

Abstract
Antiviral CD8+ T cells are a key component of the adaptive immune system against hepatitis C virus (HCV). For the development of immune therapies, it is essential to understand how CD8+ T cells contribute to clearance of infection and why they fail so often. A mechanism for secondary failure is mutational escape of the virus. However, some substitutions in viral epitopes are associated with fitness costs and often require compensatory mutations. We hypothesized that compensatory mutations may point toward epitopes under particularly strong selection pressure that may be beneficial for vaccine design because of a higher genetic barrier to escape. We previously identified two HLA-B*15-restricted CD8+ epitopes in NS5B (LLRHHNMVY2450-2458 and SQRQKKVTF2466-2474), based on sequence analysis of a large HCV genotype 1b outbreak. Both epitopes are targeted in about 70% of HLA-B*15-positive individuals exposed to HCV. Reproducible selection of escape mutations was confirmed in an independent multicenter cohort in the present study. Interestingly, mutations were also selected in the epitope flanking region, suggesting that compensatory evolution may play a role. Covariation analysis of sequences from the database confirmed a significant association between escape mutations inside one of the epitopes (H2454R and M2456L) and substitutions in the epitope flanking region (S2439T and K2440Q). Functional analysis with the subgenomic replicon Con1 confirmed that the primary escape mutations impaired viral replication, while fitness was restored by the additional substitutions in the epitope flanking region. We concluded that selection of escape mutations inside an HLA-B*15 epitope requires secondary substitutions in the epitope flanking region that compensate for fitness costs.
INTRODUCTION
Antiviral CD8+ T cells are one of the key components of the adaptive immune system against hepatitis C virus (HCV). During acute infection, HCV-specific CD8+ T cells are activated in the majority of patients (24, 38). Nevertheless, viral persistence is observed in 50 to 80% of patients. These patients with chronic HCV infection are at risk of progressive liver disease and liver cancer. For the development of immune therapies against HCV, it is essential to understand how CD8+ T cells contribute to clearance of infection and why they fail so often. Different mechanisms for CD8+ T cell failure have been described. The proposed mechanisms include primary failure by lack or impairment of priming of specific CD8+ T cells upon HCV infection, as well as secondary failure after the initial priming (reviewed in reference 40). Studies of patients with acute HCV infection suggest that CD8+ T cells are activated in most patients, irrespective of the outcome of infection (6), which supports an important role for secondary failure of HCV-specific CD8+ T cells.
Upon transition from acute to chronic HCV infection, the frequency of specific CD8+ T cells declines (6). Moreover, CD8+ T cells from patients with chronic HCV infection produce fewer cytokines and partially lose their capacity to proliferate upon exposure to their cognate antigen (55). This “exhausted,” dysfunctional phenotype of CD8+ T cells is associated with increased expression of inhibitory surface receptors such as PD-1, 2B4, CD160, CTLA-4, and TIM-3 (3, 15, 28, 37). Interestingly, not all virus-specific CD8+ T cells show this dysfunctional phenotype during chronic infection (3, 4, 20). An alternative mechanism for secondary failure of HCV-specific CD8+ T cells is mutational escape of the virus. Numerous studies have highlighted that during acute HCV infection, mutations are selected in targeted CD8+ T cell epitopes of the virus (7, 23, 48, 50). As a consequence, presentation of the variant epitope or contact with the T cell receptor is impaired. Some of these escape mutations in viral epitopes are highly reproducible in patients sharing the same HLA class I alleles, suggesting reproducible selection pressure. At the population level, these reproducible escape mutations are visible as “footprints” of the corresponding HLA allele in the viral genomes of circulating isolates (12, 29, 39, 41, 51).
Clearly, the space in which HCV can evolve is limited. Negative selection by functional constraints strongly contributes to viral evolution (39). For HCV infection, reversion of escape mutations in the absence of immune pressure has been described, indicating that some mutations are associated with fitness costs (39, 50). Indeed, impairment of viral replication by CD8+ T cell escape mutations was documented in vitro (42, 53). We hypothesized that selection of compensatory mutations may point toward epitopes under particularly strong selection pressure. Such epitopes could be suitable as templates for vaccine design because of a higher genetic barrier to escape. We previously identified two novel HLA-B*15-restricted CD8+ epitopes in NS5B, based on sequence analysis of a large single-source outbreak of HCV infection (41). Interestingly, we have now identified, in an extended data set, a residue in the N-terminal flanking region (position K2440) of one of the epitopes where mutations are selected in the presence of HLA-B*15. Covariation analysis of sequences from the database revealed that a K2440Q mutation as well as a secondary S2439T mutation is associated with an H2454R mutation or M2456L mutation inside the LLRHHNMVY2450-2458 epitope. The results of functional analysis of these substitutions in a subgenomic HCV replicon support the prediction that the substitutions outside the epitope compensate for fitness costs associated with substitutions inside the epitope. Taken together, our data provide clear evidence for fitness costs of a primary escape mutation that requires compensatory substitutions outside the epitope.
MATERIALS AND METHODS
HCV sequences from the East German HCV outbreak and from an independent multicenter cohort.
Samples from the East German cohort were collected beginning in 2008, and the NS5B region was amplified and sequenced as previously described (41). Sequences from 78 patients were published previously (GenBank accession numbers HQ908282 to HQ908359), and an additional 35 sequences were included in the current study (GenBank accession numbers JN377747 to JN377781). For verification of the findings, sequences from an independent multicenter cohort obtained at the Massachusetts General Hospital/Harvard Medical School were also analyzed (22). Written informed consent was obtained from all participants, and both study cohorts were approved by the local institutional review boards (IRBs).
HLA typing.
DNA of patients was extracted from peripheral blood lymphocytes by use of spin columns (Qiagen, Hilden, Germany). HLA-A and HLA-B typing at the two-digit resolution level was performed for 311 subjects from the East German cohort, using sequence-specific primer (PCR-SSP) methodology or using sequence-specific oligonucleotides (LABType methodology) (both provided by One Lambda Inc., Canoga Park, CA) (35). The designations of HLA-B*15 alleles and their serological equivalents were assigned according to the current version of the published “HLA Dictionary” (17). In brief, the serologically defined HLA-B62, -B63, -B70, -B71, -B72, -B75, -B76, and -B77 antigens all belong to the HLA-B*15 allelic group.
Covariation analysis of mutations in HLA-B*15-restricted CD8+ epitopes.
All available HCV genotype 1b NS5B sequences were retrieved from the public sequence database and aligned. Identical or phylogenetically closely related sequences were removed from the alignment to exclude multiple entries or clonal sequences of the same isolate. A total of 332 sequences were included in the covariation analysis. For each escape variant of the HLA-B*15-restricted epitope LLRHHNMVY2450-2458, a covariation analysis with the first 100 residues of NS5B (residues 2421 to 2520 of the polyprotein) was performed. This region was chosen based on a preliminary analysis of the NS5B structure in which this region includes the epitope. For each position, the numbers of sequences with and without the consensus residue in the presence and absence of each escape variant in the HLA-B*15-restricted epitope LLRHHNMVY2450-2458 were counted in a 2 × 2 table and subjected to Fisher's exact test. To correct for multiple comparisons, a P value of <10−5 was considered significant.
Analysis of replication in the HCV genotype 1b subgenomic replicon Con1.
The bicistronic subgenomic HCV replicon Con1/ET (pFK PI-luc/NS3-3′/Con1/ET) was described previously (26) and served as a backbone for generation of a series of variant replicons. Mutations were introduced by site-directed mutagenesis using a QuikChange II XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The correct sequence of the mutated plasmid was confirmed. Plasmid DNA was restricted with AseI and ScaI (New England BioLabs), and after purification by phenol-chloroform extraction and ethanol precipitation, the DNA pellet was dissolved in RNase-free water. In vitro transcription reaction mixtures contained 80 mM HEPES (pH 7.5), 12 mM MgCl2, 2 mM spermidine, 40 mM dithiothreitol (DTT), a 3.125 mM concentration of each nucleoside triphosphate, 1 U of RNasin (Promega, Mannheim, Germany)/μl, 0.1 μg of plasmid DNA/μl, and 0.6 U of T7 RNA polymerase (Promega)/μl. After 2 h at 37°C, an additional 0.3 U of T7 RNA polymerase/μl was added, and the reaction mixture was incubated for another 2 h. Transcription was terminated by the addition of 1.2 U of RNase-free DNase (Promega) per μg of plasmid DNA and a 30-min incubation at 37°C. After extraction with acidic phenol and chloroform, RNA was precipitated with isopropanol and dissolved in RNase-free water. Huh7-Lunet cells were transfected as previously described (23). Briefly, 4 × 106 cells were suspended in a Cytomix buffer (54), mixed with 5 μg of in vitro-transcribed RNA, and pulsed at 270 V and 960 μF with a Gene Pulser apparatus (Bio-Rad). Transfected cells were resuspended in complete Dulbecco's modified Eagle medium (DMEM) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U penicillin per ml, 100 μg streptomycin per ml, 10 mM HEPES buffer, and 10% fetal calf serum and then seeded in a 12-well plate. For assay of the luciferase activity after 4, 24, 48, 72, and 96 h, cells were washed once with phosphate-buffered saline (PBS), lysed directly in the plate with 1 ml ice-cold lysis buffer (0.1% Triton X-100, 25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, and 1 mM DTT; pH 7.8), and frozen. Upon thawing, lysates were resuspended by pipetting, and 100 μl was mixed with 360 μl of assay buffer (25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, 2 mM ATP, 15 mM K2PO4; pH 7.8) and, after the addition of 200 μl of a luciferin solution (200 μM luciferin, 25 mM glycylglycine; pH 8.0), measured in a luminometer (Lumat LB9507; Berthold, Freiburg, Germany) for 20 s. All luciferase assays were done at least in duplicate. A competition assay between a replicon with the consensus genotype 1b sequence in the epitope region and a replicon with the H2454R substitution was performed. RNAs were prepared as described earlier, and consensus and variant RNAs were mixed at a 1:10 ratio of consensus to variant RNA before transfection. After 4 h, 7 days, and 18 days, RNA was extracted from cell lysates, and a 1,230-bp fragment (Con1 nucleotides [nt] 7050 to 8280) covering the region of interest was amplified and cloned into pCR4-TOPO TA (Invitrogen). Multiple clones were sequenced to determine the relative frequencies of the consensus and variant replicons in culture.
Expansion of antigen-specific CD8+ T cells and ICS.
Peripheral blood mononuclear cells (PBMCs) (4 × 106) were resuspended in 1 ml of complete medium (RPMI 1640 containing 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μM HEPES buffer) and stimulated with peptide LLRHHNMVY or SQRQKKVTF (10 μg/ml) and anti-CD28 (0.5 μg/ml; BD Pharmingen). On days 3 and 7, 1 ml of complete medium and recombinant interleukin-2 (IL-2) (20 U/ml; Hoffmann-La Roche) were added. On day 10, the cells were tested for gamma interferon (IFN-γ) secretion after 5 h of stimulation with a prototype or variant peptide, using intracellular cytokine staining (ICS) as previously described (50).
RESULTS
Mutations are selected in the region of two HLA B*15-restricted CD8+ T cell epitopes in NS5B.
We previously identified two novel HLA-B*15-restricted T cell epitopes based on sequence analysis of 78 isolates from a large single-source outbreak of HCV infection (41). Here we extended the analysis to 113 sequences from the single-source outbreak and 145 unrelated sequences from a large multicenter cohort (22). Figure 1 shows the sequences for 15 HLA-B*15-positive individuals from the East German cohort and 29 HLA-B*15-positive individuals from the multicenter cohort. As previously described for a smaller data set from the East German cohort (41), sequence polymorphisms were selected reproducibly in the epitope regions in patients carrying the HLA-B*15 allele, suggesting mutational escape from the corresponding T cell response. In the East German cohort, 7 of 15 (46.7%) HLA-B*15-positive patients had selected substitutions in the LLRHHNMVY2450-2458 epitope, and 7 of 15 patients (46.7%) had substitutions in the SQRQKKVTF2466-2474 epitope. Only three patients showed substitutions in both epitopes. Both regions were highly conserved in HLA-B*15-negative patients (data not shown). All positions under significant selection pressure (P < 0.05; Fisher's exact test) in the presence of HLA-B*15 are shaded in gray in Fig. 1. Even though there was overlap between the selected sites in the LLRHHNMVY2450-2458 epitope, there were different sites under selection pressure in the SQRQKKVTF2466-2474 epitope in the East German cohort and the multicenter cohort. In the East German cohort, a K2470R polymorphism was selected most reproducibly, whereas a Q2467L/R polymorphism was selected most reproducibly in the multicenter cohort. Interestingly, we now identified in this larger data set of sequences from the East German cohort one site under selection pressure in the presence of HLA-B*15 which is located outside both epitopes (K2440S/Q). No HLA-B*15 binding motif was identified in this region by use of an epitope prediction tool (http://tools.immuneepitope.org). Importantly, this polymorphism was observed in three of four individuals carrying the H2454R polymorphism, suggesting that position K2440 covaries with residue H2454 inside the epitope. Note that the single HLA-B*15-positive patient with the H2454R polymorphism from the multicenter cohort also carries the K2440Q polymorphism. Figure 1 also shows the consensus sequences of HCV genotypes 1a and 3a. In both genotypes, the M2456L substitution is already present in the consensus sequence of the LLRHHNMVY2450-2458 epitope, whereas in the second epitope, an S2466C substitution is already present in genotype 1a only.
Fig 1.

Alignment of a region covering the HLA B*15-restricted CD8+ T cell epitopes LLRHHNMVY2450-2458 and SQRQKKVTF2466-2474. Sequences from 15 HLA-B*15-positive patients from the East German anti-D cohort as well as 29 HLA-B*15-positive patients from an independent multicenter cohort infected with HCV genotype 1b are shown. Parts of the sequences from the East German cohort were published previously (41). Positions with a significant association between the frequency of sequence polymorphisms and the presence of HLA-B*15 are shaded in gray (Fisher's exact test P value of <0.05). Also aligned are consensus sequences for HCV genotypes 1a and 3a, obtained from the HCV database.
The HLA-B*15-restricted epitopes LLRHHNMVY2450-2458 and SQRQKKVTF2466-2474 are frequently targeted in HLA-B*15-positive patients.
To address how frequently both HLA-B*15-restricted T cell epitopes are targeted, we tested 17 HLA-B*15(B62)-positive patients from a cohort with injection drug use, including 11 viremic patients and 6 HCV RNA-negative patients who were positive for anti-HCV (Table 1). This cohort is characterized by a very heterogeneous distribution of infecting genotypes, and the risk behavior and immune responses in these patients suggest that multiple exposures to different genotypes may occur (13). The CD8+ T cell response after 10 days of antigen-specific expansion was determined by intracellular IFN-γ staining and fluorescence-activated cell sorter (FACS) analysis. Both epitopes were targeted at a high frequency. A response against the LLRHHNMVY2450-2458 epitope was detected in 13 of 17 patients (67.5%; 6/6 RNA-negative patients and 7/11 RNA-positive patients). A response against the SQRQKKVTF2466-2474 epitope was detected in 11 of 17 patients (64.7%; 6/6 RNA-negative patients and 5/11 RNA-positive patients). Overall, in 14 of the 17 HLA-B*15(B62)-positive patients, at least one of the two epitopes was targeted. There was no reproducible hierarchy between the magnitudes of the responses against both epitopes. However, in almost all patients, the autologous sequences differed from the putative epitope sequence, irrespective of the detected magnitude of the corresponding CD8 T cell response. Some may represent selected immune escape mutations (e.g., in patient IDU147), but in many cases there are genotype-specific sequence differences that are unlikely to be products of immune selection. All genotype 1a and 3a sequences differed from the genotype 1b consensus in the LLRHHNMVY2450-2458 epitope. The putative M2456L escape mutation in genotype 1b already represents the predominant consensus sequence in genotypes 1a and 3a (Fig. 1). Nevertheless, some patients showed immune responses against the prototype 1b epitope sequence (e.g., patients IDU229 and IDU235). Similarly, in the SQRQKKVTF2466-2474 epitope, all genotype 1a sequences showed the typical S2466C variant, including one patient (IDU204) with 16.4% of CD8+ T cells targeting the prototype 1b sequence after 10 days of antigen-specific expansion. In contrast, the genotype 3a consensus sequence of this second epitope is identical to the genotype 1b sequence (Fig. 1). Interestingly, there was only one patient with a detectable CD8 T cell response (IDU210) and both epitopes conserved.
Table 1.
CD8 responses in HLA-B*15(B62)-positive patients
| Patient | Virus genotype | Viral load (IU/ml) | LLRHHNMVY variation | % IFN-γ+ CD8+ cells | SQRQKKVTF variation | % IFN-γ+ CD8+ cells |
|---|---|---|---|---|---|---|
| IDU141 | <10 | 25.8 | 8.3 | |||
| IDU193 | <10 | 19.2 | 16.0 | |||
| IDU205 | <10 | 1.1 | 0.50 | |||
| IDU213 | <10 | 0.88 | 9.2 | |||
| IDU224 | <10 | 1.3 | 0.18 | |||
| IDU225 | <10 | 5.7 | 12.6 | |||
| IDU113 | 3a | 2,258,000 | ------L-- | Not detected | Not detected | |
| IDU147 | 1b | 585,600 | -------I- | 0.7 | ----RR--- | 9.4 |
| IDU176 | 3a | 924,700 | ------L-- | Not detected | ----R---- | Not detected |
| IDU191 | 4d | 36,790 | Not done | 3.2 | Not done | Not detected |
| IDU199 | 1a | 388,800 | ------L-- | Not detected | C-------- | Not detected |
| IDU204 | 1a | 18,470 | -------I- | Not detected | CL------- | 16.4 |
| IDU210 | 1b | 4,615,000 | --------- | 0.65 | --------- | 0.74 |
| IDU223 | 1a | 801 | Not done | 1.6 | Not done | 1.7 |
| IDU226 | <615 | Not done | 0.28 | Not done | 0.18 | |
| IDU229 | 3a | 189,600 | ------L-- | 3.4 | -L------- | Not detected |
| IDU235 | 1a | 2,793,000 | ------L-- | 0.60 | C-------- | Not detected |
Next, we addressed if the selected mutations in the HLA-B*15-restricted epitopes have an impact on recognition by CD8+ T cells. Therefore, antigen-specific CD8+ T cells were expanded from PBMCs of two different HLA-B*15(B62)-positive individuals and restimulated with the prototype or variant peptides. Representative results for the LLRHHNMVY2450-2458 epitope and the SQRQKKVTF2466-2474 epitope are shown in Fig. 2. Patient IDU229 showed a strong response against the LLRHHNMVY2450-2458 epitope, with 3.4% of the cells secreting IFN-γ upon restimulation with the prototype. The number of IFN-γ-positive cells was markedly reduced when the cells were restimulated with the variant peptide containing the H2454R or M2456L substitution, and IFN-γ secretion was completely abolished when the variant peptide with both substitutions combined was used. The most frequent substitution in patients of the multicenter cohort was an H2453Y polymorphism. Again, IFN-γ secretion was completely abolished upon restimulation with the corresponding variant peptide. Importantly, patient IDU229 is infected with a genotype 3a virus, and the autologous sequence harbors the M2456L substitution, which represents the genotype 3a consensus sequence. We therefore also tried to expand specific CD8 T cells in the presence of the variant peptide LLRHHNLVY, but only a few specific T cells were expanded, which were again directed predominantly against the genotype 1b prototype sequence (Fig. 2B). The same results were obtained when cells were expanded from PBMCs of patient IDU235, infected with genotype 1a (data not shown). Patient IDU193 showed a strong response against the SQRQKKVTF2466-2474 epitope, with 16.0% of the cells secreting IFN-γ upon restimulation with the prototype peptide, but no IFN-γ was secreted upon restimulation with the variant peptide harboring the K2470R substitution. Taken together, these data support the conclusion that the selected mutations in the LLRHHNMVY2450-2458 epitope and the SQRQKKVTF2466-2474 epitope functionally act as escape mutations from an HLA-B*15-restricted CD8+ T cell response. The presence of strong T cell responses against the genotype 1b prototype sequence, which is not cross-reactive with the genotype 1a/3a sequence in patients infected with genotype 1a or 3a, may represent memory responses against a previously resolved infection.
Fig 2.

Impact of selected mutations in the LLRHHNMVY2450-2458 and SQRQKKVTF2466-2474 epitopes on recognition by CD8+ T cells. (A) Specific CD8+ T cells were expanded from PBMCs of patient IDU229 in the presence of the prototype peptide LLRHHNMVY and then restimulated after 10 days with the variant peptides indicated in the figure, followed by ICS for IFN-γ. (B) Specific CD8+ T cells were expanded from PBMCs of patient IDU229 in the presence of the variant peptide LLRHHNLVY and then restimulated after 10 days with the peptides indicated in the figure, followed by ICS for IFN-γ. (C) Specific CD8+ T cells were expanded from PBMCs of patient IDU193 in the presence of the prototype peptide SQRQKKVTF and then restimulated after 10 days with the peptides indicated in the figure, followed by ICS for IFN-γ. In all experiments, irrelevant peptides served as a negative control.
Covariation of escape mutations inside the LLRHHNMVY2450-2458 epitope with residues outside the epitope region.
Based on our observation that a K2440S/Q polymorphism is linked to the H2454R polymorphism inside the LLRHHNMVY2450-2458 epitope, we hypothesized that there is covariation between substitutions inside and outside the epitope. To address this more specifically, a detailed covariation analysis of all residues inside the LLRHHNMVY2450-2458 epitope and residues 2421 to 2520 in the polyprotein (residues 1 to 100 in NS5B) was performed utilizing 332 genotype 1b sequences from the public database (Fig. 3). Significant covariation with escape mutations was detected at two positions of the epitope in this analysis. The H2454R mutation was significantly associated with a substitution at position 2440 (P = 1.12 × 10−8) (Fig. 3A), and the M2456L polymorphism was significantly associated with a substitution at position 2439 (P = 3.66 × 10−8) (Fig. 3B). All 4 sequences with the H2454R substitution also carried a substitution at position K2440 (3 with the K2440Q substitution and 1 with the K2440A substitution). In contrast, there was only 1 sequence with a K2440E substitution among the remaining 328 sequences. The M2456L polymorphism is more frequent in the public database: 22 sequences (6.6%) carry the M2456L substitution, and 13 of these 22 sequences (59.1%) also carry an S2439T/A substitution. In contrast, the S2439A/T substitution is 10 times less frequent in the absence of the M2456L substitution (18 of 310 sequences [5.8%]). The covariant sites are located 14 and 17 amino acids upstream of the primary escape mutations. Figure 4 depicts the details of the NS5B structure in which the epitope is located. It is remarkable that residues S2439 and K2440 are in close proximity to the epitope between residues 2450 and 2458, with a minimum distance of about 3 Å, based on NCBI Protein Data Bank (PDB) entry 1C2P (strain HCV-BK; subtype 1b) (Fig. 4). This could indicate a direct physical compensatory mechanism. Specifically, the distance between the C-beta atoms of the covarying residues is 7.9 Å for residues 2454 and 2440 and 12.8 Å for residues 2456 and 2439. Notably, the correlated mutations of the first residue pair (H2454R and K2440A/Q/S) seem to function to conserve a positive charge. While H2454 is probably electrically neutral in this specific nonpolar to positively charged environment, the H2454R mutation clearly introduces a positive charge. Conversely, the correlated mutation K2440A/Q/S removes a positive charge. The long side chains of K and R—about 5 Å for the C-beta–amino distance in K and 5 to 6 Å for the C-beta–guanidino distance in R—allow for configurations where the positive charges carried by these two residues occupy the same space, i.e., direct charge compensation. For the second and physically more distant pair of residues, residues 2456 and 2439, a compensatory mechanism is less obvious, and its analysis may require a more detailed mechanistic understanding of NS5B function. These data collectively suggest that escape substitutions at position H2454 inside the LLRHHNMVY2450-2458 epitope are linked to substitutions outside the epitope, at position K2440, and that substitutions at position M2456 are possibly linked to substitutions at position S2439.
Fig 3.

Covariation analysis of selected mutations in the LLRHHNMVY2450-2458 epitope. For each position (2421 to 2520) of the polyprotein (residues 1 to 100 in NS5B), a P value was calculated for the association between sequence polymorphisms and the presence of escape mutations inside the epitope. (A) Association between the escape mutation H2454R and polymorphisms at other sites. A significant association at position K2440 is marked with an arrow. (B) Association between the escape mutation M2456L and polymorphisms at other sites. A significant association at position K2439 is marked with an arrow. The epitope regions of both HLA B*15-restricted epitopes are indicated with boxes.
Fig 4.

Locations of mutations in the NS5B structure. The structure of NS5B (NCBI PDB entry 1C2P; strain HCV-BK, subtype 1b) is shown, with the LLRHHNMVY2450-2458 epitope in red and covarying positions 2439 and 2440 in blue. Note that despite the sequence separation of 10 amino acids, the covarying positions physically touch the epitope (3.2-Å distance). The compensatory mutation K2440Q removes one positive charge introduced by the escape mutation H2454R, so the net charge of this spatial region remains unchanged.
Functional analysis of covariant sites suggests a compensatory effect.
Based on the covariation analysis, we hypothesized that the K2440Q and S2439T mutations compensate for fitness costs associated with mutations inside the HLA B*15-restricted epitope LLRHHNMVY2450-2458. To address this, we used the subgenomic genotype 1b replicon Con1 for functional analyses (Fig. 5). The Con1 sequence already harbored the S2439T and M2456L substitutions, which according to our hypothesis represents the compensated form of the LLRHHNMVY escape variant (Fig. 5A). Replication was essentially unchanged when both substitutions were removed from the replicon to yield the HCV genotype 1b consensus sequence. However, the variant with the M2456L escape mutation in the epitope without the S2439T mutation showed a reduced replication level (56% compared to that with the consensus at 48 h). When the H2454R escape mutation was introduced into the consensus sequence as a single mutation, replication was markedly reduced (24% compared to that with the consensus at 48 h), consistent with the observation that this single mutation is not observed in any isolates from the database. After the putative compensatory mutation K2440Q was added to this variant, replication was rescued to an intermediate level (67% compared to that with the consensus at 48 h). We next addressed if a replicon with the consensus sequence in the epitope has a replication advantage over the H2454R variant in a direct competition assay. RNAs were prepared from the consensus replicon and the variant replicon and mixed at a 1:10 ratio of consensus to variant RNA. The mixed RNA was transfected into Huh7 cells, and the clonal frequencies of the consensus and variant replicons were determined over a period of 18 days. After 4 h, only one of seven clonal sequences (14.3%) was the consensus sequence (Fig. 5D). Importantly, after 7 days, the consensus replicon already completely outcompeted the variant replicon, with 16 of 16 clones bearing the consensus sequence. The same result was observed after 18 days (9 of 9 clones carried the consensus sequence). Taken together, the analyses of the different variants in the subgenomic replicon support the hypothesis that the escape mutations H2454R and M2456L in the HLA-B*15-restricted CD8+ epitope LLRHHNMVY2450-2458 are associated with fitness costs and that substitutions at position K2440 and S2439 functionally act as compensatory mutations.
Fig 5.

Impacts of escape mutations inside the LLRHHNMVY2450-2458 epitope and at covariant sites on HCV replication. The impacts of different mutations were determined in a transient replication assay utilizing the HCV genotype 1b subgenomic replicon Con1. (A) Alignment of the different variants that have been tested. Note that the prototype sequence of Con1 already contains two substitutions compared to the genotype 1b consensus sequence (M2456L and S2439T). (B) Luciferase activities normalized to the 4-h result for each construct over a time course of 96 h. (C) Luciferase activities normalized to the results for the consensus sequence at 48 h (peak of replication) and 96 h (end of experiment). Data in panels B and C are mean values for three independent repetitions, each performed in duplicate, with standard errors of the means. RLU, relative light units. (D) Competition assay between a replicon with the consensus genotype 1b sequence in the epitope region and a replicon with the H2454R substitution. Consensus and variant RNAs were mixed at a ratio of 1:10 before transfection. After 4 h, 7 days, and 18 days, multiple clones of the replicons were sequenced to determine the relative frequencies of the consensus and variant replicons.
DISCUSSION
Mutational escape from CD8+ T cell responses is well described for patients with acute HCV infection. However, not all escape mutations are neutral for viral replication. Indeed, it has been shown that some escape mutations are associated with fitness costs (42, 53). We previously demonstrated that escape mutations are selected inside two HLA-B*15-restricted epitopes in NS5B (41). We now have found evidence for selection of a sequence polymorphism (K2440Q/S) upstream of one of the epitopes in the presence of HLA-B*15. No HLA-B*15 binding motif was identified in the region of this polymorphism. It was noticed that the polymorphism outside the epitope was linked to an H2454R polymorphism inside the LLRHHNMVY2450-2458 epitope. We therefore hypothesized that the polymorphism represents a compensatory mechanism for fitness costs associated with the primary substitution inside the epitope. A detailed covariation analysis of sequences from the public database revealed significant covariation between two residue pairs in this region. The H2454R and M2456L polymorphisms inside the LLRHHNMVY2450-2458 epitope were significantly associated with K2440Q/S/A and S2439T polymorphisms, respectively. The impact of the different variants was studied by use of the subgenomic HCV genotype 1b replicon Con1. Even though this system does not support analysis of the full replication cycle, at present it is the only robust system for functional analysis of replication in a genotype 1b sequence background. Our data support the conclusion that the secondary mutations are needed to maintain sufficient levels of RNA replication. Moreover, in a direct competition assay between a replicon with the consensus sequence and a replicon with the H2454R variant, the consensus replicon rapidly outcompeted the variant, supporting selection of the consensus sequence and consequent reversion in the absence of immune pressure. Even the compensatory substitution K2440Q/S/A only partially rescued replication, to intermediate levels. This explains the complete absence of the H2454R variant in its uncompensated state and the lack of this variant in the absence of HLA-B*15 in our analysis. The NS5B gene encodes the RNA-dependent RNA polymerase of HCV, which is one of the more conserved proteins of HCV (52). In the protein structure, the HLA-B*15 epitope is located on a long N-terminal loop originating from the “finger” region of the enzyme and forming extended contacts with the “thumb” (25). A more detailed look at the 3-dimensional locations of the compensatory mutations revealed a spatial proximity between the primary escape mutations and the covariant sites. At least the link between the H2454R substitution and the K2440Q/S/A polymorphism could be explained by requirements of a certain net charge in this specific environment, although this was not analyzed in detail. The mechanism of the link between the M2456L substitution and the S2439A/T polymorphism is unclear. Notably, the association between these substitutions seems less strict, as both are individually present in isolates in the database as well as in our data sets.
An alternative reason for selection of polymorphisms outside the HLA-B*15-restricted epitope could be impairment of antigen processing. Selection of mutations in the epitope flanking region that block antigen processing and presentation has been described for HIV-1 (1, 9, 30). In HCV, a Y-to-F polymorphism directly following the C-terminal residue of the HLA-A*02-restricted HCV epitope NS31073-1081 was previously demonstrated to prevent presentation of the epitope (46). Note that the authors did not find statistical support for selection of this processing mutation in the presence of HLA-A*02, which we confirmed in our previous analysis of sequences from the East German cohort (41). In the present study, the putative compensatory residues were located more distant from the epitope, 14 and 17 amino acids upstream of the primary escape substitutions. This large linear distance is unusual compared to previous examples of processing mutations that are described in the literature, but we did not formally exclude the possibility that the observed polymorphisms in the epitope flanking region affect antigen processing. Clearly, all tested substitutions inside both HLA-B*15-restricted epitopes impair recognition by CD8+ T cells. We did not find a clear hierarchy between the CD8+ T cell responses against one or the other epitope. Surprisingly, in some cases, we detected strong responses in patients infected with genotype 1a and 3a viruses. The consensus sequence of both genotypes already harbors the putative escape substitution M2456L inside the LLRHHNMVY2450-2458 epitope. Notably, both consensus sequences also harbor substitutions at the putative compensatory site, suggesting that similar functional constraints to those seen in genotype 1b may act across genotypes (Fig. 1). We tried to expand CD8+ T cells specific for the genotype 1a/3a peptide from two patients infected with genotype 1a or 3a in vitro but could detect only minor frequencies, whereas strong T cell responses were detected after expansion in the presence of the genotype 1b prototype peptide for the same patients. We therefore believe that it is unlikely that the response was primed against the current autologous virus. Rather, we hypothesize that these responses represent memory CD8 T cells against a previous genotype 1b infection, which is in line with the ability to rapidly expand them from PBMCs in vitro. Immunological evidence of exposure to different HCV genotypes was previously described for such high-risk groups for HCV infection (13, 45).
Although there is a large body of evidence that escape mutations are frequently selected in highly variable RNA viruses such as HIV and HCV, their overall role in viral persistence and immune control is less clear. In HIV-1-infected individuals, the appearance of escape mutations in targeted immunodominant CD8+ T cell epitopes was originally linked to loss of immune control (10, 16). However, more detailed studies on the determinants of HIV-1 containment in the presence of “protective” HLA class I alleles, such as HLA-B*57 and HLA-B*27, have pointed out that mutations are reproducibly selected in the immunodominant epitopes (2), even when viral replication is still under control (31, 32). In fact, it seems that viral containment is even linked to these escape mutations by their strong impact on replication capacity (14, 36). Importantly, reduced fitness associated with some escape mutations can be restored when secondary mutations outside the epitope region are selected (43). At the same time, the requirement for compensatory evolution in HIV-1 infection may represent a higher genetic barrier to achieve fully functional escape from the CD8 T cell response and may consequently correlate with slower disease progression. Loss of viral control is then observed when secondary mutations develop that restore viral fitness (5, 19).
For HCV, it is less clear if selection of less-fit variants or the requirements of complex mutation patterns to achieve fully functional escape contribute to immune control. Alleles such as HLA-A*03, HLA-B*27, and HLA-B*57 have been described to be associated with spontaneous resolution of HCV infection (21, 27, 49). It has been highlighted that these “protective” HLA class I alleles highly reproducibly select for escape mutations in the corresponding immunodominant epitopes (21, 29, 33). A study of the mechanisms of protection by HLA-B*27 pointed out that a rather complex pattern of 2, or sometimes even 3, substitutions is selected (8). In this case, multiple substitutions were required in order to achieve fully functional escape, but there was no evidence for a compensatory effect of the different polymorphisms. A recent study by Fitzmaurice et al. addressed the determinants of immune protection by HLA-A*03 in the Irish anti-D cohort (11). Again, mutations were reproducibly selected at two positions of the immunodominant epitope in NS3. In this case, one mutation represented the primary escape mutation from the immune response, and the second mutation in the neighboring position acted as a compensatory mutation for the other (11). In this first report of compensation of a CD8 escape mutation, both substitutions were located directly next to each other inside the epitope. Finally, a recent study analyzed the impact of escape substitutions selected in the presence of HLA-B*57. Similar to the case for the HLA-A*03-restricted epitope in NS3, selection of a substitution that functionally acts as a compensatory substitution was described (34). In this case, the compensatory substitution was located outside the epitope, three positions upstream of the primary escape substitution. Collectively, for all three immunodominant epitopes in HCV restricted by the “protective” HLA class I alleles HLA-A*03, -B*27, and -B*57, more complex evolutionary events are needed to achieve fully functional escape. This may support the observation that higher genetic barriers to immune escape correlate with immune control of HCV infection by CD8 T cells.
What is known about the effect of HLA-B*15 on disease outcome? An analysis of HLA frequencies in a large database of liver transplant recipients in North America revealed a protective effect of HLA-B*15(B62), supporting strong immune pressure in the presence of this HLA type (18). However, we analyzed the HLA frequencies in 214 chronically infected patients and 97 patients with spontaneously resolved infection in the East German cohort. Although the frequency of HLA-B*15 in this cohort was similar in total to that of the general German population, it was clearly not enriched in women who had spontaneously resolved infection (Fig. 6), suggesting that HLA-B*15 was neutral for disease outcome in the East German cohort. This indicates that even though there is a high genetic barrier to escape from the HLA-B*15-restricted response, requiring compensatory mutations, this did not translate into a protective effect in the East German cohort. One important aspect that distinguishes the HLA-B*15-restricted epitope in this study from the other immunodominant epitopes presented by the protective alleles HLA-A*03, HLA-B*27, and HLA-B*57 is the lower frequency of escape mutations. In about 40 to 50% of isolates, mutations are selected in the presence of HLA-B*15. In the presence of the protective alleles HLA-A*03, HLA-B*27, and HLA-B*57, more than 80% of viral isolates carry escape substitutions, suggesting that CD8+ T cell pressure against the corresponding immunodominant epitopes is more reproducible.
Fig 6.
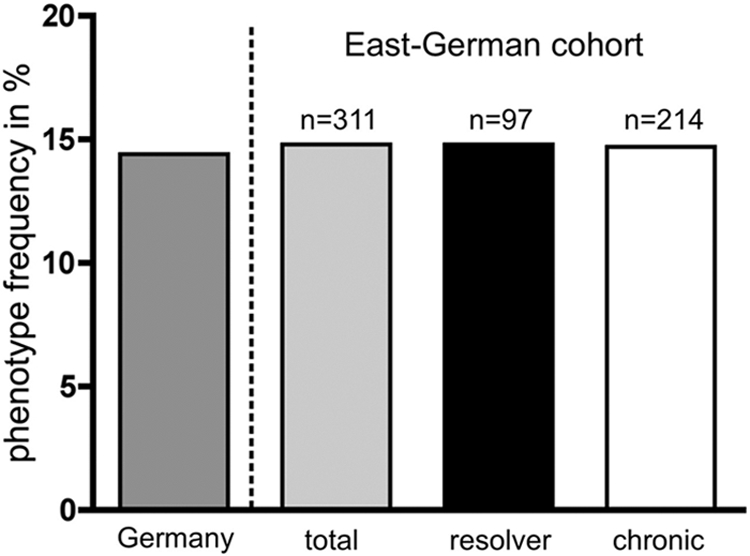
Impact of HLA-B*15 on the outcome of HCV infection in the East German cohort. A total of 311 women from the outbreak were HLA typed (97 with spontaneously resolved infection and 214 with chronic infection). Phenotype frequencies of HLA-B*15 are shown for a general German population (dark gray bar; obtained at www.allelefrequencies.net), the East German cohort in total (light gray bar), patients with resolved infection (black bar), and patients with chronic infection (white bar).
The serotype HLA-B*15(B62) belongs to the HLA class I B62 supertype (47). This serotype summarizes several HLA-B*15 subtypes (17), with HLA B*15:01 being the most prevalent in Caucasian populations (www.allelefrequencies.net). Other subtypes of the HLA-B*15 allele (serologically defined as B63, B70, B71, B72, B75, B76, and B77) show different binding motifs and are consequently distributed across a number of different HLA class I supertypes (B07, B27, B58, and B62) (47). In line with this, we failed to detect a CD8+ T cell response against either of the two epitopes (LLRHHNMVY2450-2458 and SQRQKKVTF2466-2474) in three patients with HLA-B*15(B63) and two patients with HLA-B*15(B71) or HLAB*15(B72), indicating that these epitopes may be targeted exclusively in HLA-B*15(B62)-positive patients (data not shown). However, all patients of the East German anti-D cohort without epitope variation were positive for HLA-B*15(B62), excluding the possibility that differences at the level of HLA-B*15 genetic subtypes were responsible for the lack of escape in some patients.
Apart from the effects of compensatory mutations on viral replication in individuals, there are obviously also negative aspects at the population level. Compensatory evolution may result in highly fit isolates that are possibly (at the same time) less immunogenic in a particular HLA background. Such adapted isolates could be transmitted through a population and potentially accumulate. Transmission of such isolates already harboring escape mutations may then potentially prevent early immune control. This was demonstrated for HIV-1, where compensated escape mutations were stable upon their transmission (43) and preexisting compensatory mutations facilitated the development of escape mutations associated with rapid disease progression even in the presence of protective HLA class I alleles (44). For HCV, it is unclear if the sequence of the transmitted virus also plays a role in subsequent immune control. The overall low frequency of the compensated forms of the HLA-B*15 escape variants in the public database suggests that accumulation of the escape variant is less problematic for this epitope, although this needs to be addressed in longitudinal studies.
Taken together, our results demonstrate that mutational escape in a frequently targeted CD8+ T cell epitope in NS5B requires compensatory evolution outside the epitope. This may point toward an epitope that is under particularly strong selection pressure, but in contrast to recent reports of compensatory evolution in two other immunodominant epitopes in HCV, this does not translate into a higher clearance rate in the presence of the relevant allele. The overall consequences of this compensatory evolution on HCV pathogenesis and immune control remain to be elucidated.
ACKNOWLEDGMENTS
We thank Lejla Glavinic for her excellent technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG-TRR60 and GK 1045) and by federal funds from the National Institute of Allergy and Infectious Diseases (NIAID) under grants R01-AI067926 (T.M.A.) and U19-AI082630 (T.M.A.).
Footnotes
Published ahead of print 9 November 2011
REFERENCES
- 1. Allen TM, et al. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J. Virol. 78:7069–7078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bailey JR, Williams TM, Siliciano RF, Blankson JN. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J. Exp. Med. 203:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bengsch B, et al. 2010. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 6:e1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bengsch B, et al. 2007. Analysis of CD127 and KLRG1 expression on hepatitis C virus-specific CD8+ T cells reveals the existence of different memory T-cell subsets in the peripheral blood and liver. J. Virol. 81:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brockman MA, et al. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J. Virol. 84:11937–11949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cox AL, et al. 2005. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 42:104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox AL, et al. 2005. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 201:1741–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dazert E, et al. 2009. Loss of viral fitness and cross-recognition by CD8+ T cells limit HCV escape from a protective HLA-B27-restricted human immune response. J. Clin. Invest. 119:376–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Draenert R, et al. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199:905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feeney M, et al. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term nonprogressing child. J. Virol. 78:8927–8930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fitzmaurice K, et al. 2011. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut 60:1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaudieri S, et al. 2006. Evidence of viral adaptation to HLA class I-restricted immune pressure in chronic hepatitis C virus infection. J. Virol. 80:11094–11104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giugliano S, et al. 2009. Degree of cross-genotype reactivity of hepatitis C virus-specific CD8+ T cells directed against NS3. Hepatology 50:707–716 [DOI] [PubMed] [Google Scholar]
- 14. Goepfert PA, et al. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Golden-Mason L, et al. 2007. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J. Virol. 81:9249–9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goulder PJ, et al. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212–217 [DOI] [PubMed] [Google Scholar]
- 17. Holdsworth R, et al. 2009. The HLA dictionary 2008: a summary of HLA-A, -B, -C, -DRB1/3/4/5, and -DQB1 alleles and their association with serologically defined HLA-A, -B, -C, -DR, and -DQ antigens. Tissue Antigens 73:95–170 [DOI] [PubMed] [Google Scholar]
- 18. Hraber P, Kuiken C, Yusim K. 2007. Evidence for human leukocyte antigen heterozygote advantage against hepatitis C virus infection. Hepatology 46:1713–1721 [DOI] [PubMed] [Google Scholar]
- 19. Huang KH, et al. 2011. Progression to AIDS in South Africa is associated with both reverting and compensatory viral mutations. PLoS One 6:e19018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kasprowicz V, et al. 2010. Hepatitis C virus (HCV) sequence variation induces an HCV-specific T-cell phenotype analogous to spontaneous resolution. J. Virol. 84:1656–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim AY, et al. 2011. Spontaneous control of HCV is associated with expression of HLA-B*57 and preservation of targeted epitopes. Gastroenterology 140:686–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuntzen T, et al. 2008. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naive patients. Hepatology 48:1769–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuntzen T, et al. 2007. Viral sequence evolution in acute hepatitis C virus infection. J. Virol. 81:11658–11668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lauer GM, et al. 2005. Full-breadth analysis of CD8+ T-cell responses in acute hepatitis C virus infection and early therapy. J. Virol. 79:12979–12988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lesburg CA, et al. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6:937–943 [DOI] [PubMed] [Google Scholar]
- 26. Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKiernan SM, et al. 2004. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology 40:108–114 [DOI] [PubMed] [Google Scholar]
- 28. McMahan RH, et al. 2010. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J. Clin. Invest. 120:4546–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merani S, et al. 2011. Effect of immune pressure on hepatitis C virus evolution: insights from a single-source outbreak. Hepatology 53:396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Milicic A, et al. 2005. CD8+ T cell epitope-flanking mutations disrupt proteasomal processing of HIV-1 Nef. J. Immunol. 175:4618–4626 [DOI] [PubMed] [Google Scholar]
- 31. Miura T, et al. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J. Virol. 83:2743–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miura T, et al. 2010. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J. Virol. 84:7581–7591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neumann-Haefelin C, et al. 2006. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 43:563–572 [DOI] [PubMed] [Google Scholar]
- 34. Oniangue-Ndza C, et al. 2011. Compensatory mutations restore the replication defects caused by cytotoxic T lymphocyte escape mutations in hepatitis C virus polymerase. J. Virol. 85:11883–11890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oudshoorn M, Horn PA, Tilanus M, Yu N. 2007. Typing of potential and selected donors for transplant: methodology and resolution. Tissue Antigens 69(Suppl 1):10–12 [DOI] [PubMed] [Google Scholar]
- 36. Prado JG, et al. 2010. Replicative capacity of human immunodeficiency virus type 1 transmitted from mother to child is associated with pediatric disease progression rate. J. Virol. 84:492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Radziewicz H, et al. 2007. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 81:2545–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rahman F, et al. 2004. Effects of antiviral therapy on the cellular immune response in acute hepatitis C. Hepatology 40:87–97 [DOI] [PubMed] [Google Scholar]
- 39. Ray SC, et al. 2005. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J. Exp. Med. 201:1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rehermann B. 2009. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J. Clin. Invest. 119:1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruhl M, et al. 2011. CD8(+) T-cell response promotes evolution of hepatitis C virus nonstructural proteins. Gastroenterology 140:2064–2073 [DOI] [PubMed] [Google Scholar]
- 42. Salloum S, et al. 2008. Escape from HLA-B*08-restricted CD8 T cells by hepatitis C virus is associated with fitness costs. J. Virol. 82:11803–11812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schneidewind A, et al. 2009. Transmission and long-term stability of compensated CD8 escape mutations. J. Virol. 83:3993–3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schneidewind A, et al. 2009. Maternal transmission of human immunodeficiency virus escape mutations subverts HLA-B57 immunodominance but facilitates viral control in the haploidentical infant. J. Virol. 83:8616–8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schulze Zur Wiesch J, et al. 2007. Immunologic evidence for lack of heterologous protection following resolution of HCV in patients with non-genotype 1 infection. Blood 110:1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seifert U, et al. 2004. Hepatitis C virus mutation affects proteasomal epitope processing. J. Clin. Invest. 114:250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sidney J, Peters B, Frahm N, Brander C, Sette A. 2008. HLA class I supertypes: a revised and updated classification. BMC Immunol. 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tester I, et al. 2005. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J. Exp. Med. 201:1725–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thio CL, et al. 2002. HLA-Cw*04 and hepatitis C virus persistence. J. Virol. 76:4792–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Timm J, et al. 2004. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 200:1593–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Timm J, et al. 2007. Human leukocyte antigen-associated sequence polymorphisms in hepatitis C virus reveal reproducible immune responses and constraints on viral evolution. Hepatology 46:339–349 [DOI] [PubMed] [Google Scholar]
- 52. Timm J, Roggendorf M. 2007. Sequence diversity of hepatitis C virus: implications for immune control and therapy. World J. Gastroenterol. 13:4808–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uebelhoer L, et al. 2008. Stable cytotoxic T cell escape mutation in hepatitis C virus is linked to maintenance of viral fitness. PLoS Pathog. 4:e1000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van den Hoff MJ, Christoffels VM, Labruyere WT, Moorman AF, Lamers WH. 1995. Electrotransfection with “intracellular” buffer. Methods Mol. Biol. 48:185–197 [DOI] [PubMed] [Google Scholar]
- 55. Wedemeyer H, et al. 2002. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 169:3447–3458 [DOI] [PubMed] [Google Scholar]