

Abstract
Toxoplasma gondii is a ubiquitous, obligate intracellular parasite capable of crossing the placenta to cause spontaneous abortion, preterm labor, or significant disease in the surviving neonate. Exploration of the cellular and histological components of the placental barrier is in its infancy, and both how and where T. gondii breaches it are unknown. The human placenta presents two anatomical interfaces between maternal cells and fetal cells (trophoblasts): (i) the villous region where maternal blood bathes syncytialized trophoblasts for nutrient exchange and (ii) the maternal decidua, where mononuclear, extravillous trophoblasts anchor the villous region to the uterus. Using first-trimester human placental explants, we demonstrate that the latter site is significantly more vulnerable to infection, despite presenting a vastly smaller surface. This is consistent with past findings concerning two vertically transmitted viruses and one bacterium. We further explore whether three genetically distinct T. gondii types (I, II, and III) are capable of preferential placental infection and survival in this model. We find no difference in these strains' ability to infect placental explants; however, slightly slower growth is evident in type II (Prugniaud [Pru]) parasites relative to other cell types, although this did not quite achieve statistical significance.
INTRODUCTION
Toxoplasma gondii is a ubiquitous parasite of human and veterinary medical importance. This obligate, intracellular protozoan undergoes the sexual portion of its life cycle in cats, the sole definitive host; it also has an asexual life cycle in a broad range of warm-blooded intermediate hosts, most significantly rodents, livestock, and humans. The main modes of transmission are therefore ingestion of oocysts shed in cat feces or of tissue cysts in undercooked meat from intermediate hosts. T. gondii is capable of crossing the blood-brain, blood-ocular, and maternal-fetal barriers. Thus, the vertical route from mother to fetus represents an additional mode of transmission. Fetal infection can cause spontaneous abortion, preterm labor, or significant neurological and ocular sequelae in the surviving offspring.
Primary maternal infection during pregnancy is responsible for almost all fetal infection and its frequency depends on human seroprevalence rates that range from ∼10 to 80% across nations (53). Prenatal testing for toxoplasmosis is routinely offered in many European countries, and drug therapy with spiramycin is prescribed after the diagnosis of maternal infection in order to prevent mother-to-child transmission (19). However, whether prenatal treatment has any effect on vertical transmission is unclear from the results of a systematic review of cohort studies because of numerous biases in the way these studies were designed (65). Randomized controlled clinical trials are needed to obtain evidence for the benefit of prenatal treatment.
The mechanisms by which T. gondii infects the placenta and crosses from mother to fetus are poorly understood and experimentally complicated by cross-species anatomical diversity of the placenta. The placenta is a chimeric organ that is categorized by the relationship between fetally derived trophoblasts and the maternal cells/tissues with which they come into contact. In humans and other mammals with hemochorial placentas, the maternal-fetal interface consists of (i) the syncytiotrophoblast that is bathed in maternal blood and mediates nutrient and gas exchange and (ii) the extravillous trophoblasts (EVT) that anchor the placenta in the uterine implantation site (decidua) where they are juxtaposed to maternal immune cells (Fig. 1A). The largest surface by far is the interface between the maternal blood and syncytiotrophoblast, so it is attractive to speculate that T. gondii invades the placenta directly from maternal blood. Alternatively, parasites may invade at the decidua-EVT interface, which occupies less than 5% of the maternal-fetal interface (Fig. 1B) (10).
Fig 1.

Comparison of in vivo placental structure to placenta explant model. (A) Structure and orientation of fetus and placenta in uterus at ∼6 weeks of gestation. Fetal structures are represented in shades of blue and purple while maternal structures are in shades of red. Maternal structures: MY, myometrium; SA, spiral arteries; DD, decidua (uterine lining during pregnancy); IVS, intervillous space filled with maternal blood. Fetal structures: VT, villous tree; CP, chorionic plate; UC, umbilical cord; AF, amniotic fluid. (B) Close-up view of the maternal-fetal interface as indicated by the inset in panel A. Maternal blood surrounds the villous tree composed of anchoring villi (AV) and floating villi (FV), which are covered by a syncytiotrophoblast (SYN) that is underlaid by subsyncytial cytotrophoblasts (sCTB) and a basement membrane. The sCTB layer grows increasingly discontinuous in later trimesters. Gas and nutrient exchange with the maternal blood occurs across the syncytiotrophoblast to supply fetal capillaries (not shown) in the villous stroma (STR). At the uterine wall, extravillous cytotrophoblasts (EVT) anchor the villous tree in the decidua. Some invade the decidua and move away from the tip to remodel maternal spiral arteries (not shown), with altered gene expression patterns as they move. (C) Six-week placental explant anchored in Matrigel. Bar, 1 mm. (D) Cartoon representation of the relevant structures shown in panel C. B. Mem, basement membrane.
Almost all experimental T. gondii infections of the placenta have been performed in mice (6, 24, 34, 54, 63), sheep (16), or isolated trophoblast cells (4, 7, 34, 52, 55, 56). However, none of these models accurately represent the architecture of the human placenta (reviewed in references 43 and 46). Identifying the major site of T. gondii placental invasion will improve our understanding of the barrier structure of the placenta, as well as how T. gondii disseminates through the body.
A second important question is whether some T. gondii strains are particularly predisposed to congenital infection. T. gondii strains exhibit a highly clonal population structure in which Europe and North America share three dominant serotypes with, at least at the genotype level, low intratype diversity (36). European and North African epidemiological studies have identified either type II (2, 3, 35, 36, 51) or type I or type I recombinants (13, 28) as overrepresented in congenital infection. Meanwhile, a Colombian study reports type I-like strains (29) and in Brazil, Brazilian genotype 65 is more commonly transmitted across the placenta (22). But such studies are complicated by multiple factors: bias from the regionally prevalent strain or experimental amplification in animals, sample bias against asymptomatic infections, and, in some cases, small data pools. Thus, it is unknown whether certain types are more capable of transplacental transmission.
Using first-trimester human placental explants, we have recently shown that the syncytiotrophoblast acts as a barrier for the food-borne bacterial pathogen Listeria monocytogenes (59), which preferentially colonizes the EVT instead. Two additional barriers are found in the modified vacuolar maturation of the EVT (68) and in the basement membrane that separates trophoblasts from the villous stroma that contains the fetal capillaries (59). Here we infected first-trimester human placental explants with all three well-studied T. gondii strains, both to assess strain-specific placental infection capabilities and the sites where this parasite breaches the maternal-fetal barrier. We found that, like other pathogens, T. gondii preferentially colonizes EVT over the relatively resistant syncytiotrophoblast. This is consistent with and helps to clarify pathological data from experimental animal infections (16, 24, 63). We did not detect a difference in placental infection ability across the three T. gondii strains; however, a lower rate of replication within trophoblasts was evident in the type II strain relative to growth in fibroblasts. This was not found in types I and III.
MATERIALS AND METHODS
Ethics statement.
This study was conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by the Institutional Review Board at the University of California, San Francisco, where all experiments were performed (approval no. H497-00836-28). All patients provided written informed consent for the collection of samples and subsequent analysis.
Human tissue collection and culture.
Placentas from elective terminations of pregnancy (gestational age, 4 to 8 weeks) were collected and prepared as previously described (30). Briefly, fragments from the surface of the placenta were dissected into 1- to 3-mm tree-like villi, placed on Matrigel-coated (BD Biosciences, San Jose, CA) Transwell filters (30-mm diameter, 0.4-mm pore size; Millipore, Billerica, MA) and cultured overnight before infection in Dulbecco's modified Eagle's medium-F12 medium (DMEM-F12; 1:1, vol/vol) supplemented with 20% fetal bovine serum (FBS; Fisher Scientific), 1% l-glutamine, and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA).
Removal of syncytiotrophoblast.
Syncytiotrophoblast was removed from villous trees as previously described (26). Briefly, placental explants were soaked for 5 to 15 min in a solution containing type IA collagenase (100,000 U), hyaluronidase (150,000 U), DNase (120,000 U), and 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) without divalent cations (UCSF Cell Culture Facility, San Francisco, CA). Explants were observed via a dissecting microscope, and when syncytiotrophoblast degradation was apparent, they were transferred to Matrigel and cultured overnight before infection.
T. gondii strains and growth conditions.
The T. gondii strains used in this study were green fluorescent protein (GFP)-expressing type I (RH), type II (Prugniaud [Pru]), or type III (CTG) (36, 41, 61). The parasites were maintained by serial passage in primary human foreskin fibroblasts (HFF) as described previously (60, 64) in DMEM (UCSF Cell Culture Facility) supplemented with 10% FBS and 1% penicillin-streptomycin in 25-cm2 T-flasks.
Infection of placental explants.
A confluent flask of HFF was inoculated with freshly harvested tachyzoites and incubated in DMEM with 1% FBS, 1% penicillin-streptomycin for 1 to 3 days. To collect tachyzoites, flasks were scraped and cell suspension passed through 23-gauge needles, then 27-gauge needles, 4 times each. Parasites were passed through a 5-μm filter (Whatman) to eliminate cell debris and were counted, and ∼2.5 × 105 parasites were added directly to explant cultures in 0.25 ml explant media (see above). After 5 h—enough time to allow almost all parasites to infect or die but none to replicate—explants were washed with PBS, and fresh antibiotic-containing medium was replaced until fixation in 4% paraformaldehyde (Ted Pella, Redding, CA) in PBS at 4°C overnight.
Immunofluorescence and histology.
For histological sections, fixed explants were placed into vinyl cryomolds (Ted Pella), then covered with optimal cutting temperature (OCT) media (Ted Pella) and flash-frozen. Histological sectioning (10-μm sections) was performed using a Hacker-Slee cryostat. Glass slides with sections were incubated ∼5 min in acetone at 4°C. All antibody staining was conducted at room temperature.
Sectioned slides and HFF coverslips were soaked 60 min in blocking solution (1% BSA [Sigma] in PBS), then rinsed and exposed to primary antibodies in 0.5% BSA-PBS. Slides were rinsed three times for 5 min each in 0.5% BSA-PBS, and then secondary antibodies were added at the indicated concentrations and incubated for 60 min. After three rinses, coverslips were affixed over Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA). Primary antibodies were polyclonal rabbit anti-T. gondii surface antigen 1 (anti-SAG1) antibody (1:1,000,000; gift of M. Grigg, NIH), monoclonal mouse anti-human human chorionic gonadotropin (HCG) (1:500, clone SPM105; Neomarkers, Fremont, CA), and monoclonal mouse anti-human E-cadherin (1:200, clone NCH-38; Dako). Secondary antibodies were Alexa Fluor 594 goat anti-mouse IgG (1:500) and Alexa Fluor 488 goat anti-rabbit IgG (1:1000) (both manufactured by Invitrogen).
Slides were viewed using an inverted TE2000-E microscope (Nikon, Tokyo, Japan) equipped with a 12-bit cooled charge-coupled-device (CCD) camera (Q Imaging, Surrey, Canada). Images were collected using Simple PCI software (Hamamatsu, Sewickley, PA). Counts of T. gondii localization were made by tallying every infected cell in two sections vertically separated by at least 30 μm at 100× magnification.
Confocal microscopy.
Whole-mount explants were prepared by rinsing fixed explants three times with PBS and suspending them in 1:100 Alexa Fluor 594 phalloidin and 1:100 DAPI (both manufactured by Invitrogen) for 24 h at 4°C. Explants were mounted onto glass slides in Vectashield and sealed under coverslips. Imaging was performed at the Nikon Imaging Center at UCSF using an upright Nikon C1 spectral confocal microscope equipped with 405-, 488-, and 561-nm lasers.
Quantification of invasion efficiency.
Whole-mount explants at 5 h postinfection (p.i.) were fixed overnight in 4% paraformaldehyde as described above. They were then rinsed with PBS and stained for extracellular T. gondii using the immunofluorescence protocol described above (blocking solution; primary antibody, rabbit anti-SAG1; secondary antibody, Alexa Fluor 594 goat anti-rabbit IgG) and mounted in Vectashield with DAPI. Villi were randomly selected for viewing from distal tip down (includes EVT for anchoring villi), with the only prerequisite being that they occupy >85% of the 265- by 230-μm imaging field. Parasites staining red (outside) and green (all) were enumerated; thus, total intracellular parasites are green and red. This allowed us to determine the average intracellular parasite count for three anchoring villi and three floating villi or explant body regions per placenta. This number was divided by the total viable parasites that had been added to the infection.
The number of viable parasites added to each sample was determined by plaque assay in confluent HFF grown in T-25 flasks. A volume equivalent to 250 to 500 parasites as determined by hemocytometer was added to the flask and allowed to grow at 37°C, 5% CO2 in T. gondii culture medium (see above) for 7 to 9 days. Samples were then rinsed once with PBS, then fixed for 1 min using room-temperature methanol (Thermo Fisher Scientific, Pittsburgh, PA). A layer of crystal violet (Sigma-Aldrich) was then added, and the flask was rocked for ∼5 min, then rinsed with deionized water. Plaques were counted at 4× magnification. Each plaque equals one PFU.
For counts of parasite/vacuole (see Fig. 4), extracellular parasites were excluded by SAG1 positivity. For intracellular T. gondii growth, the number of parasites in 50 to 200 vacuoles was enumerated per sample (HFF or placenta). Average number of replications (see Fig. 4C) was calculated by:
where n is the number of parasites per vacuole (1, 2, 4, 8, or 16+), xn is the number of vacuoles with n parasites, and total is the total number of vacuoles. Vacuoles with more than 16 parasites (only found in type I) were included in n = 16, due to difficulty in determining actual numbers in large vacuoles. Further, for type I parasites, some cell lysis had occurred by 48 h, resulting in adjacent cells infected with dozens of synchronized single or double vacuoles. In these instances, each new host cell was scored as a single 1- or 2-parasite-containing vacuole to prevent their overrepresentation. Lysis was not significant in type II or III parasites by 48 h. Thus, this analysis slightly underestimates the growth of type I T. gondii relative to type II and III but not between HFF and placenta.
Fig 4.

Parasite replication in trophoblasts within placental explants relative to replication in HFF. (A) Representative examples of parasitic vacuole sizes after 24-h or 48-h infection in HFF. Green, GFP-expressing type I T. gondii; white, DNA; red (outside), anti-SAG1 (stained parasites), added without permeabilization (none visible here). Bar, 10 μm. (B) Number of parasites per vacuole was scored in placental histological sections (top) or HFF cells (bottom) at 24 and 48 h p.i. Each bar represents the average of three placentas or HFF samples. The P value (between graphs) denotes the result of t tests comparing placenta and HFF percentages of single-parasite vacuoles across the three samples. (C) The log2 transformation of parasites/vacuole yields the average number of replications per parasite in HFF and placenta samples from 24 to 48 h for each parasite type (calculated from data in panel B). Of note, panel C gives logarithmically greater weight to larger vacuoles in comparison to panel B: 1-parasite vacuoles have not replicated and thus received scores of zero, while 4-parasite vacuoles were weighted twice as heavily as 2-parasite vacuoles and 8-parasite vacuoles received three times the 2-parasite weight. Some cell lysis occurred by 48 h for type I (see Materials and Methods). Bars, SEM.
Image processing for figures.
Images were prepared using Image J, Volocity LE (Perkin-Elmer, Waltham, MA), Photoshop, and Illustrator (Adobe, San Jose, CA). RGB blue color hues were linearly adjusted for better CMYK laser and inkjet printing. For confocal reconstructions (see Fig. 5 and 6), regions of red and green overlap were intensified to enunciate syncytial puncta. All images in a given comparison set were treated equivalently. No other nonlinear alterations were performed.
Fig 5.

Dissemination of T. gondii throughout placenta. (A) AV and FV in histological sections were scored for presence/absence of T. gondii at 24, 48, or 72 h p.i. AV were scored as either colonized at the EVT-rich tip or at the tip and extending toward the fetus from the tip (Tip + Mid). FV colonization was uncommon, especially by 72 h. Bars, SEM. (B) High-magnification examination of T. gondii (here, type II) dissemination in histological sections stained with anti-hCG (syncytial marker; red) or anti-human E-cadherin (red-stained sCTB boundaries), showing that the basement membrane presents a barrier to infection of STR, where fetal vessels are found. Green, GFP-expressing T. gondii; blue, DAPI. Bar, 100 μm. (C) Whole-mount placental explants were imaged on a confocal microscope and villus cross sections reconstructed to further explore the integrity of the basement membrane barrier. In this representative 72-h-p.i. image, multiparasite vacuoles can be found ringing portions of the sCTB layer but not in STR or SYN. Dashed line, basement membrane; dotted line, probable SYN/sCTB boundary. Red, F-actin (phalloidin), which strongly stains cytotrophoblasts but weakly stains SYN; green, GFP-expressing T. gondii; blue, DAPI; yellow, autofluorescent granules found in SYN. Bar, 20 μm.
Fig 6.
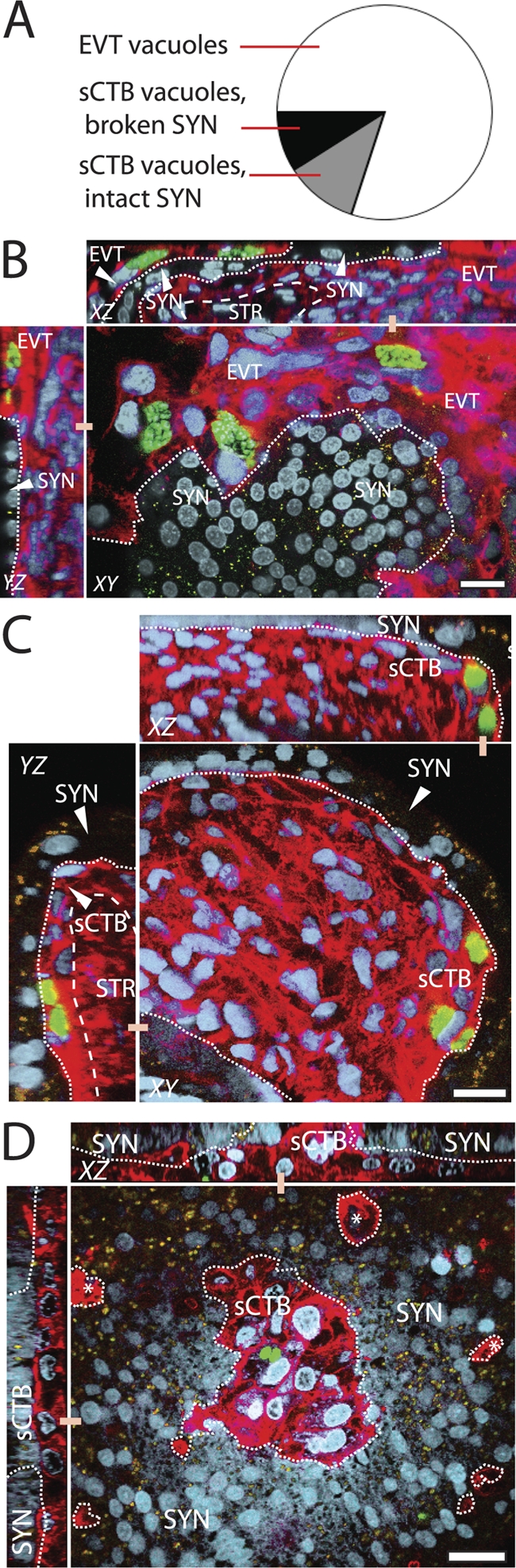
Localization of subsyncytial cytotrophoblast (sCTB) infections. (A) Multiparasite vacuole locations were scored across all three T. gondii types in whole-mount placental explants over 72 h (see Materials and Methods). Most are in EVT, while sCTB vacuoles can be found either at SYN breaks or in the apparent absence of such breaks. (B to D) Representative Z-series reconstructions via confocal imaging illustrate each of these three possibilities. Red, F-actin (phalloidin), which strongly stains cytotrophoblasts but weakly stains SYN; green, GFP-expressing T. gondii; blue, DAPI; yellow, autofluorescent granules found in SYN. Dashed line, SYN/CTB boundary; dotted line, basement membrane. Bar, 20 μm. (B) Infections of EVT in an anchoring villus at the SYN boundary, T. gondii type I at 24 h p.i. (C) Infected sCTB underlying apparently intact SYN, as in this image of T. gondii type I at 24 h p.i. (D) Vacuole in floating villus exhibiting damage in SYN, as evidenced by a discontinuous SYN layer with sCTB protruding from it. This representative image was taken 24 h p.i. with T. gondii type III. This may be the normal process of SYN renewal; however, nearby areas of discontinuity (*) suggest damage.
RESULTS
We introduced T. gondii to first-trimester human placental explants, a well-studied model system that adequately represents the in vivo architecture of the human placenta and allows the study of both sites of maternal-fetal contact: (i) the maternal blood-syncytiotrophoblast and (ii) the decidua-EVT interface (27, 48) (Fig. 1C and D). Villous trees are dissected from placentas and cultured on Matrigel, which mimics the decidual matrix sufficiently to promote EVT invasion. Many floating villi remain, and the whole of the explant is bathed in cell culture media in place of maternal blood.
As in vivo, these ex vivo explants are covered with a vast, single, multinucleated syncytiotrophoblast (Fig. 1). This overlies subsyncytial cytotrophoblasts, which in turn are supported by a basement membrane that separates trophoblasts from the villous stroma. In vivo, fibroblast-supported fetal capillaries collect here into fetal vessels that drain into the umbilical cord. Cytotrophoblasts can differentiate along one of two pathways depending on their location. In anchoring villi, which touch the uterine wall in vivo or the Matrigel here, they invade, becoming EVT. Throughout the majority of the placenta, cytotrophoblasts in floating villi can fuse with the syncytium and differentiate into syncytiotrophoblast, aiding in the growth of this continuous layer.
Extravillous trophoblasts are a major portal of entry for T. gondii.
We incubated placental explants with type I (RH), type II (Pru), or type III (CTG) T. gondii tachyzoites isolated from vacuoles in human foreskin fibroblast (HFF) cells. All strains express GFP. After 24 h, explants were fixed and mounted whole (Fig. 2A) for examination of parasite distribution in placental tissue types. Confocal scans of these explants revealed that T. gondii, indicated by a GFP signal, was primarily found in EVT of anchoring villi (Fig. 2A).
Fig 2.

T. gondii infects the placenta at extravillous cytotrophoblasts (EVT). (A) Two-dimensional (2D) projection of confocally scanned whole-mount placental explant infected for 24 h with type II T. gondii. Red, F-actin (phalloidin); green, GFP-expressing T. gondii; blue, DNA (DAPI); bar, 100 μm. Curve shows the integrated GFP signal across the image's axes, with peaks in the EVT region of anchoring villi (AV) while infectious foci in floating villi (FV) are rare. (B and E) Infected explants were sectioned at 24 h p.i. and stained with anti-hCG (syncytial marker; red) and DAPI (blue). Micrographs shown here feature type II T. gondii; bars, 100 μm. (B and C) Representative T. gondii locations at 24 h: EVT, sCTB, SYN. (C) Parasites in sCTB (arrows) were at times associated with breaks in syncytiotrophoblast (bSYN). (D) Locations of all three parasite types in two sections from each of three placentas show the majority are found in EVT at 24 h p.i. Bars, standard errors of the means (SEM). (E) Enzymatic removal of the SYN results in increased parasite infections along exposed sCTB on villus arms.
Whole mounts permit examination of the topmost syncytial layer, which has the most extensive contact with parasites in the media, but histological sections permit close examination of the precise cell types infected. When we examined histological sections of infected explants, we found ∼80% of parasite-containing vacuoles were in EVT, while the remainder was in subsyncytial cytotrophoblasts or syncytiotrophoblast (Fig. 2B to D). This is especially striking since EVT occupy less than 5% of the surface area of these explants. Syncytial infections were significantly rarer, and few parasites here had successfully replicated within their vacuoles by 24 h (Fig. 2B). Some subsyncytial parasites were located near clear sites of syncytial damage (Fig. 2C). There was no significant difference in parasite localization among the three T. gondii types (P > 0.99 by chi-square test).
Small numbers of fetal macrophages are found in the stroma of placental explants. To test whether these cells chemotax toward the parasite, we stained histological sections with anti-CD68 monoclonal antibody (clone EBM11; Dako) that should specifically stain macrophages. The results showed that the macrophages were uninfected and randomly distributed throughout the stroma over 72 h p.i., as in placental explants never exposed to T. gondii, suggesting that these macrophages do not migrate toward infected EVT (J. R. Robbins and A. I. Bakardjiev, unpublished data).
Since EVT are the only mononuclear cells of the placental explant not covered by syncytiotrophoblast, and as we sometimes observed infection near rare sites of syncytiotrophoblast damage (Fig. 2C), we hypothesized that the syncytial layer presented an obstacle to T. gondii infection, just as it does for L. monocytogenes (59), herpes simplex virus (40), and cytomegalovirus (25, 45, 47). We used enzymatic digestion to damage the explant syncytiotrophoblast and repeated the infection using type II T. gondii (Fig. 2E). Under these conditions, parasites were found in the formerly subsyncytial cytotrophoblasts lining villous arms, as well as in EVT, supporting the hypothesis that the syncytiotrophoblast represents a barrier to multiple pathogens.
Strain comparison in invasion of placental explants.
Multiple studies have explored whether specific T. gondii clonal lineages are overrepresented in congenital infections, which might suggest a strain-specific tropism toward placental and/or fetal infection, but definitive conclusions have been difficult to reach (2, 3, 13, 22, 28, 29, 35, 36, 51). We compared the ability of the three types to infect the human placenta by adding a known number of viable parasites to Transwell filters containing placental explants. After 5 h p.i., before replication had occurred, we fixed the explants and then applied an antibody that binds the Toxoplasma tachyzoite surface antigen (SAG1). No permeabilization was used; thus, the antibody stained only extracellular parasites while all parasites expressed GFP (Fig. 3A). We then used this stain to differentiate intracellular from extracellular parasites. Because the shapes and sizes of placental explants vary considerably, we counted three randomly selected fields occupied by EVT-containing anchoring villi and three fields in syncytially covered areas (floating villi) for explants from each of three placentas per T. gondii strain. This total was divided by the total viable parasites added (as determined by plaque formation in HFF monolayers) to yield relative invasion efficiency.
Fig 3.
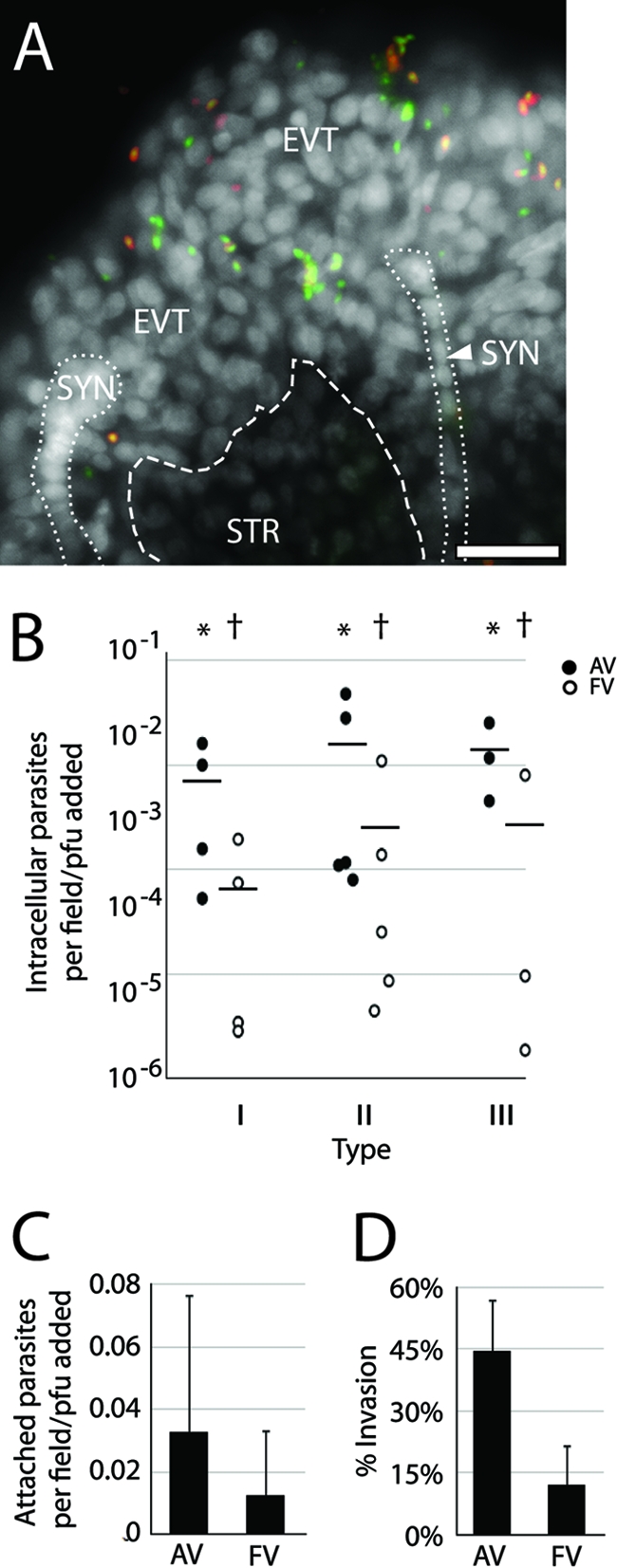
Comparison of T. gondii strains' invasion of placental explants. (A) Representative whole-mount placental explant infected for 5 h with type III T. gondii and stained with anti-SAG1 (red-stained parasites) without permeabilization. All living or recently dead parasites express GFP and are therefore green; extracellular parasites also stained red. This “inside-out” stain reveals parasites predominantly localized to EVT, SYN, or STR. White, DAPI. Bar, 100 μm. (B) Intracellular parasites were counted in AV and/or FV and normalized to the infectious dose as determined by PFU. Each dot represents the average of three fields per one placenta. * versus † denotes statistically similar groups (P < 0.04). Bars, SEM. (C) Average counts of extracellular attached parasites in each region (AV or FV) across all strains indicate a slight but not statistically significant pathogen preference for AV. (D) Counts of intracellular parasites as a fraction of total parasites found in AV and FV regions across all strains show invasion is significantly more likely (P > 0.0005) in AV. (C and D) Three AV or FV fields in each of three placentas per each of three strains. Bars, standard deviations (SD).
Invasion efficiency was variable, exhibiting an ∼100-fold range both within and across strains (Fig. 3B). No statistical difference was observed from one strain to the next (P = 0.69 and 0.58 for anchoring villi and floating villi, respectively, by analysis of variance [ANOVA]). Enumerating all parasites associated with each region, intracellular and extracellular, also revealed no difference among strains (P > 0.49 by ANOVA). If differential placental affinity exists across T. gondii types, its effect was too small to be detected by this assay.
These results do, however, show that anchoring villi are roughly 10 times more likely to contain intracellular parasites than syncytially covered floating villi across all parasite types (Fig. 3B) (P = 0.02 by Student's t test). This was not entirely due to increased ability of parasites to attach to anchoring villi, as the total number of parasites in all strains associated with each region was only slightly but not significantly different (Fig. 3C) (P = 0.16 by Student's t test). However, bound parasites of all types were significantly more likely to invade anchoring villi, which, unlike floating villi, are not covered by syncytiotrophoblast and contain extravillous trophoblasts (Fig. 3D) (P < 0.005 by Student's t test). Taken together, these results show that the syncytiotrophoblast does not appear to be a barrier to attachment but is a significant obstacle to T. gondii invasion.
T. gondii replication.
Studies differ on the question of whether T. gondii replication is inhibited in trophoblast cells using the BeWo choriocarcinoma cell line (5, 52, 56). We have previously shown that L. monocytogenes growth is impaired in EVT (though not BeWo cells) due to altered vacuole maturation (68). Thus, we wanted to assess T. gondii replication in trophoblasts within placental explants. Because of the morphological variability across explants, it was not possible to assay T. gondii raw numbers over time by plaque assay or PCR. Therefore, we enumerated the number of parasites per vacuole at 24 and 48 h compared to HFF cells (Fig. 4). In order to avoid counting extracellular parasites, a SAG1 antibody was applied extracellularly before counting as shown in Fig. 3.
By 24 h, viable parasites should have replicated at least once. In type II T. gondii, we observed an ∼2-fold increase in the number of parasites that had not replicated (single-parasite-containing vacuoles) relative to HFF at both 24 and 48 h p.i. (Fig. 4B) (P = 0.06 and 0.07, respectively, by Student's t test). By 48 h p.i., type I parasites also showed a slight, but not statistically significant, delay in replication in the primary trophoblasts relative to HFF by both visual inspection and arithmetic quantification (P = 0.15 by Student's t test). Type III placental growth closely resembled growth in HFF (P > 0.44 by Student's t test). As expected, the virulent type I strain replicated more quickly than type II or III in both cellular environments.
To further clarify the parasite growth over time, we also calculated the average replications per parasite in each condition (Fig. 4C) (see Materials and Methods). This confirmed that the greatest differences in replication rate between in situ primary trophoblasts and HFF could be found in type II parasites, but this again did not quite reach the level of statistical significance (P = 0.06 and 0.07 at 24 and 48 h p.i., respectively). It is important to note that this method can only account for live parasites; if some T. gondii die within trophoblasts as L. monocytogenes does (68), we cannot detect them. When an antibody to SAG1 was used in permeabilized sections, we frequently observed multiple SAG1-positive puncta within a vacuole-sized area in the cytoplasm of extravillous cytotrophoblasts at 24 h p.i. even though whole parasites were not apparent, suggesting the presence of degraded parasites (Robbins and Bakardjiev, unpublished).
Basement membrane acts as a barrier to fetal dissemination.
EVT is the major site of placental colonization for T. gondii, but does EVT infection lead to fetal infection? We have previously shown that L. monocytogenes spreads from EVT along subsyncytial cytotrophoblasts and eventually into the villous stroma that contains fetal capillaries, suggesting that this is the pathway of dissemination to the fetus (59). To quantify T. gondii progress down villous arms toward the fetus, or into the villous stroma, we tallied the percentage of infected anchoring and floating villi per explant over 72 h (Fig. 5A). The results showed no increase in villus colonization; in fact, the percentage of floating villi that were colonized actually decreased slightly over this period, particularly for type II T. gondii. This indicates that, unlike L. monocytogenes (59), T. gondii does not effectively disseminate through the tissue within this time frame. However, the intercellular spread rate for T. gondii (estimated to be at least 48 h) (11) is significantly greater than that of L. monocytogenes (∼5 h) (58, 66). Doubling time is likewise slower. Explant structural integrity, particularly syncytiotrophoblast, degrades with time (Robbins and Bakardjiev, unpublished), and we could not be confident of results past 72 h.
However, we did observe that parasite escape into the villous stroma, where fetal capillaries are located in vivo, was too rare to be enumerated, despite abundant adjacent infections in subsyncytial cytotrophoblasts (Fig. 5B). This can be clearly seen in sagittal reconstructions of the explant generated by confocal microscopy (Fig. 5C). A basement membrane separates trophoblasts from villous stroma, and host cells are scarcer within the stromal matrix. One or both of these factors may restrict parasite dissemination toward the fetus.
Infections of floating villi.
As noted above (Fig. 2D), parasites were detected in both the syncytiotrophoblast and subsyncytial cytotrophoblasts, albeit in fewer numbers than in EVT. If the syncytiotrophoblast acts as a barrier, how does T. gondii reach subsyncytial trophoblasts in the absence of nearby EVT?
Histological sections are made through transverse planes in the center of the explant, but most parasites' initial contact with the placenta is on the undulating upper surface, >95% of which is covered by syncytiotrophoblast. Sections can reveal little about infections in this region. We therefore used confocal microscopy to closely examine vacuoles in whole-mount placental explants infected for 24, 48, or 72 h with each of the three T. gondii types. First, we enumerated multiparasite vacuoles in three villi per placenta to determine the percentage in EVT versus other parts of the placenta (82%) (Fig. 6A and B). Next, we confocally sampled 100 multiparasite vacuoles not in EVT to determine whether they were syncytial or subsyncytial and, if the latter, whether they were covered by syncytiotrophoblast.
The explants were saturated with phalloidin, which differentially outlines syncytiotrophoblast and mononuclear cytotrophoblasts. Syncytiotrophoblast can also be distinguished by its more compact, denser nuclei (15) and relatively higher proportion of autofluorescent lipid droplets (32) that emit in both green and red spectra. Especially as the explants age, patches of discontinuity become apparent in the syncytiotrophoblast, either due to syncytial atrophy or the natural renewal process of subsyncytial cytotrophoblasts fusing with the syncytiotrophoblast.
Of the 100 multiparasite vacuoles examined, none were located in syncytiotrophoblast, although single parasites were occasionally identified within this layer. These could either represent infective “dead ends” or transsyncytial transit mid-process. All multiparasite vacuoles were found in subsyncytial cytotrophoblasts. We used reconstructed sagittal sections to determine whether they were beneath or adjacent to breaks in the syncytiotrophoblast (Fig. 6C and D). In 45% of such vacuoles, we detected no syncytial interruptions near the subsyncytial vacuoles (Fig. 6A and C). It is possible that the parasites are capable of transsyncytial migration without causing cellular damage. However, we cannot exclude the possibility that breaks were too small to detect or present at earlier times and subsequently healed.
The remaining 55% of vacuoles showed clear syncytial interruptions immediately above or beside the parasites (Fig. 6A and D), further supporting the earlier finding (Fig. 2 and 3) that the syncytiotrophoblast is a barrier to infection.
DISCUSSION
The human placenta has two histologically distinct maternal-fetal interfaces: the vast blood-syncytiotrophoblast interface where nutrient exchange occurs and the uterine anchoring site where EVT invade the decidua. This is the first reported study of T. gondii infection kinetics in the human placenta and the first demonstration that syncytiotrophoblast is strongly resistant to T. gondii invasion and growth. It is also the first study to compare the three major T. gondii types in their ability to infect placental tissues, finding no significant difference in ability to invade cells but a possible difference in the ability of type II parasites to replicate within trophoblasts.
Significantly, we have shown that the syncytiotrophoblast is a rigorous barrier against T. gondii, even though it is the most available surface, leaving the parasite to preferentially colonize EVT. This was equally true for all three major T. gondii lineages and mirrors prior findings for two vertically transmitted viruses (25, 40, 45, 47) as well as the bacterium L. monocytogenes (59, 68). In fact, despite differing molecular requirements for host cell invasion, distributions of T. gondii localization were quite similar to those previously reported for L. monocytogenes (59), suggesting that vulnerable sites are inherent to the placenta, not the pathogen. The diversity of these unrelated pathogens highlights the robust generality of the placenta's maternal blood-syncytiotrophoblast barrier and suggests an adaptive function for its unusual structure.
T. gondii at the uterine interface.
Our study shows that EVT are both significantly more susceptible to T. gondii infection and more hospitable to parasite replication than the syncytiotrophoblast. Parasites were more frequently found in EVT, and multiparasite syncytial vacuoles were rare. This supports the previous finding of a reduction in type I (RH) T. gondii invasion efficiency in partially syncytialized, isolated human trophoblasts over those with no syncytialization (1). It also helps to explain histopathological results in the pregnant vesper mouse (Calomys callosus), wherein type II (ME49) T. gondii is first detected in the maternal leukocytes of the uterine decidua—not the blood-syncytiotrophoblast interface—and progresses from there to the trophoblast giant cells (analogous to human EVT) (24). The labyrinthine region, where nutrient exchange with maternal blood occurs in mice (analogous to the human villous region), is infected secondarily and concurrent with fetal infection (24).
Data from other animals further argue that the initial infection begins in the uterus, not the blood. Shiono et al. reported an 8-day gap between the initial detection of type II (Fukaya) T. gondii in the placenta and in the fetus in laboratory mice, although they did not investigate specific parasite location (63). Further, in experimentally infected sheep, the initial necrotic foci were uterine, with first fetal infection occurring later and coincident with infection of the cotyledonary villi (analogous to the human villous region) (16).
The placentas of these various hosts are quite different, however. Humans have a multivillous placenta with only one layer of syncytiotrophoblast bathed in maternal blood, while mice have a labyrinthine placenta with two layers of syncytiotrophoblast (46). In sheep, whose trophoblasts exhibit limited uterine invasion, maternal blood does not directly contact trophoblasts (67). Rather, sheep maternal blood is separated from fetal placental tissues by maternal endothelium, connective tissue, epithelium, and then a syncytium (43). The weight of this evidence combined with the results reported here suggests that maternal blood-syncytiotrophoblast is not a significant site of T. gondii transmission. Instead, the parasite must traffic to the decidua where it can reach EVT and, later, fetal capillaries, such that infection of the nutrient exchange interface occurs at approximately the same time as fetal infection.
T. gondii at the syncytial interface.
Infection across the syncytiotrophoblast may also occur but it appears to be rarer, possibly with more rapid, severe consequences given the quick fetal dissemination from the nutrient-absorbing regions in animals (16, 24, 63). We have shown here that syncytial damage increases parasite infection of villous arms that would, in vivo, contact maternal blood. Such damage might occur in the presence of a second infectious agent or due to physical trauma. The effects may also depend on the placenta's gestational age, as the layer of subsyncytial cytotrophoblasts undergirding the syncytiotrophoblast grows thinner and partially discontinuous after the first trimester (37, 50).
Recently, de Oliveira Gomes et al. compared first- and third-trimester human placental explants infected with type I (RH) T. gondii (18). Cultured placentas past gestational week 8 do not invade or develop EVT; explants in that study were cultured in suspension and thus comprise only floating villi, often with “open” arms exposing fetal stroma to the surrounding media (48). Further, this report did not distinguish between syncytiotrophoblast and subsyncytial trophoblast infections. These limitations notwithstanding, however, the authors found that the number of parasites in trophoblasts and fetal stroma in third-trimester placenta is roughly double that of first, while attached, noninvasive parasites were actually less in the explants of greater gestational age. The authors attribute this to decreased accumulation of macrophage migration inhibitory factor (MIF) at the syncytiotrophoblast in third-trimester explants relative to first and linked this to thinning subsyncytial cytotrophoblasts. Given the technical differences in the approach, it is difficult to compare our results with these but both data sets are consistent with the syncytium representing a significant barrier to Toxoplasma infection.
Syncytial resistance to infection might be explained by its physical properties, especially given that it protects against other, unrelated pathogens (25, 40, 45, 47, 59, 68). The syncytial surface is covered with dense, branched microvilli (17, 62), and how this or other aspects of this physically unusual layer discourage infection merits further study. Of particular interest will be to determine whether syncytial resistance is immunological, structural, or both.
Even for parasites that successfully infected the syncytiotrophoblast, we observed little or no growth. However, parasites replicated well in underlying subsyncytial cytotrophoblasts, whether covered by syncytiotrophoblast or not. As with L. monocytogenes (59), their dissemination was then severely restricted by the basement membrane that separates trophoblasts from fetal stroma, a further barrier to reaching fetal capillaries. Due to the relatively slower replication and cell-to-cell spread rate of T. gondii, we cannot know whether focal infection in either subsyncytial cytotrophoblasts or EVT ultimately results in the majority of stromal colonization and therefore barrier crossing; however, the results from animal studies (16, 24, 63) suggest fetal transit begins at the EVT.
A Trojan horse? How might T. gondii reach the decidua and from there the EVT?
T. gondii appear capable of disseminating in maternal leukocytes (21), although their specific tropisms are not known. There is limited contact with endovascular trophoblasts at the distal termini of the maternal spiral arteries that empty into the intervillous space and abundant contact between EVT and decidual cells, including maternal leukocytes, which may even be drawn to them. Isolated EVT exhibit cytokine-mediated chemotactic abilities for leukocytes (20) and in a cell coculture system, T. gondii-infected monocytes bind trophoblast cell lines more frequently in an ICAM-1-dependent manner (55). Further, T. gondii infection induces dendritic cell (DC) migration, and when infected DCs are introduced to mice, the pathogen disseminates more rapidly than with parasite alone, including across the blood-brain barrier (42). Thus, T. gondii could travel to the decidua within maternal leukocytes predisposed to preferentially bind EVT; then, after lysing their “Trojan horse” host, they could invade the fetal placenta.
Leukocytes circulating in maternal blood may also bind syncytiotrophoblast. Stimulation of human placental explants with T. gondii antigen increases monocyte adhesion (23), although it is unknown whether the effect is differential for EVT versus syncytiotrophoblast. We have found that L. monocytogenes containing phagocytes are more likely to bind EVT than syncytiotrophoblast (59). Profuse microvilli at the syncytiotrophoblast surface (17, 62) may obstruct tight leukocyte adherence. Finally, the data presented here demonstrate that the deficiency in syncytial invasion and replication cannot be explained by decreased parasite adhesion to the syncytial surface; presumably parasites escaping a lysed leukocyte at the syncytial surface would face the same obstacles as a parasite binding that surface from the extracellular environment, as in our work.
It remains possible that placental colonization is incidental to, but does not cause, fetal infection. Maternal leukocytes do infrequently traffic from the decidua to the fetus (49) and might plausibly ferry T. gondii within.
T. gondii type differences.
Finally, we also assayed differential placental infection abilities among T. gondii types I, II, and III. Multiple epidemiological studies have sought a connection between parasite strain and preferential congenital infection but no clear consensus has emerged (reviewed in reference 12). Human placental explants are nonuniform in size, shape, and EVT density, resulting in high variability of pathogen invasion frequency (Fig. 3) (59). Limited extracellular T. gondii viability during the infection process introduces further variation. However, when controlled for placental surface area and live pathogens (via PFU in HFF monolayers), we found no significant difference in parasite attachment or invasion efficiency among types. Although it is feasible that our system is insufficiently sensitive to detect a difference, it is certainly possible that all strains might indeed be equally capable of direct placental infection.
Even if no difference in trophoblast invasion exists, this does not rule out the possibility that one T. gondii type may preferentially cause congenital infection over others. Others have proposed that extended dissemination capabilities might bias toward various sequelae (8). Survival in leukocytes that traffic to the decidua or increased replication in decidual cells may increase transmission frequency. It is also possible that weakly virulent strains go undetected in congenital infections due to lack of symptoms, and it is notable that our data show the relatively avirulent (based on studies in mice) and poorly studied type III T. gondii exhibit the least difference in intraplacental growth relative to other cell types.
T. gondii strains may also differ in their ability to survive within the placenta. Replication rates of type I (RH) T. gondii in BeWo choriocarcinoma cells are controversial, with one report asserting slower parasite replication (56) and others reporting normal growth relative to other cell types (5, 52). It is unclear, however, how well this cancerous cell line represents primary trophoblasts, as we have shown that BeWo cells do not restrict growth of L. monocytogenes, while primary isolated cytotrophoblasts and explant EVT do (68). Further work will be necessary to clarify the fate of T. gondii in trophoblasts as well as the roles of cytokines and promoters of apoptosis.
Host cell death is also a possibility. Angeloni et al. found that BeWo choriocarcinoma cells were more likely to apoptose in the presence of type II (ME49) T. gondii than the more virulent type I strain, suggesting that type I parasites better inhibit host cell death (4). We observed no microscopically visible signs of apoptosis over 72 h (e.g., decreased cell density, nuclear fragmentation), but further studies using primary trophoblasts may be of future value.
Differential strain abilities also merit deeper consideration. Both the small replication defect and the decline in colonized villi over time, particularly among type II T. gondii, suggest parasite death may be occurring. While type II parasites have often been implicated at higher rates in epidemiological studies of congenital infections, type II is also the prevailing strain in most of the regions where such studies have been done (2, 3, 35, 36, 51). We have previously shown that EVT, particularly those most distally located within the maternal decidua, are bactericidal to L. monocytogenes (68). Given the predisposition for pathogens to infect at the EVT, it is possible that their modified vacuolar maturation pathway restricts multiple pathogens. Some T. gondii strains may be able to overcome this better than others.
Placental pathogen barrier.
The placenta is often presented in seemingly contradictory terms: an immunoprivileged organ that nonetheless presents a serious obstacle to pathogens. Relatively few microbes cross the placental barrier (31, 57a), and most that can do so at relatively low frequencies. They exhibit at least partially intracellular lifestyles, and we suggest that this is due at least in part to the advantage of first colonizing the EVT in the decidua, which are not easily accessed from the maternal blood. This has been illustrated for diverse pathogens in human placental explants (25, 40, 45, 47, 59, 68), as well as in experimental infections of mice with T. gondii (24), L. monocytogenes (44, 57), Chlamydophila psittaci (14), Coxiella burnetii (9), Fusobacterium nucleatum (33), and Brucella abortus (39).
We further propose that infection at the maternal blood-syncytiotrophoblast interface is possible but requires overcoming the syncytial barrier. Human placental villitis associated with multiple pathogens (including T. gondii) induces syncytial expression of ICAM-1 and monocyte binding to the syncytial surface with inflammatory syncytial damage (38). Although this occurs as a consequence of infection, not a cause, infection with one pathogen may predispose to infection by another via degradation of the syncytiotrophoblast. Each pathogen's interaction with the placenta may be unique, but the outcome is surprisingly general, suggesting we still have much to learn about the physical and molecular nature of both maternal-fetal barriers.
ACKNOWLEDGMENTS
This research was supported by NIH grants R01 AI84929 (to A.I.B.) and AI73756 (to J.C.B.).
We are grateful to Susan Fisher and the members of the Fisher lab for their expertise and insight. We thank all of the donors who provided their informed consent. We thank Joanne Engel for use of her TE2000-E microscope, Manuel Amieva for use of his confocal microscope, and Kurt Thorn and the Nikon Imaging Center at UCSF for use of a confocal microscope.
Footnotes
Published ahead of print 14 November 2011
REFERENCES
- 1. Abbasi M, et al. 2003. Infection of placental trophoblasts by Toxoplasma gondii. J. Infect. Dis. 188: 608–616 [DOI] [PubMed] [Google Scholar]
- 2. Abdel-Hameed DM, Hassanein OM. 2008. Genotyping of Toxoplasma gondii strains from female patients with toxoplasmosis. J. Egypt Soc. Parasitol. 38: 511–520 [PubMed] [Google Scholar]
- 3. Ajzenberg D, et al. 2002. Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis, and correlation with clinical findings. J. Infect. Dis. 186: 684–689 [DOI] [PubMed] [Google Scholar]
- 4. Angeloni MB, et al. 2009. Apoptosis and S phase of the cell cycle in BeWo trophoblastic and HeLa cells are differentially modulated by Toxoplasma gondii strain types. Placenta 30: 785–791 [DOI] [PubMed] [Google Scholar]
- 5. Barbosa BF, Silva DA, Costa IN, Mineo JR, Ferro EA. 2008. BeWo trophoblast cell susceptibility to Toxoplasma gondii is increased by interferon-gamma, interleukin-10 and transforming growth factor-beta1. Clin. Exp. Immunol. 151: 536–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barbosa BF, et al. 2007. Susceptibility to vertical transmission of Toxoplasma gondii is temporally dependent on the preconceptional infection in Calomys callosus. Placenta 28: 624–630 [DOI] [PubMed] [Google Scholar]
- 7. Barragan A, Brossier F, Sibley LD. 2005. Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell. Microbiol. 7: 561–568 [DOI] [PubMed] [Google Scholar]
- 8. Barragan A, Sibley LD. 2003. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol. 11: 426–430 [DOI] [PubMed] [Google Scholar]
- 9. Baumgärtner W, Bachmann S. 1992. Histological and immunocytochemical characterization of Coxiella burnetii-associated lesions in the murine uterus and placenta. Infect. Immun. 60: 5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benirschke K, Kaufmann P, Baergen R. 2006. Pathology of the human placenta, 5th ed. Springer-Verlag, New York, NY [Google Scholar]
- 11. Black MW, Boothroyd JC. 2000. Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 64: 607–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boothroyd JC, Grigg ME. 2002. Population biology of Toxoplasma gondii and its relevance to human infection: do different strains cause different disease? Curr. Opin. Microbiol. 5: 438–442 [DOI] [PubMed] [Google Scholar]
- 13. Boughattas S, et al. 2010. Direct genotypic characterization of Toxoplasma gondii strains associated with congenital toxoplasmosis in Tunisia (North Africa). Am. J. Trop. Med. Hyg. 82: 1041–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buendía AJ, et al. 1998. Kinetics of infection and effects on placental cell populations in a murine model of Chlamydia psittaci-induced abortion. Infect. Immun. 66: 2128–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burton GJ, Jones CJ. 2009. Syncytial knots, sprouts, apoptosis, and trophoblast deportation from the human placenta. Taiwan J. Obstet. Gynecol. 48: 28–37 [DOI] [PubMed] [Google Scholar]
- 16. Buxton D, Finlayson J. 1986. Experimental infection of pregnant sheep with Toxoplasma gondii: pathological and immunological observations on the placenta and foetus. J. Comp. Pathol. 96: 319–333 [DOI] [PubMed] [Google Scholar]
- 17. Cantle SJ, Kaufmann P, Luckhardt M, Schweikhart G. 1987. Interpretation of syncytial sprouts and bridges in the human placenta. Placenta 8: 221–234 [DOI] [PubMed] [Google Scholar]
- 18. de Oliveira Gomes A, et al. 2011. Effect of macrophage migration inhibitory factor (MIF) in human placental explants infected with Toxoplasma gondii depends on gestational age. Am. J. Pathol. 178: 2792–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Desmonts G, Couvreur J. 1974. Congenital toxoplasmosis—a prospective study of 378 pregnancies. N. Engl. J. Med. 290: 1110–1116 [DOI] [PubMed] [Google Scholar]
- 20. Drake PM, et al. 2001. Human placental cytotrophoblasts attract monocytes and CD56(bright) natural killer cells via the actions of monocyte inflammatory protein 1alpha. J. Exp. Med. 193: 1199–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fadul CE, Channon JY, Kasper LH. 1995. Survival of immunoglobulin G-opsonized Toxoplasma gondii in nonadherent human monocytes. Infect. Immun. 63: 4290–4294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferreira IM, et al. 2011. Toxoplasma gondii isolates: multilocus RFLP-PCR genotyping from human patients in Sao Paulo State, Brazil identified distinct genotypes. Exp. Parasitol. 129: 190–195 [DOI] [PubMed] [Google Scholar]
- 23. Ferro EA, et al. 2008. Macrophage migration inhibitory factor is up-regulated in human first-trimester placenta stimulated by soluble antigen of Toxoplasma gondii, resulting in increased monocyte adhesion on villous explants. Am. J. Pathol. 172: 50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferro EA, Silva DA, Bevilacqua E, Mineo JR. 2002. Effect of Toxoplasma gondii infection kinetics on trophoblast cell population in Calomys callosus, a model of congenital toxoplasmosis. Infect. Immun. 70: 7089–7094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fisher S, Genbacev O, Maidji E, Pereira L. 2000. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J. Virol. 74: 6808–6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fisher SJ, et al. 1989. Adhesive and degradative properties of human placental cytotrophoblast cells in vitro. J. Cell Biol. 109: 891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher SJ, Leitch MS, Kantor MS, Basbaum CB, Kramer RH. 1985. Degradation of extracellular matrix by the trophoblastic cells of first-trimester human placentas. J. Cell Biochem. 27: 31–41 [DOI] [PubMed] [Google Scholar]
- 28. Fuentes I, Rubio JM, Ramirez C, Alvar J. 2001. Genotypic characterization of Toxoplasma gondii strains associated with human toxoplasmosis in Spain: direct analysis from clinical samples. J. Clin. Microbiol. 39: 1566–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gallego C, Saavedra-Matiz C, Gomez-Marin JE. 2006. Direct genotyping of animal and human isolates of Toxoplasma gondii from Colombia (South America). Acta Trop. 97: 161–167 [DOI] [PubMed] [Google Scholar]
- 30. Genbacev O, Schubach SA, Miller RK. 1992. Villous culture of first trimester human placenta—model to study extravillous trophoblast (EVT) differentiation. Placenta 13: 439–461 [DOI] [PubMed] [Google Scholar]
- 31. Givens MD, Marley MS. 2008. Infectious causes of embryonic and fetal mortality. Theriogenology 70: 270–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haigh M, Chawner LE, Fox H. 1984. The human placenta does not contain lipofuscin pigment. Placenta 5: 459–464 [DOI] [PubMed] [Google Scholar]
- 33. Han YW, et al. 2004. Fusobacterium nucleatum induces premature and term stillbirths in pregnant mice: implication of oral bacteria in preterm birth. Infect. Immun. 72: 2272–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He N, et al. 1997. Parasite load in pregnant mice infected by Toxoplasma gondii assayed by quantitative competitive-PCR. Parasitol. Int. 46: 143–147 [Google Scholar]
- 35. Howe DK, Honore S, Derouin F, Sibley LD. 1997. Determination of genotypes of Toxoplasma gondii strains isolated from patients with toxoplasmosis. J. Clin. Microbiol. 35: 1411–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howe DK, Sibley LD. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis. 172: 1561–1566 [DOI] [PubMed] [Google Scholar]
- 37. Jones CJ, Harris LK, Whittingham J, Aplin JD, Mayhew TM. 2008. A re-appraisal of the morphophenotype and basal lamina coverage of cytotrophoblasts in human term placenta. Placenta 29: 215–219 [DOI] [PubMed] [Google Scholar]
- 38. Juliano PB, Blotta MH, Altemani AM. 2006. ICAM-1 is overexpressed by villous trophoblasts in placentitis. Placenta 27: 750–757 [DOI] [PubMed] [Google Scholar]
- 39. Kim S, et al. 2005. Interferon-gamma promotes abortion due to Brucella infection in pregnant mice. BMC Microbiol. 5: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koi H, et al. 2002. Syncytiotrophoblast is a barrier to maternal-fetal transmission of herpes simplex virus. Biol. Reprod. 67: 1572–1579 [DOI] [PubMed] [Google Scholar]
- 41. Koshy AA, et al. 2010. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat. Methods 7: 307–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. 2006. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell. Microbiol. 8: 1611–1623 [DOI] [PubMed] [Google Scholar]
- 43. Leiser R, Kaufmann P. 1994. Placental structure: in a comparative aspect. Exp. Clin. Endocrinol. 102: 122–134 [DOI] [PubMed] [Google Scholar]
- 44. Le Monnier A, Join-Lambert OF, Jaubert F, Berche P, Kayal S. 2006. Invasion of the placenta during murine listeriosis. Infect. Immun. 74: 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maidji E, et al. 2010. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am. J. Pathol. 177: 1298–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maltepe E, Bakardjiev AI, Fisher SJ. 2010. The placenta: transcriptional, epigenetic, and physiological integration during development. J. Clin. Invest. 120: 1016–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McDonagh S, Maidji E, Chang HT, Pereira L. 2006. Patterns of human cytomegalovirus infection in term placentas: a preliminary analysis. J. Clin. Virol. 35: 210–215 [DOI] [PubMed] [Google Scholar]
- 48. Miller RK, et al. 2005. Human placental explants in culture: approaches and assessments. Placenta 26: 439–448 [DOI] [PubMed] [Google Scholar]
- 49. Mold JE, et al. 2008. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 322: 1562–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mori M, et al. 2007. The cytotrophoblast layer of human chorionic villi becomes thinner but maintains its structural integrity during gestation. Biol. Reprod. 76: 164–172 [DOI] [PubMed] [Google Scholar]
- 51. Nowakowska D, et al. 2006. Genotyping of Toxoplasma gondii by multiplex PCR and peptide-based serological testing of samples from infants in Poland diagnosed with congenital toxoplasmosis. J. Clin. Microbiol. 44: 1382–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oliveira JG, et al. 2006. BeWo trophoblasts are unable to control replication of Toxoplasma gondii, even in the presence of exogenous IFN-gamma. Placenta 27: 691–698 [DOI] [PubMed] [Google Scholar]
- 53. Pappas G, Roussos N, Falagas ME. 2009. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int. J. Parasitol. 39: 1385–1394 [DOI] [PubMed] [Google Scholar]
- 54. Pezerico SB, Langoni H, Da Silva AV, Da Silva RC. 2009. Evaluation of Toxoplasma gondii placental transmission in BALB/c mice model. Exp. Parasitol. 123: 168–172 [DOI] [PubMed] [Google Scholar]
- 55. Pfaff AW, et al. 2005. Toxoplasma gondii regulates ICAM-1 mediated monocyte adhesion to trophoblasts. Immunol. Cell Biol. 83: 483–489 [DOI] [PubMed] [Google Scholar]
- 56. Pfaff AW, Villard O, Klein JP, Mousli M, Candolfi E. 2005. Regulation of Toxoplasma gondii multiplication in BeWo trophoblast cells: cross-regulation of nitric oxide production and polyamine biosynthesis. Int. J. Parasitol. 35: 1569–1576 [DOI] [PubMed] [Google Scholar]
- 57. Redline RW, Lu CY. 1988. Specific defects in the anti-listerial immune response in discrete regions of the murine uterus and placenta account for susceptibility to infection. J. Immunol. 140: 3947–3955 [PubMed] [Google Scholar]
- 57a. Robbins JR, Bakardjiev AI. Pathogens and the placental fortress. Curr. Opin. Microbiol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Robbins JR, et al. 1999. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J. Cell Biol. 146: 1333–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robbins JR, Skrzypczynska KM, Zeldovich VB, Kapidzic M, Bakardjiev AI. 2010. Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes. PLoS Pathog. 6: e1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roos DS, Donald RG, Morrissette NS, Moulton AL. 1994. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 45: 27–63 [DOI] [PubMed] [Google Scholar]
- 61. Saeij JP, Boyle JP, Grigg ME, Arrizabalaga G, Boothroyd JC. 2005. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73: 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schiebler TH, Kaufmann P. 1981. In Becker V, Schiebler TH, Kubli F. (ed), Reife Placenta. Thieme, Stuttgart, Germany [Google Scholar]
- 63. Shiono Y, et al. 2007. Maternal-fetal transmission of Toxoplasma gondii in interferon-gamma deficient pregnant mice. Parasitol. Int. 56: 141–148 [DOI] [PubMed] [Google Scholar]
- 64. Striepen B, He CY, Matrajt M, Soldati D, Roos DS. 1998. Expression, selection, and organellar targeting of the green fluorescent protein in Toxoplasma gondii. Mol. Biochem. Parasitol. 92: 325–338 [DOI] [PubMed] [Google Scholar]
- 65. Thiébaut R, Leproust S, Chene G, Gilbert R. 2007. Effectiveness of prenatal treatment for congenital toxoplasmosis: a meta-analysis of individual patients' data. Lancet 369: 115–122 [DOI] [PubMed] [Google Scholar]
- 66. Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109: 1597–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wimsatt WA. 1950. New histological observations on the placenta of the sheep. Am. J. Anat. 87: 391–457 [DOI] [PubMed] [Google Scholar]
- 68. Zeldovich VB, Robbins JR, Kapidzic M, Lauer P, Bakardjiev AI. 2011. Invasive extravillous trophoblasts restrict intracellular growth and spread of Listeria monocytogenes. PLoS Pathog. 7: e1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]