

Abstract
Linker histone (H1) and heterochromatin protein 1 (HP1) are essential components of heterochromatin which contribute to the transcriptional repression of genes. It has been shown that the methylation mark of vertebrate histone H1 is specifically recognized by the chromodomain of HP1. However, the exact biological role of linker histone binding to HP1 has not been determined. Here, we investigate the function of the Caenorhabditis elegans H1 variant HIS-24 and the HP1-like proteins HPL-1 and HPL-2 in the cooperative transcriptional regulation of immune-relevant genes. We provide the first evidence that HPL-1 interacts with HIS-24 monomethylated at lysine 14 (HIS-24K14me1) and associates in vivo with promoters of genes involved in antimicrobial response. We also report an increase in overall cellular levels and alterations in the distribution of HIS-24K14me1 after infection with pathogenic bacteria. HIS-24K14me1 localization changes from being mostly nuclear to both nuclear and cytoplasmic in the intestinal cells of infected animals. Our results highlight an antimicrobial role of HIS-24K14me1 and suggest a functional link between epigenetic regulation by an HP1/H1 complex and the innate immune system in C. elegans.
INTRODUCTION
Histone H1 and heterochromatin protein 1 (HP1) are conserved chromosomal proteins that promote the formation of condensed chromatin (22, 44). H1 and HP1 act as both repressors and activators of transcription, depending on their nuclear distribution and cellular context (11, 14, 23, 50).
Linker histones represent the most heterogeneous class of histone proteins and are characterized by a highly conserved central globular domain. Interestingly, the C and N termini exhibit high variability, which may contribute to the multifunctionality of these proteins (44).
Genetic experiments in several organisms including Saccharomyces cerevisiae, tobacco, and C. elegans have shown that H1 influences development and differentiation processes (12, 27, 39). Moreover, deficiency of triple H1 isoforms (H1c, H1d, and H1e) as well as loss of HP1 mammalian isoforms causes embryonic lethality in mice (15, 18, 48).
Numerous experiments performed in vitro have demonstrated that the interaction of H1 with nucleosomes stabilizes higher-order chromatin structure, thereby influencing transcription and replication (44). Furthermore, H1 restricts nucleosome mobility, inhibits the action of chromatin-remodelling complexes, and modulates the ability of regulatory factors to access their chromatin targets (15). Recent studies revealed that chromatin is considerably more dynamic than previously imagined and that histones, particularly H1, are continuously exchanged among chromatin binding sites (30).
Heterochromatin protein 1 is a regulatory nonhistone protein which is recruited to chromatin through histone H3 di- or trimethylation at lysine 9 (H3K9me2,3). In addition, histone H1.4 (H1.b) dimethylation at lysine 26 (H1.4K26me2), other nonhistone proteins, and RNA components have also been shown to recruit HP1, depending on the chromatin context (7, 10, 25). While the HP1-H3K9me2,3 interaction plays a fundamental role in the formation and maintenance of heterochromatin (19), the biological significance of the HP1-H1.4K26me2 interaction remains unknown (10). It has been suggested that the posttranslational modifications of H1 regulate its function in chromatin condensation and in the recruitment of chromatin-specific proteins (59). While the covalent posttranslational modifications of core histones and the regulatory proteins which recognize these modifications have been extensively studied, little is known about the H1 linker histone code and its effects on cellular processes. Recent publications on H1 have mainly focused on the mapping of methylation of a single lysine residue in the N-terminal tail of the human H1 variants H1.2 and H1.4 (10, 56).
C. elegans possesses eight linker histone H1 variants and two HP1 homologues, HPL-1 and HPL-2. We previously showed that one of the eight C. elegans H1 variants, HIS-24, promotes germ line development and silences extrachromosomal arrays in the germ line (27). HPL-2 influences vulval cell fate specification by acting in the Rb-related synMuv (synthetic multivulva) pathway (8). In addition, lack of HPL-2 activity leads to desilencing of extrachromosomal arrays in the germ line, growth defects, and sterility at 25°C (8). The interaction with H3K9me2/3 appears to be conserved in HPL-2 (57). In contrast to the temperature-sensitive hpl-2 phenotypes, hpl-1 loss of function has no visible effect on C. elegans development even at higher temperatures. However, hpl-1 acts redundantly with hpl-2 to control larval growth, development of the somatic gonad, and vulval cell fate determination (47).
Given the role of HIS-24 and HPL in chromatin silencing and gene regulation, we decided to study the physiological role of these proteins and their function in transcriptional regulation in the roundworm C. elegans. Here, we show that the histone H1 variant HIS-24 and the HP1-related proteins HPL-1 and HPL-2 are specifically required for the transcriptional regulation of many immune-relevant proteins. HPL-1 interacts with HIS-24 at monomethylated lysine 14 (HIS-24K14me1), and both proteins associate in vivo with the promoters of genes involved in antimicrobial response. Using stable-isotope labeling by amino acids (SILAC)-based analysis of liquid worm cultures, we show that lack of HIS-24 leads to induction of infection-inducible proteins. Interestingly, following infection with Bacillus thuringiensis, monomethylation of HIS-24K14 is enhanced, and HIS-24::GFP (where GFP is green fluorescent protein) localization in intestinal cells changes from being mostly nuclear to both nuclear and cytoplasmic. We propose that the physical interaction of HIS-24K14me1 with HPL-1, combined with HIS-24K14me1 release in the cytoplasm, may be part of a specific state of the innate immune system in C. elegans.
MATERIALS AND METHODS
Strains.
Maintenance, culturing, and genetic manipulation of C. elegans were carried out according to standard procedures (5). Bristol strain (N2) was used as the wild type. Strains with the following genotypes were obtained from the Caenorhabditis Genetics Center (CGC): his-24(ok1024)(X), hil-3(ok1556)(X) (outcrossed one time), hpl-1(tm1624)(X) (outcrossed four times), hpl-2(tm1489)(III) (outcrossed four times), and hpl-2(tm1489)(III); hpl-1(n4317)(X). The his-24(ok1024) mutant strain was backcrossed eight times with the wild type to reduce genetic variability. hpl-1::gfp, his-24::gfp (stable integrated EC602 strain [26]), gfp::his-24 K14A, and hpl-2::gfp transgenic strains were crossed with the his-24(ok1024)(X) or hpl-1(tm1624)(X) mutant. The double mutants hpl-1(tm1624) his-24(ok1024)(X) and hpl-2(tm1489)(III); his-24(ok1024)(X) as well as the triple mutant strain hpl-2(tm1489)(III); hpl-1(tm1624) his-24(ok1024)(X) were generated by crossing. A PCR-based analysis was used to identify these mutant alleles. Worms were grown at 15°C, 21°C, or 25°C on nematode growth medium (NGM) agar plates seeded with Escherichia coli strain OP50. To analyze the synthetic multivulva (synMuv) or the everted vulva (evl) phenotype, five adult hermaphrodites were grown on plates incubated at 15°C. Next, only the progeny (embryos or early larval stage 4 [L4]) were shifted to 25°C and scored for sterility or synMuv or evl phenotype.
Generation of gfp::his-24K14A transgenic strain.
his-24::gfp plasmid DNA containing the promoter and the complete genomic coding sequence of his-24 translationally fused to gfp served as a template for the site-directed mutagenesis by PCR. The same plasmid had previously been used for the generation of the his-24::gfp reporter strain (26) The lysine residue (K) 14 was mutated to an alanine residue (A) using primers MJ224 (CGCGCCAGAGGTCCCAAAGGCTAAGGC) and MJ225 (GGGACCTCTGGCGCGACAGCGGC). The site-directed mutagenesis was performed according to the QuikChange site-directed mutagenesis protocol (Stratagene). gfp::his-24K14A plasmid DNA was coinjected with a myo-3::pmcherry marker into both gonad arms of the his-24−/− null mutant strain.
SILAC method.
Lysine auxotroph E. coli strain (DSM1099) was obtained from the DSMZ (www.dsmz.de). Bacteria were labeled with [13C6]lysine (Silantes) as described previously (54). L1 larvae were used for the inoculation of liquid culture containing S-basal medium (per liter, 10 mM K-citrates, pH 6, 10 ml of trace metals [×100], 4 mM CaCl2, 3 mM MgSO4, 10 ml of penicillin-streptomycin-neomycin [PSN] [×100] [Gibco], and 10 ml of nystatin [×100] [Sigma]). Every second day “light” (labeled with [12C6, 14N2]Lys) E. coli were fed to the wild-type and the his-24(ok1024) mutant worms. In parallel wild-type worms were SILAC-labeled by feeding with “heavy” (labeled with [13C6, 12N2]Lys) E. coli for 2 weeks to obtain incorporation rates of approximately 90% . (see Fig. 1 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html) Every single day the worms were scored for growth, fertility, and presence of food. For the SILAC experiment, light wild-type and light his-24(ok1024) mutant animals were cultured in liquid medium in triplicates.
Frozen worms were lysed in a buffer containing 4% SDS, 100 mM Tris-HCl, pH 7.6, and 0.1 M dithiothreitol (DTT) as described previously (58). Briefly, protein lysates were incubated at 95°C for 5 min and sonicated and after centrifugation, and protein concentration was measured by a Bradford assay. According to the protein concentration, equal amounts from the light and heavy samples were mixed. In-solution digest with the protease LysC was performed as described previously (1). Samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a linear trap quadrupole (LTQ)-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray source (Proxeon) and coupled online to a nanoflow high-performance liquid chromatograph (HPLC) (Proxeon) (35). Up to 15 of the most intense ions in each full MS scan were fragmented and analyzed in the linear ion trap (38). Raw data were processed by MaxQuant, version 14.10 (9), with a maximum mass deviation of 7 ppm for MS scans and 0.5 Da for MS/MS scans. For protein identification, the data were searched by the MASCOT search engine (version 2.2.2) against the NCBI database of C. elegans containing forward and reversed protein sequences. On protein and peptide levels, the maximum false discovery rates (FDRs) were set to 1%. Quantification and statistical analysis were performed with Perseus (version 1.1.1.9).
Protein extraction, purification, and identification of HIS-24K14me1.
Frozen animals were extracted with 5% (vol/vol) HClO4, and the proteins were precipitated with 33% Cl3CCOOH as described previously (59). Proteins were separated by SDS-PAGE, using NuPAGE Novex Bis-Tris 4 to 12% gels (Invitrogen, Carlsbad, CA), and the gel was stained with Coomassie blue using a Colloidal Blue staining kit (Invitrogen). Protein bands were subjected to a standard in-gel trypsin digestion protocol (51). The resulting peptide mixtures were desalted using in-house made C18 Stage tips (42), vacuum-dried, and reconstituted in 0.02% trifluoroacetic acid (TFA) prior to the analysis. Peptide mixtures were separated by online reversed-phase nanoscale capillary liquid chromatography and analyzed by electrospray tandem mass spectrometry using an Orbitrap instrument as described previously (59). The data were searched against Wormbase 200 using the MASCOT search engine (Matrix Science, United Kingdom). The maximal mass deviation of parental ions was set to 7 ppm, and that for fragment ions was 0.5 Da. The maximum peptide and protein false discovery rates were set to 0.01.
Generation of a specific antibody directed against HIS-24K14me1.
The synthetic peptide HIS-24-K14me1 [N-CSDSAVVAAAVEP-K(me)-VPKAKAAK-C] was chemically synthesized and coupled with sulfo-maleimidobenzoyl-N-hydroxysuccinimide ester (Squarix biotechnology GmbH, Marl, Germany) to bovine serum albumin (BSA). Five hundred micrograms of antigen was used for immunization of three rabbits in a series of three injections. Antigen injections and resulting antiserum collections were performed by Charles River.
Western blot and dot blot analyses.
C. elegans lysates were prepared and analyzed by Western blotting as previously described (26, 27). For dot blots synthetic HIS-24 peptides monomethylated at K14 (residues 2 to 22) or unmethylated (residues 2 to 22) were used. A HIS-24 peptide spanning amino acids 196 to 210 (AAKKAAKPAAKA) and BSA served as a control. Membranes were incubated with anti-HIS-24 antibodies directed against the C terminus at 1:10,000 and anti-HIS-24K14me1 at 1:1,000 dilution.
Expression of recombinant HPL-1 and HPL-2 proteins.
The 6×His-HPL-1, 6×His-HIS-24, and pGEX HPL-2a plasmids were expressed in E. coli BL21(DE3), and the recombinant proteins were used for the peptide pulldown assay and/or Western blot analysis.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR).
RNA was isolated as previously described (7). The cDNA was amplified from total RNA of the wild type and the Plag-2::GFP::unc-54 3′ UTR strain (where UTR is untranslated region) in a his-24−/− mutant background using reverse transcriptase SuperScript III (Invitrogen), according to the manufacturer's instruction. Quantification was normalized to actin (act-4) RNA levels, and the sequence of the primers was obtained from previously published data (40, 41, 62).
Microarray analysis and quantitative PCR (qPCR).
In brief, for the microarray studies, 80 to 100 L4 and young adult worms raised at 21°C were used. Duplicate biological replicates in TRIzol were quickly sonicated and RNA was extracted using the standard TRIzol method. Microarray analysis was carried out using the a Low RNA Input Linear Amplification Kit Plus, One Color protocol (catalog number 5188-5339; Agilent Technologies, Inc.). RNA was labeled (monocolor experiment) and hybridized to the C. elegans 4-by-44,000 design array from Agilent Technologies (015061). Quantity and Cy dye incorporation rates of the generated target material were measured using a NanoDrop ND-100 spectrophotometer. Washing and staining of the arrays were done according to the manufacturer's recommendation. Cy3 intensities were detected by one-color scanning using an Agilent DNA microarray scanner (G2505B) at 5-μm resolution. Scanned image files were visually inspected for artifacts and then analyzed. Intensity data were extracted using Agilent's Feature Extraction (FE) software, version 9.5, and analyzed using the Limma package (53) of Bioconductor (21).
All data discussed in this paper were generated conforming to the MIAME guidelines and have been deposited in NCBI's Gene Expression Omnibus.
The microarray data analysis consisted of the following steps: (i) between-array normalization, (ii) global clustering and principal-component analysis (PCA), (iii) fitting the data to a linear model, (iv) detection of differential gene expression, and (v) overrepresentation analysis of differentially expressed genes. Quantile normalization was applied to the log2-transformed intensity values as a method for between-array normalization to ensure that the intensities had similar distributions across arrays (24). For cluster analysis we used a hierarchical approach with the average linkage-method. Distances were measured by the calculation 1 − Pearson's correlation coefficient. To estimate the average group values for each gene and assess differential gene expression, a simple linear model was fitted to the data, and group-value averages and standard deviations for each gene were obtained. To find genes with significant expression changes between groups, empirical Bayes statistics were applied to the data by moderating the standard errors of the estimated values (3). P values were obtained from the moderated t statistic and corrected for multiple testing with the Benjamini-Hochberg method (4). The P value adjustment guarantees a smaller number of false-positive findings by controlling the false discovery rate (FDR). For each gene, the null hypothesis, that there is no differential expression between degradation levels, was rejected when its FDR was lower than 0.05. To find overrepresented functions (as represented by Gene Ontology terms [3] and Wormbase), we used DAVID (http://david.abcc.ncifcrf.gov). qPCR was performed using an iCycler iQ Multi-Color real-time PCR detection system (Bio-Rad) cycler. The data of qPCR were normalized to act-1. Primer sequences are available on request.
Immunoprecipitation.
Mixed populations of L4 and adult wild-type worms carrying the hpl-1::gfp transgene and wild-type or hpl-1 mutant animals carrying the hpl-2::gfp transgene were homogenized (6). About 1.5 mg of total precleared protein was incubated with the specific GFP-Trap A beads (Chromotek, Germany) at 4°C overnight. Next, the complexes were washed six times with PD150 buffer for 5 min at 4°C (20 mM HEPES [pH 7.9], 150 mM KCl, 0.2% Triton X-100, 1× protease inhibitor [Roche], 20% glycerol). Finally, the immunoprecipitated proteins were resolved on gradient Bis-Tris gels (4 to 12%; Bio-Rad) and subjected to Western blotting with antibodies against HIS-24K14me1 (1:10,000 dilution), H3 (1:20,000 dilution) or HPL-1 and HPL-2 (1:2,000 dilution).
Immunofluorescence analysis.
Wild-type worms and his-24(ok1024) mutant worms were fixed with 1.85% paraformaldehyde and stained as previously described (26). The dissected gonads and worms were stained with the specific antibody raised against HIS-24K14me1 diluted 1:500 or 1:100. Alexa Fluor 488–goat anti-rabbit (Molecular Probes) or Chromeo488 (Active Motif Chromeon GmbH) secondary antibodies attached to a secondary antibody (goat anti-rabbit IgG; Dianova) were used at dilutions 1:500. The samples were then mounted with Vectashield and analyzed using a Leica SP5 laser scanning microscope. Images were acquired as a series of 0.5-μm-thick optical sections, processed, and then merged. The samples for STED imaging were mounted with Mowiol. Simulated emission depletion (STED) microscopy of embryos and adult hermaphrodites of wild-type worms was performed using a custom-build fast-beam-scanning setup based on a continuous-wave (CW) fiber laser (34).
Immunoelectron microscopy analysis.
Ultrathin cryosections stained with HIS-24K14me1 at a the dilution 1:80 were prepared as previously described (57) and examined with a Philips CM120 electron microscope and a TVIPS charge-coupled-device camera system.
ChIP-qPCR.
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (13) with some modifications. Briefly, L4 worms and adults were homogenized in ice-cold lysis buffer with protease inhibitors and 0.1% Triton X-100 using an equal volume of cubic zirconium beads (11079105z; BioSpec Products). The mixture was vortexed for 10 min at 4°C and then sonicated using a Branson 250 Sonifier. To ca. 1.8 mg/ml of total protein, determined by the Bradford assay, ca. 50 μl of a slurry of GFP-Trap A beads (Chromotek, Germany) was added. ChIP assays were quantified by real-time PCR using SYBR green PCR Master Mix (Applied Biosystems) and an iCycler iQ Multi-Color real-time PCR detection system (Bio-Rad). All reactions were analyzed in triplicate. Primer sequences are available on request.
Peptide pulldowns from total worm lysates.
Peptide pulldown assays were performed as previously described (61). For H3 and HIS-24K14 peptide binding experiments, 10 μg of the biotinylated peptide was first coupled to streptavidin-agarose beads (Pierce). The H3 peptides were methylated at K9 (amino acids [aa] 1 to 20) and HIS-24 peptides monomethylated at K14 (residues 2 to 22) or unmethylated (residues 2 to 22). The peptides were generated by Squarix (Germany). Worm lysates were incubated overnight with the beads, washed seven times with PD-150 buffer (20 mM HEPES, pH 7.9, 150 mM KCl, 0.2% Triton X-100, 1× protease inhibitor [Roche], 20% glycerol), and bound proteins were separated on SDS-PAGE gels in Laemmli buffer.
Infection with Pseudomonas aeruginosa (PA14).
A total of 90 worms of each strain at the L4 stage (30 L4 worms per plate) were infected at 25°C with P. aeruginosa PA14. The surviving worms were counted every day and every day seeded on new plates under a dissecting microscope. Plates with OP50 bacteria were used as a control.
Survival assay and infection with B. thuringiensis.
A total of 120 worms of each strain at the L4 stage (20 L4 worms per plate) were transferred onto small NGM agar plates (without peptone) and infected at 21°C with B. thuringiensis (BT-18679, a very pathogenic strain). B. thuringiensis stock was used at a 1:50 dilution in an E. coli OP50 solution with final concentration of 1 ×109 E. coli cells/ml. Survival was scored after 24 h as the number of animals responding to touch. Plates with a nonpathogenic strain of B. thuringiensis (DSM-350) served as a control. Statistical significance was determining using a log rank test.
For Western blot analysis a total of 60 worms of each strain BT-18679 and DSM-350 12 h and 24 h after infection were collected and analyzed using antibodies against HIS-24K14me1 and against H1 variants (for more information see reference 27), HPL-1, HPL-2, and H3 (Abcam). For the analysis of GFP levels before and after infection, a total of 100 transgenic worms carrying the his-24::gfp transgene in the his-24−/− mutant background were collected and analyzed using an antibody against GFP diluted 1:20,000 (Roche). For the GFP induction assay, 120 worms at the L4 stage were infected and analyzed 12 h later for GFP signal changes under a 40× objective using a Leica DMI 6000B fluorescence microscope. ChIP-qPCR was performed using 120 infected or uninfected worms.
Thermotolerance assay.
A total of 100 L4-day-old, synchronous, adult hermaphrodites of each strain were transferred to small prewarmed (35°C) NGM agar plates and incubated at 35°C for 13 h. Survival was scored every hour as the number of animals that responded to touch. For analysis of survival, worms were shifted to 22°C. Worms that did not respond and failed to display motility or pharyngeal pumping were scored as dead. Statistical significance between strains was determining using a log rank test.
Osmotic stress assay.
Sixty L4 stage, synchronous worms of each strain were placed on small NGM agar plates with a high concentration of salt (500 mM NaCl). High-salt plates were seeded with OP50 bacteria 1 day prior to the experiment to eliminate an increase of the salt concentration due to evaporation. Animals were assayed by touch during a 10-min interval. Worms failing to respond and to show pharyngeal pumping were scored as dead. Statistical significance between strains was determined using a two-tailed t test.
Generation of EC673.
The his-24 gene was amplified by PCR from cosmid clone M163 (kindly provided by Alan Coulson, The Sanger Center, Cambridge, United Kingdom) with the primers MJ05 (GGGGTACCCCCAGCCACCCACCGC) and MJ08 (GGATTCGTCTAGCGCACGCCTCATTTG).
The PCR was done with a proofreading enzyme (Expand High Fidelity PCR System; Boehringer). The EcoRI-StuI cut restriction fragment of this PCR product was inserted into pECFP-N1 (Clontech) using the EcoRI and SmaI restriction sites of the polylinker. The resulting construct contained 5 kb of his-24 5′ noncoding sequence and represents a fusion of full-length his-24 coding sequence to the N terminus of the enhance cyan fluorescent protein (ECFP) protein. An extrachromosomal array was created by germ line transformation using a previously described technique (33). his-24::CFP plasmid DNA with a concentration of 20 ng/μl was injected into the wild-type strain N2. An integrated transgene was derived by exposure to 38-Gy, 100-keV X-ray using a 0.5-mm thick copper filter and subsequent clonal selection and outcrossing.
The hpl-1 gene was amplified by PCR from cosmid clone K08H2.6 using the primers ESMG57 (GGGGTACCTCAATAAAGCGACGACAGATGTAAACA) and ESMG59 (CGGGATCCGCGCTCATTCCTCCTGGGATGGTTGG). The resulting 4.6-kb PCR product was cut with KpnI and BamHI and cloned into pEYFP-N1 (Clontech). The resulting plasmid was supplemented with the C. elegans wild-type unc-119 gene that was obtained as an XbaI-HindIII fragment from plasmid pMM016 and inserted into the NheI and HindIII sites. This plasmid was integrated into the C. elegans strain DP38 unc-119(ed3) using particle bombardment. Both independently integrated transgenes were merged by crossing, resulting in EC673 eeIs611[unc-119(+) hpl-1::eyfp]; eeIs009[his-24::cfp].
FRET analysis.
Fluorescent resonance energy transfer (FRET) microscopy of the strain EC673 eeIs611[unc-119(+) hpl-1::eyfp]; eeIs009[his-24::cfp] was performed with a Zeiss LSM510-META inverted confocal laser scanning microscope using the spectral detector in a virtual filter mode. CFP emission was detected from 471 to 503 nm; FRET emission and enhance yellow fluorescent protein (EYFP) emission were detected from 524 to 567 nm. We used 457-nm excitation for CFP and FRET and 514-nm excitation for EYFP in a two-track configuration. A C-Apochromat 63×/1.2W objective was used. Spectral references for filter bleed-through were taken from myo-2::cfp and myo 2::eyfp transgenic C. elegans strains, respectively. FRET was calculated with the PixFRET plugin (17) of ImageJ (W. S. Rasband, U. S. National Institutes of Health, Bethesda, MD [http://rsb.info.nih.gov/ij/]) using a Gaussian blur of 1.0, and a threshold of 0.8. BTdon (BTacc) was determined as 0.485 (0.742).
Microarray data accession number.
The microarray data were deposited in the Gene Expression Omnibus (GEO) of NCBI under accession number GSE 25714.
RESULTS
Lack of HIS-24 leads to induction of infection-inducible proteins.
Worms lacking HIS-24 display a mild phenotype resulting in increased embryonic lethality and reduced fertility (27). As the linker histone variants H1c, H1d, and H1e compensate the loss of H1(0) in mice, we decided to investigate possible compensatory effects between the linker histone HIS-24 and the other seven H1 variants in C. elegans using a SILAC approach (16, 36, 52).
We prepared SILAC samples by mixing equal amounts of wild-type worms bearing the heavy isotope label [13C6]lysine with unlabeled his-24(ok1024) and unlabeled control animals (data not shown), enabling us to directly compare protein levels in both samples.
During the observation period of 2 weeks at 21°C (two worm generations), the labeling efficiency of L4 and adult worms was about 90% (see Fig. 1B at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). All animals developed normally, and SILAC labeling had no obvious effects on growth, behavior, or fertility. Thus, SILAC labeling with [13C6]lysine bacteria does not lead to abnormalities in C. elegans growth.
In total we quantified 1,217 proteins which were identified by at least two peptides. Of these, 376 proteins were differentially regulated in his-24(ok1024) animals (P < 0.05). The absence of HIS-24 did not affect protein levels of the other histone variants (Fig. 1A).
Fig 1.

HIS-24 regulates expression of infection-inducible genes. (A) Volcano plot shows protein expression between the wild type and his-24 null mutant. Log2 protein ratios are plotted against negative log10 P values performed from triplicates. Red dots represent selected infection response proteins (Table 1). The green dot represents an absence of HIS-24 in null mutant animals. (B) Venn diagrams showing the number of genes regulated by HIS-24 and HPL using a whole-genome microarray. (C) Pie charts representing the predicted biological functions of the overlapping genes. (D) Heat maps. Each column is a sample showing significant coordinate induction (red) or repression (blue) of genes regulated by HIS-24 or HPL, respectively (FDR < 0.05). The values are z-score scaled over the rows. WT, wild type.
Consistent with the his-24 phenotype (27), SILAC quantification revealed differential expression of proteins involved in embryonic development, metabolic processes, and reproduction. In addition, we observed differential expression of proteins linked to the innate immune response (Fig. 1A; Table 1). Strikingly, 31% (117 of 376) of the regulated proteins in his-24(ok1024) mutant animals are predicted to contribute to antimicrobial defense (37, 40, 41, 49, 60, 62). We identified common response proteins including metabolic enzymes such as the alcohol dehydrogenase SODH-1, the fatty acid-coenzyme A (CoA) synthetase ACS-11,-17, and the fatty acid/retinol binding protein FAR-7, as well as proteases, defense-related proteins (MAOC-1) and heat shock proteins (HSP-1, -3, -4, -7, and DAF-21/HSP90) (Table 1). Furthermore, we observed the induction of oxidative stress proteins such as glutathione S-transferases (GST) and C-type lectin CLEC-63 (Table 1), which are infection-inducible genes in C. elegans (60).
Table 1.
Selected functional infection-inducible proteins regulated by HIS-24a
| Group and Wormbase cosmid ID | Gene symbol | Description | Expression site(s) | P value (SILAC) | Fold induction (microarray) [FDR < 0.05]) | Pathogen(s)b |
|---|---|---|---|---|---|---|
| Upregulated proteins | ||||||
| Y48E1B.10 | gst-20 | Encodes a predicted glutathione S-transferase | Muscle, hypodermis, neurons | 0.04044 | 1.0 | E. carotovora, P. luminescens |
| F11G11.2 | gst-7 | Intestine | 0.00276 | 0.4 | P. aeruginosa | |
| Y39B6A.20 | asp-1 | Aspartyl protease | Intestine | 0.00162 | 0.5 | E. carotovora, E. faecalis, P. luminescens |
| T18H9.2 | asp-2 | NDd | 0.00294 | 0.0 | E. carotovora, E. faecalis, P. luminescens | |
| K09F5.3 | spp-14 | Saposin-like protein | ND | 0.04032 | 0.5 | E. carotovora, P. luminescens |
| F35C5.6 | clec-63 | C-type lectin | ND | 0.00068 | 0.0 | M. nematophilum, E. carotovora, E. faecalis, P. luminescens |
| ZC434.8 | NAc | Probable kinase | ND | 0.00171 | 0.2 | S. marcescens |
| ZK1055.7 | NA | NA | Intestine | 0.00238 | 0.4 | P. aeruginosa |
| E04F6.3 | maoc-1 | MAO-C-like dehydratase domain | Intestine | 0.00620 | 0.5 | P. aeruginosa, E. carotovora, E. faecalis, S. marcescens |
| ZK6.10 | daf-19 | Abnormal Dauer formation | Muscles, intestine | 0.00895 | 1.2 | P. aeruginosa |
| C47E8.5 | daf-21/ hsp90 | Intestine | 0.00783 | 0.5 | P. aeruginosa | |
| Downregulated proteins | ||||||
| F41C3.3 | acs-11 | Fatty acid-CoA synthetase | Hypodermis, intestine | 0.01479 | −0.8 | ND |
| C46F4.2 | acs-17 | Hypodermis, intestine | 0.00342 | −1.0 | P. aeruginosa | |
| K05B2.3 | if-a4 | Intermediate filament protein | Rectum, some neurons, excretory cell | 0.00622 | −1.4 | M. nematophilum |
| K01A2.2a | far-7 | Fatty acid/retinol binding protein | Hypodermis, excretory cells | 0.00727 | −1.3 | M. nematophilum |
| K12G11.3 | sodh-1 | Sorbitol/alcohol dehydrogenase | ND | 0.02172 | −2.5 | M. nematophilum, P. aeruginosa |
| C05E4.9 | gei-7 (icl-1) | Isocitrate lyase/malate synthase | ND | 0.00419 | −1.1 | E. carotovora, E. faecalis |
| F54D8.3 | alh-1 | Aldehyde dehydrogenase | Intestine, nervous system | 0.00017 | −0.3 | P. aeruginosa, E. carotovora, E. faecalis, S. marcescens |
| F36H1.6 | alh-3 | ND | 0.00259 | −0.1 | P. aeruginosa, E. carotovora, E. faecalis, S. marcescens | |
| T08B1.3 | alh-5 | ND | 0.04017 | −1.9 | P. aeruginosa | |
| F43E2.5 | msra-1 | Methionine sulfoxide-S-reductase | Intestine, hypodermis, neurons | 0.02069 | −2.2 | P. aeruginosa, E. carotovora, E. faecalis, S. marcescens |
| T10H9.5 | pmp-5 | Peroxisomal membrane protein | Intestine, hypodermis | 0.02343 | −2.6 | P. aeruginosa |
| F52H3.7 | lec-2 | Galectin family member | Muscle | 0.01086 | −0.8 | E. faecalis |
| Y38F1A.6 | NA | Probable alcohol dehydrogenase | Muscle, intestine | 0.00423 | −1.2 | P. aeruginosa, E. carotovora, E. faecalis, S. marcescens |
| C55F2.1b | NA | Probable AICAR transformylase | ND | 0.00090 | −0.8 | P. aeruginosa, E. faecalis, S. marcescens |
Brief annotation and expression information about representative response proteins detected in the his-24(ok1024) mutant strain using the SILAC approach are given. The fold induction of genes obtained using microarray analysis is also given.
Pathogens (39, 41, 42, 49, 55, 60, 62) that influence the regulation of the given genes (taken from WormBase) are listed. E. carotovora, Erwinia carotovora; E. faecalis, Enterococcus faecalis; P. luminescens, Photorhabdus luminescens; P. aeruginosa, Pseudomonas aeruginosa; M. nematophilum, Microbacterium nematophilum; S. marcescens, Serratia marcescens.
NA, not available.
ND, not determined.
HIS-24, HPL-1, and HPL-2 coregulate the expression of genes linked to immune response.
To analyze whether HIS-24 and the two HP1 variants HPL-1 and HPL-2 regulate the expression of common genes, we carried out whole-genome expression profiling of each single null mutant strain. Among the ∼16,000 target probes assayed, we identified a set of 273 genes commonly regulated by HIS-24, HPL-1 and HPL-2 (Fig. 1B and C). Remarkably, significantly more genes are commonly downregulated than upregulated in the absence of HIS-24 and HPL (1.1% of 15,612 and 0.6% of 15,287, respectively; FDR < 0.05) (Fig. 1B and C). Surprisingly, among these regulated genes we find a significant number of genes induced by infection with Pseudomonas aeruginosa, Microbacterium nematophilum and Drechmeria coniospora, including glycine- and tyrosine-rich antimicrobial peptides (AMPs) and members of the (neuropeptide-like proteins (NLPs) and Caenacin (CNC) families, as well as fungus-induced protein (fip-1), FIP-related proteins (fipr-17), lysozymes (lys), C-type lectins (clec), thaumatins (thn), metridin with an ShK domain, glutathione S-transferase (gst), abf-2 (Ascaris suum antibacterial factor related [ASABF]), genes with a CUB domain, and genes involved in lipid metabolism and stress response (37, 40, 41, 49, 60, 62) (see Tables 1 and 2 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html).
The hpl-2(tm1489) transcriptome deviates most from the transcriptional profile of his-24(ok1024) and hpl-1(tm1624) single mutants (Fig. 1D). Using quantitative PCR (qRT-PCR) analysis, we confirmed the differential expression of genes selected to represent functional infection response groups identified by data mining (see Fig. 2 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). A good correlation between microarray data and qRT-PCR expression values was observed in all microarray sets.
By SILAC, we also detected significant changes in the protein level of representative infection response proteins, including DAF-19, ACS-11, IFA-4, FAR-7, MSRA-1, PMP-5, SODH-1, and GEI-7, in animals lacking HIS-24. This independently verifies the microarray data (Table 1).
The relatively small number of proteins detected by SILAC (169 of 376; P < 0.05) and regulated at the transcriptional level in the microarray analysis of the his-24(ok1024) mutant (169 of 579; FDR < 0.05) can be explained by the fact that only a small fraction of mRNA is translated into protein. In addition, microarray measurements are not strictly quantitative (Fig. 2). Strikingly, 50 (30%) of the 169 proteins found in the SILAC and regulated at the transcriptional level in the microarray studies are induced by infection (see Table 3 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html).
Fig 2.
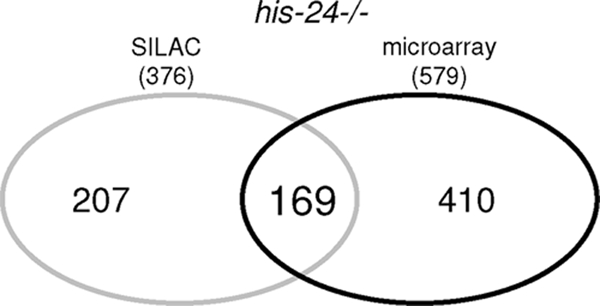
Venn diagram representing overlap between data obtained from SILAC and microarray analysis. A total of 169 gene products are represented in both groups (P < 0.05); of these 50 are genes induced by infection (see Table 3 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html).
HIS-24 recruits HPL proteins to the promoters of genes involved in the defense response.
Next, we tested directly whether HIS-24 binds in vivo and recruits HPL proteins to the promoters of antimicrobial genes to influence their transcription. In our microarray and SILAC analyses, maoc-1 and daf-21/hsp90 genes are upregulated in his-24 and hpl mutant backgrounds. Both proteins are involved in the defense response. MAOC-1 is predicted to function in peroxisomal fatty acid beta-oxidation (49). Using ChIP-qPCR analysis, we observed that HIS-24 binds directly to the promoters and gene bodies of the maoc-1 and daf-21/hsp-90 genes (Fig. 3A). No binding was detected on the promoter of the thn-5 gene, which belongs to pathogenesis-related proteins. The enrichment of HIS-24 was detected before infection and strongly reduced after infection. Interestingly, the absence of his-24 activity led to a decrease in HPL-1 and HPL-2 levels on the maoc-1 gene promoter, suggesting that a linker histone and HP1 family proteins may cooperate in coordinate regulation of the gene transcription (Fig. 3B).
Fig 3.

HIS-24 and HPL associate with promoters of antimicrobial genes. (A) Quantitative ChIP using extracts prepared from his-24::gfp transgenic worms before and after B. thuringiensis infection. Primer sets were directed to the maoc-1 and daf-21/hsp90 genes detected in SILAC and microarray analyses (Fig. 1 and Table 1). The thn-5 promoter serves as a negative control. (B) ChIP assay was carried out to assess HPL::GFP in his-24+/+ (HIS-24 present) and his-24−/− (HIS-24 absent) animals at the maoc-1 promoter. All results are quantified by real-time PCR and normalized to binding by control IgG antibody. Error bars indicate standard deviations (n = 2).
C. elegans HPL-1/HP1 interacts with HIS-24 when monomethylated at lysine 14.
To verify and visualize the interaction of HIS-24 with HPL proteins in C. elegans, we generated his-24::cfp; hpl-1::eyfp double transgenic worms, and performed FRET analysis (Fig. 4A and B; see Fig. 3 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). We observed an interaction between HPL-1 and HIS-24 during embryonic development and in adult worms.
Fig 4.

HIS-24K14me1 interacts with HPL-1. (A) FRET analysis of HIS-24::CFP and HPL-1::YFP in somatic nuclei. (B) FRET efficiency. Interaction between HIS-24 and HPL-1 is shown in red; lack of interaction is shown in blue. (C and D) Distribution of HIS-24K14me1 in embryonic nuclei (C) and in the polyploidal cell of the intestine (D) using STED. Scale bar, 2 μm. (E to G) Electron micrograph of HIS-24K14me1 in somatic nuclei. The right panel shows two different images of two nuclei. Arrows point to HIS-24K14me1 labeling (10-nm gold) of euchromatic regions (eu; less-dense regions); arrowheads indicate the positions of HIS-24K14me1 in the nucleolus. he, heterochromatin (electron-dense and dispersed regions); env, nuclear envelope; nu, nucleolus. Scale bar, 1 μm.
As previously reported (10, 29), human H1 is posttranslationally modified at lysine position K26 and interacts in a methylation-dependent manner with HP1. Consistent with this model, we mapped new modification sites of H1 variants in C. elegans using mass spectrometry and identified a single methylation site of HIS-24 at lysine (K) position 14 (see Fig. 4A and B at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). Moreover, HIS-24 is the only one of the eight linker histones in C. elegans that exclusively possesses a methylation site.
For immunodetection of HIS-24K14me1 within the nuclear interior with high resolution, we used STED (simulated emission depletion) microscopy, which revealed structures not clearly observable with standard confocal microscopy (Fig. 4C; see Fig. 4E at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). We identified HIS-24K14me1 in foci located close to the nuclear membrane and the nucleolus. To gain more insight into the localization of HIS-24K14me1 within chromatin domains, we performed immunoelectron microscopy. HIS-24K14me1 was localized to electron-dense regions (heterochromatin) as well to less-electron-dense regions (euchromatin) (Fig. 4E to G). These observations are consistent with the HIS-24 dual binding behavior (activation and/or repression) that we documented using microarray or SILAC approaches.
The HIS-24K14me1 antibody specifically recognizes native, but not bacterially expressed, HIS-24, suggesting that methylation of HIS-24K14 occurs in vivo (Fig. 4C and D at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). Antibodies raised against HIS-24K14me1 were able to pull down native HPL-1 and HPL-2 (Fig. 5A and B). To ask whether HPL proteins directly interact with HIS-24K14me1, we expressed HPL-1 and HPL-2 in E. coli and performed a peptide pulldown assay using an unmodified synthetic peptide of HIS-24 spanning amino acids 2 to 22 and the same peptide monomethylated at lysine 14. We detected specific binding of bacterially expressed HPL-1 to the HIS-24K14me1 synthetic peptide but not to the unmodified peptide (Fig. 5F). In contrast, HPL-2 failed to bind either modified or unmodified peptides (Fig. 5E). Consistent with microscopic observations showing a partial overlap between HPL-1 and HPL-2 foci in live worms (47), HPL-1 is able to pull down HPL-2 (Fig. 5C). Furthermore, we failed to detect HPL-2::GFP binding to HIS-24K14me1 in a hpl-1(tm1624) mutant background, indicating that HPL-2, physically associates with HIS-24K14me1 through its association with HPL-1 (Fig. 5D).
Fig 5.

HPL-1 physical interacts with HIS-24 when monomethylated at lysine 14. (A and B) HPL-1 and HPL-2 coprecipitate HIS-24K14me1. An enrichment relative to input of methylated HIS-24 compared to total HIS-24 is observed for HPL-1 but not HPL-2 (no enrichment). (C and D) HPL-1 coprecipitates HPL-2, but HPL-2 does not pull down HIS-24 in an hpl-1(tm1624) mutant background. (E and F) Recombinant HPL-1 protein specifically recognizes the HIS-24K14me1 region but neither H3K9me2 nor H3K9me3 in peptide pulldown assays. Recombinant HPL-2 does not interact with the HIS-24K14me1 peptide. α, anti; IP, immunoprecipitation.
Decreased resistance of his-24(ok1024) animals to Gram-positive bacterial infection.
In order to investigate the functional impact on immune-relevant gene regulation by HPL proteins and HIS-24, we tested the resistance of hpl-1(tm1624), his-24(ok1024), and hpl-2(tm1489) single mutants, hpl-1(tm1624) his-24(ok1024) and hpl-2(tm1489); his-24(ok1024) double mutants, and hpl-2(tm1489); hpl-1(tm1624) his-24(ok1024) triple mutants to infection with the Gram-negative bacterium P. aeruginosa (PA14) and the Gram-positive bacterium B. thuringiensis. hpl-1, hpl-2, and his-24 double and triple mutant combinations resulted in temperature-sensitive nonlethal defects. his-24(ok1024) mutant animals showed increased sensitivity to B. thuringiensis infection (Fig. 6A). This effect is specific to the HIS-24 linker histone variant since inactivation of an additional linker histone, hil-3, had no effect on survival following infection. These results suggest a critical role for linker histone HIS-24 in the innate immune response. Surprisingly, hpl-1(tm1624) and hpl-2(tm1489) single mutants survived significantly longer on B. thuringiensis plates than the wild type (Fig. 6A). In the case of PA14 infection, we did not observe any significant changes in the survival of his-24(ok1024), hpl-1(tm1624), or hpl-2(tm1489) single mutant strains compared to that of the wild type (data not shown). Finally, the hpl-2(tm1489); his-24(ok1024) and hpl-2(tm1489); hpl-1(n4317) double and hpl-2(tm1489); hpl-1(tm1624) his-24(ok1024) triple mutant animals showed increased resistance to B. thuringiensis infection (Fig. 6A). The absence of HPL-2 protein in a his-24(ok1024) mutant neutralized the negative effect of HIS-24 deficiency in the double and triple mutants during B. thuringiensis infection, suggesting that additional factors or mechanisms must influence the interplay between HPL-2 and HIS-24.
Fig 6.

Direct role of HIS-24 and HPL in control of microbial resistance. (A) Survival assay of mutant animals in infection experiments using the Gram-positive bacterium B. thuringiensis. (B) Survival assays of transgenic worms carrying HIS-24::GFP in the his-24+/+ or his-24−/− background. The presence of the modified form of HIS-24::GFP (HIS-24A14::GFP) did not influence the survival of his-24−/− mutant animals on B. thuringiensis plates. Data are mean survivals (± standard errors of the mean) relative to the wild type. *, P < 0.0001, versus the wild type.
Overexpression of HIS-24 increases the resistance to B. thuringiensis infection.
We next asked whether B. thuringiensis infection influences the survival of transgenic animals overexpressing gfp-tagged hpl-1, hpl-2, or his-24. We did not observe significant changes in the survival of hpl-1::gfp and hpl-2::gfp strains compared to wild type animals (Fig. 6B) or any changes in the GFP localization pattern (data not shown). However, absence of HIS-24 in hpl-1:.gfp and hpl-2::gfp transgenic worms slightly decreased survival (Fig. 6B). Remarkably, we observed that his-24::gfp transgenic worms showed a significant increase in resistance to bacterial-infection. This effect is dependent on K14 methylation, as gfp::his-24K14A his-2414K-A::gfp transgenic animals in which this residue has been mutated to a nonmethylatable alanine residue showed decreased resistance (Fig. 6B). Notably, the expression of transgenic HIS-24::GFP in the presence of endogenous HIS-24 had a significantly stronger effect on survival than the expression of HIS-24::GFP in the absence of endogenous HIS-24 (his-24−/− mutant background) (Fig. 6B).
We next asked whether HIS-24 localization changes upon infection. In intestinal cells of uninfected animals, HIS-24::GFP was diffusely distributed in the nucleus (Fig. 7A). Following infection, in 45% of worms (n = 120) HIS-24::GFP was found to be present in both the nucleus and cytoplasm as well as in the cytosol (20% of 120 infected worms) (Fig. 7B and C). The observed changes in cellular localization depend on HIS-24 methylation since the subcellular localization of HIS-24K14A::GFP was unaltered upon infection (Fig. 7D and E). Using antibodies specific to HIS-24K14me1, we observed that the levels of this modified form significantly increased upon infection in extracts prepared from his-24::gfp; his-24−/− animals (Fig. 7F). In contrast, using GFP antibodies, we failed to detect any changes in the expression levels of HIS-24::GFP. Altogether, these results suggest that the cytoplasmic pool of HIS-24::GFP detected in intestinal cells following infection consists mostly of the modified form of HIS-24 (HIS-24K14me1). We speculate that this form may be released into the cytoplasm to provide protection against penetration by microorganisms into intestinal cells (Fig. 7F).
Fig 7.

Subcellular localization of HIS-24::GFP in his-24−/− mutant background after B. thuringiensis (BT) infection. (A) Expression of HIS-24::GFP in intestinal nuclei before infection. (B) After 12 h of infection HIS-24::GFP was observed in both the cytoplasm and the nucleoplasm of intestinal cells. (C) HIS-24::GFP signal was detected in cytosol of the intestine (cytosolic signal). (D and E) Nuclear expression of HIS-24K14A::GFP in a B. thuringiensis mutant background before and after infection. (F) B. thuringiensis infection induces enrichment of HIS-24K14me1. Immunoblotting was performed with antibodies directed against HIS-24K14me1 and GFP as a loading control using his-24::gfp lysates in a his-24−/− mutant background before and after infection. Infection specifically induces enrichment of exogenous HIS-24K14me1::GFP but not GFP expression levels. (G) Immunoblots with antibodies directed against HIS-24K14me1 and H3 as a loading control. After infection with a very pathogenic strain, the level of endogenous HIS-24K14me1 is strongly increased. (H) Quantification of the HIS-24K14me1 and H3 signals on the Western blots, respectively. (I) The levels of other H1 variants as well as HPL-1 and HPL-2 did not increase during infection with B. thuringiensis. Histone H1 (named HIL) proteins are detected with the anti-H1 antibody in total lysate of C. elegans. The antibody has a strong preference for HIL-4 (H1.4) but also cross-reacts with other linker histone variants (HIL) (26). OD, optical density.
Bacterial infection enhances HIS-24K14 monomethylation.
We next sought to determine whether infection could affect the methylation of endogenous HIS-24. Notably, the bacterial pathogen Listeria monocytogenes and the Shigella flexneri protein effector OspF induce gene-specific epigenetic modifications at the promoters of an essential subset of genes involved in innate immune response (2, 46).
We found that B. thuringiensis infection enhances considerably the formation of monomethylated histone HIS-24 (2-fold after 12 h and 4-fold after 24 h), whereas infection with the nonpathogenic strain of B. thuringiensis (DSM-350) had almost no effect (Fig. 7G and H). Therefore, bacterial infection strongly influences linker histone methylation. The levels of other linker histone variants as well as HPL-1 and HPL-2 after B. thuringiensis infection were not increased, suggesting a specific role of HIS-24K14me1 in the innate immune response (Fig. 7I).
HIS-24 and HPL proteins regulate stress response.
As the innate immune response is modulated by stress and as HIS-24 and HPL proteins regulate genes predicted to protect from oxidative and other forms of stress (e.g., cyp genes, the mtl-1 metallothionein gene, gst glutathione S-transferase genes, small heat-shock genes, and the mitochondrial sod-3 superoxide dismutase gene), we asked whether his-24 and hpl loss of function leads to a general stress response (see Fig. 5 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html). We exposed mutant strains to osmotic stress (500 mM NaCl) and thermal stress (30°C) (Fig. 8A and B). Tests on hpl-1(tm1624), hpl-2(tm1489), and his-24(ok1024) single mutants showed a significant reduction in survival compared to the wild type after either type of stress. We observed that double and triple mutants were more sensitive to both types of stress than wild-type worms (Fig. 8). However, the absence of hpl-1 in an his-24(ok1024) mutant background seemed to slightly neutralize the negative effect of HIS-24 deficiency. In light of these findings, it is possible that the innate immune response can be also modulated by stress (43), and stress itself may also affect expression of immunity related antimicrobial genes as a side effect.
Fig 8.

Direct role of HIS-24 and HPL in stress response. (A) Correlation between the wild type and mutants in an osmotic stress assay. Mean survivals (± standard errors of the means) relative to the wild type. *, P < 0.0001, versus the wild type. (B) Survival of the mutant animals after heat shock. Mean survivals (± standard errors of the means) relative to the wild type. *, P < 0.0001, versus the wild type.
DISCUSSION
Model of transcriptional regulation by a HPL-1/HIS-24k14me1 complex.
Linker histone H1 is subject to a variety of posttranslational modifications including methylation, which is specifically recognized by the chromodomain of HP1 (10, 29, 59). The exact biological role of H1 methylation and its promiscuous binding to HP1 is poorly understood. Our results suggest that a physical interaction between C. elegans HIS-24K14me1 and the HP1 homologue HPL-1 may contribute to defining a specific state of the innate immune system. However, we cannot exclude that additional global changes in chromatin structure and/or chromatin stability may also affect the expression of genes involved in the stress response, thereby contributing to the altered susceptibility to infection. Our results suggest that linker histone H1 and HP1 family proteins may cooperate in coordinately regulating the innate immune response in metazoans (Fig. 9A). Chromatin-bound HIS-24K14me1 and HPL-1 could regulate chromatin compaction, resulting in a chromatin structure more accessible to trans-acting proteins which may play a causal role in gene expression.
Fig 9.

Simplified functional model of HP1/H1 interaction (C. elegans HIS-24/HPL-1) before and after infection. (A) HIS-24 and HPL-1 bind to the promoters of antimicrobial genes in the wild type (no infection). During infection, levels of HIS-24K14me1 increase, resulting in release of this variant into the cytosol as a part of the immune defense. HPL-1 levels do not change. (B) Multiple sequence alignment (ClustalX) of the N termini of HIS-24 and human linker histones H1.b (H1.2) and H1.d (H1.4).
The cytoplasmic expression of HIS-24K14me1 in intestinal cells after infection suggests that the posttranslationally modified form of HIS-24 may represent an important innate antimicrobial defense against bacteria in the C. elegans intestine. Interestingly, previous studies have suggested a role for the cytoplasmically expressed linker histone H1.2 variant in innate antimicrobial defense in the human gastrointestinal tract (45). Therefore, the function of linker histone as an antimicrobial protein acting in innate defense may be evolutionarily conserved. Despite the fact that at the sequence level linker histones have been shown to evolve relatively fast through evolution, this conserved role in innate immunity may have important implications in evolutionary adaptation to different microbial species. Further studies are clearly needed to better understand the mechanism of action of the modified linker histone variants in the defense against bacterial infection.
The relatively small number of genes identified in our expression profiling of his-24, hpl-2, and hpl-1 single mutant animals suggests that HIS-24 and HPL proteins do not have a major effect on global transcription but instead appear to specifically control through concerted action two different subsets of immune-relevant genes. This may be achieved through interaction with tissue-specific transcription factors to regulate specific genes during infection. A similar mechanism may exist in human CD4+ CD25+ regulatory T (Treg) cells, where the forkhead transcription factor FoxP3 interacts with linker histone H1.5 to modulate interleukin-2 (IL-2) gene expression in the Treg cells (32). To summarize, it appears that H1 histones, including their variants and associated posttranslational modifications, in conjunction with their reader molecules, have a more specialized function than initially presumed.
HPL-1 selectively binds to monomethylated HIS-24K14.
While the interaction of HIS-24/H1 with HPL-1/HP1 is evolutionarily conserved (Fig. 9B), it is considerably different from the well-studied binding of human HP1 to H1K26me2. HPL-1 binding to HIS-24K14me1 may appear somewhat surprising as HP1 family proteins are not known to bind to monomethylated lysines (25). However, significant differences of histone modification patterns among species and potentially species-specific histone modifications as well as novel histone modifications have been observed (20).
HPL-1 not only differs from human HP1 at the sequence level (see Fig. 5 at http://www.mpibpc.mpg.de/home/jedrusik-bode/pub/index.html) but also binds the monomethylated linker histone within the sequence motif of HIS-24K14 (AVEPKVPK) which lacks any homology within the surrounding sequence to either human H1K26 (KARKSAGA) (29) or H3K9 (QTARKSTGG) (25). Although we currently cannot explain why C. elegans HP1 has this unusual binding behavior, our data show that the interaction between methylated histone H1 and HP1 is conserved in metazoans and plays an important role in innate immune gene repression.
Additional layers of HPL complexity.
The observed opposite effects on survival following infection between hpl-2(tm1489) and his-24(ok1024) single mutant worms may arise from the fact that HPL-2 interacts with the H3K9me2,3 chromatin mark, which serves as a bridge for the recruitment of specific corepressors and/or activators (31) (Fig. 5E). Thus, the multiple effector genes whose expression is coordinated by the HPL-2/H3K9me2,3 complex may play an opposite role to HIS-24 to influence immunity. Furthermore, HPL-1 and HPL-2 have redundant functions in postembryonic development (47). A compensatory mechanism dependent on HPL-2, HPL-1, or the HPL-2/HPL-1 complex and independent of HIS-24 could potentially mask HIS-24 function, as in the case of B. thuringiensis infection, where hpl-2(tm1489); his-24(ok1024) double mutant animals unexpectedly show a higher survival rate than the corresponding single his-24(ok1024) mutant strain. It is known that HP1 family proteins participate in the regulation of specific genes via methylation-dependent and independent mechanisms (31). It is conceivable that HPL-1 and HPL-2, which differ in their transcriptional profile (Fig. 1D) as well as in nuclear and tissue specific distribution (47), may be targeted differentially to specific promoters dependent on environmental cues (fungal or bacterial infection, stress), thereby differentially regulating the microbial infection response.
Furthermore, the adaptiveness of the hpl-2(tm1489) mutant animals to infection may also be influenced by transposon spreading, which may allow hpl-2(tm1489) mutant worms to adapt to changes in the environment. Transposons are implicated in protection against acute stress conditions (i.e., response to heat stress and oxidative stress, defense and humoral immune response, and receptors and regulators of transcription by modulation of gene expression) (28). Remarkably, in our microarray analysis we detected upregulation of transposases (F46F2.4, log 2 fold change [log 2 FC] = 1.3; Y17G9B.2, log 2 FC = 1.5) as well as transposons (R03G8.2 and T23B12.10) in animals lacking HPL-2.
Whether the results reported here concerning C. elegans HIS-24 reflect a general mechanism for innate gene regulation through H1 and a HP1/H1 complex in higher eukaryotes remains to be established.
ACKNOWLEDGMENTS
We thank Masoud Bahrami for integrated his-24::cfp transgene, Rania Nakad (Kiel, Germany) for the BT cultures and her help in the establishing the BT-procedure in our group, Albert Rosenberger (Göttingen, Germany) for the bioinformatics, Alejandro Vaquero (Barcelona, Spain), Hinrich Schulenburg, Katja Dierking (Kiel, Germany), Sigrid Hoyer-Fender, Mary Osborn, and Helena Miletic (Göttingen, Germany) for critically reading the manuscript, Gregor Eichele (Göttingen, Germany) for support, and Gunther Jansen (Kiel, Germany) for the help at work with PA14.
Some C. elegans strains used were obtained from the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources. The his-24(ok1024) deletion mutant was provided by the C. elegans Gene Knockout Consortium, which is publicly funded. This work was supported by the German National Funding Agency (JE 505/1-3 to M.J.-B.) and the Max Planck Society (M.J.-B.).
We declare that we have no competing financial interests.
Footnotes
Published ahead of print 14 November 2011
REFERENCES
- 1. Andersen JS, et al. 2005. Nucleolar proteome dynamics. Nature 433:77–83 [DOI] [PubMed] [Google Scholar]
- 2. Arbibe L, et al. 2007. An injected bacterial effector targets chromatin access for transcription factor NF-κB to alter transcription of host genes involved in immune reponses. Nat. Immunol. 8:47–56 [DOI] [PubMed] [Google Scholar]
- 3. Beissbarth T, Speed TP. 2004. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 20:1464–1465 [DOI] [PubMed] [Google Scholar]
- 4. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 57:289–300 [Google Scholar]
- 5. Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheeseman IM, Desai A. 2005. A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci. STKE 266:pl1. [DOI] [PubMed] [Google Scholar]
- 7. Coustham V, et al. 2006. The C. elegans HP1 homologue HPL-2 and the LIN-13 zinc finger protein form a complex implicated in vulval development. Dev. Biol. 297:308–322 [DOI] [PubMed] [Google Scholar]
- 8. Couteau F, Guerry F, Muller F, Palladino F. 2002. A heterochromatin protein 1 homologue in Caenorhabditis elegans acts in germline and vulval development. EMBO Rep. 3:235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367–1372 [DOI] [PubMed] [Google Scholar]
- 10. Daujat S, Zeissler U, Waldmann T, Happel N, Schneider R. 2005. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J. Biol. Chem. 280:38090–38095 [DOI] [PubMed] [Google Scholar]
- 11. De Lucia F, Ni JQ, Vaillant C, Sun FL. 2005. HP1 modulates the transcription of cell-cycle regulators in Drosophila melanogaster. Nucleic Acids Res. 33:2852–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Downs JA, Kosmidu E, Morgan A, Jackson SP. 2003. Suppression of homologous recombination by the Saccharomyces cerevisiae linker histone. Mol. Cell 11:1685–1692 [DOI] [PubMed] [Google Scholar]
- 13. Ercan S, et al. 2007. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nat. Genet. 39:403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fan Y, et al. 2005. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123:1199–1212 [DOI] [PubMed] [Google Scholar]
- 15. Fan Y, et al. 2003. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol. Cell Biol. 23:4559–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan Y, Sirotkin A, Russell RG, Ayala J, Skoultchi AI. 2001. Individual somatic H1 subtypes are dispensable for mouse development even in mice lacking the H1(0) replacement subtype. Mol. Cell Biol. 21:7933–7943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feige JN, Sage D, Wahli W, Desvergne B, Gelman L. 2005. PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc. Res. Tech. 68:51–58 [DOI] [PubMed] [Google Scholar]
- 18. Filesi I, et al. 2002. Loss of heterochromatin protein 1 (HP1) chromodomain function in mammalian cells by intracellular antibodies causes cell death. J. Cell Sci. 115:1803–1813 [DOI] [PubMed] [Google Scholar]
- 19. Font-Burgada J, Rossell D, Auer H, Azorín F. 2008. Drosophila HP1c isoform interacts with the zinc-finger proteins WOC and relative-of-WOC to regulate gene expression. Genes Dev. 22:3007–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia BA, et al. 2007. Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 282:7641–7655 [DOI] [PubMed] [Google Scholar]
- 21. Gentleman RC, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hediger F, Gasser SM. 2006. Heterochromatin protein 1: don't judge the book by its cover! Curr. Opin. Genet. Dev. 16:143–150 [DOI] [PubMed] [Google Scholar]
- 23. Hellauer K, Sirard E, Turcotte B. 2001. Decreased expression of specific genes in yeast cells lacking histone H1. J. Biol. Chem. 276:13587–13592 [DOI] [PubMed] [Google Scholar]
- 24. Irizarry RA, et al. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264 [DOI] [PubMed] [Google Scholar]
- 25. Jacobs SA, Khorasanizadeh S. 2002. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295:2080–2083 [DOI] [PubMed] [Google Scholar]
- 26. Jedrusik MA, Schulze E. 2007. Linker histone HIS-24 (H1.1) cytoplasmic retention promotes germ line development and influences histone H3 methylation in Caenorhabditis elegans. Mol. Cell Biol. 27:2229–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jedrusik MA, Schulze E. 2001. Single histone H1 isoform (H1.1) is essential for chromatin silencing and germline development in Caenorhabditis elegans. Development 128:1069–1080 [DOI] [PubMed] [Google Scholar]
- 28. Krasnov A, Koskinen H, Afanasyev S, Mölsä H. 2005. Transcribed Tc1-like transposons in salmonid fish. BMC Genomics 6:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. 2004. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell 14:183–193 [DOI] [PubMed] [Google Scholar]
- 30. Lever MA, Th'ng JP, Sun X, Hendzel MJ. 2000. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 408:873–876 [DOI] [PubMed] [Google Scholar]
- 31. Li Y, Kirschmann DA, Wallrath L. 2002. Does heterochromatin protein 1 always follow code? Proc. Natl. Acad. U. S. A. 99:16462–16469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mackey-Cushman SL, et al. 2011. FoxP3 interacts with linker histone H1.5 to modulate gene expression and program Treg cell activity. Genes Immun. doi:10.1038/gene.2011.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mello CC, Kramer JM, Stinchcomb D, Ambros V. 1991. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10:3959–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moneron G, et al. 2010. Fast STED microscopy with continuous wave fiber lasers. Opt. Express 18:1302–1309 [DOI] [PubMed] [Google Scholar]
- 35. Olsen JV. 2009. A dual pressure linear ion trap orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8:2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ong S, et al. 2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1:376–386 [DOI] [PubMed] [Google Scholar]
- 37. O'Rourke D, Baban D, Demidova M, Mott R, Hodgkin J. 2006. Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res. 16:1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567 [DOI] [PubMed] [Google Scholar]
- 39. Prymakowska-Bosak MM, et al. 1999. Linker histones play a role in male meiosis and the development of pollen grains in tobacco. Plant Cell 11:2317–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pujol N, et al. 2008. Antifungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 4:e1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pujol N, et al. 2008. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr. Biol. 18:481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization nanoelectrospray and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670 [DOI] [PubMed] [Google Scholar]
- 43. Reference deleted.
- 44. Robinson PJ, Rhodes D. 2006. Structure of the ‘30 nm’ chromatin fibre: a key role for the linker histone. Curr. Opin. Struct. Biol. 16:336–343 [DOI] [PubMed] [Google Scholar]
- 45. Rose FR, et al. 1998. Potential role of epithelial cell-derived histone H1 proteins in innate antimicrobial defense in the human gastrointestinal tract. Infect. Immun. 66:3255–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmeck B, Gross R, Dje N′Guessan P, Hocke AC. 2005. Histone acetylation and flagellin are essential for Legionella pneumophila-induced cytokine expression. J. Immunol. 175:2843–2850 [DOI] [PubMed] [Google Scholar]
- 47. Schott S, Coustham V, Simonet T, Bedet C, Palladino F. 2006. Unique and redundant functions of C. elegans HP1 proteins in post-embryonic development. Dev. Biol. 298:176–187 [DOI] [PubMed] [Google Scholar]
- 48. Schotta G, et al. 2004. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18:1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shapira M, et al. 2006. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103:14086–14091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen X, Gorovsky MA. 1996. Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell 86:475–483 [DOI] [PubMed] [Google Scholar]
- 51. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 52. Sirotkin AM, et al. 1995. Mice develop normally without the H10 linker histone. Proc. Natl. Acad. Sci. U. S. A. 92:6434–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smyth GK. 2004. Linear models and empirical bayes methods for assessing di erential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article 3. doi:10.2202/1544–6115.1027 [DOI] [PubMed] [Google Scholar]
- 54. Soufi B, et al. 2010. Stable isotope labeling by amino acids in cell culture (SILAC) applied to quantitative proteomics of Bacillus subtilis. J. Proteome Res. 9:3638–3646 [DOI] [PubMed] [Google Scholar]
- 55. Tenor JL, Aballay A. 2008. A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep. 9:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weiss T, et al. 2010. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 3:7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wirth M, et al. 2009. HIS-24 linker histone and SIR-2.1 deacetylase induce H3K27me3 in the Caenorhabditis elegans germ line. Mol. Cell. Biol. 29:3700–3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wisniewski JR, Zougman A, Nagarjuna N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat. Methods 6:359–362 [DOI] [PubMed] [Google Scholar]
- 59. Wisniewski JR, Zougman A, Kruger S, Mann M. 2007. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell Proteomics 6:72–87 [DOI] [PubMed] [Google Scholar]
- 60. Wong D, Bazopoulou D, Pujol N, Tavernarakis N, Ewbank JJ. 2007. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol. 8:R194 http://genomebiology.com/2007/8/9/R194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wysocka J. 2006. Identifying novel proteins recognizing histone modifications using peptide pull-down assay. Methods 40:339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zugasti O, Ewbank JJ. 2009. Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-beta signaling pathway in Caenorhabditis elegans epidermis. Nat. Immunol. 10:249–256 [DOI] [PubMed] [Google Scholar]