

Abstract
Meiosis divides the chromosome number of the cell in half by having two rounds of chromosome segregation follow a single round of chromosome duplication. The first meiotic division is unique in that homologous pairs of sister chromatids segregate to opposite poles. Recent work in budding and fission yeast has shown that the cell cycle kinase, Cdc7-Dbf4, is required for many meiosis-specific chromosomal functions necessary for proper disjunction at meiosis I. This work reveals another role for Cdc7 in meiosis as a gene-specific regulator of the global transcription factor, Ndt80, which is required for exit from pachytene and entry into the meiotic divisions in budding yeast. Cdc7-Dbf4 promotes NDT80 transcription by relieving repression mediated by a complex of Sum1, Rfm1, and a histone deacetylase, Hst1. Sum1 exhibits meiosis-specific Cdc7-dependent phosphorylation, and mass spectrometry analysis reveals a dynamic and complex pattern of phosphorylation events, including four constitutive cyclin-dependent kinase (Cdk1) sites and 11 meiosis-specific Cdc7-Dbf4-dependent sites. Analysis of various phosphorylation site mutants suggests that Cdc7 functions with both Cdk1 and the meiosis-specific kinase Ime2 to control this critical transition point during meiosis.
INTRODUCTION
Sexual reproduction requires that specialized gametes be formed in which the chromosome number of the organism is reduced in half, thereby restoring the diploid chromosome number when fertilization occurs. The specialized cell division that accomplishes this task is meiosis, where a single round of chromosome duplication is followed by two rounds of chromosome segregation. Unique to meiosis is the first division (MI), where homologous pairs of sister chromatids segregate to opposite poles (referred to as reductional segregation) (60). In contrast, the second meiotic division (MII) is like mitosis, where sister chromatids separate.
For reductional segregation, several meiosis-specific events must occur (44). First, during premeiotic S phase, cohesin complexes containing a meiosis-specific subunit are used to hold sister chromatids together. These meiotic cohesin complexes enable cohesion to be lost in two steps: arm cohesion is removed prior to MI and centromere cohesion is then lost at MII (10, 36). Second, homologous chromosomes are physically connected by a combination of reciprocal crossovers and sister chromatid cohesion. These connections enable proper alignment on the MI metaphase plate. Third, sister kinetochores are modified so that they exhibit monopolar orientation—i.e., they attach to microtubules from only one spindle pole. In budding yeast, this is accomplished by a multisubunit complex called monopolin that binds to kinetochores (63, 82). How these events are coordinated to occur in a specific order is not well understood.
A key regulatory protein in meiosis is the highly conserved kinase Cdc7-Dbf4. Similar to cyclin-dependent kinases, kinase activity requires a catalytic subunit called Cdc7 and a regulatory subunit called Dbf4 (for simplicity, this complex will here be referred to as Cdc7) (70). Cdc7 plays a key role in the initiation of DNA replication in mitotically dividing cells by phosphorylation of the replicative helicase Mcm2-7 (22, 45, 64, 71). Due to its crucial role in DNA replication, CDC7 is essential for growth. Genetic studies in yeast to look at the function of CDC7 in meiosis have therefore used conditional alleles, including temperature-sensitive alleles (cdc7ts) (69), transcriptional shutoff of DBF4 prior to the onset of meiosis (84), or an analog-sensitive allele (cdc7-as) in which kinase activity is specifically inhibited by the addition of an inhibitor, PP1, to the medium (88, 89). In addition, the replication defect conferred by deletion of CDC7 can be bypassed by a point mutation in one of the replicative helicase subunits called bob1 (21).
Abrogation of Cdc7 kinase activity under certain genetic conditions during meiosis results in the production of two diploid, nonrecombinant cells (packaged into spores), similar to mitosis. This is because Cdc7 is critical for all of the unique meiotic processes that allow for reductional segregation: Cdc7 facilitates premeiotic S phase (84, 89), the time during which meiotic cohesion complexes generate sister chromatid cohesion. Cdc7 is required for making the double-strand breaks (DSBs) that initiate recombination (46, 56, 68, 88). In a normal meiosis, DSBs are timed to occur after DNA replication, so that recombination is not initiated until sister chromatids are present (7, 54). The requirement for Cdc7 for both premeiotic DNA replication and meiotic recombination points to Cdc7 as part of the mechanism by which these two events may be coupled (53). Cdc7 is required for the recruitment of monopolin to kinetochores, thereby allowing mono-orientation of pairs of sister chromatids (41, 46). Finally, Cdc7 is important for the regulation of cleavage of meiotic cohesion complexes at meiosis I (33).
Microarray analyses have revealed that there are waves of transcription that are temporally regulated after the induction of meiosis in budding yeast (14, 62). These induced genes have been divided up into classes: early, middle, and late, based on the timing of their transcription. Transcription of early genes is dependent on Ime1; these genes encode proteins needed in meiotic prophase such as IME2, a protein kinase needed for premeiotic S phase (6), and they also include DSB formation genes such as SPO11 (34) and synaptonemal complex genes such as HOP1, RED1, and ZIP1 (73, 78). Middle gene expression is induced by a transcriptional activator called Ndt80 (14, 62). Ndt80 is required for expression of, among other genes, the polo-like kinase gene, CDC5, which allows Holliday junction resolution and synaptonemal complex disassembly; CLB1, which forms part of the cyclin-dependent kinase (Cdk1) that allows entry into MI; and SMK1, a kinase important for spore formation (11, 37, 75). As a result, deletion of NDT80 results in a pachytene arrest with unresolved recombination intermediates. NDT80 induction therefore serves as a key transition point in meiotic prophase at which cells are committed to complete the meiotic divisions (3, 26, 92).
The promoters of many early genes contain a specific sequence called URS1 that is bound by Ume6 (9, 86). Ume6 recruits the Sin3-Rpd3 histone deacetylase complex, as well as the Isw2 chromatin remodeling complex, to repress early gene transcription during vegetative growth (20, 32, 66, 76). When MATa/MATα strains are transferred to sporulation medium, the transcriptional activator Ime1 is recruited to URS1 sites through its interaction with Ume6, thereby allowing transcription (65, 87). Whether Ime1 is tethered to the promoter via interaction with Ume6 or results in Ume6 destruction is controversial (43).
NDT80 contains two URS1 elements in its promoter, and its initial transcription is dependent on Ime1. However, NDT80 is a “delayed early” gene because its expression occurs later than that of other Ime1-dependent early genes (14). This is because of a second level of regulation exerted at the NDT80 promoter by a repressor complex comprised of Sum1, Rfm1, and a histone deacetylase, Hst1 (48, 91). Sum1 is a sequence-specific DNA binding protein that binds to midsporulation elements (MSEs) present both in the NDT80 promoter and in the promoters of other genes in the Ndt80 regulon (24, 31, 61). Deletion of SUM1 or HST1 results in middle gene expression in vegetative cells, although not all Sum1-repressed genes require HST1 (48, 91). For Ime1 to activate expression of NDT80, Sum1-mediated repression must first be removed. This loss of repression requires the meiosis-specific Ime2 kinase, as well as cyclin-dependent kinase (Cdk1) activity (57, 72). Phosphorylation of Sum1 by these kinases promotes a loss of Hst1 activity at the promoter and the removal of Sum1 that in turn allows for Ime1-dependent transcription of NDT80 (2, 72). The delay in Ime1-mediated expression of NDT80 can therefore be explained by the fact that IME2 must first be transcribed and translated before Ime1-dependent transcription can occur. Ndt80 competes with Sum1 for binding to MSEs, and therefore, after Ime1-dependent transcription has resulted in some Ndt80 protein, Ndt80 is able to replace Sum1 at MSEs to activate its own transcription, as well as the transcription of middle genes (15, 57, 61). Activation of Ndt80 for this second wave of transcription is the target of the meiotic recombination checkpoint that arrests cells prior to MI in response to unrepaired recombination intermediates or incomplete synapsis (15, 25, 40, 58, 83).
Inactivation of Cdc7 in meiosis using either cdc7ts, cdc7-as plus inhibitor, or cdc7Δ bob1 results in a meiotic arrest prior to MI due to a lack of NDT80 transcription (68, 69, 89). Lo et al. (41) showed that this arrest can be suppressed by ectopic expression of NDT80 using basal transcription from the CUP1 promoter (PCUP1-NDT80 NDT80 cdc7-as diploids are referred to as cdc7-asNDT80). The fact that NDT80 under the control of a different promoter is no longer subject to regulation by Cdc7 indicates that Cdc7 must target a factor(s) that functions in the NDT80 promoter and suggests a new role for Cdc7 as a gene-specific transcriptional regulator. The work described below examines the mechanism by which Cdc7 exerts this regulation and shows that the function of Ime2 and Cdk1 phosphorylation of Sum1 is to allow Cdc7-dependent phosphorylation of the repressor, thereby promoting removal of the Sum1/Rfm1/Hst1 complex and allowing Ime1-dependent transcription of NDT80.
MATERIALS AND METHODS
Plasmids.
An NDT80 URA3 integrating plasmid was generated by subcloning a 4.3-kb ClaI fragment containing 2.1 kb upstream and 350 bp downstream of the NDT80 open reading frame (ORF) from pNKY1212 (92) into ClaI-digested pRS306 to make pHL8. Codon 177 was changed from CGC (arginine) to GCC (alanine) by site-directed mutagenesis using the QuikChange kit (Stratagene) and PCR to make pHL8-R177A. The pHL8-R177A plasmid can be targeted to integrate at ura3 by digestion with NsiI. To make the HST1-5HA URA3 and HST1 URA3 integrating plasmids, a 2.4-kb SacI fragment from pJR2289 and a 2.2-kb SalI fragment from pJR2288 (provided by Laura Rusche, Duke University) (67) were subcloned into SacI-digested pRS306 to create pHL16 and pHL17, respectively. Digestion with StuI targets pHL16 and pHL17 to integrate at ura3. The H310Y mutation was introduced into pHL16 and pHL17 by site-directed mutagenesis in which codon 310 was changed from CAC to TAC.
To introduce alanine substitutions simultaneously for 12 putative Cdc7 phosphorylation sites, the entire SUM1 ORF along with 500 bp of upstream and 300 bp of downstream sequence was synthesized by Genewiz, Inc. (South Plainfield, NJ). The codons for the following amino acids were changed to GCA: S62, S278, S381, S385, T392, S393, S651, S655, S657, T717, T815, and T1032. In addition, there are several polymorphisms between the SUM1 sequence present in the Saccharomyces Genome Database and the SK1 SUM1 gene (72). The following SK1-specific codons were therefore used in the synthesis: D87 (GAC), V146 (GTG), A223 (GCG), L241 (CTC), P410 (CCA), T419 (ACA), I524 (ATT), L615 (CTG), S638 (TCA) R699 (AGA), I748 (ATA), and E773 (GAA). As a control, the wild-type allele of SUM1 containing the SK1 codons was also synthesized. BamHI and SalI sites were engineered onto the ends of the fragments so that the genes could be cloned into pRS306 to generate plasmids pRS306-SUM1 and pRS306-SUM1-12A, respectively.
The sum1-ci allele was introduced into a URA3 integrating plasmid by substituting a 2.6-kb HindIII/EcoRI fragment from pMES71 (generously provided by Edward Winter, Thomas Jefferson University) for the corresponding fragment in pRS306-SUM1. The presence of the mutations was confirmed by DNA sequencing. The sum1-c4i allele was created using site-directed mutagenesis of pRS306-SUM1-T306A to simultaneously change codons T313 (ACA), S379 (TCT), S738 (TCA), and T817 (ACT) to GCA (alanine) (QuikChange multisite-directed mutagenesis kit; Agilent Technologies). All point mutant alleles were sequenced in their entirety by the Stony Brook University DNA Sequencing Facility to confirm that no additional mutations are present. The pRS306-SUM1-based plasmids can be targeted to integrate into the ura3 locus by digestion with PstI.
Plasmids used for generating probes for Northern blots are pHL8 (NDT80), p18 (SPS1) (59), and p4LE159 (SPS4) (24). For a loading control, pC4/2 was used to detect an unidentified gene whose expression is constant in vegetative cells and during sporulation (39). All of the plasmids used to generate probes for Northern blots were generously provided by Jacqueline Segall (University of Toronto).
Strains.
All strains used in this study are derived from the SK1 background. Genotypes can be found in Table 1. cdc7-as indicates the cdc7-as3-9myc allele which contains the L120A and V181A mutations and nine Myc epitopes (89). Genes were deleted using either natMX4 or kanMX6 and confirmed by yeast colony PCR (42, 81). Details of the strain constructions are available upon request. The cdc7-as PCUP1-NDT80 genotype is abbreviated cdc7-asNDT80, and the construction of NH452F::CUP1-NDT80 is described in reference 41.
Table 1.
Saccharomyces cerevisiae strains
| Strain | Genotype | Reference or source |
|---|---|---|
| NH144-32aF | MATaleu2Δ::hisG his4-x hoΔ::LYS2 lys2 cdc7-as3-myc9 ura3 | 89 |
| NH144-33bF | MATα leu2-K arg4-Nsp hoΔ::LYS2 lys2 cdc7-as3-myc9 ura3 | 89 |
| NH452F | MATaleu2Δ::hisG his4-x ARG4 hoΔ::LYS2 lys2 cdc7-as3-myc9 ura3 | 89 |
| MATα leu2-K HIS4 arg4-Nsp hoΔ::LYS2 lys2 cdc7-as3-myc9 ura3 | ||
| NH452F::CUP1-NDT80 | Same as NH452F except ura3::PCUP1-NDT80-3HA::URA3 | 41 |
| NH932 | Same as NH452F except ndt80Δ::natMX4 | This work |
| ndt80Δ::natMX4 | ||
| NH932::pHL8-R177A | Same as NH452F except ndt80Δ::natMX4 ura3::ndt80-R177A::URA3 | This work |
| ndt80Δ::natMX4 ura3::ndt80-R177A::URA3 | ||
| NH788 | Same as NH452F except sum1Δ::natMX4 | This work |
| sum1Δ::natMX4 | ||
| NH788-XC | Same as NH452F except sum1Δ::kanMX6 | This work |
| sum1Δ::kanMX6 | ||
| NH1061 | Same as NH452F except hst1Δ::kanMX6 | This work |
| hst1Δ::kanMX6 | ||
| NH2056 | Same as NH452F except rfm1Δ::kanMX6 | This work |
| rmf1Δ::kanMX6 | ||
| NH1068 | Same as NH452F except ndt80Δ::natMX4 SUM1-3Flag::kanMX6 | This work |
| ndt80Δ::natMX4 SUM1-3Flag::kanMX6 | ||
| NH1078 | MATaleu2 his4-x lys2 ho::ΔLSY2 cdc7-as3-myc9 ura3::HST1-5HA::URA3 | This work |
| MATα leu2 his4-x lys2 ho::ΔLSY2 cdc7-as3-myc9 ura3::HST1-5HA::URA3 | ||
| hst1Δ::kanMX6 SUM1-3Flag::kanMX6 | ||
| hst1Δ::kanMX6 SUM1-3Flag::kanMX6 | ||
| NH1080 | MATaleu2 his4-x lys2 ho::ΔLSY2 cdc7-as3-myc9 ura3::HST1-5HA::URA3 | This work |
| MATα leu2 his4-x lys2 ho::ΔLSY2 cdc7-as3-myc9 ura3::HST1-5HA::URA3 | ||
| hst1Δ::kanMX6 ndt80Δ::natMX4 SUM1-3Flag::kanMX6 | ||
| hst1Δ::kanMX6 ndt80Δ::natMX4 SUM1-3Flag::kanMX6 | ||
| YRH38 | MATα leu2-3,112 ade2-1 can1-100 his3-11,15 trp1-1 ura3-1 SUM1-1 sir2::his5+hst1ΔkanMX6 | 77 |
Time courses.
Diploid cells were sporulated in 2% potassium acetate at a concentration of 3 × 107 cells/ml and shaken at 30°C as described in reference 16. To monitor meiotic progression, 450 μl of cells collected at various time points was mixed with 50 μl 37% formaldehyde and incubated overnight at 4°C. The cells were washed with 1× phosphate-buffered saline (PBS) two times and resuspended in 50 μl 1× PBS. Six microliters of cell suspension was dropped on a slide and mixed with 50% mounting medium with DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories). The number of nuclei in each cell was determined using fluorescence microscopy. [4-Amino-l-tert-butyl-3-(p-methylphenyl)pyrazolo[3,4-d]pyrimidine] (PP1) was generously supplied by Kevan Shokat. PP1 was resuspended in dimethyl sulfoxide (DMSO) to make a 10 mM stock solution and kept at −20°C. PP1 was added immediately after cells were transferred to Spo medium to a final concentration of 15 μM. All time course assays were performed at least three times. Due to variations that occur between experiments, representative time courses are shown without error bars, as is standard for the field.
Quick sporulation assays.
Sporulation in the presence or absence of PP1 was monitored using a quick sporulation protocol. Individual transformants were inoculated into 2 ml yeast extract-peptone-dextrose (YPD) and grown overnight on the roller at 30°C. The cells were pelleted, washed with water, and resuspended in 2 ml 2% potassium acetate (Spo medium). The cells were divided into 1-ml aliquots, and 1.5 μl 10 mM PP1 (15 μM final concentration) was added to one of the tubes. After incubation overnight at 30°C on the roller, the cells were counted using phase-contrast light microscopy. At least three independent colonies were sporulated for each strain.
RNA preparation and Northern blots.
Ten milliliters of sporulating cells was collected at indicated time points and washed once with cold water. RNA was prepared using the RiboPure-Yeast kit (Ambion). Northern blot assays were performed according to the instructions of the NorthernMax-Gly kit (Ambion), and all the buffers were supplied with the kit. Fourteen micrograms of total RNA for each sample was used in Northern blot assays. The gene-specific probes were generated as follows. For NDT80, a 1.2-kb AfeI/BamHI fragment from pHL8 was used. For IME2, a 948-bp fragment was amplified using genomic DNA from the SK1 strain NH144 (27) as the template and the primers 5′ AGACACAAGGTGTAGTGGCTATAAA 3′/5′ GTTCTGTATTAGTCACATTGCCTCT 3′. For SPS1, the probe was obtained by amplifying a 940-bp PCR fragment using p18 and the primers 5′ TATAAAGCAGTGGATAGAGTTACGC 3′/5′ GAGAGTCTTGTGTAATGGGAGATAA 3′. For SPS4, an 863-bp PCR product was generated using p4LE159 and the primers 5′ CAGACACAAGAAGCAGTTACAGAA 3′/5′ CTAAACAAACTTCTATCGGTGACAG 3′. The pC4/2 plasmid was digested with HindIII, and the whole digest was used as a template to make random probes. The radioactive probes were generated using the Prime-it II random primer labeling kit (Agilent) and 25 ng DNA.
Chromatin immunoprecipitation (ChIP) and quantitative PCR (q-PCR).
One liter of log-phase cells was grown in yeast extract-peptone-acetate (YPA) to an optical density at 660 nm (OD660) of 1.4. The cells were pelleted, washed once with water, and resuspended in ∼750 ml of Spo medium at a density of 3 × 107 cell/ml. After removal of 250 ml for the 0-h time point, the remaining 500 ml was divided in half. PP1 was added to one flask to a final concentration of 15 μM, and the cells were placed on a 30°C shaker for 8 h. At each time point, formaldehyde was added to 250 ml cells to a final concentration of 1% and the cells were incubated for 20 min at room temperature (RT) with occasional shaking to allow cross-linking. Cross-linking was stopped by addition of 37.5 ml 3 M glycine-20 mM Tris and incubation at RT for 5 min. Cross-linked cells were washed twice with 200 ml cold TBS (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) by pelleting in a Sorvall centrifuge in an SS-34 rotor at 5,000 rpm. The pellets were then resuspended in 10 ml cold FA-0.1% SDS (FA is 50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) and transferred to a 15-ml Falcon tube. The cells were pelleted and stored at −80°C until use.
To prepare chromatin, aliquots of frozen cells were thawed and resuspended in 1 ml FA-0.5% SDS. All of the FA solutions used were ice cold. The cells were lysed by bead beating with 1.5 ml 0.5-mm glass beads (Biospec) for 8 cycles: 30 s of vortexing at maximum speed and 30 s on ice. The bottom of each tube was pierced with a red-hot 22-gauge needle, and the tubes were inserted into plastic Nalgene 30-ml centrifuge tubes. After the addition of 6.5 ml FA-0.1% SDS, the lysates were collected by centrifugation at 1,000 rpm for 5 min at 4°C. The lysates were transferred to 10.4-ml Beckman polycarbonate ultracentrifuge tubes and spun at 45,000 rpm for 20 min in an ultracentrifuge using a Ti-50 rotor at 4°C. The supernatants were decanted, the pellets were resuspended with 8 ml FA-0.1% SDS, and the high-speed spin was repeated. The pellets were then resuspended in 1.5 ml FA-0.1% SDS and transferred to 2-ml cryotubes (Sarstedt). The tubes were placed on ice, and the resuspended pellets were sonicated for 5 pulses on setting 5 using a W-385 ultrasonic processor (Heat Systems; Ultrasonics, Inc.): 20 s on and 20 s off. The sonicated chromatin was then transferred to the 10.4-ml ultracentrifuge tubes, 6.5 ml ice-cold FA-0.1% SDS was added, and the samples were centrifuged at 45,000 rpm in a Ti-50 rotor for 20 min at 4°C. Supernatants containing sheared chromatin were collected, and 800-μl aliquots were transferred to microcentrifuge tubes for storage at −80°C or for use immediately in the following immunoprecipitation (IP) procedure.
For each IP, 20 μl 5 M NaCl was added to an 800-μl aliquot. Seventy microliters was transferred to a new tube to be used as the input control. Five microliters of anti-Flag M2 mouse monoclonal antibody (Sigma) was added to the remaining 750 μl. After rocking overnight at 4°C on a nutator, 50 μl of Dynabeads protein G beads (Invitrogen) equilibrated in FA-0.1% SDS was added to each sample and the IP mixtures were rocked for an additional 1.5 h at 4°C. The pellets were then washed sequentially with the following buffers: (i) FA-0.1% SDS-275 mM NaCl, (ii) FA-0.1% SDS-500 mM NaCl, (iii) 10 mM Tris-HCl–0.25 M LiCl–1 mM EDTA–0.5% NP-40–0.5% sodium deoxycholate, and (iv) 1× Tris-EDTA (TE), pH 8.0. Each wash was performed by adding 1 ml of wash buffer and nutating the mixture at RT for 4 min. After each wash, the beads were separated from the wash solution using a magnet (Invitrogen) for 1 min. To elute the chromatin off the magnetic beads, the beads were resuspended in 250 μl elution buffer (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 1% SDS), incubated at 65°C for 10 min, and separated from the supernatant on a magnet for 1 min. The supernatant containing the chromatin was moved to a new tube, and the beads were washed once with 250 μl 1× TE, pH 8.0, which was then pooled with the first supernatant for a total volume of 500 μl. To reverse the cross-links, 20 μl 20-mg/ml pronase (Roche) was added and the chromatin was incubated for 1 h at 42°C followed by 4 h at 65°C. Fifty microliters of 4 M LiCl was added to each tube, and the samples were vortexed and then extracted once with 400 μl phenol-chloroform-isoamyl alcohol (25:24:1) and once with 300 μl chloroform. The DNA was precipitated by adding 1 μl 20-mg/ml glycogen (Roche) and 1 ml ice-cold 100% ethanol and incubating the mixture at −80°C overnight. The DNA was pelleted by centrifugation at 13,000 rpm for 10 min at RT. The pellet was washed once with 1 ml RT 100% ethanol and dried for 5 min at RT. The DNA was resuspended in 200 μl TE, pH 8.0, for q-PCR.
For the input control, the 70 μl of sonicated chromatin was diluted with 180 μl 1× TE, pH 8.0, and processed from the pronase step forward along with the immunoprecipitated chromatin. The only difference is that half of the volume of pronase and LiCl was used for preparing the input DNA.
Each q-PCR mixture contained 4 μl DNA, 5 μl Lightcycler 480 SYBR green I master mix (Roche), and 0.5 μl of each 10 μM primer. Reaction mixtures were contained in a 96-well Twin Tec real-time PCR plate (Eppendorf). For the NDT80 promoter, the following primers were used: 5′ GTATATGTGACTTTACATTG/GAAAGGTTAGTAAACTTTC 3′ (−320 to −301/−56 to −38). Coordinates are relative to the ATG. For the SMK1 promoter, the following primers were used: 5′ GTGATTCGAAAAGTATCGCGC/GCGCCGAATTCTACCCTCA 3′ (−135 to −115/−103 to −85). Primers located internal to the CIT2 gene were used as negative controls: 5′ TGGACCCAAATGCCGATTATG/AGCCAACCCGTTCAAACCTGATG 3′ (+665 to +685/+845 to +867). Each reaction was performed in triplicate using an Eppendorf Realplex 2 PCR machine with the following parameters: step 1, 95°C for 10 min; step 2, 95°C for 15 s, 55°C for 15 s, and 72°C for 25 s (repeat for 40 cycles). To calculate the percentage of immunoprecipitated DNA relative to the input, the following formula was used: 1/(10.71 × 2CTIP −CTinput), where 10.71 is the volume of chromatin used for the IP (750 ml)/volume of input chromatin (70 ml) and CT represents the average number of cycles required for the fluorescent signal to exceed background levels.
The percent input DNA obtained from the IPs without antibody was subtracted from the percent input DNA obtained with the IPs using the Flag antibody for each pair of primers. The fold increase is the ratio of the precipitated DNA with the test primers (NDT80 or SMK1) to the negative-control CIT2 primers.
Western blot assays.
For Fig. 1D, 4A, and 6B and C, 8 ml sporulating cells taken at the indicated time points was pelleted, resuspended with 8 ml 5% trichloracetic acid (TCA), and incubated at 4°C for 10 min with rocking. The cells were pelleted and washed with acetone once, and the pellet was air dried for 2 h. All lysis buffers were made fresh. Cells were broken using 100 μl glass beads in 150 μl lysis buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 27.5 mM dithiothreitol [DTT], 11 mM phenylmethylsulfonyl fluoride [PMSF], 2× concentrated EDTA-free complete protease inhibitor cocktail tablets [Roche]) by vortexing at 4°C for 15 to 25 min until at least 70% of cells were lysed. Seventy-five microliters 3× SDS sample buffer was then added to each lysate, which was boiled for 5 min before fractionation using an 8% SDS polyacrylamide gel. For Fig. 3B and 4B, 40 ml of vegetative and sporulating cells, respectively, was removed at various time points and 100 mM PMSF in DMSO was added to a final concentration of 2 mM. Cells were pelleted and washed once with cold water containing 2 mM PMSF. The cell pellet was resuspended in 500 μl B70 buffer (50 mM HEPES-KOH, pH 7.4, 70 mM potassium acetate, 20 mM β-glycerophosphate, 5 mM Mg-acetate, 10% glycerol, 1 mM DTT, 0.5 mM PMSF, 2× concentrated EDTA-free complete protease inhibitor cocktail tablets [Roche]) and transferred to a 2-ml microcentrifuge tube, and 500 μl of glass beads was added. Cells were lysed by vortexing for 20 min at 4°C. NP-40 was added to crude cell extracts to a final concentration of 0.1%. Lysates were incubated on ice for 10 min and cleared by centrifugation at 13,000 rpm for 10 min at 4°C. Supernatants were transferred to new microcentrifuge tubes, and protein concentrations were determined using the Bio-Rad protein assay reagent. One hundred fifty micrograms soluble extract was loaded onto an 8% SDS polyacrylamide gel and run at 100 V for 16 h. Proteins were transferred to nitrocellulose or polyvinylidene difluoride (PVDF) using a Bio-Rad semidry transfer apparatus at 2 mA/cm2 for 1 h. Ndt80 protein was detected by anti-Ndt80 rabbit polyclonal serum (6) (a gift from Michael Lichten) at a 1:10,000 dilution, Sum1-3Flag was detected by mouse monoclonal anti-Flag M2 antibody at 1:5,000 (Sigma; F1804), Sum1 was detected by anti-Sum1(yN-20) goat polyclonal antibody at 1:100 (Santa Cruz; sc-26441), and Hst1-5HA was detected by mouse monoclonal antihemagglutinin (anti-HA) 12CA5 antibody at 1:10,000.
Fig 1.

Early and middle meiotic gene expression in various cdc7-as diploids in the absence or presence of PP1. Diploids were transferred to Spo medium in the absence or presence of 15 μM PP1, and cells from the indicated time points were analyzed by Northern blot assays (A to C) or immunoblot assays (D). (A) cdc7-as (NH452F). (B) cdc7-as ndt80-R177A (NH932::pHL8-R177A). (C) cdc7-as sum1Δ (NH788). (D) Total cell extracts were prepared from the cdc7-as, cdc7-as sum1Δ, cdc7-as hst1Δ (NH1061), and cdc7-asNDT80 (NH452F::CUP1-NDT80) diploids at the indicated time points and probed with anti-Ndt80 antibodies.
Fig 4.

Analysis of Sum1-3Flag, Hst1-5HA, and Ndt80 proteins in various cdc7-as strains without and with PP1. (A) A cdc7-as SUM1-3Flag HST1-5HA diploid (NH1078) was transferred to Spo medium in the absence or presence of 15 μM PP1, and total cell extracts were prepared at the indicated time points. Sum1-3Flag, Hst1-5HA, and Ndt80 were analyzed using immunoblot assays with anti-Flag, anti-HA, and anti-Ndt80 antibodies, respectively. (B) A cdc7-as ndt80Δ SUM1-3Flag diploid (NH1068) was transferred to Spo medium and incubated for 8 h without or with 15 μM PP1. Sum1-3Flag was detected on immunoblots using anti-Flag antibodies. (C) Sum1-3Flag protein was immunoprecipitated from NH1068 incubated after transfer to Spo medium for 0 h, 8 h, or 8 h with 15 μM PP1. Half of the immunoprecipitates were treated with λ protein phosphatase as indicated and then probed with anti-Flag antibodies.
Fig 6.

Meiotic progression, Sum1 phosphorylation, and NDT80 expression in cdc7-as diploids containing various alleles of SUM1. The cdc7-as sum1Δ diploid, NH788-XC, containing two copies of either pRS306, SUM1, sum1-12A, or sum1-c4i, was transferred to Spo medium and analyzed at different times. (A) Meiotic progression was monitored by DAPI staining of cells sporulated in the absence or presence of 15 μM PP1. (B) Immunoblots of cdc7-as SUM1, cdc7-as sum1-c4i, and cdc7-as sum1-12A strain time courses probed with anti-Sum1 and anti-Ndt80 antibodies as indicated. (C) Side-by-side comparison of Sum1 and Ndt80 mobilities in the cdc7-as SUM1, cdc7-as sum1-c4i, and cdc7-as sum1-12A diploids from the 6-h time point.
Fig 8.

Model for Cdc7 regulation of NDT80 transcription during meiosis. This model is based on models presented in references 57 and 2. Nucleosomes are depicted as blue ovals, and phosphorylation is shown by white circles with the kinase responsible indicated by italics. (A) In vegetative growth, Cdk1-phosphorylated Sum1 is bound to MSEs in the NDT80 promoter. (Binding to MSE-1 is alone shown for simplicity and because MSE-2 is downstream of the TSS; however, Xie et al. [91] have reported that MSE-2 can function as a Sum1-repressive element as well.) Ume6 bound to URS1 elements in the promoter (again only one is shown for simplicity) recruits the Isw2/Sin3-Rpd3 repression complex. (B) After the induction of meiosis, Ime1 is recruited to the NDT80 promoter, where it is initially prevented from activating transcription of NDT80 by the presence of the Sum1/Rfm1/Hst1 complex. Phosphorylation by Ime2 and Cdk1 recruits Cdc7 to the promoter, allowing Cdc7-dependent phosphorylation of Sum1. (C) This phosphorylation may promote dissociation of Hst1 and Sum1 as well as affecting the affinity of Sum1 for the MSE. In the absence of the deacetylase, nucleosomes near Sum1 become acetylated (represented by a yellow star), which acts to weaken Sum1's repressive function. Weakened repression then allows Ime1-dependent NDT80 transcription. (D) Ndt80 protein replaces Sum1 on the MSEs, thereby starting the positive feedback loop.
Fig 3.

Phenotypic analysis of an hst1 catalytic mutant in suppressing SUM1-1 in vegetative cells and the meiotic arrest conferred by cdc7-as plus PP1. (A) A MATα sir2Δ SUM1-1 hst1Δ LYS1 haploid strain (YHR38) was transformed with pRS306, HST1-5HA, hst1-H310Y-5HA, HST1, or hst1-H310Y. Different transformants were patched onto SD-Ura plates and then replica plated to a lawn of MATa lys1 cells. Diploids were selected on SD plates to test for mating. (B) Total extracts from log-phase cells of a cdc7-as hst1Δ (NH1061) diploid transformed with either pRS306, HST1-5HA (pHL16), or hst1-H310Y-5HA (pHL16-H310Y) were probed with anti-HA antibodies. As a loading control, anti-Myc antibodies detected Cdc7-as-9myc. (C) cdc7-as hst1Δ, cdc7-as HST1-5HA, cdc7-as hst1-H310Y-5HA, cdc7-as HST1 (pHL17), and cdc7-as hst1-H310Y (pHL17-H301Y) cells were subjected to the “quick sporulation” protocol in the absence or presence of 15 μM PP1, and ascus formation was monitored using phase-contrast microscopy. Two hundred cells were counted for each culture for each strain, and at least three independent colonies were monitored for each strain. Error bars indicate the standard deviations.
Immunoprecipitation and phosphatase treatment.
Sum1-Flag was immunoprecipitated using cells from 40 ml vegetative and sporulating cultures. Soluble cell lysates were generated as described for Fig. 4B. Four hundred microliters of extract containing 8 mg soluble protein was incubated with 5 μl anti-Flag M2 antibody (Sigma) at 4°C with rocking for 1 h. The protein-antibody complexes were captured by adding 50 μl (1.5 mg) Dynabeads protein A slurry (30 mg/ml; Invitrogen) and rocked at 4°C overnight. The beads were washed with B70 buffer three times and λ protein phosphatase buffer (New England BioLabs; 50 mM HEPES, pH 7.5, 100 mM NaCl, 2 mM DTT, 0.01% Brij 35, 1 mM MnCl2) two times. The immunoprecipitates were resuspended in 50 μl λ phosphatase buffer and split into two tubes. A 1.5-μl portion of λ phosphatase (New England BioLabs) was added to one tube, and both tubes were incubated at 30°C for 30 min. The samples were boiled for 5 min after adding 7 μl 5× SDS loading buffer and then resolved on an 8% SDS-polyacrylamide gel, transferred to nitrocellulose, and probed with the anti-Flag M2 antibody.
Protein purification of Sum1-3Flag.
Four hundred milliliters of cells (NH1068) was harvested after 0 or 8 h after transfer to Spo medium with or without 15 μM PP1 and washed once with cold water. Each pellet was resuspended in 10 ml lysis buffer (50 mM Tris, pH 8.0, 10 mM EDTA, pH 8.0, 600 mM KCl, 0.5 mM DTT, 5% glycerol, 1 mM PMSF, 2× concentrated EDTA-free complete protease inhibitor cocktail tablets [Roche], phosphatase inhibitor cocktails 1 and 2 [Sigma]), and 1-ml aliquots were placed in 10 2-ml microcentrifuge tubes. Cells were pelleted and resuspended in 500 μl lysis buffer with 500 μl 0.5-mm glass beads. Cells were broken by vortexing for 20 min at 4°C. Triton X-100 was added to the crude cell lysates to a final concentration of 1%, and lysates were incubated on ice for 10 min and cleared by centrifugation at 13,000 rpm for 10 min. Sum1-3Flag protein was purified using anti-Flag M2 affinity gel (Sigma), eluted from the affinity gel using 0.1 M glycine, pH 2.5, and precipitated with 100% TCA as described in reference 55. TCA-precipitated Sum1-3Flag protein was resuspended in 10 μl 2× SDS sample buffer. Five microliters protein was resolved in a precast NuPAGE 3% to 8% Tris-acetate gel (Invitrogen) at 150 V for 90 min. The gel was stained with GelCode blue stain reagent (Thermo Scientific) according to instructions. The Sum1-3Flag bands were sliced out of gels and subjected to mass spectrometry (MS) analysis.
Mass spectrometry analysis of Sum1-3Flag protein.
Proteins were processed for MS as described in reference 55. Tandem MS (MS/MS) spectra were searched using the SEQUEST algorithm (18) against a database containing the Sum1-3Flag sequence and its reverse complement. Database search criteria are as follows: two missed cleavages, a precursor mass tolerance of 3 Da, no enzyme, static modification including alkylation of cysteine (57.02146 Da), and the following variable modifications—oxidation of methionine (15.99491 Da), deamidation of asparagine and glutamine (0.98401), and phosphorylation on tyrosine, threonine, and serine (79.96533 Da).
The target-decoy strategy was used to distinguish correct and incorrect spectral identifications (17), and the peptide-level false discovery rate was restricted to <2% by using linear discriminant analysis based on several different SEQUEST parameters, including an Xcorr of ≥1.0, ΔXcorr, charge state, and a minimum peptide length of 7 amino acids (29). Further peptide processing restricted the peptide spectral matches to ±5 ppm. Phosphorylation site localization was determined using Ascore, an algorithm for probability-based phosphorylation site localization (5).
RESULTS
Cdc7 kinase activity promotes Ime1-dependent NDT80 transcription.
NDT80 is transcribed in two steps: first Ime1-dependent expression occurs and the resulting Ndt80 protein then binds upstream of the NDT80 gene to activate its own transcription (57). To determine whether Cdc7 regulates Ime1-driven transcription of NDT80, Ndt80-mediated expression was prevented using a DNA-binding-deficient mutant of Ndt80. Based on the crystal structure of the DNA binding domain of Ndt80 in complex with an MSE sequence, as well as in vitro DNA binding assays, arginine 177 (R177) was found to play a key role in binding to the MSE (19, 38, 51). Changing R177 in the Ndt80 protein to alanine should therefore have no effect on transcription dependent on Ime1, while the inability to bind MSEs should prevent Ndt80-R177A from activating transcription of both itself and other members of the Ndt80 regulon. Consistent with this idea, IME2 and ndt80-R177A transcripts were observed in the absence of inhibitor in the cdc7-as ndt80-R177A strain, while the middle gene SPS1 was not expressed and the diploid failed to sporulate (Fig. 1B). This is in contrast to the cdc7-as strain, where middle gene expression occurred and 86% of the cells formed asci (Fig. 1A). Detectable ndt80-R177A expression was delayed compared to the NDT80 diploid. This delay may be because ndt80-R177A transcription is dependent only on Ime1, whereas, in the cdc7-as diploid, Ndt80-mediated transcription can also occur, leading to a quicker accumulation of NDT80 message. The ndt80-R177A mutation can therefore separate Ime1-dependent transcription from Ndt80-activated expression.
Inactivation of Cdc7-as by PP1 reduced and delayed transcription of NDT80, SPS1, and SPS4, as well as the production of Ndt80 protein, consistent with previous results (Fig. 1A and D) (41). Transcription of ndt80-R177A was abolished by addition of PP1 (Fig. 1B). Cdc7 kinase activity therefore promotes Ime1-dependent transcription of NDT80.
Cdc7 regulation of NDT80 transcription occurs through SUM1.
The initial phase of NDT80 transcription requires both the presence of the Ime1 activator and the removal of Sum1-mediated repression (57). Inhibition of Cdc7 has no effect on Ime1-dependent expression of early sporulation genes, suggesting that Cdc7 acts to abolish Sum1 repression (Fig. 1A) (41). If this hypothesis is correct, deletion of SUM1 should allow expression of NDT80 and middle sporulation genes even when Cdc7-as is inactivated by PP1, thereby enabling the cells to proceed through the meiotic divisions and sporulate. When Cdc7-as was active, cdc7-as sum1Δ cells prematurely expressed NDT80 and entered MI ∼1 hour earlier than cdc7-as cells did (Fig. 1C and D and 2A). Inhibition of Cdc7-as arrested cdc7-as cells in prophase, whereas cdc7-as sum1Δ cells proceeded through a single meiotic division to produce binucleate cells (Fig. 2A). Consistent with this fact, sum1Δ allowed downstream targets of Ndt80 to be expressed as well (Fig. 1C).
Fig 2.

Meiotic progression and ascus formation in various cdc7-as diploids in the absence or presence of PP1. (A) Diploids containing cdc7-as, cdc7-as sum1Δ, cdc7-as hst1Δ, and cdc7-asNDT80 were transferred to Spo medium in the absence or presence of 15 μM PP1. At various times, cells were fixed and the nuclei were stained with DAPI to monitor meiotic progression. Two hundred cells were counted for each time point. (B) Sporulation is presented as the average number of asci observed from at least three independent time courses determined by phase-contrast microscopy. For the cdc7-as rfm1Δ diploid (NH2056), the quick sporulation method was used. Error bars indicate the standard deviations. Two hundred cells were counted from each culture. In the presence of PP1, no asci with greater than two spores were observed.
Bypass of the NDT80 transcriptional block by ectopic expression of NDT80 (cdc7-asNDT80) does not suppress the need for Cdc7 kinase activity for either recombination or reductional segregation at meiosis I. As a result, the asci that form are nonrecombinant, diploid dyads (41). The same is true when the requirement for Cdc7 activity for NDT80 expression is bypassed by sum1Δ. Ascus formation was increased in cdc7-as sum1Δ cells treated with PP1 (cdc7-as sum1Δ +PP1) cells to 32% compared to 2% for cdc7-as +PP1 cells, and the majority of asci formed in the cdc7-as sum1Δ +PP1 diploids were dyads (the remainder being monads) (Fig. 2B). Cdc7 therefore promotes Ime1-mediated NDT80 transcription by relieving Sum1 repression.
Deletion of RFM1 and HST1 bypasses the requirement for Cdc7 to relieve Sum1-mediated repression at the NDT80 promoter.
Sum1 recruits Hst1 to the promoter of NDT80 via a bridging protein called Rfm1 to transcriptionally repress NDT80 in vegetative and meiotic cells (48, 72, 91). If Cdc7 is necessary to counteract Hst1 activity at the NDT80 promoter, then deletion of HST1, like sum1Δ, should allow NDT80 expression and suppress the meiotic progression defect of cdc7-as +PP1 cells. Consistent with this idea, cdc7-as hst1Δ + PP1 cells exhibit Ndt80 protein and progress through a single meiotic division to make dyad asci with kinetics similar to those of cdc7-asNDT80 +PP1 cells, supporting the idea that Cdc7 function is required to antagonize Hst1 function at the NDT80 promoter (Fig. 1D and 2). As expected, deletion of RFM1 similarly suppresses the sporulation defect of cdc7-as +PP1 cells to produce dyads (Fig. 2B). It should be noted that suppression in cdc7-as +PP1 cells of both production of Ndt80 and meiotic progression by either hst1Δ or ectopic expression of NDT80 is considerably delayed compared to when SUM1 itself is deleted (Fig. 1D and 2A). This fact suggests that Cdc7 may have additional functions in NDT80 expression beyond antagonizing Hst1 (see below).
Hst1 shares a high degree of conservation with the histone deacetylase Sir2 and exhibits deacetylase activity in vitro (8, 77) One proposal is that Hst1 deacetylation of histones or other proteins at the NDT80 promoter creates a chromatin configuration which inhibits Sum1 removal (2, 72). This model assumes that the catalytic activity of Hst1 is important for repressing Ime1-mediated transcription of NDT80, but this idea has not been tested. We therefore constructed a catalytically inactive version of Hst1 using Sir2 as a paradigm. In Sir2, alteration of a highly conserved histidine at position 364 to tyrosine abolishes deacetylase activity in vitro and silencing in vivo (8, 79, 80). This mutation has no effect on the ability of Sir2 to form silencing complexes, suggesting that it abolishes specifically the catalytic activity of the enzyme, leaving the structure of the protein intact (79). In Hst1, the corresponding amino acid is histidine 310. Before testing the putative hst1-H310Y catalytic mutant in meiosis, a genetic experiment was performed to confirm that hst1-H310Y is defective in deacetylase activity.
To test whether hst1-H310Y is defective in deacetylase activity in vivo, we took advantage of a genetic condition by which the deacetylase activity of Hst1 substitutes for that of Sir2 to repress transcription at the HMR silent mating type locus. SUM1-1 is a dominant, single-amino-acid mutation that was originally identified by its ability to suppress the mating defect of sir2 mutants (12). Sum1-1, but not Sum1, is recruited to origin recognition complexes bound at HMR, which in turn recruits Hst1. The histone deacetylase activity of Hst1 then substitutes for that of Sir2, thereby allowing silencing (67, 77). If the H310Y mutation abolishes the catalytic activity of Hst1, then a MATα sir2 SUM1-1 hst1-H310Y haploid should fail to mate. Two versions of HST1 were used: a tagged version, where the protein could be monitored by immunoblot assays, and an untagged version, in case the tag has subtle phenotypic effects. The H310Y mutation was introduced into HST1-5HA and HST1, and the plasmids were transformed into a MATα sir2 SUM1-1 hst1Δ strain. Whereas both HST1-5HA and HST1 allowed mating with a MATa haploid, no mating was observed with the hst1 alleles containing the H310Y mutation (Fig. 3A). No differences in steady-state protein levels were observed between Hst1-5HA and Hst1-H310Y-5HA, further supporting the idea that the catalytic activity of the enzyme and not its stability is affected by the mutation (Fig. 3B).
cdc7-as diploids carrying either hst1Δ, HST1, or HST1-5HA sporulated well, producing both dyads and tetrads (Fig. 3C). The cdc7-as hst1-H310Y diploids did not sporulate as well as the wild type but still produced tetrads (Fig. 3C). Inactivation of Cdc7-as with PP1 prevented sporulation in the cdc7-as HST1 diploids. In contrast, the cdc7-as hst1-H310Y mutants sporulated to form dyads, similarly to cdc7-as hst1Δ cells (Fig. 3C). These experiments support the idea that Hst1 deacetylase activity is required to prevent Ime1-dependent transcription of NDT80 and that Cdc7 kinase activity is necessary to counteract this activity.
Cdc7 kinase activity regulates phosphorylation of Sum1 during meiosis.
A simple hypothesis is that Cdc7 abolishes Sum1 repression by phosphorylation of either Sum1 or Hst1. Sum1-3Flag and Hst1-5HA proteins were therefore analyzed in meiotic time course assays in the presence or absence of Cdc7 kinase activity to look for changes in protein mobility that may be indicative of phosphorylation. No mobility shift was observed for Hst1 either during meiosis or when Cdc7 was inactivated (Fig. 4A). In contrast, slower-migrating species of Sum1-3Flag were detected specifically during meiosis in the absence of PP1 (Fig. 4A). This shift was eliminated by inactivation of Cdc7-as, indicating that Sum1 is phosphorylated in a meiosis-specific, Cdc7-dependent fashion (Fig. 4A). The presence of slower-migrating species of Sum1-3Flag correlates with expression of NDT80, consistent with the idea that Cdc7-dependent modification of Sum1 protein is important for NDT80 expression. While steady-state levels of Sum1 decreased as cells proceeded through meiosis, consistent with previous observations, Sum1-3Flag protein accumulated in the presence of PP1 (2) (Fig. 4A). This result suggests that Cdc7 kinase activity is necessary for Sum1 degradation, either indirectly due to the induction of NDT80 or because phosphorylation of Sum1 or some other protein influences Sum1 protein stability.
cdc7-as diploids arrest in prophase in the presence of PP1 but not in its absence, raising the possibility that Sum1 modification occurs downstream of Ndt80 activation. This idea was ruled out by looking at Sum1-3Flag in an isogenic diploid homozygous for ndt80Δ, where cells arrest independently of PP1 (92). A meiosis-specific Cdc7-dependent mobility shift was observed in this strain as well (Fig. 4B). The mobility shift observed after 8 h in Spo medium was removed by phosphatase treatment, confirming that the Cdc7-dependent shift is due to phosphorylation (Fig. 4C).
Identification of in vivo phosphorylation sites on Sum1 in vegetative and meiotic cells.
To map phosphorylation sites on Sum1, Sum1-3Flag protein was purified from cdc7-as ndt80Δ cells and analyzed by mass spectrometry (MS). Three conditions were examined: 0 h after transfer to Spo medium (i.e., vegetative cells), 8 h in Spo, and 8 h in Spo plus PP1. ndt80Δ was included so that the cultures would arrest prior to MI independently of Cdc7 activity. The Cdc7-dependent, meiosis-specific mobility shift was detectable on the GelCode blue-stained proteins (Fig. 5A). The total peptide coverage for Sum1-3Flag protein from three conditions (T = 0, 8, and 8+PP1) was 71% (peptide coverage for the individual proteins is shown in Fig. S1 and the distribution of each peptide under the different conditions is provided in Table S1, both in the supplemental material). Phosphorylation sites are indicated in Fig. 5B. This MS analysis indicates that the pattern of Sum1 phosphorylation is dynamic, includes both constitutive and meiosis-specific Cdc7-dependent and -independent sites, and likely results from the action of several kinases under vegetative and meiotic conditions.
Fig 5.

Mapping phosphorylation sites on Sum1 under vegetative and meiotic conditions. A cdc7-as ndt80Δ SUM1-3Flag diploid (NH1068) was transferred to Spo medium, and Sum1-3Flag was purified after 0 h, 8 h, or 8 h with 15 μM PP1. (A) GelCode blue staining of Sum1-3Flag proteins used for MS analysis. (B) The Sum1-3Flag protein sequence is shown with phosphorylated amino acids indicated by larger letters. Circles below each phosphorylated amino acid indicate the three different conditions, with the bottom circle representing 0 h, the middle circle representing 8 h with PP1, and the top circle representing 8 h in Spo medium. Absence of a circle means that the peptide containing that phosphorylated residue was not detected under that condition. Open circles mean no phosphorylation. The circles are color coded based on Ascore value, which represents the degree of confidence that a particular amino acid is phosphorylated. Red, Ascore value >19, indicating 99% certainty; green, Ascore value <19 and >13, indicating 95% certainty; blue, Ascore value <13, indicating less than 95% certainty. Red letters at the end of the protein indicate the three Flag epitopes.
Cdc7-independent phosphorylation sites.
Sum1 T306 is phosphorylated by Ime2 in vitro and in vivo (2, 52). Detection of T306 phosphorylation using phosphospecific antibodies demonstrated that T306 phosphorylation is dynamic and is lost by 8 h after induction of meiosis (2). We were unable to detect phosphorylation of T306 in Sum1 from meiotic cells. This is not surprising for the protein derived from cells where Cdc7 is active, since T306 phosphorylation is no longer detectable by 8 h, the time point used for this analysis. It may also be that this phosphorylation is not stable during the arrest induced by cdc7-as plus PP1. Another explanation for not detecting phosphorylation at this site may be because T306 is surrounded by several other potential phosphorylation sites, thereby making assignment of phosphates to a particular serine or threonine by MS difficult.
Shin et al. (72) identified 11 putative minimal Cdk1 sites in Sum1. We were unable to determine the phosphorylation state of five of these sites, S242, S409, S512, S616, and T697, as these peptides were not detected by MS under any conditions (72) (see Fig. S1 in the supplemental material). Four of these sites, S313, S379, S738, and T817, exhibited constitutive phosphorylation (i.e., present in both vegetative and meiotic cells) that was independent of Cdc7 (Fig. 5B). Phosphorylation of the remaining two sites, S315 and S318, was observed only in cdc7-as cells arrested with PP1. This phosphorylation is therefore meiosis specific and may be removed as cells progress through meiosis so that the phosphates are gone by 8 h. In contrast, the cdc7-as +PP1 cells are arrested prior to MI, perhaps allowing phosphorylation to be maintained. It should be noted that, with the exception of S379 and S738, which have previously been shown to be phosphorylated by Cdk1 in vegetative cells (28), the possibility that some or all of these sites are phosphorylated by a proline-directed kinase other than Cdk1 cannot be ruled out (49).
By combining alanine substitutions for the 11 putative Cdk1 sites with one at the Ime2 T306 site, Shin et al. created an allele, sum1-ci, that fails to sporulate due to an inability to eliminate Sum1 repression (72). To determine whether phosphorylation of the four constitutive Cdk1 sites, in combination with T306 phosphorylation, is sufficient to promote NDT80 expression and allow sporulation, S313, S379, S738, and T817 were changed to alanine along with T306A to make sum1-c4i. Two copies of sum1-c4i were introduced into a cdc7-as sum1Δ diploid, and SUM1 and sum1-ci diploids were included as controls. Compared to wild type, which exhibited 87.5% sporulation (±5.1, n = 3), a reduction in sporulation was observed for both sum1-ci and sum1-c4i (43.8% ± 10.7% and 29.7% ± 5.0%, respectively). This sum1-ci phenotype is not as strong at that reported by Shin et al., where <2.5% sporulation was observed (72). For reasons that are not clear, the sum1-c4i phenotype is stronger than the sum1-ci diploid. sum1-ci suppression is very sensitive to the levels of sum-ci as well as NDT80, raising the possibility that Sum1 protein may be more limiting in our strains than in those of Shin et al. The SUM1, sum1-ci, and sum1-c4i alleles were therefore transferred into high-copy-number plasmids and tested for sporulation. The high-copy-number plasmids containing sum1-ci and sum1-c4i exhibited reductions in sporulation similar to those of the diploids with two integrated copies (SUM1, 79.0 ± 2.0; sum1-ci, 52 ± 5.4; and sum1-c4i, 24.3 ± 4.5; n = 3). The fact that the sporulation defect was not exacerbated by overexpression of the sum1 mutants suggests that limiting Sum1 protein is not responsible for the higher level of sporulation that we observed compared to that observed by Shin et al.
The sum1-c4i defect was even more apparent when meiotic progression was analyzed, with entry into the meiotic divisions delayed by 6 h compared to cdc7-as SUM1 (Fig. 6A). Consistent with the meiotic progression delay observed with active Cdc7, Ndt80 protein appeared 4 h later than in the cdc7-as SUM1 diploid. The cdc7-as sum1-c4i diploid arrested in prophase when Cdc7-as was inactivated by PP1, indicating that the alanine substitutions in the Sum1-c4i protein do not compromise its ability to repress transcription (Fig. 6A). The meiosis-specific mobility shift of Sum1-c4i was greatly reduced and resembled that observed when Cdc7 was inactivated by PP1 (Fig. 6B and C). These results suggest that the combined phosphorylation of Ime2 T306 with one or more of the four constitutively phosphorylated putative Cdk1 sites promotes the phosphorylation of Sum1 by Cdc7.
In addition to the Ime2 and putative Cdk1 sites, a number of phosphorylated amino acids were observed in the presence of PP1, indicating that they are independent of Cdc7 (Fig. 5B). The kinases responsible for these phosphorylation events remain to be determined.
Cdc7-dependent phosphorylation sites.
Amino acids whose phosphorylation is dependent upon Cdc7 in meiotic cells can be inferred from a pattern in which phosphorylation is absent when Cdc7-as is inactivated by PP1. If one assumes that Cdc7 is also required for vegetative phosphorylation of these amino acids, the Cdc7-dependent sites can be divided into constitutive and meiosis-specific groups. The constitutive sites are S20, S311, S378, and T1032. The MS analysis detected nine sites that may be meiosis-specific, Cdc7-dependent sites (i.e., they are not phosphorylated in vegetative cells or in meiosis in the presence of PP1): S62, S278, S381, S385, T392, S651, S655, S657, and T815. (S393 and T717 may also belong to this class, although because no peptides containing these amino acids were observed for the 0 h and 8 h plus PP1 conditions, respectively, this cannot be determined definitively from this analysis.) These 11 sites include ones where the Ascore value is low and their phosphorylation status is therefore questionable (Fig. 5B). Cdc7 prefers to phosphorylate amino acids directly upstream of a negative charge that can be provided either by aspartic or glutamic acid or by phosphorylation (13, 50, 64, 88). Of the 11 Cdc7-dependent phosphorylation sites, S62, S278, T392, and S655 meet one of these criteria.
To test whether any of these sites is responsible for the meiosis-specific, Cdc7-dependent Sum1 mobility shift, an allele of SUM1, sum1-12A, was synthesized in which the meiosis-specific, Cdc7-dependent phosphorylated amino acids were mutated to alanine (S62, S278, S381, S385, T392, S393, S651, S655, S657, T717, T815, and T1032, a constitutive Cdc7-dependent site which was originally classified as meiosis specific). The 12 alanine substitutions do not compromise Sum1's ability to repress NDT80 transcription, as evidenced by the fact that cdc7-as sum1-12A +PP1 cells arrest prior to MI, similar to cdc7-as SUM1 +PP1 cells (Fig. 6A). If the mutated amino acids are the sole targets of Cdc7 with regard to Sum1 regulation, then the sum1-12A diploid should arrest even when Cdc7 is active due to a failure to induce NDT80 transcription. This was not the case, as sporulation was unaffected in the cdc7-as sum1-12A strain (83% ± 4.8%). However, meiotic progression was delayed in the mutant approximately 4 h compared to the cdc7-as SUM1 strain (Fig. 6A), indicating a functional role for one or more of these Cdc7-dependent phosphorylated amino acids. Furthermore, the mobility shift of the Sum1-12A protein is delayed compared to Sum1, as is NDT80 production (Fig. 6B and C). These results show that some of the amino acids mutated in the sum1-12A mutant are functionally important, but clearly, more meiosis-specific, Cdc7-dependent phosphorylation sites remain to be identified.
Cdc7 promotes NDT80-independent removal of Sum1 at the NDT80 and SMK1 promoters.
Ndt80 and Sum1 are believed to compete for binding to overlapping DNA sequences in MSEs in vivo as evidenced by the fact that overexpression of NDT80 can overcome Sum1 repression of the NDT80 promoter and because the two proteins compete in in vitro binding assays (41, 61). However, recent work from the Winter lab has demonstrated that Sum1 can be removed from MSE-containing promoters in the absence of NDT80 and that this removal is dependent upon Ime2 or Cdk1 (2, 72). Given that Ime2 and Cdk1 phosphorylation of Sum1 is required for efficient phosphorylation by Cdc7, the prediction is that Cdc7 kinase activity should also be required for removal of Sum1 in the absence of NDT80. To test this hypothesis, chromatin immunoprecipitation (ChIP) experiments were performed by precipitating Sum1-3Flag from a cdc7-as ndt80Δ diploid under three conditions: asynchronously growing vegetative cells (0 h) or 8 h after transfer to Spo medium in the absence or presence of PP1. IPs from a cdc7-as ndt80Δ diploid containing untagged SUM1 were used to confirm the specificity of the IP while IPs with no antibodies were used for normalization as described in Materials and Methods. Primers flanking the MSEs in the NDT80 promoter were used for real-time PCRs and normalized to negative control CIT2 primers. Consistent with published results, Sum1 is bound to the NDT80 promoter in vegetative cells and this binding is greatly reduced in meiotic cells when Cdc7 is active (Fig. 7A) (2). Inactivation of Cdc7 resulted in a higher fraction of Sum1 remaining bound to the NDT80 promoter in meiosis. This pattern is lost in the IPs from the untagged Sum1 diploid (Fig. 7B). This regulation is not limited to NDT80, as a similar pattern was observed for SMK1, a gene in the Ndt80 regulon that is repressed by Sum1 (Fig. 7C) (2). Cdc7 is therefore likely promoting Sum1 removal from the promoters of a number of NDT80-regulated genes.
Fig 7.
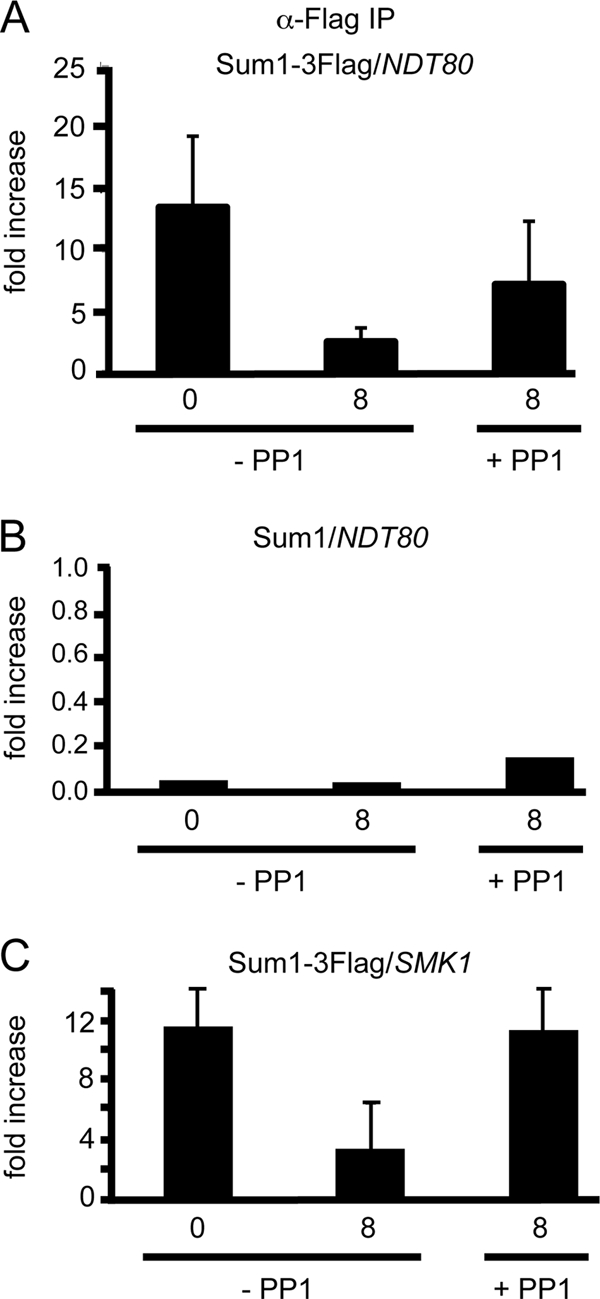
Chromatin immunoprecipitation of Sum1-3Flag at the NDT80 and SMK1 promoters in the absence or presence of Cdc7 kinase. ChIP was performed using anti-Flag antibodies on chromatin isolated from cdc7-as ndt80Δ diploids carrying either Sum1-3Flag (NH1080) or untagged Sum1 (NH932) under three conditions: 0 or 8 h after transfer to Spo medium in the absence or presence of 15 μM PP1. Three independent experiments were performed for each strain, and the standard deviations are indicated by error bars. (A) q-PCR results from Sum1-3Flag diploid using primers flanking the MSEs in the NDT80 promoter normalized to the negative-control primers. (B) q-PCR results from the untagged diploid using primers flanking the MSEs in the NDT80 promoter normalized to the negative-control primers. Note the change in scale. (C) q-PCR results from Sum1-3Flag diploid using primers flanking the MSEs in the SMK1 promoter normalized to the negative-control primers.
DISCUSSION
While a role for the essential cell cycle kinase, Cdc7, in the initiation of DNA replication has long been known, recent studies on meiosis in budding and fission yeast have revealed that Cdc7 is required for numerous additional chromosomal processes, including the initiation of meiotic recombination, the mono-orientation of homologous chromosomes at MI, and the regulated removal of meiosis-specific cohesion complexes (33, 41, 46, 54, 68, 88). This work has identified a previously unknown function of Cdc7 during meiosis as a gene-specific transcriptional regulator. Cdc7 has previously been implicated in heterochromatin-mediated transcriptional silencing around centromeres observed in vegetatively growing fission yeast cells (4). This effect is regional, however, and does not work by targeting the promoters of specific genes. In contrast, Cdc7 is required to activate transcription of NDT80 and does so by promoting the removal of the Sum1 repressor from a specific sequence in the promoter. The finding that Sum1 removal at the SMK1 promoter also requires Cdc7 indicates that Cdc7 is involved in gene-specific regulation of the Ndt80 regulon and potentially other classes of genes as well.
CDC7 control of NDT80 transcription is upstream of checkpoint activation.
That Cdc7 regulates the Ime1-dependent transcription of NDT80, and not the Ndt80-mediated autoregulatory loop, is based in part on the fact that a low level of ectopically expressed NDT80 is sufficient to suppress the sporulation defect conferred by inactivation of Cdc7-as (41). The amount of Ndt80 produced by basal transcription from the CUP1 promoter is not sufficient to complement an ndt80Δ diploid but is sufficient to bind the promoters of endogenous NDT80 genes to jumpstart the autoregulatory loop. The Ndt80 produced in the absence of Cdc7 kinase activity must therefore be functional for the Ndt80-mediated transcription of the NDT80 regulon, including NDT80 itself (41). This result rules out Cdc7 as one of the kinases that is required for the activation of Ndt80 (6, 74, 83). More directly, activation of NDT80 transcription by Ime1 and Ndt80 was uncoupled using a DNA-binding-defective mutant of NDT80. Transcripts observed in this mutant must be due to Ime1, and these transcripts are eliminated by inhibition of Cdc7 kinase activity.
In contrast to work from several labs using different strain backgrounds and methods of Cdc7 inactivation where a prophase arrest was observed (68, 69, 84, 89; this work), Matos et al. observed robust expression of NDT80 and high levels of sporulation using both cdc7ts and cdc7Δ bob1 diploids (46). These authors proposed that this discrepancy was due to “checkpoint activation in response to replication defects.” This explanation is unlikely for several reasons. First, it is not clear why the cdc7ts and cdc7Δ bob1 diploids used by other labs would trigger a checkpoint response while the same mutants in the Matos et al. paper would not. Second, Lo et al. showed that mutations that abrogate the S-phase checkpoint, DNA damage checkpoint, and meiotic recombination checkpoint do not suppress the cdc7-as +PP1 cell arrest (41). Finally, the meiotic recombination checkpoint blocks meiotic progression by preventing Ndt80 protein that has been generated using Ime1-dependent transcription from participating in the autoregulatory loop (15, 40, 58). As mentioned above, Cdc7 activity is not necessary once a priming amount of Ndt80 is generated, ruling out a requirement in Ndt80 activation. Instead, this posttranscriptional regulation is due in part to sequestration of Ndt80 protein in the cytoplasm under checkpoint-induced conditions (90). In contrast, Cdc7 is required not for Ndt80 activation but for Ime1-mediated expression of NDT80; therefore, the arrest is not checkpoint related. A more likely explanation for the lack of arrest observed by Matos et al. is that their cdc7 strains harbor a natural variant of a gene involved in the transcriptional induction of NDT80, such as SUM1, RFM1, HST1, or even NDT80 itself.
Cdc7 antagonizes Hst1 deacetylase activity to promote the removal of Sum1 repression of NDT80 transcription.
That Hst1 functions as a deacetylase in vivo has been demonstrated in vegetative cells both at the HMR locus in SUM1-1 strains and at a subset of origins of replication (30, 67). Deletion of HST1 suppresses the sporulation defect of the sum1-ci allele, suggesting that phosphorylation of Sum1 by Cdk1 and Ime2 antagonizes Hst1 function, but this experiment does not address whether it is the deacetylase, or some other unidentified function of the protein, that is being affected (72). The finding that a catalytically inactive mutant of HST1 is able to suppress the sporulation defect conferred by inactivation of Cdc7 suggests that allowing acetylation of histones (or other proteins) is a key first step in removing Sum1 repression (Fig. 8). Cdc7 could affect the catalytic activity of Hst1 directly, although no evidence that Cdc7 phosphorylates Hst1 was observed. Given that Ime2 and Cdk1 phosphorylation promotes Cdc7 phosphorylation of Sum1, we propose a modified version of a model by Shin et al. in which Cdc7 phosphorylation of Sum1 allows dissociation of Hst1 (and perhaps Rfm1) from the NDT80 promoter (see below) (72).
The assumption is that loss of Hst1 from the promoter allows acetylases to modify histones to “create a chromatin state in meiotic cells that is permissive for Sum1 removal” (2). Exactly what this change in chromatin state involves is not yet clear, however. Fine mapping studies of nucleosome positions have revealed that the −1 nucleosome in the NDT80 promoter is located just upstream of MSE-1, the MSE shown by Pak and Segall to be necessary for Sum1-mediated repression of an NDT80 minigene (57). MSE-2, which is capable of Sum1-mediated repression of a reporter plasmid in vegetative cells but was not observed to be necessary for NDT80 minigene repression, is located downstream of the transcription start site (TSS), in the nucleosome-depleted region (47, 91). Seven hours after transfer to Spo medium, the −1 nucleosome transiently moves approximately 20 base pairs away from the transcription start site (93). While this does not represent a dramatic remodeling, this movement could potentially weaken the interaction between Sum1 and MSE-1, allowing a low level of Ime1-dependent transcription to occur (Fig. 8). Alternatively, given that histone acetylation promotes binding by bromo-domain-containing proteins; perhaps recruitment of another protein(s) to the acetylated histones results in weakening Sum1 repression (85).
For many meiotic genes, deletion of HST1 is not sufficient to derepress them to the sum1Δ levels in vegetative cells (48). This has led Ahmed et al. to propose that Sum1 removal requires an additional factor (2). Cdc7 may be that factor. When Cdc7 is active, meiotic progression of cdc7-asNDT80, cdc7-as sum1Δ, and cdc7-as hst1Δ cells occurs with similar kinetics while slightly faster than cdc7-as cells, due to premature expression of NDT80. However, when Cdc7-as is inhibited by PP1, NDT80 expression and meiotic progression occur 2 h faster in the cdc7-as sum1Δ bypass strain than in the cdc7-as hst1Δ or cdc7-asNDT80 bypass diploids (Fig. 1 and 2A). Inactivation of Cdc7 therefore phenotypically differentiates between loss of the Sum1 protein from the promoter (sum1Δ) and the loss of the histone deacetylase (hst1Δ). The delay indicates that in addition to antagonizing Hst1 function, Cdc7 is required for some other step that fully removes Sum1 from the promoter. For example, Cdc7/Ime2/Cdk1 phosphorylation could disrupt the interaction between Sum1 and Rfm1/Hst1 and, in addition, Cdc7 phosphorylation could affect the binding affinity of Sum1 for the MSE (Fig. 8). The observation that Sum1 removal by the NDT80-independent pathway from the NDT80 and SMK1 promoters requires Cdc7 is consistent with this idea. Once a small amount of NDT80 is transcribed, Ndt80 can compete off any remaining Sum1 and the positive feedback loop begins.
Sum1 is regulated by a dynamic and complex set of phosphorylation events.
Mobility shift analysis demonstrated that Sum1 undergoes meiosis-specific, Cdc7-dependent phosphorylation. Whether this phosphorylation is direct or not is unclear. MS analysis of Sum1 from vegetative and meiotic cells with or without Cdc7 kinase activity revealed a number of meiosis-specific, Cdc7-dependent phosphorylated amino acids. However, many of these sites are not upstream of a negative charge (conferred by aspartic or glutamic acid or phosphorylation) and therefore do not conform to the Cdc7 consensus (13, 49, 50). Mutation of 11 of these Cdc7-dependent sites results in a delay in Sum1 phosphorylation, NDT80 expression, and meiotic progression. Therefore, some subset of these sites is important for function. Further study is required to determine if the critical amino acids are direct targets of Cdc7 or of a Cdc7-dependent protein kinase.
Why does sum1-12A fail to phenocopy the complete arrest of cdc7-as +PP1 cells? One possibility is that not all of the Cdc7-dependent sites on Sum1 were mutated. Cdc7 phosphorylation often occurs redundantly on proteins, with a cloud of negative charge being more important than the specific amino acids that are mutated (64, 68, 71). In fact, no MS data were obtained for ∼30% of the Sum1 protein, so several sites may have been missed. Alternatively, Cdc7-dependent phosphorylation may need to occur on more than one protein. For example, if the mechanism by which Cdc7 antagonizes Hst1 is to disrupt the interaction between Sum1 and Rfm1/Hst1, then Cdc7-dependent phosphorylation of either Rfm1 or Hst1 may be sufficient to dissociate the complex even in the absence of Sum1 phosphorylation. Studies of Cdc7 phosphorylation of the Mcm2-7 helicase found that mutation of the Cdc7 sites on MCM4 or MCM6 alone was not sufficient to create a growth defect, but combining the mcm4 and mcm6 phosphomutants killed the cells (64).
Both Cdk1 and Cdc7 are present in vegetative cells and yet do not interfere with Sum1 repression. Given that the four Cdk1 sites important for relieving Sum1 repression are constitutively phosphorylated, it is Cdc7 phosphorylation that is specifically promoted during meiosis. The fact that Ime2 is meiosis specific and that the combined phosphorylation of Ime2 and Cdk1 sites promotes meiosis-specific, Cdc7-dependent phosphorylation of Sum1 indicates that signals from these two kinases are integrated by Cdc7. One possibility is that Ime2 and Cdk1 phosphorylation primes Cdc7 phosphorylation of the immediately adjacent upstream amino acids, similar to what has been observed for proteins involved in meiotic recombination and DNA replication (64, 88). An argument against this idea, however, is the lack of observed Cdc7-dependent phosphorylation sites immediately upstream of the Cdk1 and Ime2 sites. An alternative hypothesis is that the cumulative phosphorylation by Cdk1 and Ime2 enhances recruitment of Cdc7 to Sum1 at promoters, thereby enabling Cdc7-dependent Sum1 phosphorylation (Fig. 8).
Conclusions.
Cdc7 is a key regulator of meiosis because of its role in promoting meiosis-specific chromosomal processes such as recombination, mono-orientation of homologous chromosomes, and meiotic cohesion cleavage. This work adds another role that Cdc7 plays in the cell—that of a gene-specific regulator of transcription. The function of Cdc7 as a gene-specific regulator may be conserved. In fission yeast, mutation of the Cdc7 ortholog, hsk1, results in a meiotic arrest with normal early gene transcription (56). Perhaps Hsk1 regulates Mei4, a transcription factor analogous to Ndt80 (1). Furthermore, reduced levels of Cdc7 kinase activity result in defects in spermatogenesis prior to MI in mammalian cells (35). Whether this is due to transcriptional effects is not yet known.
Whether Cdc7 is directly involved in coordinating different meiotic processes is not yet clear. There is not an absolute dependence on a specific order of events. For example, when the meiotic arrest due to Cdc7 inactivation is suppressed by sum1Δ, meiotic progression occurs even though recombination and mono-orientation have not. Still it is intriguing to speculate that in a normal meiosis, Cdc7 helps to ensure that a linear progression of events occurs. One way in which this could be regulated is by different processes requiring increasing amounts of Cdc7 activity, similar to what has been proposed for Cdk1-Clb5 in meiosis (23). Further studies are needed to determine whether and how Cdc7 controls the various chromosomal events of meiosis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Huei-Mei Chen, Bruce Futcher, Ed Luk, Aaron Neiman, Jacqueline Segall, and Rolf Sternglanz for helpful discussions. Bruce Futcher, Aaron Neiman, and Rolf Sternglanz provided helpful comments on the manuscript. Michael Lichten, Laura Rusche, Jacqueline Segall, Rolf Sternglanz, and Edward Winter generously provided antibodies, strains, or plasmids. We are grateful to Chao Zhang and Kevan Shokat for PP1. Thanks go to Rachel Salatka for constructing the H310Y hst1 alleles.
This work was supported by NIH grants R01GM50717 and 5P01GM088297 to N.M.H. and HG3456 to S.P.G.
Footnotes
Published ahead of print 21 November 2011
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Abe H, Shimoda C. 2000. Autoregulated expression of Schizosaccharomyces pombe meiosis-specific transcription factor Mei4 and a genome-wide search for its target genes. Genetics 154:1497–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmed NT, Bungard D, Shin ME, Moore M, Winter E. 2009. The Ime2 protein kinase enhances the disassociation of the Sum1 repressor from middle meiotic promoters. Mol. Cell. Biol. 29:4352–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allers T, Lichten M. 2001. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106:47–57 [DOI] [PubMed] [Google Scholar]
- 4. Bailis JM, Bernard P, Antonelli R, Allshire RC, Forsburg SL. 2003. Hsk1-Dfp1 is required for heterochromatin-mediated cohesion at centromeres. Nat. Cell Biol. 5:1111–1116 [DOI] [PubMed] [Google Scholar]
- 5. Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. 2006. A probability-based approach for high throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24:1285–1292 [DOI] [PubMed] [Google Scholar]
- 6. Benjamin KR, Zhang C, Shokat KM, Herskowitz I. 2003. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev. 17:1524–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borde V, Goldman AS, Lichten M. 2000. Direct coupling between meiotic DNA replication and recombination initiation. Science 290:806–809 [DOI] [PubMed] [Google Scholar]
- 8. Brachmann CB, et al. 1995. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 9:2888–2902 [DOI] [PubMed] [Google Scholar]
- 9. Buckingham LE, et al. 1990. Nucleotide sequence and promoter analysis of SPO13, a meiosis-specific gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 87:9406–9410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buonomo SB, et al. 2000. Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 103:387–398 [DOI] [PubMed] [Google Scholar]
- 11. Carlile TM, Amon A. 2008. Meiosis I is established through division-specific translational control of a cyclin. Cell 133:280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chi MH, Shore D. 1996. SUM1-1, a dominant suppressor of SIR mutations in Saccharomyces cerevisiae, increases transcriptional silencing at telomeres and HM mating-type loci and decreases chromosome stability. Mol. Cell. Biol. 16:4281–4294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho WH, Lee YJ, Kong SI, Hurwitz J, Lee JK. 2006. CDC7 kinase phosphorylates serine residues adjacent to acidic amino acids in the minichromosome maintenance 2 protein. Proc. Natl. Acad. Sci. U. S. A. 103:11521–11526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chu S, et al. 1998. The transcriptional program of sporulation in budding yeast. Science 282:699–705 (Erratum, 282:1421.) [DOI] [PubMed] [Google Scholar]
- 15. Chu S, Herskowitz I. 1998. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol. Cell 1:685–696 [DOI] [PubMed] [Google Scholar]
- 16. de los Santos T, Hollingsworth NM. 1999. Red1p: a MEK1-dependent phosphoprotein that physically interacts with Hop1p during meiosis in yeast. J. Biol. Chem. 274:1783–1790 [DOI] [PubMed] [Google Scholar]
- 17. Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4:207–214 [DOI] [PubMed] [Google Scholar]
- 18. Eng JK, McCormack AL, Yates JR., III 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989 [DOI] [PubMed] [Google Scholar]
- 19. Fingerman I, Sutphen K, Montano SP, Georgiadis MM, Vershon AK. 2004. Characterization of critical interactions between Ndt80 and MSE DNA defining a novel family of Ig-fold transcription factors. Nucleic Acids Res. 32:2947–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldmark JP, Fazzio TG, Estep PW, Church GM, Tsukiyama T. 2000. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103:423–433 [DOI] [PubMed] [Google Scholar]
- 21. Hardy CF, Dryga O, Seematter S, Pahl PM, Sclafani RA. 1997. mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc. Natl. Acad. Sci. U. S. A. 94:3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heller RC, et al. 2011. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146:80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Henderson KA, Kee K, Maleki S, Santini P, Keeney S. 2006. Cyclin-dependent kinase directly regulates initiation of meiotic recombination. Cell 125:1321–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hepworth SR, Ebisuzaki LK, Segall J. 1995. A 15-base-pair element activates the SPS4 gene midway through sporulation in Saccharomyces cerevisiae. Mol. Cell. Biol. 15:3934–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hepworth SR, Friesen H, Segall J. 1998. NDT80 and the meiotic recombination checkpoint regulate expression of middle sporulation-specific genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 18:5750–5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hollingsworth NM. 2008. Deconstructing meiosis one kinase at a time: polo pushes past pachytene. Genes Dev. 22:2596–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hollingsworth NM, Ponte L, Halsey C. 1995. MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes Dev. 9:1728–1739 [DOI] [PubMed] [Google Scholar]
- 28. Holt LJ, et al. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325:1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huttlin EL, et al. 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143:1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Irlbacher H, Franke J, Manke T, Vingron M, Ehrenhofer-Murray AE. 2005. Control of replication initiation and heterochromatin formation in Saccharomyces cerevisiae by a regulator of meiotic gene expression. Genes Dev. 19:1811–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jolly ER, Chin CS, Herskowitz I, Li H. 2005. Genome-wide identification of the regulatory targets of a transcription factor using biochemical characterization and computational genomic analysis. BMC Bioinformatics 6:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kadosh D, Struhl K. 1998. Targeted recruitment of the Sin3-Rpd3 histone deacetylase complex generates a highly localized domain of repressed chromatin in vivo. Mol. Cell. Biol. 18:5121–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katis VL, et al. 2010. Rec8 phosphorylation by casein kinase 1 and Cdc7-Dbf4 kinase regulates cohesin cleavage by separase during meiosis. Dev. Cell 18:397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keeney S. 2001. Mechanism and control of meiotic recombination initiation. Curr. Top. Dev. Biol. 52:1–53 [DOI] [PubMed] [Google Scholar]
- 35. Kim JM, Takemoto N, Arai K, Masai H. 2003. Hypomorphic mutation in an essential cell-cycle kinase causes growth retardation and impaired spermatogenesis. EMBO J. 22:5260–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klein F, et al. 1999. A central role for cohesins in sister chromatid cohesion, formation of axial elements and recombination during meiosis. Cell 98:91–103 [DOI] [PubMed] [Google Scholar]
- 37. Krisak L, et al. 1994. SMK1, a developmentally regulated MAP kinase, is required for spore wall assembly in Saccharomyces cerevisiae. Genes Dev. 8:2151–2161 [DOI] [PubMed] [Google Scholar]
- 38. Lamoureux JS, Stuart D, Tsang R, Wu C, Glover JN. 2002. Structure of the sporulation-specific transcription factor Ndt80 bound to DNA. EMBO J. 21:5721–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Law DT, Segall J. 1988. The SPS100 gene of Saccharomyces cerevisiae is activated late in the sporulation process and contributes to spore wall maturation. Mol. Cell. Biol. 8:912–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lindgren A, et al. 2000. The pachytene checkpoint in Saccharomyces cerevisiae requires the Sum1-transcriptional repressor. EMBO J. 19:6489–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lo H-C, Wan L, Rosebrock A, Futcher B, Hollingsworth NM. 2008. Cdc7-Dbf4 regulates NDT80 transcription as well as reductional segregation during budding yeast meiosis. Mol. Biol. Cell 19:4956–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Longtine MS, et al. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961 [DOI] [PubMed] [Google Scholar]
- 43. Mallory MJ, Cooper KF, Strich R. 2007. Meoisis-specific destruction of the Ume6p repressor by the Cdc20-directed APC/C. Mol. Cell 27:951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marston AL, Amon A. 2004. Meiosis: cell-cycle controls shuffle and deal. Nat. Rev. 5:983–997 [DOI] [PubMed] [Google Scholar]
- 45. Masai H, et al. 2006. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281:39249–39261 [DOI] [PubMed] [Google Scholar]
- 46. Matos J, et al. 2008. Dbf4-dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135:662–678 [DOI] [PubMed] [Google Scholar]
- 47. Mavrich TN, et al. 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 18:1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCord R, et al. 2003. Rfm1, a novel tethering factor required to recruit the Hst1 histone deacetylase for repression of middle sporulation genes. Mol. Cell. Biol. 23:2009–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mok J, et al. 2010. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 3:ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Montagnoli A, et al. 2006. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 281:10281–10290 [DOI] [PubMed] [Google Scholar]
- 51. Montano SP, et al. 2002. Crystal structure of the DNA-binding domain from Ndt80, a transcriptional activator required for meiosis in yeast. Proc. Natl. Acad. Sci. U. S. A. 99:14041–14046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Moore M, et al. 2007. Ar-Pro-X-Ser/Thr is a consensus phosphoacceptor sequence for the meiosis-specific Ime2 protein kinase in Saccharomyces cerevisiae. Biochemistry 46:271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murakami H, Keeney S. 2008. Regulating the formation of DNA double-strand breaks in meiosis. Genes Dev. 22:286–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Murakami H, Nurse P. 2001. Regulation of premeiotic S phase and recombination-related double-strand DNA breaks during meiosis in fission yeast. Nat. Genet. 28:290–293 [DOI] [PubMed] [Google Scholar]
- 55. Niu H, et al. 2009. Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol. Cell 36:393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ogino K, et al. 2006. Hsk1 kinase is required for induction of meiotic dsDNA breaks without involving checkpoint kinases in fission yeast. Proc. Natl. Acad. Sci. U. S. A. 103:8131–8136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pak J, Segall J. 2002. Regulation of the premiddle and middle phases of expression of the NDT80 gene during sporulation of Saccharomyces cerevisiae. Mol. Cell. Biol. 22:6417–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pak J, Segall J. 2002. Role of Ndt80, Sum1, and Swe1 as targets of the meiotic recombination checkpoint that control exit from pachytene and spore formation in Saccharomyces cerevisiae. Mol. Cell. Biol. 22:6430–6440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Percival-Smith A, Segall J. 1986. Characterization and mutational analysis of a cluster of three genes expressed preferentially during sporulation of Saccharomyces cerevisiae. Mol. Cell. Biol. 6:2443–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Petronczki M, Siomos MF, Nasmyth K. 2003. Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell 112:423–440 [DOI] [PubMed] [Google Scholar]
- 61. Pierce M, et al. 2003. Sum1 and Ndt80 proteins compete for binding to middle sporulation element sequences that control meiotic gene expression. Mol. Cell. Biol. 23:4814–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Primig M, et al. 2000. The core meiotic transcriptome in budding yeasts. Nat. Genet. 26:415–423 [DOI] [PubMed] [Google Scholar]
- 63. Rabitsch KP, et al. 2003. Kinetochore recruitment of two nucleolar proteins is required for homolog segregation in meiosis I. Dev. Cell 4:535–548 [DOI] [PubMed] [Google Scholar]
- 64. Randell JC, et al. 2010. Mec1 is one of multiple kinases that prime the Mcm2-7 helicase for phosphorylation by Cdc7. Mol. Cell 40:353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rubin-Bejerano I, Mandel S, Robzyk K, Kassir Y. 1996. Induction of meiosis in Saccharomyces cerevisiae depends on conversion of the transcriptional repressor Ume6 to a positive regulator by its regulated association with the transcriptional activator Ime1. Mol. Cell. Biol. 16:2518–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rundlett SE, Carmen AA, Suka N, Turner BM, Grunstein M. 1998. Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4 by RPD3. Nature 392:831–835 [DOI] [PubMed] [Google Scholar]
- 67. Rusche LN, Rine J. 2001. Conversion of a gene-specific repressor to a regional silencer. Genes Dev. 15:955–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sasanuma H, et al. 2008. Cdc7-dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev. 22:398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schild D, Byers B. 1978. Meiotic effects of DNA-defective cell division cycle mutations of Saccharomyces cerevisiae. Chromosoma 70:109–130 [DOI] [PubMed] [Google Scholar]
- 70. Sclafani RA. 2000. Cdc7p-Dbf4p becomes famous in the cell cycle. J. Cell Sci. 113:2111–2117 [DOI] [PubMed] [Google Scholar]
- 71. Sheu YJ, Stillman B. 2006. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24:101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shin ME, Skokotas A, Winter E. 2010. The Cdk1 and Ime2 protein kinases trigger exit from meiotic prophase in Saccharomyces cerevisiae by inhibiting the Sum1 transcriptional repressor. Mol. Cell. Biol. 30:2996–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith AV, Roeder GS. 1997. The yeast Red1 protein localizes to the cores of meiotic chromosomes. J. Cell Biol. 136:957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sopko R, Raithatha S, Stuart D. 2002. Phosphorylation and maximal activity of Saccharomyces cerevisiae meiosis-specific transcription factor Ndt80 is dependent on Ime2. Mol. Cell. Biol. 22:7024–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sourirajan A, Lichten M. 2008. Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev. 22:2627–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Strich R, et al. 1994. UME6 is a key regulator of nitrogen repression and meiotic development. Genes Dev. 8:796–810 [DOI] [PubMed] [Google Scholar]
- 77. Sutton A, et al. 2001. A novel form of transcriptional silencing by Sum1-1 requires Hst1 and the origin recognition complex. Mol. Cell. Biol. 21:3514–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sym M, Engebrecht J, Roeder GS. 1993. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72:365–378 [DOI] [PubMed] [Google Scholar]
- 79. Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. 1999. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99:735–745 [DOI] [PubMed] [Google Scholar]
- 80. Tanny JC, Moazed D. 2001. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: evidence for acetyl transfer from substrate to an NAD breakdown product. Proc. Natl. Acad. Sci. U. S. A. 98:415–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tong A, Boone C. 2005. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae. Methods Mol. Biol. 313:171–192 [DOI] [PubMed] [Google Scholar]
- 82. Toth A, et al. 2000. Functional genomics identifies monopolin: a kinetochore protein required for segregation of homologs during meiosis i. Cell 103:1155–1168 [DOI] [PubMed] [Google Scholar]
- 83. Tung KS, Hong EJ, Roeder GS. 2000. The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis-specific transcription factor Ndt80. Proc. Natl. Acad. Sci. U. S. A. 97:12187–12192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Valentin G, Schwob E, Della Seta F. 2006. Dual role of the CDC7-regulatory protein DBF4 during yeast meiosis. J. Biol. Chem. 281:2828–2834 [DOI] [PubMed] [Google Scholar]
- 85. Venters BJ, Pugh BF. 2009. How eukaryotic genes are transcribed. Crit. Rev. Biochem. Mol. Biol. 44:117–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vershon AK, Hollingsworth NM, Johnson AD. 1992. Meiotic induction of the yeast HOP1 gene is controlled by positive and negative regulatory elements. Mol. Cell. Biol. 12:3706–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vershon AK, Pierce M. 2000. Transcriptional regulation of meiosis in yeast. Curr. Opin. Cell Biol. 12:334–339 [DOI] [PubMed] [Google Scholar]
- 88. Wan L, et al. 2008. Cdc28-Clb5 (CDK-S) and Cdc7-Dbf4 (DDK) collaborate to initiate meiotic recombination in yeast. Genes Dev. 22:386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wan L, Zhang C, Shokat KM, Hollingsworth NM. 2006. Chemical inactivation of Cdc7 kinase in budding yeast results in a reversible arrest that allows efficient cell synchronization prior to meiotic recombination. Genetics 174:1767–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang Y, Chang CY, Wu JF, Tung KS. 2011. Nuclear localization of the meiosis-specific transcription factor Ndt80 is regulated by the pachytene checkpoint. Mol. Biol. Cell 22:1878–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xie J, et al. 1999. Sum1 and Hst1 repress middle sporulation-specific gene expression during mitosis in Saccharomyces cerevisiae. EMBO J. 18:6448–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. 1995. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol. Cell. Biol. 15:6572–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang L, Ma H, Pugh BF. 2011. Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res. 21:875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.