

Abstract
Glioblastoma-derived stem cells (GSCs) are responsible for the cancer resistance to therapies. We show here that GSC-enriched neurospheres are resistant to the treatment of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) due to the insufficient expression of the death receptor DR4 and DR5 and the overexpression of cellular Fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein (c-FLIP). However, treatment with cisplatin leads to the upregulation of DR5 and downregulation of c-FLIP and restores TRAIL apoptotic pathway in the neurospheres. This study suggests that the combined treatment of TRAIL and cisplatin can induce apoptosis in GSCs and thus provide an effective treatment of glioblastomas.
Keywords: apoptosis, cancer stem cell, cisplatin, glioblastoma, TRAIL
INTRODUCTION
Glioblastoma is the most common and lethal brain tumor and current standard therapies including surgery, chemotherapy and radiation provide no curative treatments (1). Recent studies have identified cancer stem cells or tumor-initiating cells in glioblastomas (2–4) and shown that glioblastoma-derived stem cells (GSCs) are responsible for the tumor resistance to radiation (5), chemotherapy (6) and chemoradiotherapy (7). Cancer stem cells are defined as a minority of undifferentiated cell populations that are able to self-renew, differentiate to multiple cell lineages and initiate tumor xenografts (8). These properties of cancer stem cells have been demonstrated in neurospheres (9–11) and cell populations positive for the stem cell surface markers, CD133 (3–5, 12) and CD15 (13). These GSC-enriched neurospheres, CD133+ and CD15+ cell populations isolated from glioblastomas have been shown to retain the genomic properties of the original tumors (11, 14) and they are responsible for the tumor progression (15). Further studies of these GSC-enriched models have elucidated signal pathways that are activated in GSC and shown that targeting of the GSC signal pathways inhibits the cancer growth and progression (16–19). These studies have established glioblastoma-derived neurospheres and CD133+/CD15+ cell populations as GSC-enriched models for studying of signal pathways and screening of therapeutic agents in treating the cancer (20). This has prompted us to examine whether and how GSC responds to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL).
TRAIL is a member of tumor necrosis factor (TNF) ligand family that is normally expressed in immune system and plays a role in immunosurveillance against the tumorigenesis and tumor progression (21). Studies of glioblastoma-derived cell lines have shown that TRAIL can trigger apoptosis in some of the cell lines (22–24) and thus suggested that targeting TRAIL apoptotic pathway may provide novel therapeutic approach in treating glioblastomas (25). TRAIL-induced apoptosis occurs through its binding of the cell surface death receptor 4 and 5 (DR4, DR5), which in turn recruit intracellular Fas-associated death domain (FADD) and caspase-8 for the assembly of a death-inducing signaling complex (DISC) (26). In the DISC, caspase-8 is enzymatically activated and thus initiates apoptotic cascade through cleavage of downstream effector caspase-3 and caspase-7 and activation of the mitochondrial apoptosis pathway (27). However, the majority of glioblastoma-derived cell lines are resistant to TRAIL-induced apoptosis in part due to the expression of cellular FADD-like interleukin-1β-converting enzyme-inhibitory protein (c-FLIP) (28). The c-FLIP protein exists in two isoforms: a short form (c-FLIPS) consisting of two death effector domains (DEDs) and a long form (c-FLIPL) composed of two DEDs and a caspase-like domain that lacks catalytic activity (29, 30). Through DEDs, c-FLIPL and c-FLIPS interact with FADD and caspase-8 and inhibit caspase-8 activation and TRAIL-induced apoptosis (26, 28, 31).
While TRAIL apoptotic pathway has been extensively studied, phase I and II clinical trials of TRAIL apoptotic pathway-targeted therapies have shown that the vast majority of human cancers are resistant to the treatments (32). In examining the molecular mechanisms of the glioblastoma resistance to TRAIL treatment, we show here that GSC-enriched neurospheres are resistant to TRAIL-induced apoptosis due to the lack of DR4 and DR5 expression and the overexpression of c-FLIPL and c-FLIPS. In searching of the therapeutic modalities that can overcome the TRAIL resistance in GSC, we show that chemotherapeutic agent, cisplatin can up-regulate DR5 but down-regulate c-FLIPL and c-FLIPS, thus release their inhibition of caspase-8 and thereby restore TRAIL apoptotic pathway in GSC. This study suggests that the combination therapy of cisplatin and TRAIL can drive GSC into self-destructive process and therefore provide effective treatment for glioblastomas.
MATERIALS AND METHODS
Glioblastoma-derived Neurospheres, Serum-Grown Cultures and Cell Lines
Glioblastoma tissues were collected in accordance with protocols approved by the Emory University Institutional Review Boards and enzymatically dissociated based on the procedure as reported (9). The dissociated cells were submitted for neurosphere and serum-grown culture according to the protocol as earlier reported (11). For neurosphere culture, the dissociated cells were plated in uncoated plastic flasks at a clonal density of 3,500 cells/cm2 in neurobasal medium (Invitrogen), B27 (0.5x; Invitrogen), 1 mM L-Glutamine, epidermal growth factor (EGF; 20 ng/ml; Peprotech Inc, Rocky Hill, NJ) and fibroblast growth factor 2 (FGF2; 20 ng/ml; Peprotech Inc). The cultures were fed every 7 days by changing half of the medium and, once neurospheres reached approximately 200–300 cells, they were dissociated by repeatedly triturating and grown at the clonal density in the same medium again for passage. For serum-grown culture, the dissociated cells were plated in coated plastic dishes at a density of 5x104 cells per cm2 and grown as a monolayer in DMEM/F-12 (Invitrogen) supplemented with 10% fetal bovine serum (FBS). Glioblastoma cell lines LN18 and U87MG were reported previously and grown in DMEM containing 10% FBS (22).
Neurosphere formation assay
Dissociated cells from neurospheres and serum-grown cultures were dissociated and plated at 200 cells per well in 24-well plates, treated or untreated as described in the Results, and grown in the neurosphere culture conditions as described above for 7 days. The neurospheres formed were counted and presented as the percentage of the neurosphere forming cells over the total 200 cells planted.
Flow Cytometry
The cell surface expression of CD133, CD15, DR4 and DR5 was examined by flow cytometry based on the protocol modified from our report (27). In brief, the neurospheres were dissociated by repeatedly triturating and serum-grown monolayers were harvested by trypsin treatment and 2μg/ml of phycoerythrin (PE)-conjugated anti- CD133 and FITC-conjugated CD15 (Miltenyi, Auburn, CA), and PE-conjugated DR4 and DR5 (mouse IgG1; eBioscience, San Diego, CA) or mouse IgG1 (BD PharMingen, San Diego, CA), a negative control was added to 106 cells in 200 μl of immunofluorescence buffer (PBS containing 2% FBS and 0.02% sodium azide). After 1 hour of incubation in the dark at 4°C, 10,000 cells were analyzed using a Becton and Dickinson FACScan™ (Mountain View, CA). The results were processed by using Flowjo software as the percentage of positive cells.
Mouse Intracranial Xenograft
The neurospheres were dissociated and stereotactically injected into the striatum of the right side of brains of five to six weeks old female NOD-SCID mice (National Cancer Institute). Mice were anesthetized and immobilized on a stereotactic frame. A small incision was made in the middle of head skin and 5μl of serum-free culture medium containing 5 x 104 dissociated cells were introduced in the right striatum. The mice were maintained for the development of orthotopic xenografts. The mice were sacrificed and brains were removed and submerged in 10% neutrally buffered formalin and paraffinized. Five-micron think paraffin section were deparaffinized, rehydrated and stained with hematoxylin and eosin (H&E) according to the standard procedure at the Emory Medical Laboratory. The animal studies were approved by the Emory University Institutional Animal Care and Use Committee.
Cell Death Assay
The dissociated cells from neurospheres and serum-grown cultures were plated in 96-well plates at 5 x 103 cells per well. The cells were untreated or treated with TRAIL and cisplatin, alone or in combination for 24 hours, in the doses as indicated in the RESULTS. The treated or untreated cells were then analyzed using Cell Titer-glo Luminescent Cell Viability Assay kit according to the manufacture’s protocol (Promega, Madison, WI). Cell death was calculated based on the formula: 1 − (luminescent density of cells treated/luminescent density of cells untreated) x 100.
Caspase Enzymatic Activity assay
Caspase-8, -3 and -7 activities were examined using the Caspase-Glo®8 and Caspase-Glo®3/7 kits according to the manufacturer’s protocol (Promega,). In brief, dissociated cells from neurospheres and serum-grown cultures were plated and grown overnight in 96-well plates (5 x 103 cells/well) and untreated or treated with with TRAIL and cisplatin for 24 hours in the doses as indicated in the RESULTS. The cells were then incubated with 100μl of Caspase-Glo®8 or Caspase-Glo®3/7 reagent per well at room temperature for one hour. The caspase activity was determined by caspase substrate luminescence as recorded by a Dynex MLX® luminometer.
c-FLIP siRNA
Small interfering RNA (siRNA) duplexes specific to nucleotides 535–555 of the c-FLIP gene (Gene Bank accession number U97074) were synthesized through QIAGEN service (Valencia, CA). The synthetic siRNA and scramble siRNA (QIAGEN) were transfected using HiPerfect transfection reagent (QIAGEN) based on the manufacturer’s protocol. Transfected cells were allowed to grow for 72 h and then experimentally treated as outlined in the RESULTS.
Western Blotting
For protein expression, neurosphere and serum-grown culture, treated or untreated as outlined in the RESULTS, were lysed in a cell lysis buffer (50 mM Tris, 150 mM NaCl, 2mM EDTA, 10% glycerol, 1% Triton X-100, 1% protease inhibitor mixture and 1mM PMSF). Fifty micrograms of total protein from each lysate were separated through SDS–PAGE and transferred to nitrocellulose membranes. The membranes were incubated with the primary antibodies against FADD and caspase-8 (Medical & Biological Laboratories, Nagoya, Japan), and c-FLIP (NF6 clone from Alexis). After wash, the membranes were incubated for 1 hour with HRP-conjugated goat anti-mouse and anti-rabbit antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) and developed by chemiluminescence (Thermo Scientific).
RESULTS
Glioblastoma-derived neurospheres maintain the GSC properties
Glioblastoma-derived neurospheres have been shown to maintain the properties of cancer stem cells (9–11) and retain the genomic properties of the parental tumors (11, 14). In contrast, however, glioblastoma cells under traditional serum-grown culture condition do not recapitulate the genomic properties of the tumors (11). To examine how glioblastoma stem cells respond to TRAIL, we generated neurosphere culture GSC091106 and GSC091112 and matched serum-grown culture SC091106 and SC091112 from the same glioblastoma tissues surgically removed from patients (Fig. 1A). Neurosphere formation assay showed that approximately 10% cells in each of the neurospheres were able to form neurospheres; in contrast, few if any neurospheres were found in SC091106 and SC091112 under the same neurosphere culture condition (Fig. 1B). These results indicate the neurospheres but not serum-grown cultures retain the self-renewal capacity of cancer stem cells, keeping in line with earlier reports (10, 11). To further examine the stemness of neurospheres, we analyzed the expression of the stem cell surface markers in these cultures. Flow cytometry identified approximately 18% CD133+ cells in GSC091106, 51% CD133+ cells in GSC091112 (Fig. 1C), 12% CD15+ cells in GSC091106 and 14% CD15+ cells in GSC091112 (Fig. 1D). In contrast, approximately 1–2% CD133+ and CD15+ cells were detected in the matched serum grown SC091106 and SC091112.
Fig. 1.

Generation of GSC-enriched neurospheres. (A) Phase contrast imagings of GSC-enriched neurosphere GSC091106 and GSC091112 and matched serum-grown culture SC091106 and SC091112. (B) Neurosphere formation assay of the neurospheres and serum-grown cultures after seven days under the neurosphere culture conditions. Data are mean ± SEM (n=6). (C) Flow cytometry analysis of the cells surface expression of CD133 in the neurospheres and serum-grown cultures. CD133+ cells were presented as percentage of total cells. (D) Flow cytometry detection of the CD15 on the surface of the neurospheres and serum-grown cultures.
To further confirm that the neurospheres maintain the properties of cancer stem cells, we examined the tumorigenic capacity of GSC091106 and GSC091112. The neurospheres were dissociated by repeatedly triturating and 5 x 104 cells were injected in NOD-SCID mouse brains at the right side. The mice were observed and sacrificed once the xenografts were detected by MRI (data not shown). Histological examination of the whole mounted brains identified the xenografts on the right side of the brains injected with GSC091106 and GSC091112 (Fig. 2A), as compared to normal SCID mouse brain (Fig. 2B). Further microscopic examination of the H&E sections revealed the histological features of glioblastoma such as diffuse infiltration through the cerebral tissues (Fig. 2C) and mitotic activities in the xenograft (Fig. 2D). These results demonstrate that glioblastoma-derived neurospheres maintain the properties of cancer stem cells and provide GSC models for studying glioblastoma signal pathways and screening cancer therapeutic agents.
Fig. 2.
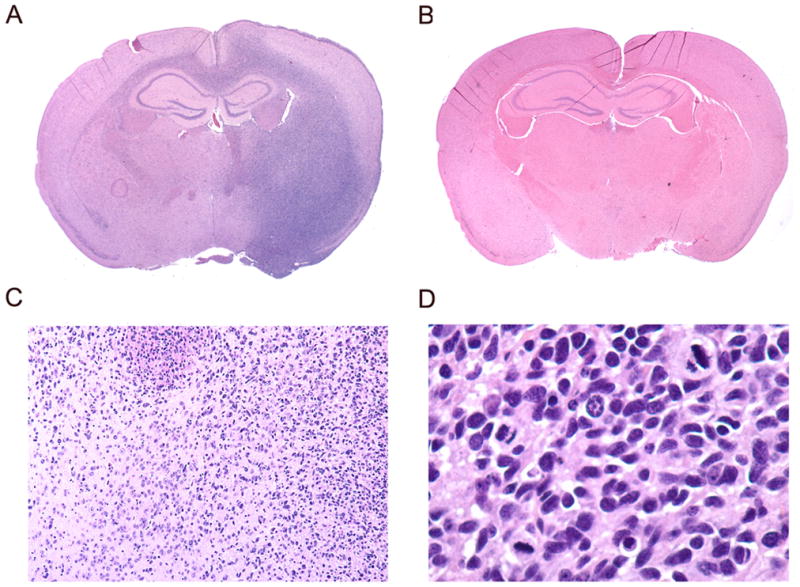
Xenograft formation of GSC091112 in SCID mouse brain. (A) GSC091112-derived xenograft was observed on this H&E section in the right side of the mouse brain (10 X). (B) Normal H&E section of SCID mouse brain (10 X). (C) The xenograft tumor cells diffusely infiltrate through the brain (100 X). (D) The high magnification H&E section identifies three mitotic features (400 X).
GSC-enriched neurospheres are resistant to TRAIL-induced apoptosis
To determine whether and how the neurosphere and matched serum-grown cultures respond to TRAIL treatment, we examined these cultures for TRAIL-induced apoptosis. GSC091106 and GSC091112 and matched SC091106 and SC091112 were dissociated, plated in 96-well plates, grown under the neurosphere culture condition in the presence of 300 ng/ml TRAIL for 16 hrs. The TRAIL treated cells were then examined for TRAIL-induced apoptosis in the following three assays. First, cell death analysis revealed that TRAIL treatment results in the significant cell death in the serum-grown SC091106 and SC091112 but not the matched neurosphere GSC091106 and GSC091112 (Fig. 3A). This suggests that neurospheres but not matched serum grown cells are resistant to TRAIL. Then, we examined the activation of the apoptosis initiating caspase-8 and showed the enzymatic activity of caspase-8 in the TRAIL treated serum-grown but not neurosphere cultures (Fig. 3B). Last, we examined the activation of caspase-8 downstream effectors and showed the enzymatic activities of caspase-3/-7 in the TRAIL-treated SC091106 and SC091112 but not GSC091106 and GSC091112 (Fig. 3C). These results indicate TRAIL-induced cell death occurs through caspase-8-initiated caspase cascade in serum-grown cultures. In contrast, however, GSC-enriched neurospheres are resistant to TRAIL-induced apoptosis.
Fig. 3.

TRAIL-induced apoptosis in the serum-grown cultures but not neurospheres. (A) Cell death analysis detected the cytotoxicity of TRAIL to the serum-grown cultures but not matched neurospheres after 16 hrs treatment with 300 ng/ml TRAIL. (B) Caspase-8 enzymatic activity and (C) caspase-3 and -7 activities were detected in the TRAIL-treated serum-grown cultures but not the neurospheres. Data are mean ± SEM (n+6).
Death receptors are less but c-FLIP is highly expressed in GSC-enriched neurospheres
TRAIL-induced apoptosis occurs through its binding of the surface DR4 and DR5 for the recruitment of intracellular FADD and caspase-8 in the formation of a DISC (32). To examine the molecular basis of TRAIL resistance in GSC, we analyzed the expression of the key DISC proteins in the neurospheres. Flow cytometry analysis showed the lack of the cell surface expression of DR4 and DR5 as approximately 1% DR4+ and 2% DR5+ cells were detected in GSC091106 and 0% DR4+ and 1% DR5+ cells were seen in GSC091112; in contrast, 66% and 51% DR5+ cells were observed in SC091106 and SC091112 (Fig. 4A). This suggests that the lack of DR4 and DR5 expression may result in the GSC resistance to TRAIL treatment.
Fig. 4.

Expression of the key DISC proteins. (A) Flow cytometry analyzed of the cell surface expression of DR4 and DR5 in GSC091106 and GSC091112 (top panel) and matched SC091106 and SC091112 (bottom panel). (B) Western blots detected the expression of caspase-8 (Casp-8), FADD, c-FLIPL and c-FLIPS with actin included as loading control. The proteins were indicated to the left and the molecular weights were indicated to the right of the panel.
Our earlier studies clarified glioblastoma-derived cell lines into TRAIL-sensitive and resistant phenotype (22) and showed that both c-FLIPL and c-FLIPS are highly expressed in the resistant such as U87MG as compare to the sensitive lines such as LN18 and the overexpression of c-FLIP proteins inhibit caspase-8 cleavage and thereby TRAIL-induced apoptosis in the resistant cell lines (28). To test this in the stem cells, we first examined the expression of intracellular FADD and caspase-8 protein by western blot in the neurospheres, together with TRAIL sensitive LN18 and resistant U87MG as controls. The results showed that FADD was expressed consistently in the neurospheres and cell lines, whereas caspase-8 was expressed more in GSC091112 and U87MG but less in GSC091106 and LN18, without the correlation to the TRAIL sensitivity of these cultures (Fig. 4B). Finally, we analyzed the expression of c-FLIPL and c-FLIPS, caspase-8 inhibitors (28) and showed that c-FLIPL protein was highly expressed in GSC091106 and both c-FLIPL and c-FLIPS were highly expressed in GSC091112 and U87MG as compared to TRAIL sensitive line LN18 (Fig. 4B). These results demonstrate that there is a much less expression of TRAIL death receptors on the cell surface but a much more expression of the intracellular caspase-8 inhibitors, c-FLIPL and c-FLIPS in GSC-enriched neurospheres.
c-FLIP inhibits TRAIL-induced apoptosis in GSC-enriched neurosphere
To define the role of c-FLIPL and c-FLIPS in TRAIL resistance, we transfected siRNA specific to c-FLIP nucleotides 535–555 nucleotides shared by c-FLIPL and c-FLIPS to inhibit the expression of both c-FLIPL and c-FLIPS (28). GSC091112 was disassociated, transfected with c-FLIP siRNA for 48 or 72 hrs and grown under the neurosphere culture condition. Western blot detected the knockdown of both c-FLIPL and c-FLIPS after 72 hrs of transfection (Fig. 5A). The transfected or untransfected GSC091112 was treated or untreated with 300 ng/ml TRAIL for 16 hrs. Cell death analysis showed that the knockdown of c-FLIP proteins converted the neurosphere from TRAIL resistant to sensitive phenotype (Fig. 5B). This study defines the role of c-FLIP in the inhibition of TRAIL-induced apoptosis in GSC-enriched neurospheres.
Fig. 5.

Transfection of c-FLIP siRNA in GSC091112 neurospheres. (A) Western blots detected the knockdown of both c-FLIPL and c-FLIPS in the neurospheres after transfected with c-FLIP siRNA for 72 hrs. (B). The c-FLIP siRNA transfected GSC09112 neurospheres were treated with 300 ng/ml TRAIL for 16 hrs and examined by cell death assay. Data are mean ± SEM (n+6).
Cisplatin upregulates DR5, downregulate c-FLIP in eliminating TRAIL resistance in GSC
Chemotherapy has been shown to enhance TRAIL-induced apoptosis in various cancer cell lines through upregulation of DR5 (33, 34) and inhibition of c-FLIP (35, 36); thus, we sought first to determine whether chemotherapy can overcome TRAIL resistance and then examine these molecular mechanisms of chemotherapy action on GSC-enriched neurospheres. GSC091106 and GSC091112 were dissociated and treated with a series of dilutions of cisplatin. Cell death assay revealed that GSC091106 was resistant but GSC091112 was partially sensitive to cisplatin (Fig. 6A). The experiment was repeated in the presence of 300 ng/ml TRAIL and the results revealed a synergistic effect of cisplatin on TRAIL killing of both GSC091106 and GSC091112 (Fig. 6A). GSC091106 and GSC091112 were then treated with 300 ng/ml TRAIL in the combination of 5 μg/ml cisplatin for 16 hrs; caspase activity assay detected the enzymatic activities of caspase-8 and caspase-3/-7 in both the treated neurospheres (Fig. 6B). These results indicate that cisplatin synergistically enhances TRAIL-induced apoptosis in GSC-enriched neurospheres.
Fig. 6.

Cisplatin synergistic effects on TRAIL-induced apoptosis. (A) Cell death analysis of GSC091106 and GSC091112 after treated for 16 hrs with cisplatin in the doses as indicated in the bottom of the panels. Data are mean + SEM (n=6). (B) The enzymatic activities of caspase-8 (left) and caspase-3/-7 (right) were detected in the TRAIL treated GSC091106 and GSC091112. Data are mean + SEM (n=6). (C) Flow cytometry analysis of the cell surface expression of DR4 and DR5 in GSC091106 and GSC091112 after treated with cisplatin for 16 hrs. (D) Western blot analysis of the intracellular expression of DR5 and c-FLIP isoforms in the neurospheres after treated with cisplatin for 8 and 16 hrs. Actin was included as protein loading control.
To examine the molecular mechanisms of the synergistic effect of cisplatin on TRAIL-induced apoptosis, we examined cisplatin action on GSC-enriched neurospheres. GSC091106 and GSC091112 were treated with 5 μg/ml cisplatin for 16 hrs. Flow cytometry analysis detected a dramatic increase of the cell surface expression of DR5 from 2% in untreated (Fig. 4A) to 55% in treated GSC091106 (Fig. 6C) and 1% in untreated (Fig. 4A) to 50% in treated GSC091112 (Fig. 6C). In contrast, the increase in the DR4 expression in the cisplatin treated neurospheres was insignificant at 1–2% (Fig. 6C). Western blot demonstrated an increase of DR5 protein in both GSC091106 and GSC091112 following the cisplatin treatment (Fig. 6D). In addition, western blot further showed a significant decrease of c-FLIPL in GSC091106 and a reduced c-FLIPL and c-FLIPS expression in GSC091112 (Fig. 6D). Taken together, these results indicate that cisplatin treatment leads to the up-regulation of TRAIL receptor, DR5 and down-regulation of caspase-8 inhibitor, c-FLIP and thus overcome the resistance of GSC-enriched neurospheres to TRAIL treatment.
The combination treatment of cisplatin and TRAIL eliminates neurosphere formation
GSCs are defined as undifferentiated cells that have the self-renewal capability for regeneration of cancers (8) and the GSC self-renewal ability can be tested by neurosphere formation assay (9–12). We therefore performed a neurosphere formation assay to determine the effect of the combination treatment of cisplatin and TRAIL on the GSC self-renewal. GSC091106 and GSC-01112 were plated at 200 cells per well in 24-well plates and grown in the neurosphere culture conditions for 7 days in the presence of cisplatin (5 μg/ml) and TRAIL (300 ng/ml), alone or in combination. Cisplatin treatment alone reduced the number of neurospheres in GSC091112 but not GSC091106 (Fig. 7A), consistent with cell death analysis that showed GSC091112 was sensitive to cisplatin (Fig. 6A,B). TRAIL treatment alone did not reduce the number (Fig. 7A) and size (Fig. 7B) of either GSC091112 or GSC091106 neurospheres. In contrast, however, the combination treatment of cisplatin and TRAIL completely eliminated the formation of GSC091112 and GSC091106 neurospheres. Taken together, these studies have demonstrated that the combination treatment of cisplatin and TRAIL induces apoptosis (Fig. 6) and thus eliminates the self-renewal ability of glioblastoma derived stem cells (Fig. 7).
Fig. 7.
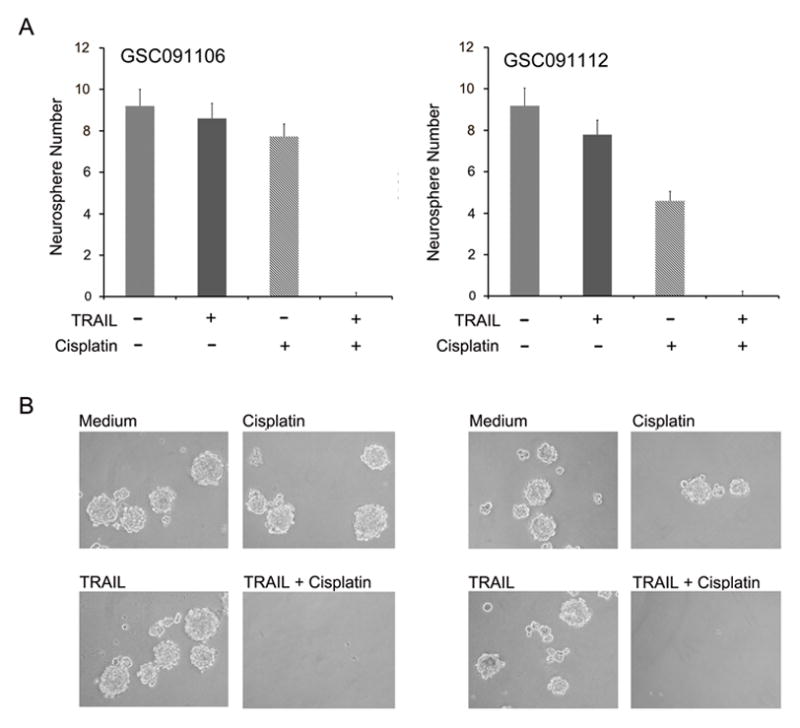
Effects of the combination treatment of cisplatin and TRAIL on neurosphere formation. (A) Neurosphere formation assay and (B) phase contrast imagings of GSC-enriched neurosphere GSC091106 and GSC091112 after 7 days in neurosphere culture condition in the presence of cisplatin and TRAIL. Data are mean ± SEM (n=6).
DISCUSSION
In the last decade, extensive studies of glioblastoma-derived cell lines have established a novel therapeutic approach in treating glioblastomas by targeting TRAIL apoptotic pathway (25). The cytotoxicity of TRAIL to glioblastoma cell lines and cell lines-derived xenografts was first reported by Ashkenazi and Weller’s groups (37, 38) and then identified as an apoptotic cell death by Ashkenazi’s (24), Hawkins’ (23) and Hao’s groups (22), while Zhou’s group reported that this apoptotic pathway can be therapeutically targeted by an agonistic DR5 antibody (39). Further studies have resulted in the generation of this apoptotic pathway targeted therapeutic agents such as recombinant human TRAIL (rhTRAIL) and agonistic DR4/5 antibodies (32). While clinical trails have proved that these TRAIL pathway-targeted therapeutic agents are safe in patients, only a limited antitumor activity has been observed (32). This raises the question whether traditional serum-grown cell lines provide proper models for screening therapeutic agents. Here, we show for the first time the different responses of GSC-enriched neurospheres and matched serum-grown cell cultures generated from the same glioblastoma tissues to TRAIL treatment in that serum-grown cells undergo apoptosis under TRAIL treatment whereas GSC-enriched neurospheres are resistant to the treatment.
Neurospheres were first generated from human glioblastoma tissues in Steindler’s group in 2002 (2) in following the experimental approach first reported by Weiss’ group in the generation of neural stem cells (40). Vescovi’s (9) and Yu’s groups (10) identified the properties of cancer stem cells in glioblastoma-derived neurospheres and Fine’s group showed that neurospheres retain the genomic properties of the original tumors (11). Dirks’ group first isolated CD133− cells through cell sorting from glioblastoma tissues and identified the stem cell properties of these CD133+ cells (3, 4). Recently, Fine’s group established the cancer stem cell properties in CD15+ cell populations isolated from glioblastoma tissues (13). Consistent with these reports, we show here that a subset population of neurospheres but not matched serum-grown cells generated from the same glioblastoma tissues express CD133 and CD15 on the cell surface and generate xenografts in mouse brains. The GSC-enriched neurospheres but not serum-grown cultures are resistant to TRAIL treatment, keeping in line with clinical study that has correlated neurosphere formation with chemoresistance (41).
While traditional serum-grown cell lines may not be the proper models for screening cancer drugs, studies of glioblastoma cell lines have shed light on TRAIL signal pathways (25). TRAIL-induced apoptosis occurs through its binding of DR4 and DR5 and the recruitment of intracellular FADD and apoptosis-initiating protease caspase-8, leading to the caspase-8-initiated caspase cascade (42). These studies suggest that the caspase-8 activation in the DISC is a critical step in TRAIL-induced apoptosis. The data presented here show that FADD and caspase-8 are expressed in GSC-enriched neurospheres, but the expression of DR4 and DR5 is not sufficient enough for TRAIL-induced apoptosis in GSC-enriched neurospheres.
Studies of cell lines have also shown that TRAIL apoptotic pathway can be inhibited by the adaptor proteins that can inhibit caspase-8 in the DISC (42). These adaptor proteins include the proteins that contain death domain (DD) such as receptor-interacting protein (RIP) (28) and that have DED such as phosphorprotein enriched in astrocytes-15 kDa/diabetes (PEA-15/PED) (26) and c-FLIP (26, 31, 43). These DD and DED-containing proteins can be recruited through DD and DED interaction with DR4/DR5 and FADD to the DISC where they interact with caspase-8 and interrupt caspase-8 dimerization, cleavage and activation (25). In examining the molecular mechanisms of TRAIL resistance in GSC, we show here for the first time that c-FLIPL and c- FLIPS are highly expressed in GSC-enriched neurospheres and responsible for the resistance of GSC to TRAIL treatment
Chemotherapy is a major clinical treatment of glioblastomas. Chemotherapeutic agent can interact with DNA and form intra-strand cross-links that activate intracellular signaling pathways for cancer treatment (44). However, the majority of glioblastomas are resistant to chemotherapy (45). In the meantime, chemotherapeutic agents have been shown to sensitize glioblastoma cell lines to TRAIL-induced apoptosis through regulation of the key proteins in TRAIL signal pathways (25). Cisplatin and etoposide were first reported for this synergetic effect on TRAIL through up-regulation of DR5 (46). 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU), temozolomide and topotecan were then shown to have the synergistic cytotoxicity with TRAIL on glioblastoma cell lines (47). Cisplatin and camptothecin were further shown to inhibit the expression of c-FLIPS in glioblastoma cell lines and sensitize the cell lines to TRAIL-induced apoptosis (27). Systemic injection of TRAIL and temozolomide increases the survival of the xenograft mice (48). In this study, we show for the first time that cisplatin treatment results in up-regulation of DR5 and down-regulation of c-FLIP and leads to TRAIL-induced apoptosis in GSC-enriched neurospheres. DR5 was initially reported as a DNA damage-inducible p53-regulated death receptor gene (49) and our subsequent study of glioblastoma cell lines has indeed shown that cisplatin upregulates DR5 expression through p53 transcriptional pathway (27).
CONCLUSION
This study has shown that GSC is resistant to TRAIL-induced apoptosis due to the lack of the cell surface expression of DR4 and DR5 and the overexpression of intracellular c-FLIP proteins. Chemotherapeutic agent cisplatin can up-regulate DR5 and down-regulate c-FLIP in GSC and eliminate the resistance of GSC to TRAIL treatment. This study suggests that the combination therapy of cisplatin and TRAIL may provide a novel and effective treatment for glioblastomas.
Acknowledgments
This work was supported in part by National Institutes of Health grant CA129687 (C.H.). C.H. was a Georgia Cancer Coalition Distinguished Scholar.
Footnotes
DECLARATION OF INTEREST
The authors report no conflict of interest. The authors alone are responsible for the content and writing of this paper.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 4.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of cd133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015–24. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 8.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 9.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 10.Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann-Hogiu S, Farkas DL, Black KL, Yu JS. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 11.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bfgf and egf more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 12.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–8. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 13.Son MJ, Woolard K, Nam DH, Lee J, Fine HA. Ssea-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–52. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, Ene C, Ma H, Su Q, Zenklusen JC, Zhang W, Maric D, Fine HA. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–5. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 16.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–33. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamandis P, Wildenhain J, Clarke ID, Sacher AG, Graham J, Bellows DS, Ling EK, Ward RJ, Jamieson LG, Tyers M, Dirks PB. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat Chem Biol. 2007;3:268–73. doi: 10.1038/nchembio873. [DOI] [PubMed] [Google Scholar]
- 18.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–13. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of notch signaling in cancer. Oncogene. 2008;27:5124–31. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 20.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806–23. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 21.Johnstone RW, Frew AJ, Smyth MJ. The trail apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–98. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 22.Hao C, Beguinot F, Condorelli G, Trencia A, Van Meir EG, Yong VW, Parney IF, Roa WH, Petruk KC. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (trail) mediated apotosis in human malignant glioma cells. Cancer Res. 2001;61:1162–70. [PubMed] [Google Scholar]
- 23.Knight MJ, Riffkin CD, Muscat AM, Ashley DM, Hawkins CJ. Analysis of fasl and trail induced apoptosis pathways in glioma cells. Oncogene. 2001;20:5789–98. doi: 10.1038/sj.onc.1204810. [DOI] [PubMed] [Google Scholar]
- 24.Pollack IF, Erff M, Ashkenazi A. Direct stimulation of apoptotic signaling by soluble apo2l/tumor necrosis factor-related apoptosis-inducing ligand leads to selective killing of glioma cells. Clin Cancer Res. 2001;7:1362–9. [PubMed] [Google Scholar]
- 25.Bellail AC, Mulligan P, Hao C. Targeting of trail apoptotic pathways for glioblastoma therapies. CNS CANCER, Cancer Drug Discovery and Development. 2009:977–1009. [Google Scholar]
- 26.Xiao C, Yang BF, Asadi N, Beguinot F, Hao C. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-flip and ped/pea-15 in glioma cells. J Biol Chem. 2002;277:25020–5. doi: 10.1074/jbc.M202946200. [DOI] [PubMed] [Google Scholar]
- 27.Song JH, Song DK, Pyrzynska B, Petruk KC, Van Meir EG, Hao C. Trail triggers apoptosis in malignant glioma cells through extrinsic and intrinsic pathways. Brain Pathol. 2003;13:539–553. doi: 10.1111/j.1750-3639.2003.tb00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellail AC, Tse MC, Song JH, Phuphanich S, Olson JJ, Sun SY, Hao C. Dr5-mediated disc controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J Cell Mol Med. 14:1303–17. doi: 10.1111/j.1582-4934.2009.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular flip. Nature. 1997;388:190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 30.Scaffidi C, Medema JP, Krammer PH, Peter ME. Flice is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J Biol Chem. 1997;272:26953–8. doi: 10.1074/jbc.272.43.26953. [DOI] [PubMed] [Google Scholar]
- 31.Panner A, James CD, Berger MS, Pieper RO. Mtor controls flips translation and trail sensitivity in glioblastoma multiforme cells. Mol Cell Biol. 2005;25:8809–23. doi: 10.1128/MCB.25.20.8809-8823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. Trail agonists on clinical trials for cancer therapy: The promises and the challenges. Rev Recent Clin Trials. 2009;4:34–41. doi: 10.2174/157488709787047530. [DOI] [PubMed] [Google Scholar]
- 33.Lacour S, Hammann A, Wotawa A, Corcos L, Solary E, Dimanche-Boitrel MT. Anticancer agents sensitize tumor cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated caspase-8 activation and apoptosis. Cancer Res. 2001;61:1645–51. [PubMed] [Google Scholar]
- 34.Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–80. doi: 10.1093/jnci/djh322. [DOI] [PubMed] [Google Scholar]
- 35.Song JH, Song DK, Herlyn M, Petruk KC, Hao C. Cisplatin down-regulation of cellular fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory proteins to restore tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human melanoma cells. Clin Cancer Res. 2003;9:4255–66. [PubMed] [Google Scholar]
- 36.Wang P, Zhang J, Bellail A, Jiang W, Hugh J, Kneteman NM, Hao C. Inhibition of rip and c-flip enhances trail-induced apoptosis in pancreatic cancer cells. Cell Signal. 2007;19:2237–46. doi: 10.1016/j.cellsig.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH. Safety and antitumor activity of recombinant soluble apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roth W, Isenmann S, Naumann U, Kugler S, Bahr M, Dichgans J, Ashkenazi A, Weller M. Locoregional apo2l/trail eradicates intracranial human malignant glioma xenografts in athymic mice in the absence of neurotoxicity. Biochem Biophys Res Commun. 1999;265:479–83. doi: 10.1006/bbrc.1999.1693. [DOI] [PubMed] [Google Scholar]
- 39.Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, Zhou T. Tumoricidal activity of a novel anti-human dr5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954–60. doi: 10.1038/91000. [DOI] [PubMed] [Google Scholar]
- 40.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–10. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 41.Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, Yong WH, Vinters HV, Liau LM, Lazareff JA, Mischel PS, Cloughesy TF, Horvath S, Kornblum HI. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27:980–7. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hao C, Song JH, Vilimanovich U, Kneteman NM. Modulation of trail signaling complex. Vitam Horm. 2004;67:81–99. doi: 10.1016/S0083-6729(04)67006-3. [DOI] [PubMed] [Google Scholar]
- 43.Panner A, Murray JC, Berger MS, Pieper RO. Heat shock protein 90alpha recruits flips to the death-inducing signaling complex and contributes to trail resistance in human glioma. Cancer Res. 2007;67:9482–9. doi: 10.1158/0008-5472.CAN-07-0569. [DOI] [PubMed] [Google Scholar]
- 44.Siddik ZH. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 45.Rich JN, Bigner DD. Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov. 2004;3:430–46. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- 46.Nagane M, Pan G, Weddle JJ, Dixit VM, Cavenee WK, Huang HJ. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res. 2000;60:847–53. [PubMed] [Google Scholar]
- 47.Rohn TA, Wagenknecht B, Roth W, Naumann U, Gulbins E, Krammer PH, Walczak H, Weller M. Ccnu-dependent potentiation of trail/apo2l-induced apoptosis in human glioma cells is p53-independent but may involve enhanced cytochrome c release. Oncogene. 2001;20:4128–37. doi: 10.1038/sj.onc.1204534. [DOI] [PubMed] [Google Scholar]
- 48.Saito R, Bringas JR, Panner A, Tamas M, Pieper RO, Berger MS, Bankiewicz KS. Convection-enhanced delivery of tumor necrosis factor-related apoptosis-inducing ligand with systemic administration of temozolomide prolongs survival in an intracranial glioblastoma xenograft model. Cancer Res. 2004;64:6858–62. doi: 10.1158/0008-5472.CAN-04-1683. [DOI] [PubMed] [Google Scholar]
- 49.Wu GS, Burns TF, McDonald ER, 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry WS. Killer/dr5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–3. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]