. 2002. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog. Cardiovasc. Dis. 45: 173–202 [DOI] [PubMed] [Google Scholar]
- 24. Khallou-Laschet J, et al. 2010. Macrophage plasticity in experimental atherosclerosis. PLoS One 5: e8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurup VP, Shen HD, Banerjee B. 2000. Respiratory fungal allergy. Microbes Infect. 2: 1101–1110 [DOI] [PubMed] [Google Scholar]
- 26. Laouini D, et al. 2003. IL-10 is critical for Th2 responses in a murine model of allergic dermatitis. J. Clin. Invest. 112: 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Latge JP. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 12: 310–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lessner SM, Prado HL, Waller EK, Galis ZS. 2002. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am. J. Pathol. 160: 2145–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19: 683–765 [DOI] [PubMed] [Google Scholar]
- 30. Murdock BJ, et al. 2011. Coevolution of TH1, TH2, and TH17 responses during repeated pulmonary exposure to Aspergillus fumigatus conidia. Infect. Immun. 79: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osterholzer JJ, et al. 2009. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J. Immunol. 183: 8044–8053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rivera A, et al. 2009. Aberrant tissue localization of fungus-specific CD4+ T cells in IL-10-deficient mice. J. Immunol. 183: 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rivera A, et al. 2006. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity 25: 665–675 [DOI] [PubMed] [Google Scholar]
- 34. Romani L, et al. 1997. Neutrophil production of IL-12 and IL-10 in candidiasis and efficacy of IL-12 therapy in neutropenic mice. J. Immunol. 158: 5349–5356 [PubMed] [Google Scholar]
- 35. Romani L, et al. 1994. Neutralization of IL-10 up-regulates nitric oxide production and protects susceptible mice from challenge with Candida albicans. J. Immunol. 152: 3514–3521 [PubMed] [Google Scholar]
- 36. Rydell-Tormanen K, Johnson JR, Fattouh R, Jordana M, Erjefalt JS. 2008. Induction of vascular remodeling in the lung by chronic house dust mite exposure. Am. J. Respir. Cell Mol. Biol. 39: 61–67 [DOI] [PubMed] [Google Scholar]
- 37. Rydell-Tormanen K, Uller L, Erjefalt JS. 2008. Remodeling of extra-bronchial lung vasculature following allergic airway inflammation. Respir. Res. 9: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sellon RK, et al. 1998. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66: 5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shiang C, et al. 2009. Pulmonary periarterial inflammation in fatal asthma. Clin. Exp. Allergy 39: 1499–1507 [DOI] [PubMed] [Google Scholar]
- 40. Stampfli MR, et al. 1999. Interleukin-10 gene transfer to the airway regulates allergic mucosal sensitization in mice. Am. J. Respir. Cell Mol. Biol. 21: 586–596 [DOI] [PubMed] [Google Scholar]
- 41. Steele C, et al. 2005. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 1: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stehr M, et al. 2009. Charles River altered Schaedler flora (CRASF) remained stable for four years in a mouse colony housed in individually ventilated cages. Lab. Anim. 43: 362–370 [DOI] [PubMed] [Google Scholar]
- 43. Stevens WW, Kim TS, Pujanauski LM, Hao X, Braciale TJ. 2007. Detection and quantitation of eosinophils in the murine respiratory tract by flow cytometry. J. Immunol. Methods 327: 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swain SD, Han S, Harmsen A, Shampeny K, Harmsen AG. 2007. Pulmonary hypertension can be a sequela of prior pneumocystis pneumonia. Am. J. Pathol. 171: 790–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang PC, et al. 2008. MyD88-dependent, superoxide-initiated inflammation is necessary for flow-mediated inward remodeling of conduit arteries. J. Exp. Med. 205: 3159–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tonnetti L, et al. 1995. Interleukin-4 and -10 exacerbate candidiasis in mice. Eur. J. Immunol. 25: 1559–1565 [DOI] [PubMed] [Google Scholar]
- 47. Tozzi CA, Christiansen DL, Poiani GJ, Riley DJ. 1994. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am. J. Respir. Crit. Care Med. 149: 1317–1326 [DOI] [PubMed] [Google Scholar]
- 48. van Rijt LS, et al. 2005. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201: 981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang X, Wang S, Fan Y, Han X. 2000. IL-10 deficiency prevents IL-5 overproduction and eosinophilic inflammation in a murine model of asthma-like reaction. Eur. J. Immunol. 30: 382–391 [DOI] [PubMed] [Google Scholar]
- 50. Zelante T, et al. 2007. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur. J. Immunol. 37: 2695–2706 [DOI] [PubMed] [Google Scholar]