

Abstract
Plasmacytoid dendritic cells (pDCs) respond to viral infection by production of alpha interferon (IFN-α), proinflammatory cytokines, and cell differentiation. The elimination of hepatitis C virus (HCV) in more than 50% of chronically infected patients by treatment with IFN-α suggests that pDCs can play an important role in the control of HCV infection. pDCs exposed to HCV-infected hepatoma cells, in contrast to cell-free HCV virions, produce large amounts of IFN-α. To further investigate the molecular mechanism of HCV sensing, we studied whether exposure of pDCs to HCV-infected hepatoma cells activates, in parallel to interferon regulatory factor 7 (IRF7)-mediated production of IFN-α, nuclear factor kappa B (NF-κB)-dependent pDC responses, such as expression of the differentiation markers CD40, CCR7, CD86, and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and secretion of the proinflammatory cytokines TNF-α and interleukin 6 (IL-6). We demonstrate that exposure of pDCs to HCV-infected hepatoma cells surprisingly did not induce phosphorylation of NF-κB or cell surface expression of CD40, CCR7, CD86, or TRAIL or secretion of TNF-α and IL-6. In contrast, CpG-A and CpG-B induced production of TNF-α and IL-6 in pDCs exposed to the HCV-infected hepatoma cells, showing that cell-associated virus did not actively inhibit Toll-like receptor (TLR)-mediated NF-κB phosphorylation. Our results suggest that cell-associated HCV signals in pDCs via an endocytosis-dependent mechanism and IRF7 but not via the NF-κB pathway. In spite of IFN-α induction, cell-associated HCV does not induce a full functional response of pDCs. These findings contribute to the understanding of evasion of immune responses by HCV.
INTRODUCTION
Plasmacytoid dendritic cells (pDCs) are a highly specialized subset of dendritic cells that function as sentinels for viral infection and are responsible for production of type I interferons (IFN), proinflammatory cytokines, and antigen presentation during viral infection (15, 19, 32). pDCs are able to detect the genetic material of viruses with a subset of Toll-like receptors (TLR) localized to the endosomal compartment (10). These nucleotide-sensing TLRs include TLR7 and TLR8, which recognize single-stranded RNA, and TLR9, which recognizes DNA. TLR7 also recognizes synthetic imidazoquinoline components, for example R848 (resiquimod), whereas TLR9 recognizes synthetic CpG oligonucleotides, for example CpG-A or CpG-B. Ligation of TLR7 and TLR9 with their agonists triggers a signaling cascade, which starts with recruitment of the MyD88 adaptor molecule to the cytoplasmic domain of nucleotide-sensing TLR. This activates the assembly of a multiprotein signal-transducing complex in the cytoplasm that includes interferon-regulatory factor 7 (IRF7) (10). Activated IRF7, which is constitutively expressed in pDCs, translocates to the nucleus and initiates the transcription of type I IFN.
The elimination of hepatitis C virus (HCV) in more than 50% of chronically infected patients by treatment with alpha interferon (IFN-α) (9, 20) suggests that pDCs can play an important role in the control of HCV infection. Several reports have shown that exposure of pDCs from healthy donors to HCV particles results in no or only weak production of type I IFN and cell differentiation (4, 7, 11, 13, 31). A recent report has shown that pDCs exposed in direct cell-to-cell contact with HCV-infected hepatoma cells, unlike those exposed to cell-free HCV virions, produce large amounts of type I IFN via TLR7 signaling (35). This suggests that pDCs could be responsible for production of intrahepatic type I IFN (17, 35). Importantly, these events require viral RNA replication but not virion formation in the stimulator cells. In parallel to IRF7-mediated production of IFN-α, MyD88 signaling also leads to activation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs). Both NF-κB and MAPKs stimulate secretion of the proinflammatory cytokines interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) and stimulate expression of costimulatory molecules such as CD80 and CD86. Recent reports have identified a new signaling pathway induced by TLR7 and dependent on PI3K-p38MAPK, which stimulates the early IFN-inducible genes MxA and CXCL10 and the TNF-related apoptosis-inducing ligand (TRAIL) in the absence of type I IFN (6, 27).
To better understand the molecular mechanism of HCV sensing, we investigated whether exposure of pDCs to HCV-infected hepatoma cells induces not only IRF7 signaling but also NF-κB signaling pathways necessary for pDC functions. We demonstrate that in comparison to influenza virus or synthetic agonists of TLR7 and TLR9, HCV-infected hepatoma cells did not stimulate in pDCs phosphorylation of NF-κB and activation of NF-κB-dependent pDC responses, such as cell surface expression of differentiation markers CD40, CCR7, CD86, and TRAIL and secretion of TNF-α and IL-6. In contrast, production of TNF-α and IL-6 in pDCs exposed to the HCV-infected hepatoma cells was induced by CpG-A and CpG-B, showing that HCV-infected hepatoma cells did not actively inhibit TLR-mediated NF-κB phosphorylation. Our results suggest that cell-associated HCV signals in pDCs via an endocytosis-dependent mechanism and IRF7 and induces production of IFN-α; however, like cell-free virus, it does not induce a full functional response of pDCs. Taken together, our results are thus important for an understanding of the HCV-DC interaction and of the mechanisms leading to the establishment of chronic HCV infection.
MATERIALS AND METHODS
Isolation and culture of pDCs.
Peripheral blood mononuclear cells (PBMCs) from healthy anonymous donors were obtained from the national blood services (Etablissement Francais du Sang [EFS], Marseille, France). pDCs purified from PBMCs as described previously (4, 11, 13, 31) yielded levels of purity from 75% to 95%, with a contamination of less than 5% myeloid dendritic cells. Isolated pDCs were cultured in RPMI 1640 supplemented with 10% fetal calf serum and antibiotics. To optimize viability, recombinant IL-3 (R&D Systems Europe, Ltd., Abingdon, United Kingdom) was added to a final concentration of 10 ng/ml.
Production and purification of cell culture-derived HCVcc (JFH-1 3 M).
Cell culture-derived HCV (HCVcc) virions were prepared in Huh7.5 cells (2) (kindly provided by APATH L.L.C.) on the basis of plasmid pJFH-1 displaying mutations F172C and P173S in core, and N534K in E2 (5), as described previously (4, 11, 13, 31). The ultracentrifuged virus purified through a cushion of 20% sucrose was resuspended in RPMI 1640 medium to obtain a 1,000-fold-concentrated virus suspension.
Quantification of viral genome copies.
The titers of HCVcc genome-containing virus particles were determined routinely with quantitative reverse transcription-PCR (RT-PCR) as described previously (4, 11, 13, 31) using the primer RTU1 (25) for cDNA synthesis, primer pair UTR2 and RTU2 (25) for PCR amplification, and HCV JFH-1 RNA prepared in vitro with T7 polymerase as a standard.
Influenza virus stocks.
Influenza virus A/H3N2/Johannesburg/34/99 was produced in MDCK cells (12).
Infected and transfected cell lines.
Cells of human hepatoma cell line Huh7.5 infected with JFH-1 3 M at a multiplicity of infection of 0.01 focus-forming units (FFU)/cell were cultured under standard conditions for 1 week. The percentage of infected cells was determined by immunofluorescence staining for HCV core protein using a mouse monoclonal anti-core antibody (C7-50) (Thermofisher Scientific, Courtaboeuf, France). Monolayers containing >60% HCV core+ Huh7.5 cells were used in coculture with pDCs. Huh7.5 cells transfected with H/SG-neo (L+I) subgenomic replicon (SGR) (2) (kindly provided by C. M. Rice, The Rockefeller University, New York, NY) and virus-free Huh7.5 cells were used as controls.
In vitro pDC stimulation.
Purified pDCs were kept at a concentration of 106 cells/ml aliquoted in 100-μl quantities in 96-well round-bottom culture plates and stimulated with 10 μg/ml CpG-A (ODN 2216), CpG-B (ODN 2006), or CpG-C (ODN 2395) (InvivoGen, San Diego, CA), 5 μM R848 (InvivoGen, San Diego, CA), 10 ng/ml TNF-α (Becton Dickinson, Le Pont-De-Claix, France), HCV JFH-1 3 M virions at a multiplicity of 100 virus genome copies per cell, or Huh7.5 cells infected with HCV JFH-1 3 M or transfected with SGR at the ratio of 2 Huh7.5 cell per pDC, for 20 or 40 h in the presence of IL-3. Endocytosis and endosomal maturation/acidification was inhibited by chlorpromazine (6.25 μg/ml) or chloroquine (5 μM), both from Sigma-Aldrich.
Immunofluorescence analysis.
For analysis of cell surface marker expression, cells were incubated for 15 min at room temperature in the presence of V450-conjugated CD86, fluorescein isothiocyanate (FITC)-conjugated Lineage Cocktail 1 (CD3, CD14, CD16, CD19, CD20, CD56), PercP-Cy5.5-conjugated CD123, PE-Cy7-conjugated CCR7, APC-conjugated CD11c and APC-H7-conjugated CD40 monoclonal antibodies (MAbs) (all Becton Dickinson), and biotin-conjugated TRAIL MAb (eBioscience, Paris, France) with second-step reagent phycoerythrin (PE)-conjugated streptavidin. Live pDCs were detected by means of Vivid (Becton Dickinson). After labeling, cells were fixed with 4% paraformaldehyde and analyzed using the LSR II instrument (Becton-Dickinson, Le Pont de Claix, France) by gating on live Lin− CD11c− CD123+ cells. Data were analyzed with Flowjo software (Tree Star, Inc., Ashland, OR).
Determination of secreted IFN-α, TNF-α, and IL-6.
The quantities of total IFN-α, TNF-α, and IL-6 produced by pDCs were measured in cell-free supernatants using human enzyme-linked immunosorbent assay (ELISA) kits (IFN-α from eBiosciences, Paris, France, and TFN-α and IL-6 from Becton-Dickinson, Le Pont de Claix, France, respectively).
Determination of NF-κB p65 phosphorylation by dynamic phospho-flow cytometry.
Phospho-flow analysis of phosphorylation of NF-κB p65 at serine 536 was performed as previously described (8). Briefly, cells were fixed, permeabilized, and incubated successively with Phospho-NF-κB p65 (Ser536) (93H1) rabbit MAb (Cell Signaling, Danvers, MA) and anti-rabbit biotinylated antibodies. Finally, detection was performed with a streptavidin-phycoerythrin solution (Beckman Coulter, Paris, France).
Statistical analysis.
Quantitative variables are expressed as the means ± standard errors of the means (SEM). To compare the levels of cytokine production by pDCs, data were analyzed by means of a nonparametric Mann-Whitney test with GraphPad Prism 4 software (GraphPad Software, La Jolla, CA). All tests of significance were two-sided, and a P value of ≤0.05 was considered to be significant.
RESULTS
Exposure of pDCs to hepatoma cells infected with HCV induces production of IFN-α but not of TNF-α.
First, we compared the levels of secreted IFN-α and TNF-α induced by exposure of pDCs to Huh7.5 cells infected with HCV JFH-1 or transfected with HCV subgenomic replicon (HCV SGR) with the levels of IFN-α induced by HCV JFH-1 virions, influenza virus A/H3N2/Johannesburg (Flu), R848, CpG-A, CpG-B, and CpG-C. Stimulation of pDCs with Huh7.5 cells infected with HCV JFH-1 or transfected with SGR or stimulation with the TLR7 agonists R848 and Flu, with the TLR9 agonist CpG-A, or with the TLR7 and TLR9 agonist CpG-C strongly increased production of IFN-α (Fig. 1). In contrast, stimulation of pDC with HCV JFH-1 virions or CpG-B did not increase the production of IFN-α in comparison with nonstimulated or control Huh7.5-stimulated pDCs. Thus, consistent with previous results (35), cell-associated HCV does but cell-free HCV does not induce production of IFN-α in pDCs. In addition to IFN-α, we also investigated the release of the proinflammatory cytokine TNF-α in the same experimental system. Production of TNF-α was strongly stimulated with CpG-A, CpG-B, CpG-C, R848, and Flu. In contrast to results for IFN-α, production of TNF-α in pDCs exposed to Huh7.5 cells infected with HCV JFH-1 (P = 0.0004) or transfected with HCV SGR (P = 0.0018) was significantly reduced.
Fig 1.
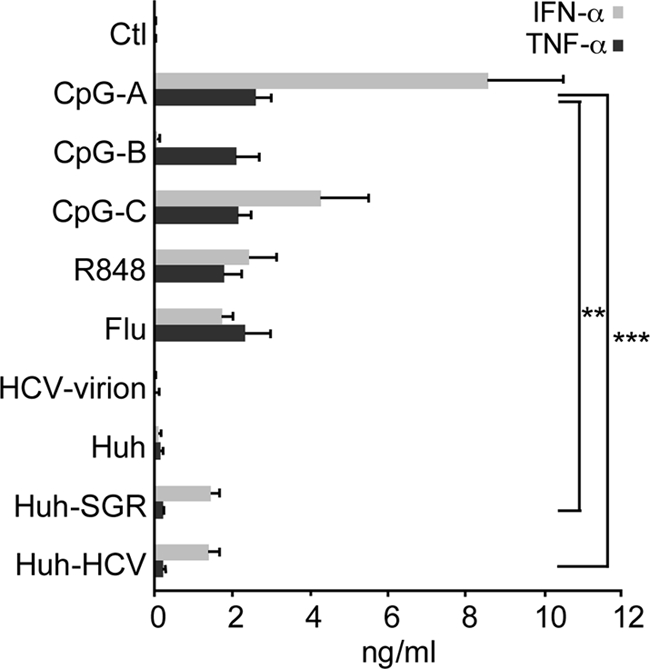
IFN-α and TNF-α production by coculture of pDCs with HCV-infected hepatoma cells. pDCs were cocultured with HCV-infected Huh7.5 cells (Huh-HCV), with Huh7.5 cells transfected with HCV subgenomic replicons (Huh-SGR) or with control Huh7.5 cells (Huh), were inoculated with HCV JFH-1 (HCV-virion) or influenza virus A/H3N2/Johannesburg (Flu), or were stimulated with CpG-A, CpG-B, CpG-C, or R848. Secretion of IFN-α (light columns) and TNF-α (dark columns) in cell-free supernatant was determined by ELISA 20 h poststimulation. The data show means and SEM of results of at least seven independent experiments with pDCs from different donors. **, 0.05 > P ≥ 0.01; ***, 0.01 > P ≥ 0.001; Mann-Whitney two-tailed nonparametric test.
Exposure to hepatoma cells infected with HCV does not induce phosphorylation of NF-κB p65 in pDCs.
The absence of TNF-α in cell-free supernatants of pDCs exposed to cell-associated HCV led us to investigate the NF-κB signaling pathway, which is known to control production of proinflammatory cytokines and differentiation of pDCs (10). First, we compared the levels of phosphorylation of the p65 subunit of NF-κB induced by Huh7.5 cells infected with HCV JFH-1 or transfected with HCV-SGR with the levels induced by R848, Flu, CpG-B, or HCV JFH-1 virions (Fig. 2). Stimulation of pDCs with R848, Flu, or CpG-B strongly increased phosphorylation of NF-κB p65. In contrast, stimulation of pDC with HCV JFH-1 virions or with Huh7.5 cells infected with HCV JFH-1 or transfected with HCV SGR revealed no increase of phosphorylated NF-κB p65 in comparison with control nonstimulated or Huh7.5-stimulated pDCs (Fig. 2A and B). These findings show that neither cell-free nor cell-associated HCV induced phosphorylation of NF-κB p65 in pDCs.
Fig 2.

Phosphorylation of NF-κB p65 in pDCs cocultured with HCV-infected hepatoma cells. (A) pDCs were cocultivated with Huh7.5 cells infected with HCV JFH-1 (Huh-HCV), with Huh7.5 cells transfected with HCV SGR (Huh-SGR), or with control Huh 7.5 cells (Huh). In parallel, pDCs were inoculated with HCV JFH-1 virions (HCV-virion) or with influenza virus A/H3N2/Johannesburg (Flu) and incubated for 60 min. In control experiments, pDCs were stimulated with CpG-B or with R848. Phosphorylation of NF-κB p65 was determined by phospho-flow cytometry. Shaded areas show nonstimulated pDC; black lines show stimulated pDCs. Data are representative of results of six independent experiments with pDCs from different donors that gave comparable results. (B) The relative mean fluorescence intensity (MFI) corresponds to the MFI of cells stimulated for 40, 60, 75, and 120 min divided by the respective MFI value of nonstimulated cells. The data show means and SEM of results of six independent experiments with pDCs from different donors.
Exposure to hepatoma cells infected with HCV does not induce differentiation of pDCs.
Then, we investigated the ability of cell-associated HCV to induce expression of cell surface molecules associated with the pDC cross-presentation function. To address this question, we compared the impact on pDC differentiation induced by Huh7.5 cells infected with HCV JFH-1 or transfected with HCV SGR with the impact of CpG-B, R848, and HCV JFH-1 virions (Fig. 3). We determined the expression of surface markers CD40, CD86, CCR7, and TRAIL in CD11c− CD123+ gated live cells (Fig. 3A). Culture of pDCs in medium supplemented with IL-3, an important factor for pDC survival, resulted in partial pDC maturation (Fig. 3B). Flow cytometry analysis of pDCs from three donors stimulated with TLR7 agonist R848 or TLR9 agonist CpG-B showed strongly increased expression of CD40, CD86, CCR7, and TRAIL (Fig. 3C). In contrast, stimulation of pDCs with Huh7.5 cells infected with HCV JFH-1 or transfected with HCV SGR did not result in an increase of expression of CD40, CD86, CCR7, and TRAIL; the same pDC phenotype was observed with HCV JFH-1 virions.
Fig 3.

Expression of differentiation markers of pDCs exposed to HCV-infected hepatoma cells. (A) Gating strategy for identification of pDCs: magnetic bead-purified pDCs were gated according to their size and then into singlets, and after exclusion of dead cells (Vivid+), live cells were gated further by exclusion of Lin1+CD11c+ cells into a CD123+ pDC population. The purity of the isolated pDCs was determined from flow cytometry analysis as a fraction of Lin1−, CD123+, and CD11c− cells. Numbers displayed in delimited areas are percentages of positive cells. (B and C) The expression of the cell surface markers CD40, CD86, and CCR7 was determined immediately after cell separation (0 h, shaded area), after 20 h culture (gray line), or after 40 h culture (black line). (B) pDC differentiation in the presence of IL-3. (C) pDCs were cocultured with Huh7.5 cells infected with HCV JFH-1 (Huh-HCV) or transfected with HCV SGR (Huh-SGR), were cocultured with control Huh7.5 cells (Huh), or were inoculated with cell-free HCV JFH-1 (HCV-virion). In control experiments, pDCs were stimulated with CpG-B or with R848. Expression of CD40, CD86, CCR7, and TRAIL was determined in Lin− CD11c− CD123+ gated live cells at 0 h (shaded area) and 40 h (black line) poststimulation. Data are representative of results of three independent experiments with pDCs from different donors that gave comparable results. (D) Kinetics of expression of differentiation markers of pDCs exposed to HCV. Percentages of stimulated CD40+, CCR7+, CD86+, and TRAIL+ cells relative to nonstimulated cells at the time zero are shown. The data show means and SEM of six (20 h) and three (40 h) independent experiments with pDCs of different donors stimulated for 0, 20 (n = 6), and 40 (n = 3) h. **, 0.05 > P ≥ 0.01.
Additional analysis of the kinetics of expression of differentiation markers of pDCs stimulated with R848 and CpG-B confirmed significant upregulation of CD40 (PR848 = 0.0022, PCpG-B = 0.0048; P values for pDCs stimulated with R848 and CpC-B, respectively, calculated for 20 h and n = 6), CD86 (PR848 = 0.0046, PCpG-B = 0.0046), CCR7 (PR848 = 0.0022, PCpG-B = 0.0048), and TRAIL (PR848 = 0.0046, PCpG-B = 0.0046) over time in the cells of all assayed donors (Fig. 3D). No significant increase (P > 0.05) of differentiation markers in comparison with negative controls was observed in pDCs exposed to HCV virions or Huh7.5 cells infected with HCV JFH-1 or transfected with HCV SGR. Taken together, these findings show that neither cell-free HCV nor cell-associated HCV induces differentiation of pDCs.
Exposure of pDCs to HCV-infected hepatoma cells does not impair TLR-dependent or independent phosphorylation of NF-κB and subsequent production of TNF-α and IL-6.
Finally, we investigated whether HCV-infected hepatoma cells elicit in pDCs a dominant negative effect on the phosphorylation of NF-κB and subsequent NF-κB-dependent pDC responses or are just weak inducers of the NF-κB signaling pathway (Fig. 4). To address this question, we stimulated pDCs with CpG-B (via a TLR9-dependent pathway) or with TNF-α (via a TLR9-independent pathway), and we investigated the effects of HCV-infected hepatoma cells on NF-κB phosphorylation. Results of these experiments show that HCV-infected hepatoma cells blocked neither CpG-B- nor TNF-α-mediated phosphorylation of NF-κB (Fig. 4A).
Fig 4.

Effect of HCV-infected hepatoma cells on TLR-dependent and TLR-independent phosphorylation of NF-κB and on NF-κB-dependent functions. pDCs were stimulated with CpG-A, CpG-B, or TNF-α in the presence of HCV JFH-1-infected Huh7.5 cells (Huh-HCV) or control Huh7.5 cells (Huh) or in the presence of endocytosis inhibitors. (A) Phosphorylation of NF-κB p65 was determined by phospho-flow cytometry. The relative MFI corresponds to the MFI of cells stimulated for 30 min divided by the respective MFI values of nonstimulated cells. The data show means and SEM of results of five independent experiments with pDCs from different donors. (B and C) Levels of TNF-α, IL-6, and IFN-α in the cell-free supernatants of pDCs from at least six donors were determined by ELISA, either after simultaneous exposure of pDCs to Huh-HCV or Huh stimulated with CpG-A or CpG-B (B) or after priming with Huh-HCV or Huh that was followed 2 h later by stimulation with CpG-A or CpG-B (C). In a similar way production of IFN-α was determined in pDCs exposed to Huh-HCV or CpG-A that were simultaneously (D) or 2 h later (E) treated with inhibitors of endocytosis, chlorpromazine (CHP), or chloroquine (CHQ). For a better comparison of the effect of endocytosis inhibitors, IFN-α production was expressed as a percentage of the production triggered by the control agonists without inhibitors.
Furthermore, HCV-infected hepatoma cells did not impair production of TNF-α and IL-6 induced by CpG-A or CpG-B (Fig. 4B). We have previously shown that inhibition of TLR9-mediated production of IFN-α in pDCs by HCV particles is more efficient if the priming with the cell-free virus precedes stimulation with TLR9 agonist, in comparison with the simultaneous treatment (4, 7, 11, 13, 31). Therefore, we investigated the effect of priming of pDCs with HCV-infected Huh7.5 cells followed 2 h later by stimulation with CpG-A or CpG-B on production of TNF-α or IL-6 (Fig. 4C). Even under these experimental conditions, cell-associated HCV did not block production of proinflammatory cytokines TNF-α or IL-6. In a control experiment, we showed that in contrast to HCV particles (4, 7, 11, 13, 31), HCV-infected Huh7.5 cells did not inhibit production of IFN-α by pDCs stimulated simultaneously (Fig. 4 B) or 2 h later with CpG-A (Fig. 4 C). Taken together, these results suggest that cell-associated HCV fails to activate NF-κB signaling in pDCs without having a dominant negative effect on NF-κB phosphorylation induced by other activators.
Endocytosis is relevant for sensing of HCV-infected hepatoma cells by pDCs.
To further explore the mechanism of sensing of cell-associated HCV by pDCs, we investigated the functional impact of endocytosis on virus recognition. To address this question, we tested several inhibitors impairing cellular endocytosis pathways. Inhibitors were mixed with pDCs, either simultaneously with HCV-infected Huh7.5 cells (Fig. 4D) or added 2 h after the priming (Fig. 4E). Chlorpromazine (CHP), an inhibitor of clathrin-coated pit-mediated endocytosis, inhibited production of IFN-α to 6.3% when pDCs were simultaneously exposed to HCV-infected Huh7.5 cells and endocytosis inhibitor, while production of IFN-α was blocked to 10% when pDCs were exposed to CHP 2 h after exposure to HCV-infected Huh7.5 cells. Incubation of pDCs with chloroquine (CHQ), a lysosomotropic weak base that neutralizes the acidic environment of endocytic vesicles, resulted in even stronger inhibition of HCV-infected Huh7.5 cell or CpG-A-induced IFN-α secretion, showing that acidification/maturation of the endosomes is necessary for activation of pDCs. Inhibitors of endocytosis used in these experiments did not change the background levels of proinflammatory cytokines TNF-α and IL-6 induced by exposure of pDCs to HCV-infected Huh7.5 cells, although they efficiently blocked production of proinflammatory cytokines induced by CpG-A (not shown). In conclusion, our findings suggest that endocytosis of viral or cellular structures of HCV-infected Huh7.5 cells and subsequent acidification of endosomes play a role in pDC activation.
DISCUSSION
Our results demonstrate that HCV-infected hepatoma cells trigger the IRF7 signaling pathway but not signaling via NF-κB in pDCs. Despite robust production of IFN-α by pDCs exposed to HCV-infected cells, these cells did not induce significant phosphorylation of the p65 subunit of NF-κB or production of the proinflammatory cytokines TNF-α and IL-6. Moreover, HCV-infected cells did not induce a significant expression of differentiation markers CD40, CCR7, CD86, or TRAIL. These results suggest that cell-associated HCV does not stimulate a full functional response of pDCs. Our findings correspond to results obtained for HIV where CD4+ T cell-associated virus was shown to induce a strong IFN-α response in pDCs but only a weak expression of pDC activation and differentiation markers (18, 30).
While type I IFN is the master effector and regulatory molecule of antiviral responses (10), CD80 and CD86 are pivotal molecules for effective cross-presentation by pDCs (22). TRAIL plays an essential role in their cytotoxicity (3). These functions facilitate the central role of pDCs in linking innate and adaptive immune responses. Our results are consistent with observations that suggest that this link might be impaired in HCV infection (1, 37, 38). These ex vivo studies performed with patients with chronic HCV infections reported decreased numbers and impaired function of virus-specific CD4 and CD8 T cells, suggesting a defect in pDC function. Low plasma levels of IFN-α and of proinflammatory cytokines, together with a delayed systemic immune response, were also observed in acute HCV infection, in comparison with levels in human immunodeficiency virus 1 (HIV-1) infection (33). In addition, studies investigating responses of pDCs from healthy donors concluded that HCV does not induce production of proinflammatory cytokines and cell differentiation (4, 11, 31).
Exposure of pDCs from healthy donors to cell-free HCV particles (4, 7, 11, 13, 31) or cell-associated virus did not induce cell differentiation and production of proinflammatory cytokines. The major difference between both viral systems lies in the production of type I IFN, induced in pDCs exclusively by the cell-associated virus (35), and the blockade of TLR9-mediated production of IFN-α, induced only by HCV particles (4, 7, 11, 13, 31). We hypothesize that the sensing of the cell-associated virus via TLR7 (35), in contrast to the sensing of the HCV particles by regulatory receptors of pDCs, is responsible for this difference (13). A high level of variability of analyzed markers among different donors in both viral systems could reflect genetic polymorphism of the determinants of response to HCV infection. This variability could result in different outcomes of HCV infection (spontaneous resolution versus chronicity). Information on some variables, such as sex (21), age (24, 34), and race (36) that were previously shown to be important for pDC function remains undisclosed in our present study with anonymous healthy donors.
Capacity of cell-associated HCV to induce IRF7 but not NF-κB signaling pathway in pDCs is compatible with the mechanism of spatiotemporal regulation of TLR signaling. This mechanism is based on the different compartmentalization and retention time of TLR ligands within the endolysosomal pathway (14, 16, 39). According to this concept, TLR signaling in early endosomes (induced for example by A-type and C-type CpG DNA) is coupled with production of type I IFN, whereas signaling from late endosomes (induced for example by B-type and C-type CpG DNA) is coupled with cell differentiation and production of proinflammatory cytokines (14). As expected, our findings show that endocytosis and subsequent acidification of endosomes are relevant for pDC activation by HCV-infected hepatoma cells.
The concept of spatiotemporal regulation was recently modified by demonstration of bifurcation of TLR7 and TLR9 signals by selective receptor trafficking within the endosomal system (29). Whereas Flu and R848 signal, according to this model, from both early (NF-κB and IRF7) and late endosomes and stimulate production of proinflammatory cytokines, type I IFN, and cell differentiation, our data show that HCV-infected hepatoma cells signal only via IRF7 endosomes to stimulate production of type I IFN. Preincubation of pDCs with HCV-infected hepatoma cells prior to TLR9 stimulation did not alter the production of IFN-α and proinflammatory cytokines. This contrasts with the inhibitory effect of cell-free HCV on IFN-α production (11) and suggests different spatiotemporal regulation of TLR signaling employed by the two forms of virus (13). A recent report has shown that HIV particles preferentially traffic to the early IRF7 endosomes in pDCs and therefore skew pDCs toward a partially matured, persistently IFN-α-secreting phenotype (23).
The role of pDCs in pathogenesis of HCV infection is not clear. In infected liver, pDCs are exposed to both hepatocyte-associated HCV and cell-free HCV virions. pDCs exposed to hepatocyte-associated HCV are good candidates for production of intrahepatic type I IFN (17, 28, 35), which not only activates expression of interferon-stimulated genes (ISGs) but also promotes greater activation of both pDCs and myeloid dendritic cells during virus infections (26). Production of type I IFN and efficient antigen processing and presentation for specific stimulation of T cells are both necessary for clearing HCV infection. Thus, incomplete activation of pDCs during chronic hepatitis C might be a reason for their inappropriate responsiveness. We found that the HCV-infected hepatoma cells did not actively block NF-κB phosphorylation and that full response of pDCs exposed to cell-associated virus could be induced by nucleotide-sensing TLR agonists. In addition to TLR agonists, NF-κB phosphorylation in pDCs exposed to cell-associated virus can be induced also via a TLR-independent pathway, e.g., by TNF-α. The finding that CpG-A and CpG-B can restore NF-κB-dependent functions that are not induced in pDCs exposed to cell-associated HCV suggests a potential perspective of TLR agonists to improve response to antiviral therapy. In view of the central role of pDCs in regulating the immune system, our results provide insight into the role of HCV-pDC interaction in HCV immune evasion, and they are important for our understanding of the mechanisms leading to the establishment of chronic HCV infection.
ACKNOWLEDGMENTS
Our work was supported by grants from the French National Agency for AIDS Research and Viral Hepatitis (ANRS), Institut National de la Santé et de la Recherche Médicale), Institut Paoli Calmettes, and Plateform Cancer Immuno-Monitoring IBiSA and by fellowships from ANRS (R.S.) and from the French Ministry of Higher Education and Research (C.D. and J.F.).
The sponsors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank T. Wakita for providing the HCV JFH-1 clone and C. M. Rice for the H/SG-neo (L+I) subgenomic replicon.
Footnotes
Published ahead of print 16 November 2011
REFERENCES
- 1. Bain C, et al. 2001. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology 120:512–524 [DOI] [PubMed] [Google Scholar]
- 2. Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaperot L, et al. 2006. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 176:248–255 [DOI] [PubMed] [Google Scholar]
- 4. Decalf J, et al. 2007. Plasmacytoid dendritic cells initiate a complex chemokine and cytokine network and are a viable drug target in chronic HCV patients. J. Exp. Med. 204:2423–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delgrange D, et al. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J. Gen. Virol. 88:2495–2503 [DOI] [PubMed] [Google Scholar]
- 6. Di Domizio J, et al. 2009. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood 114:1794–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dolganiuc A, Szabo G. 2011. Dendritic cells in hepatitis C infection: can they (help) win the battle? J. Gastroenterol. 46:432–447 [DOI] [PubMed] [Google Scholar]
- 8. Firaguay G, Nunes JA. 2009. Analysis of signaling events by dynamic phospho-flow cytometry. Sci. Signal. 2:pl3. [DOI] [PubMed] [Google Scholar]
- 9. Fried MW, et al. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975–982 [DOI] [PubMed] [Google Scholar]
- 10. Gilliet M, Cao W, Liu YJ. 2008. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8:594–606 [DOI] [PubMed] [Google Scholar]
- 11. Gondois-Rey F, et al. 2009. Hepatitis C virus is a weak inducer of interferon alpha in plasmacytoid dendritic cells in comparison with influenza and human herpesvirus type-1. PLoS One 4:e4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Govorkova EA, Kodihalli S, Alymova IV, Fanget B, Webster RG. 1999. Growth and immunogenicity of influenza viruses cultivated in Vero or MDCK cells and in embryonated chicken eggs. Dev. Biol. Stand. 98:39–51, discussion 73-74 [PubMed] [Google Scholar]
- 13. Hirsch I, Caux C, Hasan U, Bendriss-Vermare N, Olive D. 2010. Impaired Toll-like receptor 7 and 9 signaling: from chronic viral infections to cancer. Trends Immunol. 31:391–397 [DOI] [PubMed] [Google Scholar]
- 14. Honda K, et al. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434:1035–1040 [DOI] [PubMed] [Google Scholar]
- 15. Ito T, et al. 2002. Interferon-alpha and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J. Exp. Med. 195:1507–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. 2008. UNC93B1 delivers nucleotide-sensing Toll-like receptors to endolysosomes. Nature 452:234–238 [DOI] [PubMed] [Google Scholar]
- 17. Lau DT, et al. 2008. Interferon regulatory factor-3 activation, hepatic interferon-stimulated gene expression, and immune cell infiltration in hepatitis C virus patients. Hepatology 47:799–809 [DOI] [PubMed] [Google Scholar]
- 18. Lepelley A, et al. 2011. Innate sensing of HIV-infected cells. PLoS Pathog. 7:e1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu YJ. 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23:275–306 [DOI] [PubMed] [Google Scholar]
- 20. Manns MP, et al. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965 [DOI] [PubMed] [Google Scholar]
- 21. Meier A, et al. 2009. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat. Med. 15:955–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mouries J, et al. 2008. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood 112:3713–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Brien M, et al. 2011. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-alpha-producing and partially matured phenotype. J. Clin. Invest. 121:1088–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panda A, et al. 2010. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol. 184:2518–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pham TN, et al. 2004. Hepatitis C virus persistence after spontaneous or treatment-induced resolution of hepatitis C. J. Virol. 78:5867–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phipps-Yonas H, Seto J, Sealfon SC, Moran TM, Fernandez-Sesma A. 2008. Interferon-beta pretreatment of conventional and plasmacytoid human dendritic cells enhances their activation by influenza virus. PLoS Pathog. 4:e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Riboldi E, Daniele R, Cassatella MA, Sozzani S, Bosisio D. 2009. Engagement of BDCA-2 blocks TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. Immunobiology 214:868–876 [DOI] [PubMed] [Google Scholar]
- 28. Sarasin-Filipowicz M, et al. 2008. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 105:7034–7039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sasai M, Linehan MM, Iwasaki A. 2010. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 329:1530–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. 2005. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology 343:256–266 [DOI] [PubMed] [Google Scholar]
- 31. Shiina M, Rehermann B. 2008. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology 47:385–395 [DOI] [PubMed] [Google Scholar]
- 32. Siegal FP, et al. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science 284:1835–1837 [DOI] [PubMed] [Google Scholar]
- 33. Stacey AR, et al. 2009. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 83:3719–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stout-Delgado HW, Yang X, Walker WE, Tesar BM, Goldstein DR. 2008. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J. Immunol. 181:6747–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi K, et al. 2010. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U. S. A. 107:7431–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thomas DL, et al. 2009. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461:798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Valdez H, et al. 2000. Immune responses to hepatitis C and non-hepatitis C antigens in hepatitis C virus infected and HIV-1 coinfected patients. AIDS 14:2239–2246 [DOI] [PubMed] [Google Scholar]
- 38. Wedemeyer H, et al. 2002. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 169:3447–3458 [DOI] [PubMed] [Google Scholar]
- 39. Yoshizaki M, et al. 2009. Spatiotemporal regulation of intracellular trafficking of Toll-like receptor 9 by an inhibitory receptor, Ly49Q. Blood 114:1518–1527 [DOI] [PubMed] [Google Scholar]