

Abstract
The oral bacterium Porphyromonas gingivalis is a key etiological agent of human periodontitis, a prevalent chronic disease that affects up to 80% of the adult population worldwide. P. gingivalis exhibits neuraminidase activity. However, the enzyme responsible for this activity, its biochemical features, and its role in the physiology and virulence of P. gingivalis remain elusive. In this report, we found that P. gingivalis encodes a neuraminidase, PG0352 (SiaPg). Transcriptional analysis showed that PG0352 is monocistronic and is regulated by a sigma70-like promoter. Biochemical analyses demonstrated that SiaPg is an exo-α-neuraminidase that cleaves glycosidic-linked sialic acids. Cryoelectron microscopy and tomography analyses revealed that the PG0352 deletion mutant (ΔPG352) failed to produce an intact capsule layer. Compared to the wild type, in vitro studies showed that ΔPG352 formed less biofilm and was less resistant to killing by the host complement. In vivo studies showed that while the wild type caused a spreading type of infection that affected multiple organs and all infected mice were killed, ΔPG352 only caused localized infection and all animals survived. Taken together, these results demonstrate that SiaPg is an important virulence factor that contributes to the biofilm formation, capsule biosynthesis, and pathogenicity of P. gingivalis, and it can potentially serve as a new target for developing therapeutic agents against P. gingivalis infection.
INTRODUCTION
Sialic acid is a generic term to describe a group of over 40 naturally occurring nine-carbon keto sugar acids (52, 67, 74). Among these molecules, the most abundant and best-studied sialic acid is N-acetylneuraminic acid (Neu5Ac). As such, the term “sialic acid” in the literature primarily means this particular compound. Sialic acids typically occupy the terminal positions within glycan molecules on the surfaces of eukaryotic cells, where they play important roles in a wide range of biological processes, including cell-cell interactions and small molecule-cell recognitions (70–73). Several pathogenic bacteria have evolved to utilize sialic acids either as nutrients or as decorating molecules to modify their surface-exposed macromolecules, including lipopolysaccharides (LPS) and polysialic acid (PSA) capsules (11, 53, 74). These sialic acid modifications allow bacterial pathogens to disguise themselves as host cells and thus circumvent and/or counteract the host's immune responses.
Bacteria have two primary routes to acquire sialic acids: by de novo biosynthesis and by means of a scavenger pathway (53, 72, 74). For the first route, the metabolite UDP-GlcNAc can be converted to Neu5Ac. In Escherichia coli, UDP-GlcNAc is first converted to ManNAc by NeuC (UDP-GlcNAc 2-epimerase) and then to Neu5Ac by NeuB (Neu5Ac synthase). For the second route, bacteria can directly uptake free sialic acids from the environments via transporters. They may also utilize neuraminidases to release sialic acids from a diverse range of host sialoglycoconjugates. The released sialic acids can then be transported into the cells. Neuraminidase is a family of enzymes that hydrolyze the glycosidic linkage between a terminal sialic acid and an adjacent glycosyl residue of certain glycoproteins, glycolipids, and oligosaccharides (1, 49, 50, 73). There are two types of neuraminidases. One is exo-α-neuraminidase, which hydrolyzes α-(2-3)-, α-(2-6)-, and α-(2-8)-glycosidic linkages of terminal sialic acids; the other is endo-neuraminidase, which cleaves α-(2-8)-sialosyl linkages in oligo- or poly-sialic acids.
Neuraminidases have been identified in several pathogenic bacteria (53, 73), such as Vibrio cholerae (20), Streptococcus pneumoniae (47), and Pseudomonas aeruginosa (29, 38). In these pathogens, neuraminidases are often associated with virulence. The neuraminidase of V. cholerae enhances the activity of cholera toxin (20). The two neuraminidases of S. pneumoniae contribute to the progression of infection (e.g., promoting pneumococcal brain endothelial cell invasion) (59, 69), and the neuraminidase of P. aeruginosa increases the binding of this organism to host cells (8). Neuraminidase activity has also been detected in several oral bacteria, such as Tannerella forsythia and Porphyromonas gingivalis (44, 66). T. forsythia possesses two neuraminidases, and one of them (NanH) is associated with bacterial colonization and invasion (28, 51).
P. gingivalis, a black-pigmented, asaccharolytic, Gram-negative anaerobe (4), is considered a major pathogen in human periodontitis, which is a prevalent chronic disease that affects ca. 80% of the adult population (27, 48, 58). P. gingivalis produces several virulence factors, including LPS, capsule, fimbriae and a group of proteolytic enzymes that cleave host proteins such as immunoglobulin and complement components (6, 36, 54, 57). In addition, P. gingivalis is highly invasive and is able to invade a variety of host cells (35). The de novo biosynthesis pathway of Neu5Ac is not evident in the genome of P. gingivalis (45); however, previous studies suggest that it produces neuraminidase (3, 44). For instance, Moncla et al. examined 25 P. gingivalis isolates and found that they all had neuraminidase activity, suggesting that neuraminidase commonly exists in P. gingivalis strains and that it may play a very important role in the biology and pathogenesis of P. gingivalis. In the present study, a gene encoding a neuraminidase was identified, and its enzymatic activity was analyzed. Its roles in biofilm formation, capsule synthesis, serum killing resistance, and virulence were explored using an approach consisting of biochemistry, targeted mutagenesis, cryoelectron microscopy (cryo-EM) and cryoelectron tomography (cryo-ET), and both in vitro and in vivo virulence studies.
MATERIALS AND METHODS
Ethics statement.
All animal experimentation was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. Human serum samples were collected from two healthy volunteers in the laboratory, and a written informed consent was provided by two participants. The protocols for animal studies and the use of human serum were approved by the Institutional Animal Care and Use Committee (IACUC; permit number: ORB23068Y) and the Human Subjects Review Committee (HSIRB; permit number: ORB0321006E) of the State University of New York at Buffalo, respectively.
Bacterial strains and growth conditions.
P. gingivalis W83 (Pg83) (45), ATCC 33277 (Pg33277) (26), and FDC381 (Pg381) (9) strains were grown either in Trypticase soy broth (TSB; BD Diagnostic Systems, Sparks, MD) supplemented with vitamin K (1 μg ml−1) and hemin (5 μg ml−1) or on TSB agar plates containing 5% defibrinated sheep blood, vitamin K, and hemin (PML Microbiologicals, Wilsonville, OR) at 37°C in an anaerobic atmosphere of 80% N2, 10% H2, and 10% CO2 as previously described (9). E. coli TOP10 (Invitrogen, Carlsbad, CA) was used for DNA cloning, strain DH5α was used for β-galactosidase assays, and strain M15 used for preparing recombinant proteins. E. coli strains were cultured in lysogeny broth (LB) supplemented with appropriate concentrations of antibiotics.
Reverse transcription-PCR (RT-PCR) and RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE).
Total RNA was extracted from 100 ml of exponential-phase P. gingivalis cultures using TRI reagent (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. The resultant samples were treated with Turbo DNase I (Ambion, Austin, TX) at 37°C for 2 h to eliminate genomic DNA contamination. The obtained RNA samples were then extracted using acid-phenol-chloroform, precipitated in isopropanol, and washed with 70% ethanol. Finally, the RNA pellets were resuspended in RNase-free water. RNA (1 μg) was reverse transcribed using avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI) to generate cDNA.
For RT-PCR analysis, 1 μl of cDNA was PCR amplified using Taq DNA polymerase (Qiagen, Valencia, CA). The P. gingivalis 16S rRNA gene was used as a positive control. The primers for RT-PCR are listed in Table 1. To determine the transcription start site upstream of PG0352, 5′-RACE analysis was carried out using the FirstChoice RLM-RACE Kit (Ambion), according to the manufacturer's protocol. Purified P. gingivalis RNA (10 μg) was reverse transcribed to cDNA with a 5′-RACE adapter, followed by PCR amplifications with primers P1/P2 (Table 1). The resultant PCR products were cloned into pGEM-T-easy vector (Promega) and sequenced (Roswell Park Cancer Institute DNA Sequencing Laboratory, Buffalo, NY).
Table 1.
Primers used in this study
| Primer | Orientationa | Sequence (5′–3′)b | Description |
|---|---|---|---|
| P1 | F | ACCCGGAGGAAGGGATTCACTTAA | PG0352 5′RACE inner primer |
| P2 | R | GCCCTTTGTAGTCCGGCAAGTC | PG0352 5′RACE outer primer |
| P3 | F | GAATTCCTGGTTAGTTTTTGGTTTGTG | PG0352 promoter conformation |
| P4 | R | GGATCCCATTCGAAAACAATTTTATACCG | PG0352 promoter conformation |
| P5 | F | GCTCTTTCAGCTTGGTATAGG | PG0352 upstream region |
| P6 | R | AGATCTGACATAACGTCGAGTCTTCGC | PG0352 upstream region |
| P7 | F | AGATCTACGATCTCTTCGATGTCCGGC | PG0352 downstream region |
| P8 | R | GACCTACCACGAATATCAACC | PG0352 downstream region |
| P9 | F | AGATCTAGCTTCCGCTATTGCTTT | erm cassette |
| P10 | R | AGATCTTTTATCTACATTCCCTTTAGT | erm cassette |
| P11 | F | CACCGCAAATAATACTCTTTTGGCGA | Confirmation of erm insertion |
| P12 | F | TGCCGTTGCAGAAAAGCC | Confirmation of erm location |
| P13 | R | TTGCCGGACATCGAAGAGATCGTC | Confirmation of erm insertion |
| P14 | F | TTGTCGGTTTCTGTTGGCTC | Confirmation of PG0352 deletion |
| P15 | R | AAAGCTTTGCCTCATCGC | Confirmation of PG0352 deletion |
| P16 | F | AGAGCTGTCGCCTGCCTC | PG0352 RT-PCR |
| P17 | R | CTGTCGGCTCTCCTGCCGTC | PG0352 RT-PCR |
| P18 | F | GTCGACATGGCAAATAATACTCTTTT | Overexpression of PG0352 |
| P19 | R | CTGCAGTTGCCGGACATCGAAGAGAT | Overexpression of PG0352 |
F, forward primers; R, reverse primers.
The engineered restriction enzyme sites are underlined.
β-Galactosidase activity assay.
A fragment spanning from nucleotides −425 to +3 of PG0352 was PCR amplified from Pg83 genomic DNA with primers P3/P4, generating a fragment with engineered EcoRI and BamHI cut sites at the 5′ and 3′ ends, respectively. The obtained fragment was in-frame fused to the promoterless lacZ gene in the pRS415 plasmid (56), creating a reporter vector, pRSCLPg352. The resultant plasmid was transformed into E. coli DH5α. The β-galactosidase activity was measured as previously described (25), and the activity was expressed as the average Miller units of triplicate samples from two independent experiments.
Preparations of PG0352 recombinant protein and antiserum.
The entire open reading frame of PG0352 was PCR amplified with the primers P18/P19 using Vent DNA polymerase (New England Biolabs, Ipswich, MA). The resultant PCR product was cloned into pGEM-T vector (Promega) and then subcloned into pQE30 expression vector (Qiagen) at engineered SalI and PstI cut sites. The obtained plasmid was transformed into E. coli M15 strain. The overexpression of PG0352 was induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The recombinant protein was purified using Ni-NTA agarose (Qiagen) under native conditions as previously described (65). The purified protein (rSiaPg) was dialyzed overnight and then concentrated using a 6.0-kDa molecular cutoff spectra membrane (Spectrum Laboratories Inc., Rancho Dominguez, CA). The concentration of rSiaPg was determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA). Antiserum against rSiaPg was raised as previously described (65).
Detection of neuraminidase activity.
The neuraminidase activity was detected by using a filter paper spot test as previously described (43, 44). Briefly, rSiaPg or the whole-cell lysates of P. gingivalis were coincubated with 4-methylumbelliferyl-α-d-N-acetylneuraminic acid (4-MUNANA; Sigma-Aldrich), a fluorogenic neuraminidase substrate. Fluorescence was detected using the ChemiDoc XRS System (Bio-Rad) with an excitation wavelength of 302 nm and an emission wavelength of 548 nm.
Lectin blot assay.
To determine the substrate specificity of rSiaPg, the lectin blot analysis described before (23) was carried out. Briefly, 1 μg of human α-1 acid glycoprotein (AGP) was coincubated with rSiaPg (0.6 nM) or with the Clostridium perfringens neuraminidase (NanH; Sigma-Aldrich) in a reaction buffer (10 mM potassium phosphate buffer [pH 7.0]) for 12 h. After the incubation, the samples were separated by SDS–12% PAGE and transferred to polyvinylidene difluoride membrane (Bio-Rad). A DIG glycan differentiation kit (Roche, Mannheim, Germany) was used to detect carbon hydrates within AGP according to the manufacturer's instructions. Lectins from three plants—Sambucus nigra (SNA), Maackia amurensis (MAA), and Datura stramonium (DSA)—were used to detect terminal α-(2-6)-linked sialic acid, α-(2-3)-linked sialic acid, and galactose linked to GlcNAc (14), respectively.
Constructing a P. gingivalis PG0352 deletion mutant.
To inactivate PG0352, its upstream and downstream regions were PCR amplified from Pg83 genomic DNA using the primer pairs P5/P6 and P7/P8 (see Fig. S1 in the supplemental material), respectively. A previously described ermF/AM erythromycin resistance (Ermr) cassette was PCR amplified using the primer pair P9/P10 (39). The amplicons were individually cloned into pGEM-T Easy vector (Promega). The downstream fragment was released by BglII and NdeI digestion and then ligated to the 3′ end of the upstream fragment that had been digested at the same cut sites. The Ermr cassette was then released by BglII digestion and inserted into the above obtained fragment, generating PG0352-erm plasmid in which the entire open reading frame of PG0352 was deleted and replaced with the Ermr cassette. For the allelic-exchange mutagenesis, PG0352-erm plasmid was linearized with NdeI. Approximately 10 μg of the linearized plasmid was electroporated into the Pg83 cells, and the transformed cells were selected on the blood TSB agar plates containing clindamycin (5 μg ml−1) as described previously (18).
Measuring the growth rates and biofilm formation of P. gingivalis.
To measure the growth rates, 50 μl of overnight P. gingivalis cultures (optical density at 600 nm [OD600] = 0.5) were inoculated into 5 ml of normal TSB or TSB medium supplemented with hemin (5 μg ml−1) and vitamin K (1 μg ml−1) without dextrose (BD Diagnostic Systems) and cultured at 37°C in an anaerobic chamber. The absorbance (OD600) of cultures at given times was measured using a Genesys spectrometer (Thermo Scientific, Waltham, MA). Biofilm formation was measured as previously described (24, 46, 77) with slight modifications. Briefly, Pg83 and ΔPG352 strains were grown to mid-log phase, and then the cell densities (OD600) were measured to ensure that the same amounts of Pg83 and ΔPG352 cells were used for biofilm measurements. These cultures were diluted 1:100 in phosphate-buffered saline (PBS; pH 7.4)-balanced TSB broth (50% PBS), and then 100-μl portions of the diluted cultures were added to 96-well flat-bottom polystyrene plates. The plates were then incubated anaerobically at 37°C for 3 days, allowing time for biofilms to develop. The biofilms were stained with 25 μl of 1% crystal violet for 15 min, washed with water three times, and then air dried for 30 min. To quantify the amount of biofilms, 150 μl of 95% ethanol was added to each well and shaken for 15 min. The absorbance (OD570) was measured with an xMark microplate spectrophotometer (Bio-Rad). The data are expressed as the average absorbance of 12 parallel samples. The data were statistically analyzed by one-way analysis of variance (ANOVA), followed by Tukey's multiple comparison at P < 0.01.
Cryo-EM and cryo-ET of frozen-hydrated P. gingivalis.
Cryoelectron microscopy (cryo-EM) and cryoelectron tomography (cryo-ET) were carried out as described previously (40, 83). Briefly, the P. gingivalis cultures (5 μl) were mixed with 15-nm gold particles, deposited onto a freshly glow-discharged Holey carbon grid, blotted, and rapidly frozen in liquid ethane. The frozen-hydrated specimens were imaged at −170°C using a Polara G2 electron microscope (FEI Company, Hillsboro, OR) equipped with a field emission gun and a 4K×4K charge-coupled device (TVIPS, GmbH, Germany). The microscope was operated at 300 kV, and cryo-EM images were recorded at the magnification of ×23,000 (0.77 nm/pixel). A low-dose single-axis tilt series was collected from each bacterium at −8 μm defocus with a cumulative dose of ∼100 e−/Å2 distributed over 65 images with an angular increment of 2°, covering a range from −64° to +64°. The tilt series images were aligned and reconstructed using the IMOD software package (32). In all, 20 and 10 cryo-tomograms from Pg83 and ΔPG352 were reconstructed, respectively. Tomographic reconstructions were visualized using IMOD. One representative reconstruction from Pg83 and ΔPG352 was segmented manually using Amira 3-D modeling software (Visage Imaging, San Diego, CA). The 3-D segmentations of capsule (CPS), peptidoglycan (PG), outer membrane (OM), and inner membrane (IM) were manually constructed.
India ink staining.
The staining was carried out as previously described (15) with slight modifications. P. gingivalis cells were taken from 4-day-old TSB agar plates and resuspended in 1 ml of PBS. Then, 10 μl of suspended cells was dropped on a glass slide, and a thin film was made by another slide. The film was air dried. A drop of 0.2% fuchsine (Sigma-Aldrich) was carefully added to the film and removed after 2 min by decanting. After air drying for 10 min, 10 μl of India ink (American MasterTech Scientific, Inc., Lodi, CA) was dropped on one end of the film, scraped by a clean slide to the other end, and then air dried again. A drop of 40% glycerol was added to the film and then covered with a new glass slip. Images were taken under a Zeiss Imager A2 microscope and processed using the Axiovision program (Zeiss, Germany).
Serum resistance assay.
Serum resistance assays were carried out as previously described (57). Serum samples were collected from healthy laboratory donors. After venipuncture, the blood was allowed to clot, and the serum was separated by centrifugation. Serum samples were dispensed in aliquots and frozen at −80°C within 2 h of blood collection. Each aliquot was thawed once only. Heat-inactivated serum (used as a control) was prepared by incubating aliquots of the serum at 56°C for 30 min. Overnight cultures of P. gingivalis were diluted 1:10 in fresh TSB medium and grown to early exponential phase (5 h). Aliquots containing 75-μl bacterial cultures were mixed with 25 μl of serum (for a final concentration of 25% serum) and anaerobically coincubated for 1 or 3 h. After the incubations, the samples were removed, serially diluted, and plated on blood agar plates. The plates were incubated for 6 days before the colonies were counted. The average percentage of surviving bacteria (the number of living cells in fresh serum/the number of living cells in heat-inactivated serum) was calculated from triplicate samples of two independent experiments.
Animal studies and histopathology.
Pg83, ΔPG352, and Pg381(a noncapsular strain) were tested for the virulence in a mouse model as previously described (18, 34). P. gingivalis strains were grown in TSB medium overnight, and cultures (10 ml at OD600 = 1.0) were harvested by centrifugation. The obtained cell pellets were washed twice with PBS under anaerobic conditions, counted in a Petroff-Hausser counting chamber (Hausser Scientific, Horsham, PA), and adjusted to 1011 ml−1 in PBS. Four-week-old BALB/c mice (female) were subcutaneously injected with 100 μl of bacterial suspension (∼1010 cells) on the dorsal surface. A group of mice was injected with PBS as a sham control. The general health, weight, appearance, and location of lesions were monitored daily until either the animals were dead or 10 days postinfection. Surviving mice were euthanized 10 days after infection by CO2 asphyxiation. Various tissues (skin with abscess, lung, liver, heart, spleen, and kidney) were collected and fixed in 10% neutral buffered formalin. Hematoxylin and eosin slides were prepared by standard pathological techniques. These experiments were approved by the IACUC at the State University of New York at Buffalo.
RESULTS
PG0352 is a neuraminidase.
Neuraminidase activity has been detected in different isolates of P. gingivalis (44). To identify the enzyme(s) responsible for the activity, neuraminidases from other bacterial species such as Clostridium perfringens (NanH) were used as queries to search the genome of Pg83 (45). It was found that the gene PG0352 encodes a well-conserved neuraminidase. PG0352 (hereafter designated SiaPg) consists of 526 amino acids (aa) and has a predicted molecular mass of 58.5 kDa. The N terminus (1 to 30 aa) of SiaPg contains a putative signal peptide (see Fig. S2 in the supplemental material), suggesting that this protein is probably secreted. Following the signal peptide is a peptidoglycan-binding domain (30 to 180 aa). The C terminus (180 to 526 aa) is a conserved neuraminidase domain (12, 13, 84). Sequence alignment analysis further revealed that the C terminus of SiaPg has a conserved catalytic domain of neuraminidase, which consists of one RIP motif and three Asp-box motifs (S/T-X-d-[X]-G-X-T-W/F) (Fig. 1), suggesting that SiaPg is a neuraminidase.
Fig 1.

Sequence alignment of neuraminidases. The underlined sequences represent the conserved domains identified in neuraminidases, including a RIP motif and three “Asp-box” motifs (S/T-X-D-[X]-G-X-T-W/F). Only a part of the alignment is presented. The aligned proteins include: Salmonella enterica serovar Typhimurium strain LT2 NanH (NP_459905), T. forsythia ATCC 43037 NanH (TF0035) (http://www.oralgen.lanl.gov/index.html), and P. gingivalis W83 (PG0352). The alignment was conducted using the program CLUSTAL W2.
To confirm these observations, the filter paper spot assay was carried out to test the enzymatic activity of recombinant SiaPg protein (rSiaPg). As shown in Fig. 2, rSiaPg was able to cleave the fluorogenic neuraminidase substrate, 4-MUNANA, and produced fluorescence (spot 1). This activity was completely abolished when the protein was denatured (spot 2). In addition, a neuraminidase specific inhibitor (82), 2,3-didehydro-2-deoxy-N-acetylneuraminic acid (Neu5Ac2en), inhibited the activity of rSiaPg (spots 3 and 4). Collectively, these results demonstrate that SiaPg is a neuraminidase.
Fig 2.

PG0352 (SiaPg) exhibits neuraminidase activity. (A) Preparation of recombinant PG0352 protein (rSiaPg). (B) Filter paper spot test of rSiaPg. The assay was conducted using 4-MUNANA as the substrate, and the images were processed using the ChemiDoc XRS system (Bio-Rad) with an excitation wavelength of 302 nm and an emission wavelength of 548 nm. Neu5Ac2en is an inhibitor of neuraminidase.
SiaPg has an exo-α-neuraminidase activity.
To determine whether SiaPg is an exo-neuraminidase or an endo-neuraminidase, the lectin blot assay was carried out. For this assay, C. perfringens neuraminidase (NanH, designated here as SiaCp), a well-studied exo-α-neuraminidase (33), was used as a positive control. As shown in Fig. 3, for the samples treated with PBS (lane 1), three lectins (DSA, SNA, and MMA [described in Materials and Methods]) bound to AGP. After treatment with SiaCp (lane 3), SNA and MMA failed to bind AGP, indicating that SiaCp removed the terminal α-(2-6)-linked and α-(2-3)-linked sialic acid. The treatment of rSiaPg had the same pattern as SiaCp (lane 2), showing that SiaPg is an exo-α-neuraminidase like its counterpart of C. perfringens.
Fig 3.
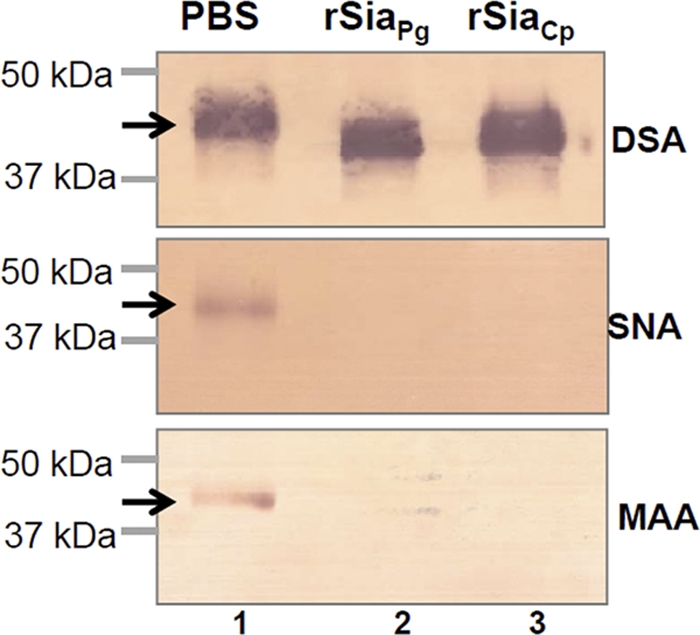
Lectin blot analysis of rSiaPg. The assay was carried out as previously described (23) using a DIG glycan differentiation kit (Roche). The DIG-labeled SNA, MAA, and DSA lectins were used to detect terminal α-(2-6)-linked sialic acid, α-(2-3)-linked sialic acid, and galactose linked to the GlcNAc of human α-1 acid glycoprotein (AGP), respectively. The recombinant neuraminidase (rSiaCp) of C. perfringens was used as a positive control for detecting the enzymatic activity (lane 3), and PBS was used as a negative control (lane 1). Arrows point to the products detected by three lectins.
Transcriptional analysis of PG0352.
The genes adjacent to PG0352 are divergently transcribed (see Fig. S1 in the supplemental material), suggesting that this gene is monocistronic. To understand its regulation, RLM-RACE analysis was conducted to determine its transcription start site. The start site was mapped to an adenosine that is 101 nucleotides from the start codon of PG0352 (Fig. 4). A promoter-like consensus was identified at the −10 (TCTATT) and −35 (TTGGGA) regions (Fig. 4A), which is similar to those of the σ70 promoter of E. coli (68), suggesting that PG0352 is probably regulated by a σ70-like promoter. The identified promoter was named P0352. Transcription analysis using lacZ gene as a reporter further showed that P0352 was functional in E. coli. The β-galactosidase activity (the average value = 5190 ± 284 Miller units) of a plasmid containing the P0352 promoter region (pRSCLP0352) is ∼100-fold greater than that of pRS415 (average activity = 54 ± 11 Miller units), a promoterless vector (56). Collectively, these results indicate that P0352 is a promoter that can be recognized by the RNA polymerases and possibly by other transcription factors of E. coli.
Fig 4.

Transcriptional analysis of the PG0352 gene. (A) Upstream region of PG0352. The underlined sequences are the −10 and −35 regions of the identified P0352 promoter, and the boldface italic sequence represents the translation start codon of PG0352. (B) 5′-RLM-RACE analysis of the PG0352 transcript. The arrow shows the sequencing direction, and an asterisk (*) indicates the transcriptional start site of the PG0352 transcript.
Isolation and characterization of PG0352 deletion mutant.
To study the function of SiaPg, the PG0352 gene was inactivated by targeted mutagenesis as illustrated in Fig. S1 in the supplemental material. Erythromycin-resistant (Ermr) colonies appeared 7 days after plating, and 10 colonies were selected and screened by PCR with specific primers to Ermr cassette. All of the examined colonies contained the Ermr cassette. One clone (ΔPG352) was further analyzed by PCR with different pairs of primers located at the flanking regions of PG0352 and the Ermr cassette, and the results showed that the PG0352 gene was deleted and replaced with Ermr cassette as expected (see Fig. S1 in the supplemental material). RT-PCR analysis showed that the PG0352 transcript was abolished in the mutant (Fig. 5A). Western blot analysis using a specific antibody against SiaPg showed that the cognate gene product was abrogated in the mutant (Fig. 5B).
Fig 5.
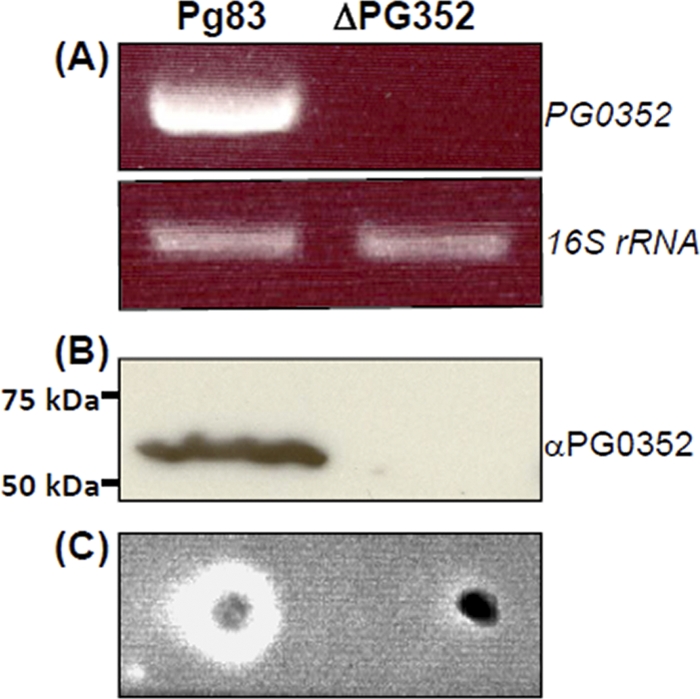
Characterization of the ΔPG352 mutant. (A) RT-PCR analysis of the ΔPG352 mutant. The transcripts of PG0352 and 16S rRNA (positive control) genes in Pg83 and the ΔPG352 mutant were detected by RT-PCR. (B) Western blotting analysis of the ΔPG352 mutant. The same amount of Pg83 and ΔPG352 whole lysates were analyzed by SDS-PAGE and then probed with a specific antiserum against PG0352. (C) Filter paper spot assay using the whole-cell lysates of Pg83 and the ΔPG352 mutant.
The filter paper spot assay was repeated using the whole-cell lysates of Pg83 and ΔPG352. As expected, the neuraminidase activity was detected in Pg83 but was not in the mutant (Fig. 5C). Collectively, these results showed that the PG0352 gene was totally disrupted in the ΔPG352 mutant. In addition, the neuraminidase activity was also detected in Pg33277 and Pg381 strains (see Fig. S3 in the supplemental material). Taken together with previous reports (44), these results suggest that neuraminidase genes are broadly distributed in P. gingivalis isolates.
Inactivation of PG0352 does not influence the planktonic growth of P. gingivalis.
To determine the influence of sialic acid on the growth of P. gingivalis, Pg83 and ΔPG352 were first cultured in normal TSB medium and then in the medium supplemented with exogenous Neu5Ac (30 μg ml−1). As shown in Fig. 6, the mutant grew at rates similar to the wild type with or without the sialic acid, indicating that SiaPg is not required for the planktonic growth of P. gingivalis. The experiment was repeated using a modified TSB medium in which dextrose was removed and supplemented with Neu5Ac (30 μg ml−1), and it was found that both Pg83 and ΔPG352 had a similar growth pattern (Fig. 6B), highlighting that P. gingivalis probably does not use sialic acid as a nutrient at the in vitro culture conditions.
Fig 6.

Growth curves of Pg83 and the ΔPG352 mutant. (A) P. gingivalis strains were cultured in normal TSB medium or in this medium supplemented with Neu5Ac (S30 represents 30 μg/ml). (B) P. gingivalis strains were cultured in dextrose-free TSB medium (dTSB) or in dTSB medium supplemented with Neu5Ac (30 μg/ml).
SiaPg influences the biofilm formation of P. gingivalis.
To test the effect of sialic acids on the biofilm growth of P. gingivalis, the normal biofilm growth medium was supplemented with four different concentrations of Neu5Ac (3, 8, 16, and 30 μg/ml). As shown in Fig. 7, for the wild type, the addition of exogenous sialic acid slightly reduced biofilm formation, but the effect was not significant. Compared to Pg83, ΔPG352 significantly decreased (∼2.5-fold reduction) the biofilm formation. Interestingly, the supplement of exogenous Neu5Ac restored the biofilm formation, and this restoration was in a dose-dependent manner (Fig. 7). For example, the addition of 3 μg of Neu5Ac/ml only partially restored the biofilm formation of the ΔPG352 mutant; however, biofilm formation in the mutant was fully restored at the concentration of 30 μg of Neu5A/ml. Taken together, these results suggest that SiaPg is involved in the biofilm formation of P. gingivalis.
Fig 7.

Biofilm formation of Pg83 and the ΔPG352 mutant. The assay was carried out in 96-well microtiter plates as previously described (24, 46). The upper panel shows representative wells of Pg83 and the ΔPG352 mutant, and the lower panel gives the average absorbances of Pg83 and the mutant. The error bars represent the standard deviation. The numbers represent the concentration of Neu5Ac in the medium. The data were statistically analyzed by one-way ANOVA, followed by Tukey's multiple comparison at P < 0.01.
The ΔPG352 mutant fails to produce an intact capsule.
To further explore the role of SiaPg, cryo-ET was used to dissect the cellular structures of P. gingivalis. As shown in Fig. 8, the P. gingivalis cell is rod-shaped, and it consists of an inner membrane (IM), a peptidoglycan layer (PG), and an outer membrane (OM). The outermost part of the cell is a thick layer of capsule-like materials (Fig. 8A and C). This layer could not be detected in Pg381, a noncapsular strain (see Fig. S4 in the supplemental material), indicating that it is capsule (CPS). The CPS layer in Pg83 has a defined shape and is evenly distributed on the cell surface. Its average width is 37 ± 2.76 nm (n = 10 cells). Compared to Pg83, the CPS layer observed in the ΔPG352 mutant was uneven and substantially thinner (average thickness = 13.72 ± 1.36 nm, n = 10 cells), and it lost the defined shape due to the reduced thickness and density (Fig. 8B and D).
Fig 8.

Cellular architectures of Pg83 and the ΔPG352 mutant revealed by cryo-EM and cryo-ET. One central slice of a tomographic reconstruction from Pg83 and ΔPG352 is shown in panels A and B, respectively. (C and D) Corresponding surface views of the reconstructions from Pg83 (A) and ΔPG352 (B), respectively. The prominent structural features include the outer membrane (OM), inner membrane (IM), peptidoglycan layer (PG), and capsular polysaccharide (CPS). Noticeably, the density corresponding to CPS is significantly weaker in ΔPG352. (E) Proteinase K treatment (200 μg/ml for 40 min at 37°C) does not remove the thin CPS layer of the ΔPG352 mutant CPS. (F) Supplementation with Neu5Ac (30 μg/ml) restored the CPS formation in the ΔPG352 mutant. The thickness of the CPS layer in the mutant is ∼30 nm, which is similar to that of Pg83 (G). The black dots are golden particles (diameter, 15 nm) which were used to calibrate the measurements of CPS thickness. The numbers represent the thickness of CPS.
To rule out the possibility that the observed CPS-like structure in ΔPG352 is another type of surface structure that is composed of proteins (e.g., fimbriae), the mutant was treated with proteinase K prior to the cryo-ET analysis. After the treatment, this layer still could be observed in the mutant (Fig. 8E), indicating that the observed structure is indeed a thin layer of capsule. In addition, when the ΔPG352 mutant was cultured in the medium supplemented with exogenous Neu5Ac (30 μg/ml), the mutant had a layer of CPS that was similar to the wild-type CPS (Fig. 8F and G), suggesting that the defective CPS in the mutant is simply due to the lack of sialic acid. India ink staining further confirmed that the remaining CPS layer in the ΔPG352 mutant was defective. As expected, the encapsulated P83 strain displayed colorless halos around individual cells, indicative the layer of CPS (Fig. 9, left panel). In contrast, the ΔPG352 mutant and Pg381 were stained by the dye (Fig. 9, middle and right panels), indicating that the mutant is not resistant to the dye due to lack of intact capsule. Collectively, these results demonstrate that SiaPg is required for the synthesis and/or assembly of capsule of P. gingivalis.
Fig 9.

India ink staining for the detection of capsule. The samples were stained with fuchsine and India ink as described in Materials and Methods. Images were taken under a Zeiss Imager A2 microscope (magnification, ×1,000).
The ΔPG352 mutant is less resistant to complement killing.
P. gingivalis is resistant to killing by host complement, and it is believed that capsule plays a central role in the resistance (57, 63). Since ΔPG352 has a defective capsule, we hypothesize that the mutant might become sensitive to the complement killing. To test this hypothesis, Pg83 and ΔPG352 were coincubated with 25% fresh human serum for 1 and 3 h. As expected, Pg83 was resistant to the killing, since its survival rate at the 1-h incubation point was almost 100%. In contrast, after a 1-h exposure to the serum, the survival rate of ΔPG352 was decreased to 22.4% (Fig. 10), indicating that the mutant is more vulnerable to the killing. Interestingly, after the 3-h exposure, the survival rate (26.2%) of the mutant was almost identical to that of the 1-h incubation (Fig. 10), suggesting that there might be another factor(s) also contributing to the serum resistance of P. gingivalis. Taken together, these results indicate that the presence of SiaPg is able to enhance the serum resistance of P. gingivalis.
Fig 10.
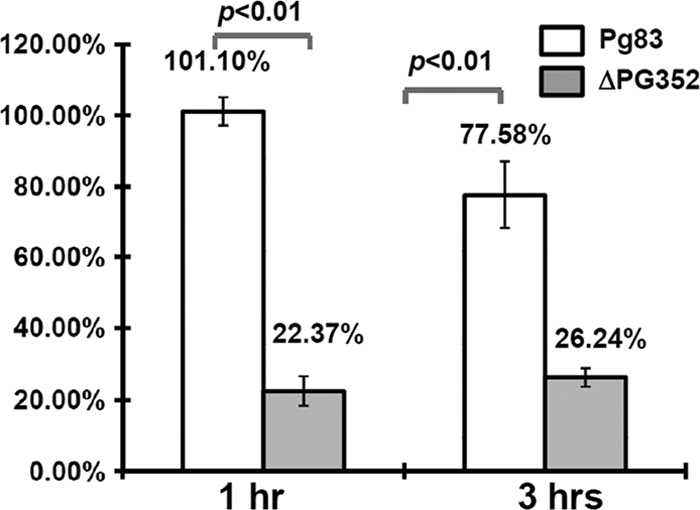
Survival rates of Pg83 and the ΔPG352 mutant. Two strains were coincubated with 25% fresh serum or heat-inactivated serum for 1 and 3 h. The survival rates were calculated as follows: the total numbers of colonies present in the samples treated with 25% serum divided by the total numbers of colonies in the samples treated with heat-inactivated serum. The results are expressed as the average survival rates of triplicates. The data were statistically analyzed by Student t test at P < 0.01.
The ΔPG352 mutant has decreased virulence in vivo.
A previously described mouse abscess model (18, 34) was used to further evaluate the role of SiaPg in the virulence of P. gingivalis. In the present study, Pg83, ΔPG352, Pg381, and a sham control (PBS) were included. Each mouse was subcutaneously injected with ∼1010 cells of P. gingivalis or an equal volume of PBS at the dorsal sites. As shown in Table 2, for the Pg83 strain, no localized abscesses were observed at the injection sites. Instead, all three mice developed secondary lesions (average size of secondary lesions was 18.7 mm2, ranging from 10 to 24 mm2) on their ventral sides. All three mice died within 6 days after the injections. In contrast to Pg83, Pg381 only induced localized abscesses at the injection sites (the average size of abscesses was 147 mm2, ranging from 60 to 260 mm2), but no secondary lesions were observed. All animals survived. Compared to these two strain, for the ΔPG352 mutant, two mice developed localized abscess at the injection sites (44.2 and 28.3 mm2), one mouse had a tiny secondary lesion (4 mm2), and all three mice survived. These results showed that the virulence of ΔPG352 was substantially reduced.
Table 2.
Virulence of Pg83 and ΔPG352 mutant in micea
| Treatment | Death | Virulence (size in mm2) |
|
|---|---|---|---|
| Localized abscess | Secondary lesion | ||
| Pg83 | 3/3 | 0/3 | 3/3 (10.0, 22.5, 24.5) |
| ΔPG352 | 0/3 | 2/3 (44.16, 28.26) | 1/3 (4.0) |
| Pg381 | 0/3 | 3/3 (263.5, 120, 60) | 0/3 |
| PBS | 0/3 | 0/3 | 0/3 |
Death in indicated as the number of animals that died/number of animals tested. Virulence is indicated as the number of animals with abscesses or lesions/the number of animals tested. Where applicable, the individual abscess or lesion size(s) is indicated in parentheses.
Histological studies confirmed that Pg83 was able to cause a spreading type of infection, which had been previously reported (30, 34). Inflammation could be observed in multiple organs, including the lungs, liver, spleen, and kidneys. The inflammation was more prominent in the lung and liver tissues of infected animals (e.g., severe intrasinusoidal inflammatory infiltrates were observed in the lung and liver tissues [Fig. 11]). In contrast, the ΔPG352 mutant failed to cause the spreading type of infection, and no obvious infiltrates were observed in the lung and liver tissues (Fig. 11). Taken together, these results indicate that the virulence of the ΔPG352 mutant is substantially reduced compared to that of the wild type, highlighting that SiaPg is essential for the pathogenicity of P. gingivalis.
Fig 11.

Hematoxylin and eosin staining of lung and liver tissues from mice infected with Pg83 and the ΔPG352 mutant. (A and C) Lung and liver tissues from a mouse infected with Pg83. (B and D) Lung and liver tissues from a mouse infected with the ΔPG352 mutant. In Pg83-infected mouse, the lung and liver both show significant inflammation (arrows indicate granulocytes), while in the mutant-infected mouse, there is minimal to no inflammation.
DISCUSSION
Is SiaPg the sole neuraminidase of P. gingivalis?
The evidence presented in this report (Fig. 1, 2, and 3) clearly demonstrates that SiaPg is a neuraminidase that is similar to its counterparts from other bacteria; inactivation of PG0352 completely abolished the neuraminidase activity (Fig. 5), suggesting that SiaPg may be the sole neuraminidase of P. gingivalis. However, Aruni et al. recently reported that in addition to PG0352, P. gingivalis has another two sialidases (PG0778 and PG1724), and the inactivation of PG0352 reduces total sialidase activity by only 5% (3). This discrepancy is most likely due to different detection methods. In the previous report, an Amplex Red neuraminidase assay kit was used to measure general sialidase and sialoglycoprotease activity; here, we used the filter paper spot assay—a specific method for testing neuraminidase activity (43, 44). PG0778 is annotated as a hypothetical protein and PG1724 as a putative DNA-binding/iron metalloprotein/AP endonuclease or O-sialoglycoprotein endopeptidase (45). BLAST analysis reveals that these two proteins do not belong to the neuraminidase Pfam (12). In addition, both PG0778 and PG1724 lack the F/YRIP motif. Crystal structural and site-directed mutagenesis analyses have shown that this motif is essential for the enzymatic activity of neuraminidases (10, 13). In this regard, it seems unlikely that PG0778 and PG1724 function as a neuraminidase. Instead, they may function as other enzymes, e.g., acting as a sialoglycoprotease, and their enzymatic activities can be defined by biochemical, genetic, and structural studies.
Does P. gingivalis utilize Neu5Ac as a nutrient?
Several bacteria use Neu5Ac as an alternative nutrient (53, 74). In this case, the mutants, which fail to synthesize or use Neu5Ac, have growth defects when the sialic acid serves as a sole carbon source. Several lines of evidence suggest that P. gingivalis probably does not use Neu5Ac as a nutrient. First, P. gingivalis, as an asaccharolytic bacterium, primarily utilizes short peptides instead of carbohydrates for energy production (4). Second, in the catabolic pathway of sialic acid, NanA first cleaves Neu5Ac to ManNAc and pyruvate. ManNAc is ultimately converted to fructose-6-phosphate and ammonia via a series of reactions catalyzed by NanK, NanE, NagB, and NagA (16, 53, 74). However, the genes encoding these enzymes have not yet been annotated in the genome of P. gingivalis (45), suggesting that the catabolic pathway of Neu5Ac most likely does not exist in P. gingivalis. Third, supplementing sialic acid did not stimulate the growth of P. gingivalis (Fig. 6A). Finally, the inactivation of PG0352 did not influence the growth of P. gingivalis (Fig. 6B).
How does SiaPg influence the biofilm formation of P. gingivalis?
The impact of neuraminidases on biofilm formation has been described in several pathogenic bacteria (47, 51, 59, 64), even though the mechanism involved remains obscure. In S. pneumoniae, the expression of nanA (encoding a neuraminidase) is upregulated when it grows under biofilm conditions and NanA inhibitors block its biofilm formation. In P. aeruginosa, the nanA deletion mutant has diminished biofilm production, and the mutant is unable to establish respiratory infection. Here, we also found that the biofilm formation was substantially reduced in the ΔPG352 mutant (Fig. 7). Interestingly, the ΔPG352 mutant restored the biofilm formation in the addition of exogenous sialic acid (Fig. 7), e.g., at the concentration of 30 μg of Neu5Ac/ml, the mutant almost produced the same amount of biofilms as Pg83. At the same condition, the ΔPG352 mutant also restored capsule synthesis (Fig. 8F). Thus, it is possible that the regaining biofilm production could be due to the restoration of capsule in the mutant. However, a recent report from Davey et al. does not support this assumption, since they found that the loss of capsule enhances the biofilm formation of P. gingivalis (15). Since sialic acids are often used to modify bacterial cell surface molecules (e.g., LPS and capsule), it is also possible that the presence of sialic acids may change the cell surface hydrophobicity and consequently influence the biofilm formation of P. gingivalis. Fimbria is required for the biofilm formation of P. gingivalis, and its expression level can be influenced by different environmental cues (80, 81). In E. coli, sialic acids often act as a signaling molecule to modulate the production of fimbriae (for recent review, see reference 74). Thus, a similar scenario can also exist in P. gingivalis, i.e., exogenous sialic acids may function as an environmental cue to regulate the fimbrial gene expression, which further influences the biofilm production of P. gingivalis. At this point, we are unable to rule out which one of these possibilities is accountable for the observed phenotype; however, the evidence described here highlights the role of neuraminidases in the biofilm formation of P. gingivalis, and it provides us a solid foundation to further unveil the potential mechanism involved.
How does SiaPg influence the capsule synthesis of P. gingivalis?
The majority of P. gingivalis isolates are encapsulated. Based on the serotypes of K antigens, P. gingivalis isolates can be divided into at least six groups (K1 to K6) (7, 34), highlighting the complexity of carbohydrates presented in P. gingivalis capsular polysaccharides. Little is known about the structure and composition of carbohydrates in the capsule of P. gingivalis. It is possible that P. gingivalis (at least Pg83 strain) utilizes sialic acid either to directly synthesize the PSA-type capsule or to modify sugar molecules on other types of capsules. P. gingivalis lacks the de novo synthesis pathway of sialic acid. For example, NeuB (Neu5Ac synthase) (53, 74), a key enzyme that converts UDP-GlcNAc to Neu5Ac, is absent in the genome of Pg83 (45). As such, the scavenge pathway mediated by Siapg might be the only route for P. gingivalis to acquire sialic acid. If this is the case, we expect that the deletion of SiaPg would completely block the scavenge route and the mutant would fail to produce the capsule due to the lack of sialic acid. This proposition is consistent with the phenotype of the ΔPG352 mutant (Fig. 8 and 9).
The E. coli K1 and K92 capsules use polysialic acid (PSA) as the nucleotide sugars (17, 78, 79). In the biosynthesis pathway of PSA capsule, NeuA (CMP-Neu5Ac synthetase) and NeuS are two signature enzymes. NeuA converts Neu5Ac to its active form CMP-Neu5Ac (31, 42), which is then added to appropriate acceptors by NeuS, a linkage-specific sialyl-transferase (60, 61). PSA is exported through the Kps system (78). However, the homologs of NeuA and NeuS are not annotated in the genome of Pg83 (45), suggesting that P. gingivalis may have a unique system to utilize sialic acids to either directly synthesize PSA-type capsule or modify its capsule via sialylation. Two recent studies reveal that capsule biosynthesis in P. gingivalis is quite unique (2, 15). The findings in the present study could provide a new angle for studying the capsule biosynthesis of P. gingivalis. In addition, sialic acids can serve as signaling molecules (74). Thus, it is also possible that the lack of sialic acids in ΔPG352 may have an impact on other factors that are required for the capsule biosynthesis and/or assembly.
Why is ΔPG352 more sensitive to the serum killing?
P. gingivalis is resistant to complement killing (63). Compared to the wild type, the ΔPG352 mutant became more vulnerable to serum killing (Fig. 9). Previous studies have suggested that the possible mechanisms for serum killing resistance of P. gingivalis include protease production and capsule formation (22, 57, 62). P. gingivalis is able to degrade the complement factors C3 and C5, and the degradation is dependent on the activity of gingipains. A recent report describes that the P. gingivalis sialidase/sialoglycoprotease activity may be involved in regulating gingipain activity (3). Thus, the loss of serum resistance in the ΔPG352 mutant could be due to the impact on the activity of gingipains. However, Slaney et al. report that gingipains are not required for the serum resistance, since the gingipain-deficient mutants are still as resistant as the wild type to serum killing. Instead, they found that the capsule plays an essential role in complement resistance. Since the ΔPG352 mutant has defected capsule (Fig. 8), it seems more likely that the vulnerability to serum killing in the mutant is ascribed to the defected capsule. In addition, previous studies from Neisseria show that sialyated surface molecules bind to factor H and enhance the complement resistance (76). Thus, it is also possible that the loss of SiaPg may influence the sialylation of other surface molecules and consequently interfere with the serum resistance. In future studies, we will attempt to pinpoint which mechanism(s) is responsible for the observed phenotype.
Role of SiaPg in the pathogenicity of P. gingivalis.
As a member of the “red-complex” bacteria, P. gingivalis primarily inhabits the gingival crevices (27, 37). The major component of crevicular fluid is plasma (41). The total sialic acid in the plasma is quite abundant (∼2 mM). Under normal physiological conditions, almost all sialic acid (99.9%) is bound to a diverse range of proteins or lipids (55, 74). In addition to the plasma, salivary mucins and fibronectins are also rich in sialic acids (19). In this unique niche, a microorganism armed with a neuraminidase will be able to acquire sialic acid from the host via the scavenge pathway and strategically position itself to compete with other organisms or to overcome host immune attacks by camouflaging their surface molecules (e.g., LPS and capsules) with sialic acids. The studies described here reveal an important role that SiaPg plays in the pathogenicity of P. gingivalis, e.g., influencing the capsule biosynthesis.
The capsule is highly associated with the virulence of P. gingivalis (34). Animal studies have demonstrated that the noncapsular (K−) P. gingivalis strains are much less virulent than the encapsulated isolates. Gonzales et al. reported that immunization with purified K1 capsule prevents mice from P. gingivalis-elicited oral bone loss (21). Slaney et al. recently revealed that the capsule plays a key role in the resistance to serum killing (57). The studies reported here show that the ΔPG352 mutant has a defective capsule. Compared to the wild-type strain, the mutant is sensitive to complement killing and much less virulent in the mouse model (Fig. 10 and 11 and Table 2). The observed phenotype is similar to that of the noncapsular P. gingivalis isolates or mutants, suggesting that SiaPg most likely contributes to the pathogenicity of P. gingivalis by influencing capsule formation. Besides being used in capsule formation, sialic acids can also be utilized to modify LPS and other virulence factors, which have been observed in Haemophilus influenzae, N. meningitidis, and several other pathogenic bacteria (5, 53, 74, 75). Thus, the observed in vivo phenotype could also be ascribed to the change of other virulence factors in the ΔPG352 mutant. The existence of this possibility cannot be ruled out until a genome-wide glycomic analysis is conducted. Nevertheless, the studies described here provide us a solid starting point to further explore the role of sialylation in the biology and pathogenicity of P. gingivalis.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Sojar, K. Homma, and A. Sharma for providing P. gingivalis strains and antibodies and E. Haase for providing pRS415 plasmid.
This study was supported by NIH/NIDCR and NIH/NIAID grants DE019667 (Chunhao Li), AI078958 (Chunhao Li), and AI087946 (J.L.), along with grant from the Welch Foundation (AU-1714 to J.L.). Maintenance of the Polara electron microscope facility is partially supported by the Structural Biology Center at the UT Houston Medical School.
Footnotes
Published ahead of print 24 October 2011
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Achyuthan KE, Achyuthan AM. 2001. Comparative enzymology, biochemistry, and pathophysiology of human exo-α-sialidases (neuraminidases). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 129: 29–64 [DOI] [PubMed] [Google Scholar]
- 2. Aduse-Opoku J, et al. 2006. Identification and characterization of the capsular polysaccharide (K-antigen) locus of Porphyromonas gingivalis. Infect. Immun. 74: 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aruni W, et al. 2011. Sialidase and sialoglycoproteases can modulate virulence in Porphyromonas gingivalis. Infect. Immun. 79: 2779–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boone D, Castenholtz RW. (ed). 2001. Bergey's manual of systematic bacteriology, 2nd ed, vol 1 Springer-Verlag, New York, NY [Google Scholar]
- 5. Bouchet V, et al. 2003. Host-derived sialic acid is incorporated into Haemophilus influenzae lipopolysaccharide and is a major virulence factor in experimental otitis media. Proc. Natl. Acad. Sci. U. S. A. 100: 8898–8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brien-Simpson NM, Veith PD, Dashper SG, Reynolds EC. 2003. Porphyromonas gingivalis gingipains: the molecular teeth of a microbial vampire. Curr. Protein Pept. Sci. 4: 409–426 [DOI] [PubMed] [Google Scholar]
- 7. Brunner J, Crielaard W, Van Winkelhoff AJ. 2008. Analysis of the capsular polysaccharide biosynthesis locus of Porphyromonas gingivalis and development of a K1-specific polymerase chain reaction-based serotyping assay. J. Periodontal Res. 43: 698–705 [DOI] [PubMed] [Google Scholar]
- 8. Cacalano G, Kays M, Saiman L, Prince A. 1992. Production of the Pseudomonas aeruginosa neuraminidase is increased under hyperosmolar conditions and is regulated by genes involved in alginate expression. J. Clin. Invest. 89: 1866–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W, Honma K, Sharma A, Kuramitsu HK. 2006. A universal stress protein of Porphyromonas gingivalis is involved in stress responses and biofilm formation. FEMS Microbiol. Lett. 264: 15–21 [DOI] [PubMed] [Google Scholar]
- 10. Chien CH, Shann YJ, Sheu SY. 1996. Site-directed mutations of the catalytic and conserved amino acids of the neuraminidase gene, nanH, of Clostridium perfringens ATCC 10543. Enzyme Microb. Technol. 19: 267–276 [DOI] [PubMed] [Google Scholar]
- 11. Comstock LE, Kasper DL. 2006. Bacterial glycans: key mediators of diverse host immune responses. Cell 126: 847–850 [DOI] [PubMed] [Google Scholar]
- 12. Copley RR, Russell RB, Ponting CP. 2001. Sialidase-like Asp-boxes: sequence-similar structures within different protein folds. Protein Sci. 10: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crennell SJ, Garman EF, Laver WG, Vimr ER, Taylor GL. 1993. Crystal structure of a bacterial sialidase (from Salmonella typhimurium LT2) shows the same fold as an influenza virus neuraminidase. Proc. Natl. Acad. Sci. U. S. A. 90: 9852–9856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crowley JF, Goldstein IJ, Arnarp J, Lonngren J. 1984. Carbohydrate binding studies on the lectin from Datura stramonium seeds. Arch. Biochem. Biophys. 231: 524–533 [DOI] [PubMed] [Google Scholar]
- 15. Davey ME, Duncan MJ. 2006. Enhanced biofilm formation and loss of capsule synthesis: deletion of a putative glycosyltransferase in Porphyromonas gingivalis. J. Bacteriol. 188: 5510–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Du J, et al. 2009. Metabolic glycoengineering: sialic acid and beyond. Glycobiology 19: 1382–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferrero MA, Aparicio LR. 2010. Biosynthesis and production of polysialic acids in bacteria. Appl. Microbiol. Biotechnol. 86: 1621–1635 [DOI] [PubMed] [Google Scholar]
- 18. Fletcher HM, et al. 1995. Virulence of a Porphyromonas gingivalis W83 mutant defective in the prtH gene. Infect. Immun. 63: 1521–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gabriel MO, Grunheid T, Zentner A. 2005. Glycosylation pattern and cell attachment-inhibiting property of human salivary mucins. J. Periodontol. 76: 1175–1181 [DOI] [PubMed] [Google Scholar]
- 20. Galen JE, et al. 1992. Role of Vibrio cholerae neuraminidase in the function of cholera toxin. Infect. Immun. 60: 406–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez D, Tzianabos AO, Genco CA, Gibson FC., III 2003. Immunization with Porphyromonas gingivalis capsular polysaccharide prevents P. gingivalis-elicited oral bone loss in a murine model. Infect. Immun. 71: 2283–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grenier D, et al. 2003. Effect of inactivation of the Arg- and/or Lys-gingipain gene on selected virulence and physiological properties of Porphyromonas gingivalis. Infect. Immun. 71: 4742–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gut H, King SJ, Walsh MA. 2008. Structural and functional studies of Streptococcus pneumoniae neuraminidase B: an intramolecular trans-sialidase. FEBS Lett. 582: 3348–3352 [DOI] [PubMed] [Google Scholar]
- 24. Haase EM, Bonstein T, Palmer RJ, Jr, Scannapieco FA. 2006. Environmental influences on Actinobacillus actinomycetemcomitans biofilm formation. Arch. Oral Biol. 51: 299–314 [DOI] [PubMed] [Google Scholar]
- 25. Haase EM, Stream JO, Scannapieco FA. 2003. Transcriptional analysis of the 5′ terminus of the flp fimbrial gene cluster from Actinobacillus actinomycetemcomitans. Microbiology 149: 205–215 [DOI] [PubMed] [Google Scholar]
- 26. Hamada N, Sojar HT, Cho MI, Genco RJ. 1996. Isolation and characterization of a minor fimbria from Porphyromonas gingivalis. Infect. Immun. 64: 4788–4794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holt SC, Ebersole JL. 2005. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol. 2000 38: 72–122 [DOI] [PubMed] [Google Scholar]
- 28. Honma K, Mishima E, Sharma A. 2011. Role of Tannerella forsythia NanH sialidase in epithelial cell attachment. Infect. Immun. 79: 393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsiao YS, Parker D, Ratner AJ, Prince A, Tong L. 2009. Crystal structures of respiratory pathogen neuraminidases. Biochem. Biophys. Res. Commun. 380: 467–47119284989 [Google Scholar]
- 30. Kesavalu L, Holt SC, Ebersole JL. 1997. Porphyromonas gingivalis virulence in a murine lesion model: effects of immune alterations. Microb. Pathog. 23: 317–326 [DOI] [PubMed] [Google Scholar]
- 31. Krapp S, et al. 2003. The crystal structure of murine CMP-5-N-acetylneuraminic acid synthetase. J. Mol. Biol. 334: 625–637 [DOI] [PubMed] [Google Scholar]
- 32. Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116: 71–76 [DOI] [PubMed] [Google Scholar]
- 33. Kruse S, Kleineidam RG, Roggentin P, Schauer R. 1996. Expression and purification of a recombinant “small” sialidase from Clostridium perfringens A99. Protein Expr. Purif. 7: 415–422 [DOI] [PubMed] [Google Scholar]
- 34. Laine ML, Van Winkelhoff AJ. 1998. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol. Immunol. 13: 322–325 [DOI] [PubMed] [Google Scholar]
- 35. Lamont RJ, et al. 1995. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 63: 3878–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lamont RJ, Jenkinson HF. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62: 1244–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lamont RJ, Jenkinson HF. 2000. Subgingival colonization by Porphyromonas gingivalis. Oral Microbiol. Immunol. 15: 341–349 [DOI] [PubMed] [Google Scholar]
- 38. Leprat R, Michel-Briand Y. 1980. Extracellular neuraminidase production by a strain of Pseudomonas aeruginosa isolated from cystic fibrosis. Ann. Microbiol. (Paris) 131B: 209–222 [PubMed] [Google Scholar]
- 39. Limberger RJ, Slivienski LL, Izard J, Samsonoff WA. 1999. Insertional inactivation of Treponema denticola tap1 results in a nonmotile mutant with elongated flagellar hooks. J. Bacteriol. 181: 3743–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu J, et al. 2009. Intact flagellar motor of Borrelia burgdorferi revealed by cryo-electron tomography: evidence for stator ring curvature and rotor/C-ring assembly flexion. J. Bacteriol. 191: 5026–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lundholm M, et al. 2010. Variation in the Cd3 zeta (Cd247) gene correlates with altered T cell activation and is associated with autoimmune diabetes. J. Immunol. 184: 5537–5544 [DOI] [PubMed] [Google Scholar]
- 42. Mizanur RM, Pohl NL. 2008. Bacterial CMP-sialic acid synthetases: production, properties, and applications. Appl. Microbiol. Biotechnol. 80: 757–765 [DOI] [PubMed] [Google Scholar]
- 43. Moncla BJ, Braham P. 1989. Detection of sialidase (neuraminidase) activity in Actinomyces species by using 2′-(4-methylumbelliferyl)α-d-N-acetylneuraminic acid in a filter paper spot test. J. Clin. Microbiol. 27: 182–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moncla BJ, Braham P, Hillier SL. 1990. Sialidase (neuraminidase) activity among gram-negative anaerobic and capnophilic bacteria. J. Clin. Microbiol. 28: 422–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nelson KE, et al. 2003. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J. Bacteriol. 185: 5591–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olczak T, Wojtowicz H, Ciuraszkiewicz J, Olczak M. 2010. Species specificity, surface exposure, protein expression, immunogenicity, and participation in biofilm formation of Porphyromonas gingivalis HmuY. BMC Microbiol. 10: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parker D, et al. 2009. The NanA neuraminidase of Streptococcus pneumoniae is involved in biofilm formation. Infect. Immun. 77: 3722–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pihlstrom BL, Michalowicz BS, Johnson NW. 2005. Periodontal diseases. Lancet 366: 1809–1820 [DOI] [PubMed] [Google Scholar]
- 49. Powell LD, Varki AP. Sialidases. Curr. Protoc. Mol. Biol. 2001 doi: 10.1002/0471142727.mb1712s27. Chapter 17:Unit 17. [DOI] [PubMed] [Google Scholar]
- 50. Roggentin P, Schauer R, Hoyer LL, Vimr ER. 1993. The sialidase superfamily and its spread by horizontal gene transfer. Mol. Microbiol. 9: 915–921 [DOI] [PubMed] [Google Scholar]
- 51. Roy S, Douglas CW, Stafford GP. 2010. A novel sialic acid utilization and uptake system in the periodontal pathogen Tannerella forsythia. J. Bacteriol. 192: 2285–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schauer R. 2009. Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 19: 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Severi E, Hood DW, Thomas GH. 2007. Sialic acid utilization by bacterial pathogens. Microbiology 153: 2817–2822 [DOI] [PubMed] [Google Scholar]
- 54. Sheets SM, Robles-Price AG, McKenzie RM, Casiano CA, Fletcher HM. 2008. Gingipain-dependent interactions with the host are important for survival of Porphyromonas gingivalis. Front. Biosci. 13: 3215–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sillanaukee P, Ponnio M, Jaaskelainen IP. 1999. Occurrence of sialic acids in healthy humans and different disorders. Eur. J. Clin. Invest. 29: 413–425 [DOI] [PubMed] [Google Scholar]
- 56. Simons RW, Houman F, Kleckner N. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53: 85–96 [DOI] [PubMed] [Google Scholar]
- 57. Slaney JM, Gallagher A, Aduse-Opoku J, Pell K, Curtis MA. 2006. Mechanisms of resistance of Porphyromonas gingivalis to killing by serum complement. Infect. Immun. 74: 5352–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Socransky SS, Haffajee AD. 2005. Periodontal microbial ecology. Periodontol. 2000 38: 135–187 [DOI] [PubMed] [Google Scholar]
- 59. Soong G, et al. 2006. Bacterial neuraminidase facilitates mucosal infection by participating in biofilm production. J. Clin. Invest. 116: 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Steenbergen SM, Vimr ER. 2003. Functional relationships of the sialyltransferases involved in expression of the polysialic acid capsules of Escherichia coli K1 and K92 and Neisseria meningitidis groups B or C. J. Biol. Chem. 278: 15349–15359 [DOI] [PubMed] [Google Scholar]
- 61. Steenbergen SM, Wrona TJ, Vimr ER. 1992. Functional analysis of the sialyltransferase complexes in Escherichia coli K1 and K92. J. Bacteriol. 174: 1099–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sundqvist G, Carlsson J, Herrmann B, Tarnvik A. 1985. Degradation of human immunoglobulins G and M and complement factors C3 and C5 by black-pigmented Bacteroides. J. Med. Microbiol. 19: 85–94 [DOI] [PubMed] [Google Scholar]
- 63. Sundqvist G, Johansson E. 1982. Bactericidal effect of pooled human serum on Bacteroides melaninogenicus, Bacteroides asaccharolyticus, and Actinobacillus actinomycetemcomitans. Scand. J. Dent. Res. 90: 29–36 [DOI] [PubMed] [Google Scholar]
- 64. Swords WE, et al. 2004. Sialylation of lipooligosaccharides promotes biofilm formation by nontypeable Haemophilus influenzae. Infect. Immun. 72: 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sze CW, Li C. 2011. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect. Immun. 79: 1270–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thompson H, Homer KA, Rao S, Booth V, Hosie AH. 2009. An orthologue of Bacteroides fragilis NanH is the principal sialidase in Tannerella forsythia. J. Bacteriol. 191: 3623–3628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Traving C, Schauer R. 1998. Structure, function, and metabolism of sialic acids. Cell Mol. Life Sci. 54: 1330–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Typas A, Stella S, Johnson RC, Hengge R. 2007. The −35 sequence location and the Fis-sigma factor interface determine σs selectivity of the proP (P2) promoter in Escherichia coli. Mol. Microbiol. 63: 780–796 [DOI] [PubMed] [Google Scholar]
- 69. Uchiyama S, et al. 2009. The surface-anchored NanA protein promotes pneumococcal brain endothelial cell invasion. J. Exp. Med. 206: 1845–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Varki A. 2007. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 446: 1023–1029 [DOI] [PubMed] [Google Scholar]
- 71. Varki NM, Varki A. 2007. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab. Invest. 87: 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vimr E, Lichtensteiger C. 2002. To sialylate, or not to sialylate: that is the question. Trends Microbiol. 10: 254–257 [DOI] [PubMed] [Google Scholar]
- 73. Vimr ER. 1994. Microbial sialidases: does bigger always mean better? Trends Microbiol. 2: 271–277 [DOI] [PubMed] [Google Scholar]
- 74. Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. 2004. Diversity of microbial sialic acid metabolism. Microbiol. Mol. Biol. Rev. 68: 132–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vogel U, Claus H, Heinze G, Frosch M. 1999. Role of lipopolysaccharide sialylation in serum resistance of serogroup B and C meningococcal disease isolates. Infect. Immun. 67: 954–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vogel U, Frosch M. 1999. Mechanisms of neisserial serum resistance. Mol. Microbiol. 32: 1133–1139 [DOI] [PubMed] [Google Scholar]
- 77. Wang BY, Kuramitsu HK. 2005. Interactions between oral bacteria: inhibition of Streptococcus mutans bacteriocin production by Streptococcus gordonii. Appl. Environ. Microbiol. 71: 354–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Whitfield C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75: 39–68 [DOI] [PubMed] [Google Scholar]
- 79. Whitfield C, Roberts IS. 1999. Structure, assembly, and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 31: 1307–1319 [DOI] [PubMed] [Google Scholar]
- 80. Xie H, Cai S, Lamont RJ. 1997. Environmental regulation of fimbrial gene expression in Porphyromonas gingivalis. Infect. Immun. 65: 2265–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xie H, Chung WO, Park Y, Lamont RJ. 2000. Regulation of the Porphyromonas gingivalis fimA (fimbrillin) gene. Infect. Immun. 68: 6574–6579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xu G, et al. 2008. Crystal structure of the NanB sialidase from Streptococcus pneumoniae. J. Mol. Biol. 384: 436–449 [DOI] [PubMed] [Google Scholar]
- 83. Xu H, Raddi G, Liu J, Charon NW, Li C. 2011. Chemoreceptors and flagellar motors are subterminally located in close proximity at the two cell poles in spirochetes. J. Bacteriol. 193: 2652–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yesilkaya H, Soma-Haddrick S, Crennell SJ, Andrew PW. 2006. Identification of amino acids essential for catalytic activity of pneumococcal neuraminidase A. Res. Microbiol. 157: 569–574 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.