

Abstract
Epstein-Barr virus (EBV) uses different virus and cell proteins to enter its two major targets, B lymphocytes and epithelial cells. The routes that the virus takes into the two cell types are also different. To determine if these differences extend to movement from the cell surface to the nucleus, we examined the fate of incoming virus. Essentially all virus that entered a B cell remained stable for at least 8 h. In contrast, up to 80% of virus entering an epithelial cell was degraded in a compartment sensitive to inhibitors of components involved in autophagy. Inhibitors of actin remodeling blocked entry into a B cell but had no effect or enhanced entry into an epithelial cell. Inhibitors of the microtubule network reduced intracellular transport in both cell types, but movement to the nucleus in an epithelial cell also required involvement of the actin cytoskeleton. Deletion of the cytoplasmic tail of CR2, which in an epithelial cell interacts with the actin nucleator FHOS/FHOD when cross-linked by EBV, had no effect on infection. However, inhibitors of downstream signaling by integrins reduced intracellular transport. Cooperation of the microtubule and actin cytoskeletons, possibly activated by interaction with integrin binding proteins in the envelope of EBV, is needed for successful infection of an epithelial cell.
INTRODUCTION
Epstein-Barr virus (EBV) is an orally transmitted human gammaherpesvirus that is carried by more than 90% of the population worldwide. Most primary infections are asymptomatic, but in those individuals in whom infection is delayed beyond childhood it is more likely to manifest as acute infectious mononucleosis (reviewed in reference 38). The virus is also associated with both lymphoid and epithelial tumors, reflecting its principal tropism for these two cell types. Current models of persistence propose that the latent reservoir of virus is in long-lived memory B cells but that amplification of virus in epithelial cells contributes to reinfection of B cells and maintenance of the reservoir and also to spread of virus to new hosts (18, 22, 47).
Cycling of EBV between B cells and epithelial cells is facilitated by the fact that different virus and cell proteins are used for entry into each. This provides a mechanism whereby modulation of the amount of a differentially used virion envelope glycoprotein during replication in one cell type can switch tropism to the other (3). After attachment to a cell, the core fusion machinery, comprised of heterodimers of glycoproteins gH and gL (gHgL) and trimers of glycoprotein gB (1), is responsible for fusion of the virus with the cell membrane (reviewed in references 19 and 43). Activation of the machinery for fusion with an epithelial cell is triggered by an interaction between integrin αvβ6 or αvβ8 and a KGD motif that is part of a prominent loop on the surface of gH (8, 29). Activation for fusion with a B cell is triggered by an interaction between a fourth glycoprotein, gp42, which forms a tripartite complex with some of the heterodimers of gHgL, and HLA class II. The presence of gp42 in a complex with gHgL occludes access of the integrin binding-loop of gHgL to its ligand and blocks epithelial cell infection. Thus, the two complexes gHgL and gHgLgp42 have mutually exclusive functions. In a B cell, some tripartite complexes are lost to the HLA class II trafficking pathway and virus emerges rich in bipartite complexes. This does not happen in epithelial cells, and virus emerges with more tripartite complexes. The progeny of each cell type is thus better placed to infect the other.
Beyond these differences in how fusion is triggered, there are also differences in the sites at which it occurs in B cells and epithelial cells. Fusion of EBV with B cells, with the apparent exception of the Burkitt's lymphoma line Raji (39, 46), occurs after endocytosis (33). It is sensitive to the effects of chlorpromazine, which, among other things, can inhibit clathrin-mediated endocytosis. Although not critical to the process, fusion occurs at an acidic pH. In contrast, fusion with an epithelial cell is not inhibited by treatment of cells with chlorpromazine, is more resistant to sodium azide treatment, and occurs at neutral pH, which has been interpreted as being consistent with fusion taking place at the cell surface (31). These striking differences suggest that there may also be differences in the subsequent fate of virus. Indeed, it has been reported that, while transport to the nucleus is efficient in a B cell, many virus particles internalized into epithelial cells fail to reach the nucleus (40).
Trafficking of EBV into the nucleus of a cell can be conveniently monitored by expression of green fluorescent protein (GFP) from a constitutively active promoter inserted in the virus genome. Judged by this criterion, initiation of infection of epithelial cells with cell-free virus in vitro is frequently less efficient than infection of B cells (4, 12, 41), despite the fact that the kinetics of fusion are identical (31) and virus binding, even in the absence of one of the EBV attachment receptors, CR2, can be quite high (4). Reproducibly high levels of infection can, however, be achieved with the SVKCR2 cell line, a simian virus 40 (SV40)-transformed keratinocyte cell line engineered to express CR2 (24). To examine the fates of EBV after fusion with B cells and epithelial cells, we thus began by exploring transport of virus to the nucleus in the Akata B cell line, where virus entry is sensitive to chlorpromazine, and in the SVKCR2 cell line, where it is not (4). Despite robust levels of infection, we found that the proportion of internalized virus that survived and reached the nucleus was lower in SVKCR2 cells than in Akata B cells and that the observation could be duplicated in primary epithelial cultures. As for many herpesviruses, an intact microtubule network was important to intracellular transport in both cell types, but the actin cytoskeleton also played a critical role. In B cells the requirement was consistent with a role for actin remodeling in endocytosis, and in epithelial cells actin remodeling was important for steps subsequent to virus entry into the cytoplasm.
MATERIALS AND METHODS
Cells.
EBV-negative Akata cells (referred to as Akata cells throughout), a Burkitt's lymphoma-derived cell line that has lost EBV episomes (42), Raji cells, EBV-positive Burkitt's lymphoma-derived cells (37), Akata cells carrying EBV in which the thymidine kinase gene is interrupted with a double cassette expressing neomycin resistance and the green fluorescent protein (EBV-GFP) (32), lymphoblastoid cell lines (LCL), and tonsil lymphocytes were grown in RPMI 1640. AGS, a CR2-negative gastric carcinoma cell line (ATCC) which had been cured of parainfluenza type 5 infection (52) by treatment with ribavirin, AGS cells carrying EBV-GFP, and 293 cells were grown in Ham's F-12 medium (Sigma). All media for Akata and AGS cells were supplemented with 10% heat-inactivated fetal bovine serum (Gibco), and cells carrying EBV-GFP were also supplemented with 500 μg/ml active G418 (Mediatech). SVK, an SV40-transformed keratinocyte cell line, SVKCR2, derived from SVK cells by stable transfection of a plasmid expressing CR2 (24) and, SVKCR2-CIITA cells expressing HLA class II as a result of stable transfection with the class II transactivator CIITA (3) were grown in Joklik's modified Eagle's medium supplemented with 10% heat-inactivated HyClone Cosmic calf serum (Sigma) and 10 ng/ml cholera toxin (Sigma). NOK cells (36), H-tert immortalized normal oral keratinocytes (a gift of Karl Munger, Harvard University), and primary tonsil epithelial cells were grown in keratinocyte SFM (Gibco). Primary tonsil epithelium was isolated from fresh tonsil tissue discarded from patients undergoing routine tonsillectomy for relief from sleep apnea. Briefly, tissue was cut into small explants and placed in Matrigel (BD Biosciences) in 24-well plates, and explants were covered in medium. Medium was changed every other day, and approximately 1 week later explants were checked. When epithelial cells appeared as a small ring around the tissue, the explant was removed and the epithelial cells were allowed to grow to subconfluence. All the epithelial cells grown from a single tonsil were pooled for use. Tonsil lymphocytes were isolated by cutting tissue into small fragments, forcing cells through a sterile mesh, and sedimenting the cell suspension over Ficoll-Paque Plus (GE Healthcare BioSciences).
Virus production.
Virus made in B cells (B-EBV-GFP) was harvested from the supernatant of Akata cells carrying EBV-GFP that had been induced by treatment with anti-human immunoglobulin as described previously (32). Virus made in epithelial cells (E-EBV-GFP) was harvested from spent culture medium of AGS cells carrying EBV-GFP 5 days after induction with 12-O-tetradecanoylphorbol-13-acetate (Sigma) and 2.5 mM sodium butyrate (Calbiochem). In both cases virus was concentrated by centrifugation as described previously (32). Previous experiments have shown that the method of induction does not influence the behavior of the virus (3). For all experiments virus was used at a copy number that resulted in approximately 50% infection of Akata or SVKCR2 cells.
Antibodies and reagents.
Monoclonal antibodies used were those to α-tubulin (Ab-2 DM1A; Neomarkers), lamin B (101-B7; Calbiochem), and CR2 (HB5; ATCC) (49). HB5 was purified by affinity chromatography on protein A coupled to agarose (Repligen). Binding to cells was visualized with F(ab′)2 fragments of goat anti-mouse antibody conjugated to phycoerythrin (PE) (Jackson ImmunoResearch), and Western blots were visualized with ECL sheep anti-mouse antibody coupled to horseradish peroxidase (GE Healthcare). Stock solutions of latrunculin A (1 mM), paclitaxel (originally named taxol) (10 mM), nocodazole (1 mM), SB203580 (2 mM), wortmannin (10 mM), PP2 (10 mM), and vinblastine (10 mM) (all from Sigma), LY294002 (25 mM; Calbiochem), jasplakinolide (10 mM; Molecular Probes), and H1152 (10 mM; Tocris) were made in dimethyl sulfoxide. Stock solutions of colchicine (20 mM; Sigma), leupeptin (10 mM; Sigma), and CT04 (0.1 mg/ml; Cytoskeleton Inc.) were made in water. Effects of nocodazole, paclitaxel, latrunculin, and jasplakinolide on SVKCR2 cells were examined by fixing cells for 10 min at room temperature with 4% paraformaldehyde, permeabilizing with 0.1% Triton X-100 for 3 min, blocking with 5% bovine serum albumin, and staining either with phalloidin-Alexa Fluor 546 (Molecular Probes) or with monoclonal antibody to α-tubulin and rabbit anti-mouse antibody conjugated to Alexa Fluor 488 (Molecular Probes) and mounted in proLong Gold Antifade with DAPI (4′,6′-diamidino-2-phenylindole) (Invitrogen). All drugs were confirmed to be nontoxic at the concentrations used by testing cells for cell viability with trypan blue. None of the drugs affected the transcription rate of actin.
Plasmids.
Full-length CR2 and CR2 lacking a cytoplasmic tail (CR2-CT) were cloned into the pCAGGS/MCS vector (a gift of Martin Muggeridge, LSU Health Sciences Center) for expression under the control of the β-actin promoter in cooperation with the human cytomegalovirus immediate-early (HCMV-IE) enhancer (34). pCAGGS-CR2 was made by PCR amplification of pBS-CR2 (a gift of John Sixbey, LSU Health Sciences Center) with primers 5′-ATGAGCTCATGGGCGCCGCGGGC-3′ and 5′-AGGCATGCTCAGCTGGCTGGGTTG-3′, cutting the amplified fragment with SacI and SphI, and cloning it into pCAGGS cut with the same enzymes. pCAGGS-CR2-CT was made by PCR amplifying pCAGGS-CR2 with primers 5′-ATGAGCTCATGGGCGCCGCGGGC-3′ and 5′-GGAGATCTTTTTGATATCACGTATAAGG-3′, cutting the amplified fragment with SacI and BglII, and cloning it into pCAGGS cut with the same enzymes. Both plasmids were confirmed to have sequences identical to those of a fragment amplified directly from the integrated cDNA copy in SVKCR2 cells.
Infection of cells expressing CR2 and CR2-CT.
Two million SVK cells were transfected using nucleofection (Nucleofected) with 1 μg pCAGGS-CR2 or pCAGGS-CR2-CT using an Amaxa Nucleofector 1 and program T20. Each sample was seeded in 2 wells of a 6-well plate. Twenty four hours later, cells in 1 well were removed by trypsinization, recovered for 45 min in a 37°C water bath, and incubated sequentially with antibody HB5 to CR2 and PE-conjugated anti-mouse antibody, and the expression of CR2 or CR2-CT was measured by flow cytometry. Cells in the second well were infected with Akata-GFP for 4 h at 37°C, at which time virus-containing medium was replaced with fresh medium. Cells were reincubated for 48 h, and infection was measured by trypsinizing cells, resuspending them in phosphate-buffered saline (PBS) at 106/ml, and subjecting them to flow cytometric analysis of GFP expression.
QPCR and RT-QPCR.
Quantitative real-time PCR (QPCR) to determine the number of virus genomes per cell was done as previously described (48) for cells that were not already carrying EBV. Briefly, a 76-bp region of the EBV EBNA1 gene in the BamHI K fragment (primers 5′-GGATGCGATTAAGGACCTTGTT-3′ and 5′-CGTCAAAGCTGCACACAGTCA-3′, base coordinates 109677 and 109753, respectively; National Center for Biotechnology Information GenBank accession no. VO1555) was amplified together with a 101-bp DNA sequence of the human C-reactive protein (CRP) gene (primers 5′-CTTGACCAGCCTCTCTCATGC-3′ and 5′-TGCAGTCTTAGACCCCACCC-3′, base coordinates 132705 and 132605, respectively; accession no. AL445528) using the TaqMan fluorogenic system (ABI Prism 7500 Fast; PE Applied Biosystems). The EBV probe (5′-CAAAGCCCGCTCCTACCTGCAATATCA-3′, base coordinate 109703) was labeled with 6-carboxyfluorescein (FAM) and the CRP probe (5′-TTTGGCCAGACAGGTAAGGGCCACC-3′, base coordinate 132682) was labeled with VIC (PE Applied Biosystems). Serial dilutions of DNA from IB4, a Burkitt's lymphoma cell line containing five copies of EBV per cell, served as a standard. To measure incoming virusom Applied Biosystems, and the probe was labeled with VIC. Each reaction included a control PCR amplification of isolated RNA to which no reverse transcriptase had been added and a water-only control.
Measurement of virus binding and internalization.
Virus was bound to cells at 4°C for 2 h, and cells were washed 3 times with phosphate-buffered saline (PBS) containing Complete Mini EDTA-free protease inhibitors (Roche) and 15 mg/ml bovine serum albumin. DNA was isolated using a QIAamp DNA Blood Mini Kit (Qiagen), and the number of virus copies bound per cell was determined by QPCR. Internalization of virus was measured by binding virus to cells for 2 h at 4°C followed by warming the cells to 37°C for various amounts of time. Cells were then placed on ice and treated with 1 mg/ml proteinase K (Roche) in PBS with 10 mM EDTA for 45 min to remove virus remaining at the cell surface. Cells were washed 3 times as before, and DNA was isolated for determination of the number of virus copies remaining per cell by QPCR.
Nuclear isolation.
Nuclei were isolated according to established protocols (20). Briefly, cells were washed with PBS and lysed in lysis buffer containing 10 mM Tris (pH 8.0), 140 mM NaCl, 1.5 mM MgCl2, and 0.5% NP-40. Nuclei were washed with lysis buffer containing 1% Tween 40 and 0.5% deoxycholic acid. Purity of nuclei was determined by boiling in sample buffer, polyacrylamide gel electrophoresis, and Western blotting with antibodies to α-tubulin and lamin B and anti-mouse antibody coupled to horseradish peroxidase. Antibody was visualized with the ECL-Plus Western blot detection system (Amersham; GE Healthcare).
RESULTS
Internalized virus DNA is stable in a B cell but not in an epithelial cell.
Infection of both EBV-negative Akata B cells and SVKCR2 epithelial cells is robust if judged in terms of the percentage of cells that express EBV nuclear antigen (24) or GFP under the control of a constitutively active promoter in the virus genome (4). If E-EBV-GFP is bound at a DNA copy number of approximately 50 per cell, then approximately 40% of B cells can be infected, and if B-EBV-GFP is bound at a DNA copy number of approximately 60 per cell to SVKCR2, cells then approximately 50% of cells can be infected. Despite this, the number of particles reaching the nucleus in an epithelial cell at 24 h postinfection has been reported to be lower than that in a B cell (40). To explore further the fate of internalized virus in the two cell types, virus was bound on ice for 2 h to SVKCR2 cells and EBV-negative Akata cells, unbound virus was removed by washing, and cells were warmed to 37°C. At intervals over 8 h cells were harvested, chilled, and treated with proteinase K to remove virus remaining at the cell surface, and the virus DNA remaining was measured on a per-cell basis by QPCR as a measure of the amount internalized. Pilot experiments indicated that at least 90% of remaining surface-bound virus was removed by this protocol. Internalization of virus into Akata B cells increased gradually over 4 h, presumably reflecting the fact that virus particles that are bound differ in the efficiency with which they are taken up. Uptake then plateaued, and the amount of internalized virus was maintained for at least 4 h more (Fig. 1) (all experiments throughout have been repeated multiple times, and results shown are representative). Incoming virus DNA was also stable in the Raji B cell line. In contrast, although the amount of virus entering SVKCR2 cells increased for up to 30 min, after this time loss exceeded uptake and the amount of internalized virus gradually decreased to a plateau at 4 h. Virus was lost in the same way in the CR2-negative AGS gastric carcinoma cell line and primary tonsil epithelial cells. Infecting B cells with B-EBV-GFP or epithelial cells with E-EBV-GFP altered the efficiency of infection but did not influence the stability of internalized virus DNA in any cell type (data not shown). The different ways in which virus fusion is triggered on a B cell and an epithelial cell also had no effect on the fate of virus. Virus DNA was lost in the same way in SVKCR2-CIITA cells, which express high levels of HLA class II, as it was in SVKCR2 cells.
Fig 1.

Virus DNA is lost following internalization into an epithelial cell but not a B cell. Virus was bound on ice for 2 h to Akata and Raji B cells, SVKCR2 and AGS epithelial cells, primary tonsil epithelial cells, or SVKCR2 CIITA cells which express HLA class II. Unbound virus was removed, and cells were warmed for the times indicated. Surface-bound virus was removed by digestion with proteinase K, and virus (BamK) and cellular (CRP) copies were measured by QPCR. Error bars are the standard deviations of triplicates.
To determine if the virus remaining in an epithelial cell had reached the nucleus, nuclei of SVKCR2 cells were separated from cell lysates by hypotonic lysis and detergent washes (Fig. 2). The amount of virus DNA in the nucleus at 4 h was slightly less than the total amount in the cell, and the total amount of virus DNA remaining in cells at 8 h was not significantly different from that associated with nuclei at 4 h, suggesting that virus DNA at the nucleus was stable.
Fig 2.

Virus DNA in the nucleus of an SVKCR2 epithelial cell remains stable. (A) Western blot of isolated nuclear and cytoplasmic fractions stained for α-tubulin or lamin B. (B) Virus was bound on ice for 2 h, unbound virus was removed, and cells were warmed for the times indicated. Surface-bound virus was removed by digestion with proteinase K. The total amount of virus DNA remaining per cell was compared with the amount in the fractionated nucleus by QPCR. Error bars are the standard deviations of triplicates.
Leupeptin increases the stability of virus in an epithelial cell.
Two ways in which virus DNA might be lost prior to reaching the nucleus of an epithelial cell were shuttling of the entire particle to a degradative compartment or premature uncoating of virus and exposure of DNA to digestion in the cytoplasm. To distinguish between these possibilities, cells were pretreated for 1 h before and 90 min after warming with 200 μM leupeptin, a serine protease inhibitor which might be expected to block digestion of the entire particle but not to influence premature uncoating. Pretreatment of SVKCR2 cells with leupeptin resulted in twice as much virus DNA remaining at 4 h after infection (Fig. 3). Although leupeptin could block DNA loss, if its effects were simply to block proteases rather than to influence trafficking to the nucleus, then the preservation of DNA would not be expected to translate into an increase in infection as judged by transcription from the incoming genome. To test for this, expression of GFP under the control of the cytomegalovirus immediate-early promoter in a cassette inserted in the nonessential thymidine kinase gene of the virus was measured by RT-QPCR at 4 h postwarming, the earliest time at which RNA transcripts could be detected. Leupeptin treatment failed to increase the level of transcription from the incoming genome suggesting, that its effects were limited to blocking degradation of virus. In contrast, pretreatment with 10 nM wortmannin or 100 μM LY294002, which inhibit class III phosphoinositide 3-kinase (PI3K) and reduce autophagy (25), significantly increased the amount of transcription, consistent with more virus being transported to the nucleus.
Fig 3.

Leupeptin stabilizes virus DNA in an SVKCR2 epithelial cell but has no effect on GFP expression, whereas inhibitors of class III phosphoinositide 3-kinase increase GFP expression. (A) Cells were pretreated with 200 μM leupeptin for 1 h, virus was bound for 2 h on ice, and cells and bound virus were warmed in the continued presence of drug for 4 h. Cells were treated with proteinase K to remove surface-bound virus, and the virus DNA copies remaining per cell were measured by QPCR. (B) Cells were pretreated with 200 μM leupeptin, 100 μM LY294002, or 10 nM wortmannin for 1 h, virus was bound for 2 h on ice, and cells and bound virus were warmed in the continued presence of drug for 4 h. The fold change in GFP transcripts was determined by RT-QPCR. Error bars are the standard deviations of triplicates.
Inhibitors of actin remodeling prevent internalization of virus into a B cell but enhance internalization of virus into an epithelial cell, and reagents that affect microtubules have no effect.
We have previously found that infection of EBV-negative Akata cells is sensitive to the effects of chlorpromazine, whereas infection of SVKCR2 cells is not. This is consistent with entry by endocytosis into a B cell and entry at the cell surface in an epithelial cell. Entry by endocytosis would probably require remodeling of the actin cytoskeleton; fusion at the cell surface might not. We therefore examined the effects of 0.5 μM jasplakinolide, which binds F-actin and stabilizes the actin cytoskeleton, and 5 μM latrunculin A, which binds G-actin and inhibits actin polymerization (13), on internalization of virus into each cell type. Cells were pretreated with drug for 1 h, virus was bound on ice for 2 h, unbound virus was removed by washing in the cold, and cells and virus were warmed for 4 h. Although chilling of cells can affect the cytoskeleton, this step was necessary to maximize the synchronicity of entry, and both treated and untreated cells were incubated in the same way. Following the 4-h warming period, cells were treated with proteinase K to remove virus remaining at the cell surface, and the amount that was internalized and protected from digestion was measured by QPCR. As anticipated, both inhibitors of actin remodeling significantly reduced the amount of virus DNA internalized on a per-cell basis into Akata B cells or LCL (Fig. 4). In contrast, jasplakinolide and latrunculin A increased or had no effect on the amount of virus that was internalized into SVKCR2, AGS, and NOK epithelial cells, despite the facts that disruption of the actin cytoskeleton by latrunculin A could be confirmed by staining actin filaments with phalloidin (Fig. 5) and successful binding of jasplakinolide to F-actin could be inferred because the drug binds competitively with phalloidin and compromises its use for actin visualization (5). Neither drug had any effect on the amount of virus initially bound to cells or on the amount that could be removed by proteinase K treatment (data not shown). As expected, neither 5 μM nocodazole, which destabilizes microtubules, nor 20 μM paclitaxel, which stabilizes microtubules and causes reorganization of the microtubule network (13), had any effect on entry. The effects of both drugs on epithelial cells were confirmed by examination of cells stained with antibody to α-tubulin (Fig. 5). Effects of nocodazole on B cells could be assumed because of its subsequent effect on GFP transcription (see below). The effects of paclitaxel on a B cell were not possible to visualize directly because of the very high nuclear-to-cytoplasmic ratio in these cells.
Fig 4.

Inhibitors of actin remodeling prevent virus internalization into a B cell but enhance internalization into an epithelial cell. Drugs that influence the microtubule network have no effect. (Top panel) Akata B cells, LCL, and SVKCR2, AGS, or NOK epithelial cells were pretreated for 1 h with 5 μM latrunculin A (Lat A) or 0.5 μM jasplakinolide (Jas), virus was bound on ice for 2 h, and cells and bound virus were warmed for 4 h in the presence of drug. Surface-bound virus was removed by proteinase K digestion, and the change in virus DNA copies remaining per cell was measured by QPCR. The horizontal line at 1 indicates the virus DNA copies remaining per cell in the absence of drug. (Bottom panels) Akata B cells or SVKCR2 epithelial cells were pretreated with 10 μM nocodazole (Noco) or 20 μM paclitaxel for 1 h, virus was bound on ice for 2 h, and cells and bound virus were warmed for 4 h in the presence of drug. Surface-bound virus was removed by proteinase K digestion, and the change in virus DNA copies remaining per cell was measured by QPCR. Error bars are the standard deviations of triplicates.
Fig 5.

Effects of drugs on the cytoskeleton of SVKCR2 cells. Cells were treated for 1 h with DMSO (A and D), 20 μM paclitaxel (B), 10 μM nocodazole (C), 5 μM latrunculin A (E), or 0.5 μM jasplakinolide (F) and then were fixed. Cells were then stained with antibody to α-tubulin and anti-mouse antibody coupled to Alexa Fluor 488 (A, B, and C) (green) or with phalloidin coupled to Alexa Fluor 546 (D, E, and F) (red). Nuclei are stained with DAPI (blue). Panels D and E are at a lower magnification.
Both inhibitors of actin remodeling and reagents that disrupt microtubules reduce transcription from the incoming virus genome.
Although nocodazole and paclitaxel had no effect on entry, it was expected, based on work with other herpesviruses (7, 26), that nuclear transport would involve the microtubule network. To examine this further, Akata B cells and SVKCR2 epithelial cells were pretreated for 1 h with 20 μM paclitaxel, 2 μM colchicine, 10 μM vinblastine, or 5 μM nocodazole, virus was bound on ice for 2 h, unbound virus was removed by washing in the cold, and cells and virus were warmed in the presence of drug for 4 h. Cells were harvested, and transcription of GFP was measured by RT-QPCR. All of the agents that disrupt microtubules inhibited GFP expression, and paclitaxel, which stabilizes microtubules, did not (Fig. 6). Transcription was slightly increased in SVKCR2 cells by paclitaxel, but the increase was only marginally significant (P = 0.038). None of the drugs used throughout this work affected the actin transcription rate, so effects on GFP could not be attributed to toxicity.
Fig 6.

Microtubule-targeted inhibitors reduce GFP expression from the incoming genome. Cells were pretreated for 1 h with 20 μM paclitaxel, 20 μM colchicine, 10 μM vinblastine, 5 μM nocodazole, or DMSO control, virus was bound on ice for 2 h, and cells and bound virus were warmed for 4 h in the presence of drug. The change in expression of GFP relative to that from the control was measured by RT-QPCR. Error bars are the standard deviations of triplicates.
A similar experiment was conducted with the inhibitors of actin remodeling. Somewhat unexpectedly, pretreatment for 1 h and maintenance of cells in 0.5 μM jasplakinolide or 5 μM latrunculin for the 4 h prior to analysis almost completely abrogated transcription of GFP in Akata B cells, tonsil B cells, and 293 and SVKCR2 epithelial cells (Fig. 7). An effect had been expected in B cells because of the effects of both drugs on actin remodeling and endocytosis of virus. An effect on epithelial cells, where uptake in the presence of drugs was increased rather than decreased, had not. Drugs were then added at different times after warming of cells and virus to determine the length of time during which transcription, as a surrogate for arrival in the nucleus, could be affected. By 5 min after warming, the effect of jasplakinolide on transcription in a B cell was already lost. In an epithelial cell its effects were still apparent if added after 5 min and only gradually diminished over time to disappear by 2 h. The effects of latruculin A were slightly different. In an epithelial cell a slight reduction was seen if drug was added after 5 min, but the effect was lost by 30 min postwarming. The effects in Akata B cells were still quite marked at 5 min but disappeared by 30 min, the approximate time at which fusion is complete in both cell types (31). Thus, although a requirement for actin remodeling for transport in an epithelial cell could be definitively concluded since drugs enhanced rather than inhibited virus uptake, it appeared that in a B cell the actin cytoskeleton might not be important for trafficking once the virus has escaped the endosome.
Fig 7.
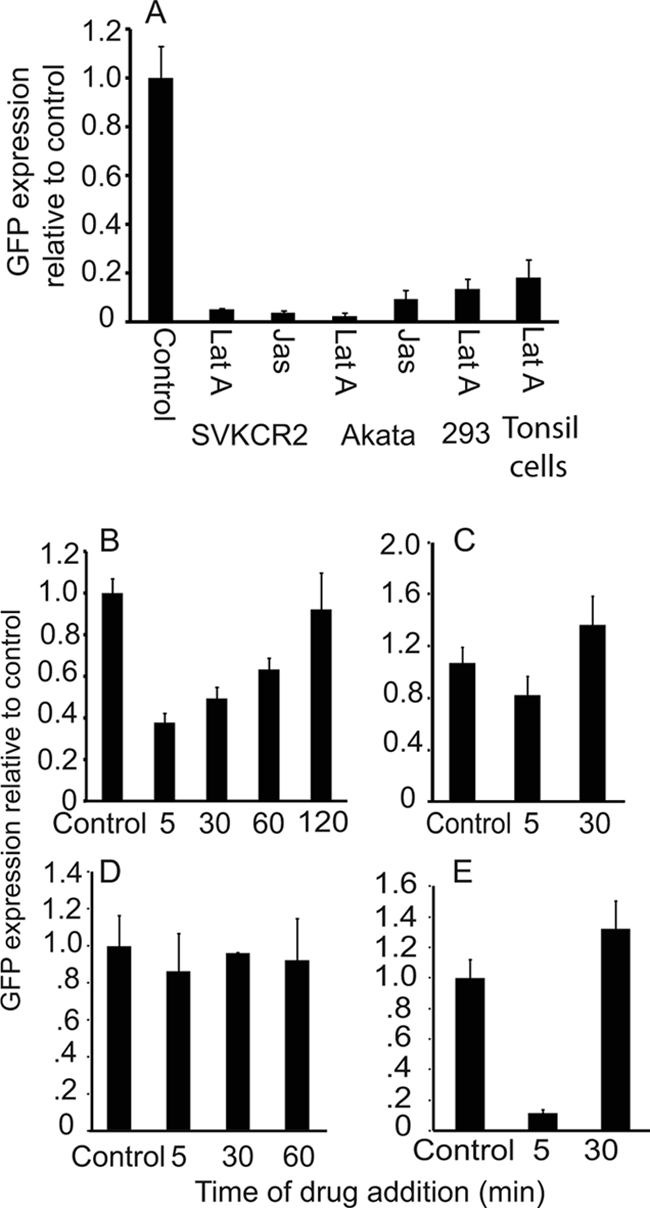
Inhibitors of actin dynamics reduce GFP expression from the incoming genome. (A) SVKCR2 epithelial cells, Akata B cells, 293 epithelial cells, or tonsil lymphocytes were preincubated for 1 h with 5 μM latrunculin A (Lat A) or 0.5 μM jasplakinolide (Jas) or DMSO (control), virus was bound to cells on ice for 2 h, and cells and bound virus were warmed for 4 h in the presence of drug. The change in expression of GFP relative to that from the control was measured by RT-QPCR. (B to E) Virus was bound on ice to SVKCR2 cells (B and C) or Akata B cells (D and E) for 2 h, and cells and bound virus were warmed for the times indicated, at which point 0.5 μM jasplakinolide (B and D) or 5 μM latrunculin A (C and E) was added. After a total of 4 h of warming, the change in expression of GFP relative to that from the control was measured by RT-QPCR. Error bars are the standard deviations of triplicates.
The cytoplasmic tail of CR2 is not required for efficient infection of SVKCR2 cells, but activities associated with integrin signaling are required.
The amount of virus that can bind to CR2-negative AGS gastric carcinoma cells is approximately 5-fold less than the amount that can bind to SVKCR2 cells, but transport of virus DNA to the nucleus and expression of GFP occur in almost 100-fold fewer cells (4). The observation that actin remodeling is essential to intracellular transport in an epithelial cell raised the possibility that CR2, which if cross-linked by EBV on an epithelial cell interacts via its cytoplasmic domain with the actin nucleator formin FHOS/FHOD, (15) influences infection as a result of effects on actin dynamics. To test this possibility, SVK cells, the parental line of SVKCR2 cells, which lack CR2 expression, were transiently transfected either with a plasmid capable of expressing full-length CR2, pCAGGS-CR2, or a plasmid expressing CR2 without its 34-residue cytoplasmic domain, pCAGGS-CR2-CT. The expression levels of CR2 and CR2-CT were similar at 24 h (Fig. 8), at which time cells were bound to virus for 2 h on ice, warmed to 37°C, and incubated for 48 h before analysis of GFP expression by flow cytometry. Rates of infection of cells were similar irrespective of the presence or absence of the cytoplasmic domain of CR2.
Fig 8.
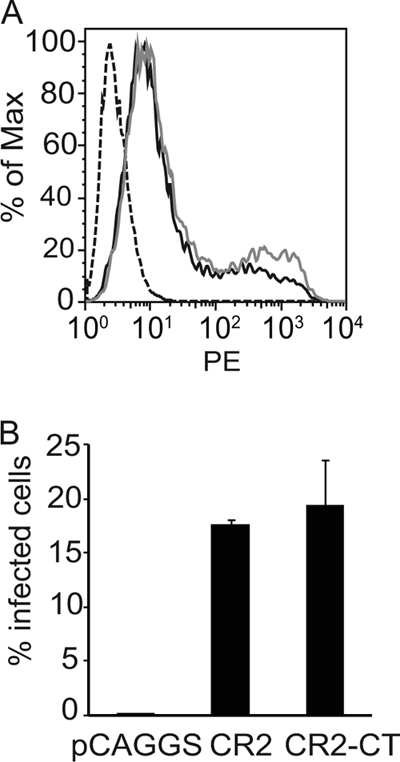
The cytoplasmic tail of CR2 has no impact on infection of epithelial cells. (A) Flow cytometric analysis of CR2 expression on SVK cells (dashed line) or SVK cells transfected with pCAGGS-CR2 (black line) or pCAGGS-CR2-CT (gray line). (B) Percentage of SVK cells transfected with pCAGGS, pCAGGS-CR2, or pCAGGS-CR2-CT that expressed GFP after infection with EBV-GFP. Infection was measured by flow cytometric analysis. Error bars are the standard deviations of triplicates.
The envelope of EBV contains two glycoproteins that include integrin binding motifs, the multispan membrane protein BMRF2 (51) and glycoprotein gH (8). Integrin signaling is also relevant to actin nucleation and actin dynamics, and players linking integrins to these events include RhoA, ROCK, p38/mitogen-activated protein kinase (MAPK), and Src. To examine the potential role of these signaling molecules, SVKCR2 or Akata cells were treated for 1 h with PP2, an Src inhibitor, Rho CTO4, which inhibits Rho, H1152, which inhibits ROCK, and SB203580, which is an inhibitor of p38/MAPK. Virus was bound to treated cells for 2 h on ice, and cells were subsequently warmed for 4 h in the continued presence of drug before measurement of GFP transcription. Inhibitors of RhoA, p38/MAPK, and Src reduced GFP transcription in both cells types. However, the inhibitor of ROCK uniquely affected SVKCR2 cells (Fig. 9).
Fig 9.
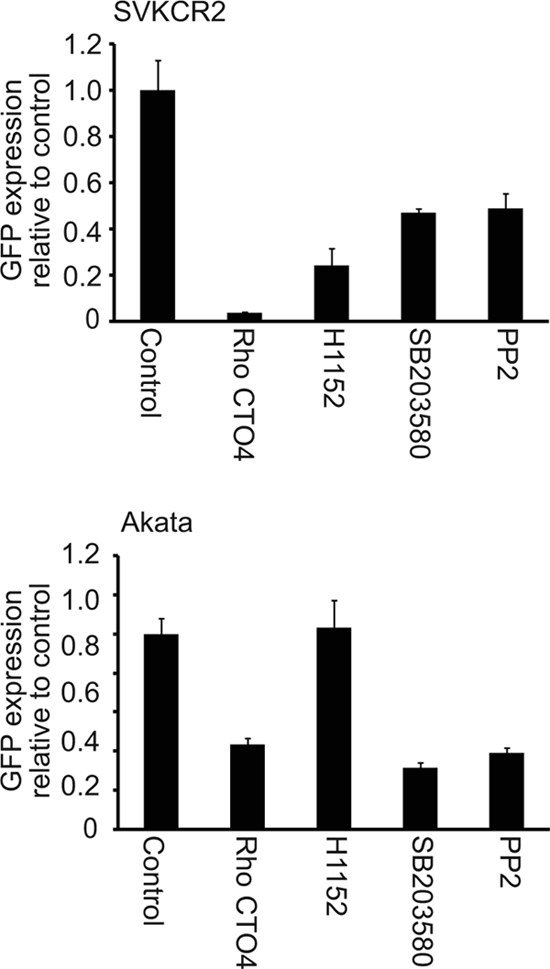
Inhibitors of Src, ROCK, Rho, and p38/MAPK have differential effects on GFP expression in Akata B cells and SVKCR2 epithelial cells. Cells were pretreated with 0.5 μg/ml Rho CTO4, 10 μM H1152, 20 μM SB203580, 20 μM PP2, or DMSO control for 1 h, virus was bound to cells for 2 h on ice, and cells and bound virus were warmed for 4 h in the presence of drug. The change in expression of GFP relative to that from the control was measured by RT-QPCR. Error bars are the standard deviations of triplicates.
DISCUSSION
Over the last several decades, the major players required for EBV attachment and fusion with B cells and epithelial cells have been identified. Less is known about the cell biology of events occurring concurrently with and prior to arrival of the virus at the nucleus. A study of virus entry into primary B cells and epithelial cells using fluorescent in situ hybridization (FISH) analysis of virus genomes in the nucleus at 24 h postinfection found that far fewer copies were visible in the nuclei of epithelial cells than in those of B cells (40). We confirmed this finding with our own FISH analysis (data not shown) and thus sought to determine the fate of virus between attachment to a cell and arrival at the nucleus. It seems clear that although, with approximately 50 particles bound to a cell, enough virus was transported to the nucleus in both cell types to produce similar numbers of infected cells, less than half of the virus that entered an epithelial cell was successfully transported to the nucleus. The remainder was fairly rapidly degraded. This loss of virus occurred whether the epithelial cells expressed or lacked CR2, expressed or lacked HLA class II, or were established cell lines or primary cultures. The identity of the compartment in which virus was degraded was not precisely identified. However, all available evidence to this point suggests that virus enters an epithelial cell by fusion at the plasma membrane (31) and direct delivery to the cytosol. Thus, delivery to a lysosome by autophagy (23) would seem more likely than delivery via an endosomal pathway. The rescue of infection with inhibitors of class III PI3Ks is at least consistent with this (25).
The differential effects of drugs that modulate actin dynamics on the initial internalization of virus into a B cell and an epithelial cell were not unexpected, given that the routes of entry into the two cell types are clearly different. The effects of chlorpromazine on infection of B cells, where fusion takes place in a low-pH compartment (4, 31), have implicated involvement of clathrin and are consistent with early studies of B cell entry by electron microscopy (33) and observations that capping of CR2 on a B cell by EBV induces endocytosis (45). Thus, the reduction in the amount of virus entering a B cell in which actin remodeling has been inhibited presumably reflects a loss of endocytic function. Actin is frequently, if not universally, required for vesicle formation (14, 30). As previously mentioned, the observation that virus fusion with an epithelial cell occurs at neutral pH is consistent with entry by direct fusion at the cell surface, and even on SVKCR2 cells there is no evidence that endocytosis occurs (4). The ability of jasplakinolide and latruculin A to enhance or have no effect on virus uptake in this cell type, then, might be interpreted as the absence of a requirement for actin involvement and perhaps even a reduction in the barrier presented by cortical actin (28).
The reductive effects of the microtubule disrupters nocodazole, vinblastine, and colchicine on intracellular transport in both B cells and epithelial cells were also not unexpected, given the well-established role for intact microtubules in trafficking of many herpesviruses (26). The lack of an effect of the microtubule stabilizer paclitaxel on B cell infection and the slight increase in infection that it effected in epithelial cells, however, suggest that, as for herpes simplex virus (27), movement is not dependent on microtubule tread milling. The effects of the inhibitors of actin remodeling latrunculin A and jasplakinolide were more surprising. The inhibitory effects of both drugs on successful intranuclear transport in a B cell could be explained at least in part by their effects on initial virus internalization. Experiments designed to determine at what time transport no longer was dependent on actin dynamics were generally consistent with an effect prior to, but not after, completion of fusion and delivery of the virus into the cytoplasm. It is not exactly clear how rapidly the drugs block actin remodeling, but the response to latruculin A is reported to occur in 5 min or less (44). Since disruption of actin dynamics failed to inhibit internalization into an epithelial cell, it seems reasonable to conclude that there is a need for the actin cytoskeleton for intracellular trafficking. The differential effects in the two cell types of the actin stabilizer jasplakinolide, which induces actin polymerization and binds F-actin, and of the actin disruptor latrunculin A, which inhibits polymerization by binding G-actin, were curious but might be explained by the relative pools of G-actin and polymerization-competent G-actin in different cell types (6). Differential effects of the two drugs have been observed in different cell types and even in the same cell type grown adherently or in suspension (14).
The presence of CR2 on an epithelial cell increases infection disproportionally to its effects on binding (4). Binding of virus cross-links CR2 and results in colocalization with the formin FHOS/FHOD1, which binds to the cytoplasmic domain of CR2 (15). Formins directly nucleate actin (17), and since actin dynamics seemed to be uniquely important to virus transport in epithelial cells, the possibility that CR2 might provide a link to cytoskeletal reorganization was very attractive. Deletion of the cytoplasmic tail of CR2, however, clearly indicates that this link is not the relevant factor. It remains to be determined whether or not there are interactions, perhaps involving the transmembrane domain of CR2, that are important to downstream events. It does, however, seem likely that integrin signaling might be an important component in efficient infection. On a B cell, CR2, cross-linked by EBV, can initiate signaling through Src (2) and in concert with CD19 may activate p38 and Vav, a guanine nucleotide exchange factor for the Rho family of GTPases (11). Inhibitors of Src, Rho, and p38/MAPK all influenced B cell infection. However, on an epithelial cell inhibitors not only of Src, Rho, and p38/MAPK but also of ROCK, all potential downstream players in integrin activation that can influence actomyosin contractility (9, 10, 35), reduced virus transport to the nucleus. Of the two known integrin binding proteins in the envelope of EBV, one, gH, is essential to virus fusion, and thus the impact that its interaction with an integrin has on subsequent intracellular transport is not possible to assess directly. However, a recombinant virus lacking the entire gHgLgp42 complex is available (32), as is a soluble form of gHgL (8), and these reagents may allow analysis of downstream signaling events. The other integrin binding protein, BMRF2, has not been described as essential to infection (50), and a recombinant virus in which the RGD motif in BMRF2 has been mutated is being constructed to determine any possible contributions that it may make.
Microtubules are generally described as being responsible for long-range transport within a cell, whereas short-range movement is thought to occur on actin filaments. However, there is clearly functional cooperation between microtubules and the actin cytoskeleton (16). In an epithelial cell EBV appears to require both transport systems for successful and productive delivery to the nucleus.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants DE016669 (to L.M.H.-F.) and DE019599 (to S.M.V.) from the National Institute of Dental and Craniofacial Research.
We thank Karl Munger for the gift of the NOK cells.
Footnotes
Published ahead of print 26 October 2011
REFERENCES
- 1. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a timeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barel M, Balbo M, Le Romancer M, Frade R. 2003. Activation of Epstein-Barr virus/C3d receptor (gp140, CR2, CD21) on human cell surface triggers pp60src and Akt-GSK3 activities upstream and downstream to PI 3-kinase, respectively. Eur. J. Immunol. 33:2557–2566 [DOI] [PubMed] [Google Scholar]
- 3. Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599 [DOI] [PubMed] [Google Scholar]
- 4. Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. 2004. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 78:5007–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. 1994. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 269:14869–14871 [PubMed] [Google Scholar]
- 6. Bubb MR, Spector I, Beyer BB, Fosen KM. 2000. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 275:5163–5170 [DOI] [PubMed] [Google Scholar]
- 7. Chandran B. 2010. Early events in Kaposi's sarcoma associated herpesvirus infection of target cells. J. Virol. 84:2188–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chesnokova LS, Nishimura S, Hutt-Fletcher L. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral proteins gHgL to integrins αvβ6 or αvβ8. Proc. Natl. Acad. Sci. U. S. A. 106:20464–20469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark EA, Brugge JS. 1995. Integrins and signal transduction pathways: the road taken. Science 268:233–239 [DOI] [PubMed] [Google Scholar]
- 10. Defilippi P, et al. 1999. Actin cytoskeleton organizaton in response to integrin-mediated adhesion. Micros. Res. Tech. 47:67–78 [DOI] [PubMed] [Google Scholar]
- 11. Fearon DT, Carroll MC. 2000. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu. Rev. Immunol. 18:393–422 [DOI] [PubMed] [Google Scholar]
- 12. Feederle R, et al. 2007. Epstein-Barr virus B95. 8 produced in 293 cells shows marked tropism for differentiated primary epithelial cells and reveals interindividual variation in susceptibility to viral infection. Int. J. Cancer 121:588–594 [DOI] [PubMed] [Google Scholar]
- 13. Fenteany G, Zhu S. 2003. Small-molecule inhibitors of actin dynamics and cell motility. Curr. Top. Med. Chem. 3:593–616 [DOI] [PubMed] [Google Scholar]
- 14. Fujimoto LM, Roth R, Heuser JE, Schmid SL. 2000. Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic 1:161–171 [DOI] [PubMed] [Google Scholar]
- 15. Gill MB, et al. 2004. EBV attachment stimulates FHOS/FHOD1 redistribution and co-aggregation with CD21:formin interactions with the cytoplasmic domain of human CD21. J. Cell Sci. 117:2709–2720 [DOI] [PubMed] [Google Scholar]
- 16. Goode BL, Drubin DG, Barnes G. 2000. Functional cooperation between the microtubule and actin cytoskeletons. Curr. Opin. Cell Biol. 12:63–71 [DOI] [PubMed] [Google Scholar]
- 17. Goode BL, Eck MJ. 2007. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 76:593–627 [DOI] [PubMed] [Google Scholar]
- 18. Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. 2009. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 7:e10000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hutt-Fletcher LM. 2007. Epstein-Barr virus entry. J. Virol. 81:7825–7832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang H-W, Wentzel EA, Mendell JT. 2007. A hexanucleotide element directs microRNA nuclear import. Science 315:97–100 [DOI] [PubMed] [Google Scholar]
- 21. Jiang R, Gu X, Nathan C, Hutt-Fletcher L. 2008. Laser-capture microdissection of oropharyngeal epithelium indicates restriction of Epstein-Barr virus receptor/CD21 mRNA to tonsil epithelial cells. J. Oral Pathol. Med. 37:626–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang R, Scott RS, Hutt-Fletcher LM. 2006. Epstein-Barr virus shed in saliva is high in B cell tropic gp42. J. Virol. 80:7281–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klionsky DJ, Emr SD. 2000. Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li QX, et al. 1992. Epstein-Barr virus infection and replication in a human epithelial system. Nature 356:347–350 [DOI] [PubMed] [Google Scholar]
- 25. Lindmo K, Stenmark H. 2006. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 119:605–614 [DOI] [PubMed] [Google Scholar]
- 26. Lyman MG, Enquist LW. 2009. Herpesvirus interactions with the host cytoskeleton. J. Virol. 83:2058–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mabit H, et al. 2002. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 76:9962–9971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marsh M, Bron R. 1997. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 110:95–103 [DOI] [PubMed] [Google Scholar]
- 29. Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mayor S, Pagano RE. 2007. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell. Biol. 8:603–612 [DOI] [PubMed] [Google Scholar]
- 31. Miller N, Hutt-Fletcher LM. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cell but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324–6332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nemerow GR, Cooper NR. 1984. Early events in the infection of human B lymphocytes by Epstein-Barr virus. Virology 132:186–198 [DOI] [PubMed] [Google Scholar]
- 34. Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199 [DOI] [PubMed] [Google Scholar]
- 35. Pellegrin S, Mellor H. 2007. Actin stress fibers. J. Cell Sci. 120:3491–3499 [DOI] [PubMed] [Google Scholar]
- 36. Piboonniyom S, Timmermann S, Hinds P, Munger K. 2002. Aberrations in the MTS1 tumor suppressor locus in oral squamous carcinoma lines preferentially affect the INK4A gene and result in increased cdk6 activity. Oral Oncol. 38:179–186 [DOI] [PubMed] [Google Scholar]
- 37. Pulvertaft RJV. 1964. Cytology of Burkitt's tumor (African lymphoma). Lancet i:238–240 [DOI] [PubMed] [Google Scholar]
- 38. Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700. In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 39. Seigneurin J-M, Villaume M, Lenoir G, deThe G. 1977. Replication of Epstein-Barr virus: ultrastructural and immunofluorescent studies of P3HR1-superinfected Raji cells. J. Virol. 24:835–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shannon-Lowe C, et al. 2009. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J. Virol. 83:7749–7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shannon-Lowe CD, Neuhierl B, Baldwin G, Rickinson AB, Delecluse H-J. 2006. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 103:7065–7070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shimizu N, Tanabe-Tichikura A, Kuroiwa Y, Takada K. 1994. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J. Virol. 68:6069–6073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spear PG, Longnecker R. 2003. Herpesvirus entry: an update. J. Virol. 77:10179–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spector I, Braet F, Shochet NR, Bubb MR. 1999. New anti-actin drugs in the study of the organization and function of the actin cytoskeleton. Microsc. Res. Technol. 47:18–37 [DOI] [PubMed] [Google Scholar]
- 45. Tanner J, Weis J, Fearon D, Whang Y, Kieff E. 1987. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping and endocytosis. Cell 50:203–213 [DOI] [PubMed] [Google Scholar]
- 46. Tedder T, Goldmacher FVS, Lambert JM, Schlossman SF. 1986. Epstein-Barr virus binding induces internalization of the C3d receptor: a novel immunotoxin delivery system. J. Immunol. 137:1387–1391 [PubMed] [Google Scholar]
- 47. Thorley-Lawson DA, Gross A. 2004. Persistence of Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 350:1328–1337 [DOI] [PubMed] [Google Scholar]
- 48. Turk SM, Jiang R, Chesnokova LS, Hutt-Fletcher LM. 2006. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol. 80:9628–9633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weis JJ, Tedder TF, Fearon DT. 1984. Identification of a 145,000 Mr membrane protein as the C3d receptor (CR2) of human B lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 81:881–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiao J, Palefsky JM, Herrera R, Berline J, Tugizov SM. 2008. The Epstein-Barr virus BMRF-2 protein facilitates attachment to oral epithelial cells. Virology 370:430–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xiao J, Palefsky JM, Herrera R, Tugizov SM. 2007. Characterization of the Epstein-Barr virus glycoprotein BMRF2. Virology 359:382–396 [DOI] [PubMed] [Google Scholar]
- 52. Young DF, Carlos TS, Hagmaier K, Fan L, Randall RE. 2007. AGS and other tissue culture cells can unknowingly be persistently infected with PIV5: a virus that blocks interferon signalling by degrading STAT1. Virology 365:238–240 [DOI] [PubMed] [Google Scholar]