

Abstract
Approximately 10% of gastric carcinomas (GC) are comprised of cells latently infected with Epstein-Barr virus (EBV); however, the mechanism by which EBV contributes to the development of this malignancy is unclear. We have investigated the cellular effects of the only EBV nuclear protein expressed in GC, EBNA1, focusing on promyelocytic leukemia (PML) nuclear bodies (NBs), which play important roles in apoptosis, p53 activation, and tumor suppression. AGS GC cells infected with EBV were found to contain fewer PML NBs and less PML protein than the parental EBV-negative AGS cells, and these levels were restored by silencing EBNA1. Conversely, EBNA1 expression was sufficient to induce the loss of PML NBs and proteins in AGS cells. Consistent with PML functions, EBNA1 expression decreased p53 activation and apoptosis in response to DNA damage and resulted in increased cell survival. In addition, EBNA1 mutants unable to bind CK2 kinase or ubiquitin-specific protease 7 had decreased ability to induce PML loss and to interfere with p53 activation. PML levels in EBV-positive and EBV-negative GC biopsy specimens were then compared by immunohistochemistry. Consistent with the results in the AGS cells, EBV-positive tumors had significantly lower PML levels than EBV-negative tumors. The results indicate that EBV infection of GC cells leads to loss of PML NBs through the action of EBNA1, resulting in impaired responses to DNA damage and promotion of cell survival. Therefore, PML disruption by EBNA1 is one mechanism by which EBV may contribute to the development of gastric cancer.
INTRODUCTION
Epstein-Barr virus (EBV) is a gammaherpesvirus whose latent infection is reported to be associated with approximately 10% of all gastric carcinomas (GC) (10, 35), although a recent study suggested that this percentage might be much higher (25). GC is the fourth most common cancer worldwide and is the second most common cause of death from cancer. GC presents itself without regional or racial differences and is a major clinical problem worldwide (2, 9). There are several lines of evidence suggesting a causative role for EBV in the onset of virus-associated GC. First, EBV is present in every tumor cell but absent in the surrounding normal epithelium (31). Second, the presence of clonal EBV in every tumor cell suggests that EBV infection preceded the final transforming event giving rise to the tumor (17). Third, EBV-associated GC has unique morphological features that distinguishes it from EBV-negative tumors, again suggesting a role for EBV in the pathogenesis of this tumor (38).
In EBV-associated GC, EBV establishes type I latency expressing the EBNA1, LMP2A, and secreted BARF1 proteins but does not express the LMP1 oncoprotein (9, 17). The lack of LMP1 expression suggests that other viral proteins likely play important roles in the development of this tumor. Although LMP2A is known to alter host DNA through hypermethylation of tumor suppressor genes (9, 10), the molecular events that lead to tumor formation still remain unclear. EBNA1 is the only viral nuclear protein expressed in GC. It is also the only viral protein required to maintain latency, due to its roles in the replication and segregation of the EBV episomes (8). However, the role of EBNA1 is not limited to EBV genome maintenance, as there is increasing evidence that EBNA1 alters the cellular environment in ways that promote genomic instability, thereby leading to tumorigenesis (11, 19, 45). For example, EBNA1 can lower p53 levels by sequestering the ubiquitin-specific protease USP7 (26). In addition, several EBV-positive cell lines have been shown to become dependent on EBNA1 expression, such that silencing of EBNA1 leads to decreased proliferation or apoptosis (16, 18, 47).
We recently demonstrated that EBNA1 disrupts PML (promyelocytic leukemia) nuclear bodies (NBs) in the context of nasopharyngeal carcinoma (NPC) cells, by inducing the degradation of the PML proteins that form the structural basis for these NBs (33). PML NBs (also called ND10s) are important for several processes associated with tumor suppression, including p53 activation, apoptosis, and DNA repair (1, 4, 7, 24, 36, 48). The PML NBs also play a role in suppressing lytic viral replication; consequently, many viruses encode proteins targeting the disruption of PML NBs, in some case by inducing the degradation of PML proteins (6). Accordingly, pml knockout mice demonstrate increased susceptibility to both malignant transformation and viral infections (37, 43). In addition, loss of PML NBs has been associated with the development of several types of human malignancies, but to date this has not been examined in gastric carcinoma (13). While our studies with NPC cells suggest that disruption of PML NBs is a mechanism by which EBNA1 may contribute to this tumor, EBNA1 was not found to alter PML NBs in HEK293 or HeLa cells (our unpublished data). Therefore, it was unclear whether this was an NPC-specific phenomenon. In addition, since most NPC tumors are EBV positive, we could not assess whether the presence of EBV and EBNA1 alters PML NBs in the context of NPC tumors.
In this study, we investigated the effect of EBNA1 on PML NBs in gastric carcinoma cell lines and demonstrated that EBNA1 dramatically affected the number and function of PML NBs by inducing degradation of PML proteins. As a result, EBNA1 expression inhibited p53 activation and apoptosis, leading to cell survival. Importantly, the availability of both EBV-positive and EBV-negative GC tumor biopsy specimens enabled a comparison of PML NBs in the primary tumors, demonstrating for the first time that EBV-positive GC tumors have reduced PML staining compared to EBV-negative samples.
MATERIALS AND METHODS
Cell lines.
The human adenocarcinoma cell line AGS has been described previously (39). AGS-EBNA1 cells stably expressing EBNA1 were generated as described by Valentine et al. (39). AGS-EBV was generated by coculturing AGS cells with Akata Burkitt's lymphoma cells carrying recombinant neomycin-resistant EBV as described previously (34). All three cell lines were cultured in RPMI 1640 (Sigma) supplemented with 10% fetal calf serum and 1% l-glutamine. The AGS-EBNA1 and AGS-EBV cells were maintained in G418 (Invitrogen; 400 μg/ml) to select for cells containing the EBNA1 expression cassette and EBV, respectively.
Transfections.
For transient EBNA1 expression, 5 × 105 AGS cells were transfected with 2 μg of pc3OriPE (46), pc3oriPEΔ395-450 (15), pc3oriPEΔ387-394 (32), or the negative-control plasmid lacking EBNA1 (pc3Orip) (46) using Lipofectamine 2000 (Invitrogen). Cells were fixed 24 h later for immunofluorescence microscopy or harvested for Western blotting as described below. AGS, AGS-EBNA1, and AGS-EBV cells were transfected with small interfering RNA (siRNA) against green fluorescent protein (GFP) or EBNA1 as described previously (33) or with AllStars negative-control siRNA (Qiagen) and analyzed by immunofluorescence microscopy (IF) and Western blotting.
Immunofluorescence microscopy.
Cells grown on coverslips were processed for imaging as described previously (33). Samples were incubated with primary antibodies against EBNA1 (R4 rabbit serum at 1:300) (15), PML (Santa Cruz PG-M3 at 1:50), p21 (Santa Cruz sc-817 at 1:50), and acetyl-p53K382 (Cell Signaling 2525S at 1:200) followed by secondary antibodies goat anti-rabbit Alexa Fluor 555 (Molecular Probes) and goat anti-mouse Alexa Fluor 488 (Molecular Probes) in 4% bovine serum albumin (BSA). Coverslips were mounted onto slides using ProLong Gold antifade medium containing DAPI (4′,6′-diamidino-2-phenylindole) (Invitrogen). Images were obtained using the 40× oil objective on a Leica inverted fluorescence microscope and processed using OpenLAB (version X.0) software. PML nuclear bodies were quantified by counting all visible PML foci in 50 to 100 cells. For detection of apoptotic bodies by microscopy, cells were treated with 10 μg/ml etoposide for 48 h and then were harvested and placed onto coverslips. Coverslips were dried in a fume hood and then mounted on slides as described above. Apoptotic bodies were quantified by counting >100 cells for each sample using multiple planes of view.
Western blots.
Cells were lysed in 9 M urea–10 mM Tris-HCl (pH 6.8) and briefly sonicated. Fifty micrograms of total protein was subjected to SDS-PAGE and transferred onto nitrocellulose. Membranes were blocked in 5% nonfat dry milk in phosphate-buffered saline (PBS) and then incubated with antibodies against PML (Bethyl A301-167A at 1:2,000), EBNA1 (OT1X at 1:2,000 or K67-3 at 1:1,000), actin (Oncogene Research Products Ab-1 at 1:10,000), p53 (Santa Cruz sc-126 at 1:2,000), acetyl-p53 K382 (Cell Signaling, 2525S at 1:2,000), p21 (Santa Cruz sc-817 at 1:500), or caspase-3 (Cell Signaling 9665 at 1:2,000). After washing, membranes were probed with goat anti-mouse peroxidase (1:3,000) or goat anti-rabbit peroxidase (1:5,000) (Santa Cruz) and then developed using chemiluminescence reagents (ECL; Perkin-Elmer). Membranes were stripped in 0.1 M glycine (pH 2.9) for 30 min, washed in PBS-Tween, blocked, and reprobed with the next antibody as described above. Where etoposide was used, cells were treated with 50 μg/ml etoposide for 5 h (see Fig. 5A and 6) or 24 h (see Fig. 5D) prior to harvesting. For acetyl-p53 and caspase-3 blots, membranes were blocked in 5% nonfat milk for 1 h, rinsed briefly with PBS-Tween, and incubated with primary antibody in 4% BSA in PBS-Tween overnight at room temperature.
Fig 5.
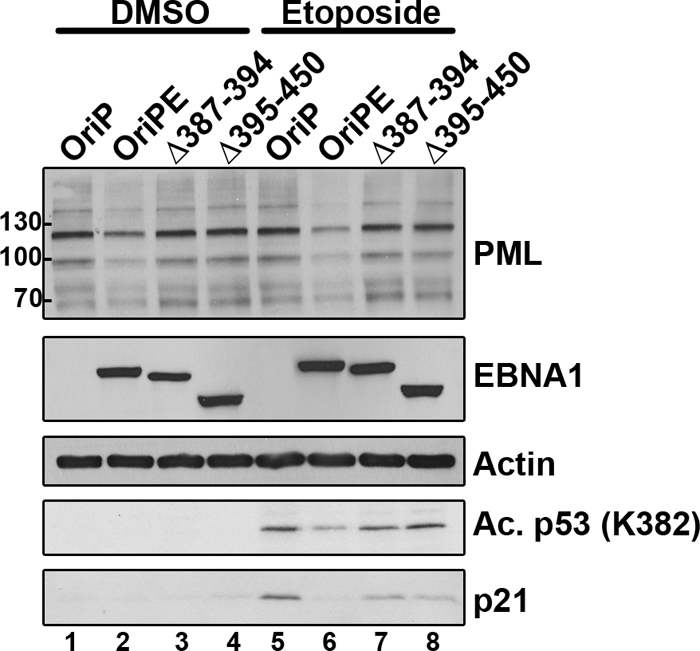
Effect of EBNA1 mutations on PML and p53 activation. AGS cells were transfected with a plasmid expressing EBNA1 (OriPE) or EBNA1 mutant Δ387-394 or Δ395-450 or with the empty plasmid (OriP) and then treated with etoposide or DMSO as for Fig. 4A. Equal amounts of cell lysates were then Western blotted for PML, EBNA1, actin, p53 acetylated on K382, and p21.
Fig 6.
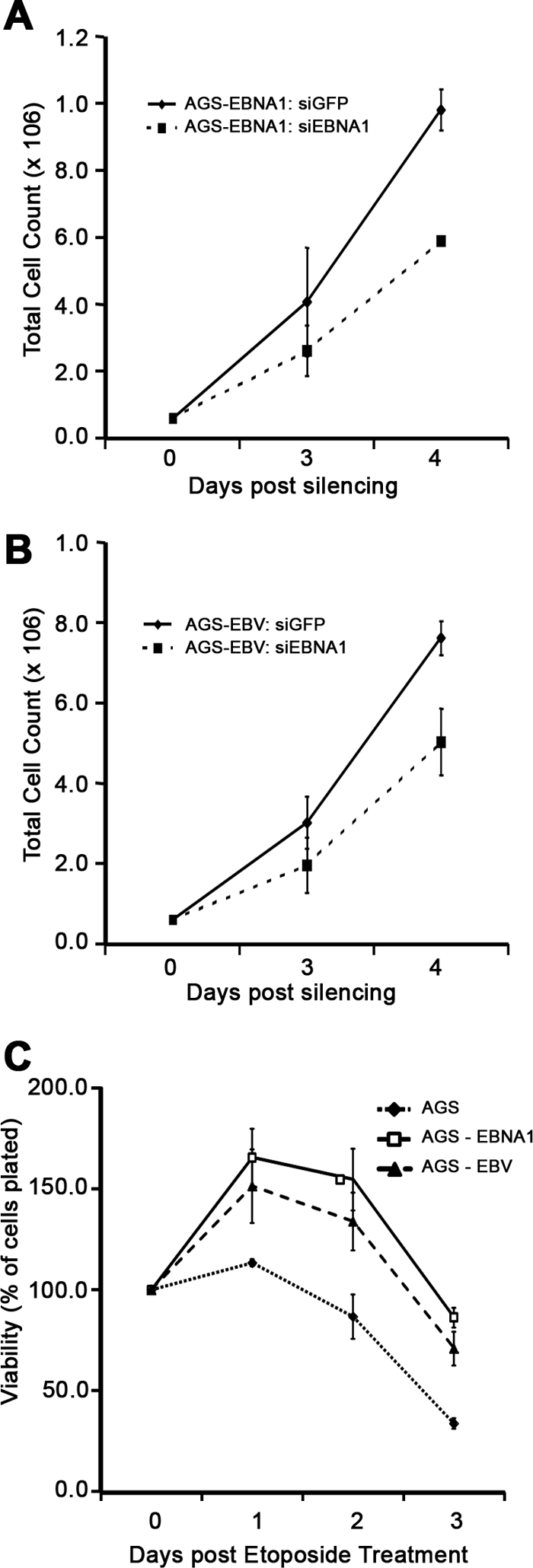
Effects of EBNA1 on cell proliferation and viability. (A and B) AGS-EBNA1 (A) and AGS-EBV (B) were treated with siRNA targeted against GFP or EBNA1. Cells were harvested immediately (day 0) or at the indicated days after silencing and counted. (C) AGS, AGS-EBNA1, and AGS-EBV cells were treated with etoposide, and cells were harvested and counted at 0, 1, 2, and 3 days posttreatment. For all experiments, average numbers with standard deviations from three separate experiments are plotted.
Cell proliferation and viability assays.
Proliferation of AGS-EBNA1 and AGS-EBV cells was monitored after transfection of 6 × 105 cells in 10-cm dishes with siRNA against GFP or EBNA1 as described previously (33). Cells were harvested and counted at 3 and 4 days after siRNA treatment. Cell viability after DNA damage was measured by plating 2 × 105 cells in 12-well plates, followed 24 h later by treatment with 50 μg/ml of etoposide for 5 h. Cells were then rinsed in PBS and grown in fresh medium for 24, 48, and 72 h prior to harvesting of floating and adherent cells. Harvested cells were stained with 0.04% trypan blue in PBS to identify dead cells, and viable (unstained) cells were counted immediately. Experiments were performed in triplicate.
Immunohistochemistry.
GC tumor biopsy specimens were obtained from the archives of the VU University Medical Center, Amsterdam, and have been described in earlier studies (40, 41). Sections of these tumors were analyzed for the presence of EBV-encoded RNAs (EBERs) by EBER in situ hybridization (EBERish) as described previously (42). Sections of the same tumors were also processed for immunohistochemical detection of PML as described previously (30). Blocking was carried out using a Dako LSAD kit (Dako catalog no. K0679) as per the manufacturer's instructions, and then samples were incubated in a diluent containing PML antibody (Bethyl A301-167A) at 1:1,000 in a humidified chamber at 4°C overnight. Slides were then rinsed and further processed according to the Dako kit. After counterstaining in hematoxylin, slides were dried, mounted using Permamount, and analyzed using the 40× objective on a LEICA DMLB microscope.
RESULTS
EBNA1 disrupts PML NBs in gastric carcinoma cells.
To determine if EBNA1 affected PML NBs in GC, we initially compared the parental gastric adenocarcinoma cell line AGS to AGS cells containing an integrated EBNA1 expression cassette (AGS-EBNA1) or AGS cells carrying a recombinant EBV (AGS-EBV). Staining for EBNA1 in both AGS-EBNA1 and AGS-EBV cells showed diffuse nuclear localization with some subnuclear foci, as reported previously for EBNA1 expression in NPC cells (33) (Fig. 1A). Comparison of the PML NBs by immunofluorescence microscopy (IF) revealed that AGS-EBV cells had significantly fewer PML NBs (on average 3 NBs per cell) than the parental AGS cells (9 NBs per cell) (Fig. 1A), suggesting that the presence of EBV in these cells led to the loss of PML NBs. Comparison of AGS-EBNA1 to AGS cells showed that EBNA1 alone was sufficient to cause this loss of PML NBs in GC cells, as AGS-EBNA1 cells had only two PML NBs per cell on average (Fig. 1A).
Fig 1.

PML NBs in GC cells are reduced by EBV and EBNA1. Log-phase cells were fixed and stained with anti-EBNA1 (red) and anti-PML (green) antibodies. PML foci were counted for 50 to 100 cells for each sample in three separate experiments, and the average number with standard deviation is shown in the histograms. Images with the same antibody treatment were captured using the same exposure times. (A) AGS cells were compared to AGS-EBNA1 and AGS-EBV cells. (B) AGS-EBNA1 and AGS-EBV cells are shown after treatment with siRNA against GFP (siGFP) or EBNA1 (siEBNA1). (C) AGS cells were treated with siRNA against GFP or EBNA1 or with AllStars negative-control siRNA.
We further verified that the decreased level of PML NBs in AGS-EBNA1 and AGS-EBV cells was due to EBNA1 expression by downregulating EBNA1 in these cells using siRNA. siRNA treatment against EBNA1 reduced EBNA1 levels, which resulted in a 4-fold increase in PML NBs in both AGS-EBNA1 and AGS-EBV cells compared to the negative-control siRNA against GFP (Fig. 1B). In the AGS-EBV cells, it was also noted that the PML NBs increased in size after EBNA1 silencing (Fig. 1B, bottom panels), suggesting that some aspect of the EBV infection induced PML protein levels and that this effect was counteracted by EBNA1. To ensure that the effect of the EBNA1 siRNA was indeed due to depletion of EBNA1, we also compared PML NBs in AGS cells (lacking EBNA1 expression) after transfection with siRNA against EBNA1, GFP, or a universal negative-control siRNA (AllStars) (Fig. 1C). As expected, there were no significant differences in PML NBs in AGS cells receiving these treatments.
We next examined whether disruption of PML NBs by EBNA1 occurred immediately or was a consequence of long-term expression. To this end, we transiently transfected AGS cells with an EBNA1 expression plasmid and examined PML NBs 24 h later (Fig. 2). EBNA1 expression lowered the number of PML NBs by 3-fold compared to that in mock-transfected cells (Fig. 2), thereby demonstrating that EBNA1 expression has an immediate effect on PML NBs.
Fig 2.

Transient EBNA1 expression induces loss of PML NBs. AGS cells were transfected with a plasmid expressing EBNA1 (OriPE) or empty plasmid (OriP) and then stained 24 h later with antibodies against EBNA1 and PML and counterstained with DAPI. Both EBNA1-expressing and nonexpressing cells are shown for the OriPE transfection. PML NBs were quantified from 3 separate experiments as for Fig. 1.
EBNA1 lowers PML protein levels.
Disruption of PML NBs can occur either through interference with PML protein-protein interactions or due to degradation of the PML proteins. The two mechanisms can be distinguished, since only the latter will reduce levels of PML proteins. We therefore determined whether EBNA1 induced loss of PML proteins by comparing the levels of PML isoforms in AGS, AGS-EBNA1, and AGS-EBV cells by Western blotting. PML is known to exist as several isoforms, each of which undergoes extensive posttranslational modifications, resulting in multiple bands migrating between 60 and 200 kDa on immunoblots (6). Cells containing EBV or expressing EBNA1 alone had decreased expression of all PML isoforms (Fig. 3A) compared to the parental AGS cells. In addition, EBNA1 depletion in AGS-EBNA1 and AGS-EBV cells increased PML proteins compared to control siGFP treatment (Fig. 3B). In particular, we observed a robust increase in all PML isoforms in AGS-EBV cells after EBNA1 depletion (Fig. 3B and C), which is consistent with the larger PML NBs previously observed with this treatment by IF (Fig. 1B). In contrast, treatment of the EBNA1-negative AGS cells with EBNA1 siRNA did not change the PML levels relative to treatment with negative-control siRNAs (GFP and AllStars) (Fig. 3C). Finally, we also observed that transient EBNA1 expression was sufficient to degrade PML proteins in AGS cells (Fig. 3D). Therefore, the data are consistent with EBNA1 disrupting PML bodies by inducing the degradation of PML proteins.
Fig 3.

EBNA1 expression decreases PML protein levels. (A) Equal amounts of cell lysates from AGS, AGS-EBNA1, and AGS-EBV cells were compared by Western blotting using an antibody that recognizes all PML isoforms. Blots for EBNA1 and actin are also shown. (B) AGS-EBNA1 and AGS-EBV cells were transfected with siRNA against EBNA1 (+) or GFP (−), and then equal amounts of cell lysate were analyzed by Western blotting as for panel A. (C) AGS or AGS-EBV cells were transfected with siRNA against GFP or EBNA1 or AllStars negative-control siRNA, and then Western blotting was performed as for panel A. (D) AGS cells were transfected with a plasmid expressing EBNA1 (OriPE) or with the empty plasmid (OriP), and 24 h later whole-cell lysates were compared by Western blotting for PML, EBNA1, and actin.
EBNA1 interferes with p53 activation and apoptosis.
PML NBs are known to be important for p53 activation by acetylation and for apoptosis (12, 24). Since both of these functions would be expected to affect GC, we examined whether EBNA1 expression inhibited p53 acetylation and apoptosis as a consequence of PML disruption. The AGS cells express wild-type p53 protein (22), which should be activated in response to DNA damage. First, we compared levels of total and acetylated (at K382) p53 in response to etoposide-induced DNA damage in AGS, AGS-EBNA1, and AGS-EBV cells. Although etoposide induced p53 stabilization in all three cell lines, Western blotting using an antibody specific for acetylated p53 showed that the presence of EBV or EBNA1 decreased p53 acetylation (Fig. 4A, second panel, compare lane 2 to lanes 4 and 6). We also observed that basal p53 levels were lower in cells expressing EBNA1 or containing EBV (Fig. 4A, top panel, compare lane 1 to lanes 3 and 5), consistent with our previous findings that EBNA1 can decrease p53 levels (26). To verify that the reduced levels of acetylated p53 detected in response to etoposide resulted in compromised p53 function, we examined p21 protein expression, which is a direct downstream transcriptional target of p53. Consistent with the effects on p53 acetylation, we observed a significant decrease in p21 induction by etoposide in AGS-EBNA1 and AGS-EBV cells compared to parental AGS cells (Fig. 4A, third panel, compare lanes 2 to lanes 4 and 6). In addition to the Western blots, such changes were also observed using IF (Fig. 4B). We found that upon induction of DNA damage, cells expressing EBNA1 or carrying EBV showed an overall decrease in staining for p53 acetylated at K382 (Fig. 4B, right panels) and p21 (Fig. 4B, middle panels).
Fig 4.

EBNA1 decreases p53 activation and apoptosis. (A and D) AGS, AGS-EBNA1, and AGS-EBV cells were treated with etoposide (+) or dimethyl sulfoxide (DMSO) (−) for 5 (A) or 48 (D) hours, and then equal amounts of cell lysates were analyzed by Western blotting using antibodies against p53 acetylated on K382, total p53, p21, actin, and caspase-3, as indicated. In panel D, the positions of molecular weight markers are shown for caspase bands. (B) Cells treated with etoposide as for panel A were stained with antibodies against p21 and acetylated p53. All images were captured at the same exposure time. (C) Cells treated with either DMSO or etoposide as for panel D were stained with DAPI to show apoptotic bodies, indicated by the arrowheads. The apoptotic bodies were quantified by counting over 300 cells in 3 separate experiments, and average numbers with standard deviations are shown in the histogram.
We also compared the induction of apoptosis in AGS, AGS-EBV, and AGS-EBNA1 cells, first by detecting apoptotic bodies after etoposide treatment by their intense DAPI staining in fluorescence microscopy (Fig. 4C). We observed that the number of apoptotic cells was decreased 5- to 10-fold in AGS-EBNA1 and AGS-EBV cells compared to parental AGS cells. These results were supported by a second assay for apoptosis, caspase-3 cleavage, where the parental AGS cells had higher levels of cleaved caspase-3 after etoposide treatment than the other two cell lines (Fig. 4D, compare lane 2 to lanes 4 and 6). Hence, all these data demonstrate that EBNA1 impairs PML function in terms of reduced p53 acetylation and apoptosis.
EBNA1 mutants defective in CK2 and USP7 binding have reduced abilities to induce PML degradation and interfere with p53 activation.
EBNA1 binds directly to two different host proteins implicated in PML regulation, CK2 kinase and the ubiquitin-specific protease USP7 (also called HAUSP) (14, 15, 26–28, 32). We have previously identified EBNA1 mutations that selectively disrupt binding to either CK2 (Δ387-394) or USP7 (Δ395-450) (15, 32) and have shown that these EBNA1 mutations fail to disrupt PML NBs or induce the degradation of PML proteins in NPC cell lines (32, 33). To determine if EBNA1-mediated disruption of PML NBs occurs by the same mechanism in GC cells, we compared PML levels in AGS cells after transient expression of EBNA1 or the EBNA1 Δ387-394 or Δ395-450 mutant. Neither of the EBNA1 mutants induced the loss of PML proteins seen with wild-type EBNA1, despite the fact that all three EBNA1 proteins were expressed at the similar levels (Fig. 5, compare lane 2 to lanes 3 and 4 and lane 6 to lanes 7 and 8). The results suggest that EBNA1 binding to CK2 and USP7 is part of the mechanism by which EBNA1 induces PML degradation. We also compared the ability of these EBNA1 proteins to interfere with p53 activation, by treating AGS cells expressing these proteins (or containing the empty expression plasmid) with etoposide and comparing levels of acetylated (at K382) p53 and p21 by Western blotting (Fig. 5, bottom 2 panels). Neither EBNAΔ387-394 nor EBNAΔ395-450 reduced p53 acetylation or p21 expression to the same degree as wild-type EBNA1, showing that PML loss correlated with p53 activation.
EBNA1 provides a survival advantage to GC cells.
We next examined whether EBNA1 had affected cell proliferation and survival that might therefore contribute to development of GC. We did not detect a difference in the basal rate of proliferation in AGS-EBNA1 or AGS-EBV cells compared to the parental AGS cells (data not shown). However, after EBNA1 silencing, proliferation of both AGS-EBNA1 and AGS-EBV cells was significantly decreased compared to that of siGFP-treated cells (Fig. 6A and B), suggesting that over time, cells expressing EBNA1 can become dependent on EBNA1 for growth. In addition, we examined whether the presence of EBNA1 affected survival after DNA damage. Both AGS-EBNA1 and AGS-EBV cells were consistently found to be more resistant to etoposide than parental cells (Fig. 6C). The data indicate that EBNA1 confers a survival advantage to AGS cells, implying an important role for EBNA1 in GC development.
Decreased PML staining in EBV-positive GC biopsy specimens.
While we have shown that EBNA1 expression can decrease PML NBs in GC cell lines, it is important to determine whether this effect translates to primary GC tissues. To this end, we examined 10 GC biopsy samples that were classified as either EBV positive (five samples) or EBV negative (five samples) based on fluorescent in situ hybridization for the EBV-expressed EBERs, resulting in red staining (Fig. 7, EBERish panels). All tumor samples were then stained for PML by immunohistochemistry, and three representative samples from each category are shown. We observed that EBV-negative biopsy specimens had intense, dark brown nuclear staining for PML for both intestinal (Fig. 7, row I), and diffuse (Fig. 7, rows II and III) types of GC (Fig. 7). However, a noticeable reduction in PML staining was observed in all of the EBV-positive biopsy specimens (three examples are in Fig. 7). The decreased PML staining was confined to the carcinoma cells, while the infiltrating lymphocytes (L) in the same tumors exhibited strong (brown) PML staining. The results demonstrate a remarkable difference in PML expression in EBV-positive versus EBV-negative GC, consistent with the EBNA1-induced loss of PML as observed in cell line studies.
Fig 7.

PML staining is diminished in EBV-positive GC biopsy specimens. Sections of EBV-positive and EBV-negative human gastric carcinoma biopsy specimens were stained for PML using standard immunohistochemistry (brown stain). Sections of the same biopsy specimens were also subjected to in situ hybridization staining for EBER (EBERish) (red stain). All images were captured at the same exposure time.
DISCUSSION
In recent years, EBV has become increasingly recognized as a causative agent in a significant proportion of GC cases. Here we provide a mechanism by which EBV could contribute to the development of this cancer mediated by its EBNA1 protein. We have shown that EBNA1 alters nuclear bodies formed by the PML tumor suppressor protein and induces the degradation of all PML isoforms in GC cells that contain EBV or just express EBNA1. Previously we reported that EBNA1 alters PML NBs in NPC, implicating EBNA1 as a contributing factor in the development of NPC. However, since virtually all undifferentiated NPCs are EBV positive, these tumors could not be used to address the important question of whether the EBNA1-induced loss of PML observed in NPC cell lines also occurred in primary NPCs. Using GC samples, we were able to compare EBV-positive and EBV-negative biopsy specimens to demonstrate that the presence of EBV leads to significantly decreased PML protein levels in these tumors.
GC can be classified into conventional gastric adenocarcinomas (intestinal type) or lymphoepithelioma-like carcinomas (diffuse type) (9, 10). We observed that the presence of EBV decreased PML staining in both types of GC. In the EBV-positive diffuse type, the EBER staining is not uniformly detected in the tumor cells but instead is observed in a percentage of the cells scattered throughout the tumor. In contrast, we observed decreased PML staining more extensively throughout the tumor (but not in the infiltrating lymphocytes). A recent study by Ryan et al. (25) indicated that EBERs were detected only in GC cells containing very high EBV loads. They detected EBV in 64% of GC samples from the United States using a quantitative PCR assay, whereas only 17% of GC scored as EB positive based on EBER staining, and those that stained positive for EBERs contained very high levels of the EBV genome. Therefore, cells in our EBV-positive tumors that were not stained by EBERish might simply have had lower levels of EBV. Since we had previously shown that even low levels of EBNA1 (such as are present in the EBV-positive NPC cell line C666-1) are sufficient to disrupt PML NBs (33), the EBER-negative cells that contain EBV may still express sufficient amount of EBNA1 to disrupt PML NBs.
PML has long been recognized as a tumor suppressor protein, stemming from the initial observations that a rearrangement of the PML gene occurs in promyelocytic leukemia, resulting in a chimeric protein that disrupts the formation of normal PML NBs (21). A correlation between loss of PML NBs and tumor development and/or progression was subsequently reported for prostate, colorectal, and breast and lung carcinomas, lymphomas, and central nervous system and germ cell tumors (13). In addition, fibroblasts from PML knockout mice grew faster as monitored by 3H-labeled thymidine incorporation, leading to enhanced colony formation (43). Furthermore, these mice had an increased frequency of tumor formation following injection with a tumor-initiating compound, dimethybenzanthacene (43). Hence, the EBNA1-mediated disruption of PML NBs that we have observed in gastric carcinoma cells would be expected to contribute to the onset and/or progression of this malignancy.
The reason that PML NBs suppress oncogenesis likely stems from their known importance in several processes that govern genomic integrity and cell survival, including apoptosis and DNA repair. Moreover, PML NBs have been found to be required for activation of p53, which becomes acetylated only after association with PML NBs (7, 24). We have shown that EBNA1 impairs p53 acetylation, p53-dependent transcription (of p21), and apoptosis and leads to cell survival following DNA damage. Moreover, EBNA1 mutants that fail to induce PML loss also fail to interfere with p53 acetylation and p21 expression, providing further evidence of the connection between PML and p53 activation. These effects indicate that the degree by which EBNA1 decreases PML proteins and NBs is sufficient to significantly impede PML function. These findings are consistent with the observations of Cheng et al. (3), who reported that EBNA1 expression reduced cisplatin-induced cell death in gastric cancer cells and enhanced tumorigenicity in LL/2 cells.
We have shown that EBNA1 mutants lacking amino acids 387 to 394 or 395 to 450 fail to induce PML degradation in GC cells. These deletions do not disrupt EBNA1 functions in EBV maintenance or gene expression but specifically abrogate binding to CK2 (Δ387-394) or USP7 (Δ395-450), implicating these two host proteins in the mechanism by which EBNA1 disrupts PML NBs in GC (15, 32). CK2 has been shown to phosphorylate PML proteins at S517, which triggers their polyubiquitylation and degradation (28, 29), and our previous studies with NPC cell lines showed that EBNA1 increased PML S517 phosphorylation by increasing the association of CK2 with PML (32). USP7 is also partly associated with PML NBs and can promote PML degradation (5, 27). In NPC cells, EBNA1 increases the association of USP7 with PML NBs and requires USP7 in order to disrupt PML NBs (32, 33). Our results with EBNA1 mutants in GC cells suggest that EBNA1-mediated disruption of PML NBs occurs through the same mechanism in NPC and GC cells, namely, by increasing the association of CK2 and USP7 with PML NBs.
Recent work by Wei et al. (44) on Helicobacter pylori provides an interesting parallel between the two major microorganisms associated with gastric carcinoma, H. pylori and EBV. They demonstrated that the H. pylori CagA protein induces the degradation of p53 in gastric cells and increases their survival even with damaged DNA. Therefore, these two very different pathogens that contribute to the development of GC both affect p53 functions. EBNA1 affects p53 in two ways: by inhibiting its activation through the disruption of PML NBs as discussed above and by lowering the steady-state levels of p53 (as seen in Fig. 4A). The lowering of the basal p53 levels is likely due to the ability of EBNA1 to block the interaction between p53 and the deubiquinating enzyme USP7, which normally stabilizes p53 (26).
We also observed that silencing of EBNA1 in AGS cells that had been stably expressing EBNA1 (AGS-EBNA1 cells) or contained EBV (AGS-EBV cells) decreased cellular proliferation. This is despite the fact that we did not observe any differences in growth rates between the parental AGS cells and AGS cells expressing EBNA1 (data not shown). These observations suggest that although EBNA1 might not be required for proliferation initially, over time cells expressing EBNA1 might become dependent on its presence. This could therefore be an example of “oncogene addiction,” as described by others, in which the continued expression of a protein changes the cellular environment such that the cell becomes dependent on that protein for proliferation (20). On that note, Oh et al. (23) reported that a spontaneous EBV-positive GC cell line was dependent on EBV for its survival. It is currently unclear whether the cellular changes induced by EBNA1 that lead to EBNA1 dependency are related to PML disruption or other effects of EBNA1.
In summary, we have shown for the first time that the EBNA1 protein increases the survival of gastric carcinoma cells, at least in part due to its ability to induce the loss of PML NBs, resulting in impairment of p53 function and apoptosis. The decreased apoptotic response in combination with the impairment of DNA repair associated with loss of PML NBs would be expected to promote the survival of cells with DNA damage, thereby contributing to the development of human gastric cancer.
ACKNOWLEDGMENTS
We thank Willa Shi for her guidance on immunohistochemistry staining.
This work was funded by an operating grant to L.F. from the Canadian Cancer Society. N.S. was supported by a Canadian Institutes for Health Research doctoral award. L.F. is a tier 1 Canada Research Chair in Molecular Virology.
Footnotes
Published ahead of print 19 October 2011
REFERENCES
- 1. Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8:1006–1016 [DOI] [PubMed] [Google Scholar]
- 2. Catalano V, et al. 2009. Gastric cancer. Crit. Rev. Oncol. Hematol. 71:127–164 [DOI] [PubMed] [Google Scholar]
- 3. Cheng TC, et al. 2010. Expression of Epstein-Barr nuclear antigen 1 in gastric carcinoma cells is associated with enhanced tumorigenicity and reduced cisplatin sensitivity. Int. J. Oncol. 36:151–160 [PubMed] [Google Scholar]
- 4. de Stanchina E, et al. 2004. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 13:523–535 [DOI] [PubMed] [Google Scholar]
- 5. Everett R, et al. 1997. A novel ubiquitin-specific protease is dyamically associted with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16:1519–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830 [DOI] [PubMed] [Google Scholar]
- 7. Fogal V, et al. 2000. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19:6185–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frappier L. 2004. Viral plasmids in mammalian cells, p 325–339 InFunnell BE, Phillips GJ. (ed), Plasmid biology. ASM Press, Washington, DC [Google Scholar]
- 9. Fukayama M. 2010. Epstein-Barr virus and gastric carcinoma. Pathol. Int. 60:337–350 [DOI] [PubMed] [Google Scholar]
- 10. Fukayama M, Hino R, Uozaki H. 2008. Epstein-Barr virus and gastric carcinoma: virus-host interactions leading to carcinoma. Cancer Sci. 99:1726–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gruhne B, et al. 2009. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. U. S. A. 106:2313–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo A, et al. 2000. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2:730–736 [DOI] [PubMed] [Google Scholar]
- 13. Gurrieri C, et al. 2004. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 96:269–279 [DOI] [PubMed] [Google Scholar]
- 14. Holowaty MN, Sheng Y, Nguyen T, Arrowsmith C, Frappier L. 2003. Protein interaction domains of the ubiqutin specific protease, USP7/HAUSP. J. Biol. Chem. 278:47753–47761 [DOI] [PubMed] [Google Scholar]
- 15. Holowaty MN, et al. 2003. Protein profiling with Epstein-Barr nuclear antigen 1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 278:29987–29994 [DOI] [PubMed] [Google Scholar]
- 16. Ian MX, Lan SZ, Cheng ZF, Dan H, Qiong LH. 2008. Suppression of EBNA1 expression inhibits growth of EBV-positive NK/T cell lymphoma cells. Cancer Biol. Ther. 7:1602–1606 [DOI] [PubMed] [Google Scholar]
- 17. Imai S, et al. 1994. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc. Natl. Acad. Sci. U. S. A. 91:9131–9135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kennedy G, Komano J, Sugden B. 2003. Epstein-Barr virus provide a survival factor to Burkitt's lymphomas. Proc. Natl. Acad. Sci. U. S. A. 100:14269–14274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu J, et al. 2011. Epstein-Barr virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 410:64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo J, Solimini NL, Elledge SJ. 2009. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136:823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. 2002. Mechanisms of action of arsenic trioxide. Cancer Res. 62:3893–3903 [PubMed] [Google Scholar]
- 22. Muller M, et al. 1998. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188:2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oh ST, Cha JH, Shin DJ, Yoon SK, Lee SK. 2007. Establishment and characterization of an in vivo model for Epstein-Barr virus positive gastric carcinoma. J. Med. Virol. 79:1343–1348 [DOI] [PubMed] [Google Scholar]
- 24. Pearson M, et al. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406:207–210 [DOI] [PubMed] [Google Scholar]
- 25. Ryan JL, et al. 2009. High levels of Epstein-Barr virus DNA in latently infected gastric adenocarcinoma. Lab. Invest. 89:80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saridakis V, et al. 2005. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 18:25–36 [DOI] [PubMed] [Google Scholar]
- 27. Sarkari F, Wang X, Nguyen T, Frappier L. 2011. The herpesvirus associated ubiquitin specific protease, USP7, is a negative regulator of PML proteins and PML nuclear bodies. PLoS One 6:e16598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scaglioni PP, et al. 2006. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126:269–283 [DOI] [PubMed] [Google Scholar]
- 29. Scaglioni PP, et al. 2008. CK2 mediates phosphorylation and ubiquitin-mediated degradation of the PML tumor suppressor. Mol. Cell. Biochem. 316:149–154 [DOI] [PubMed] [Google Scholar]
- 30. Shi W, et al. 2003. Dysregulated PTEN-PKB and negative receptor status in human breast cancer. Int. J. Cancer 104:195–203 [DOI] [PubMed] [Google Scholar]
- 31. Shibata D, Weiss LM. 1992. Epstein-Barr virus-associated gastric adenocarcinoma. Am. J. Pathol. 140:769–774 [PMC free article] [PubMed] [Google Scholar]
- 32. Sivachandran N, Cao JY, Frappier L. 2010. Epstein-Barr virus nuclear antigen 1 hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 84:11113–11123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sivachandran N, Sarkari F, Frappier L. 2008. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through the disruption of PML nuclear bodies. PLoS Pathog. 4:e1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stewart S, et al. 2004. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. U. S. A. 101:15730–15735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takada K. 2000. Epstein-Barr virus and gastric carcinoma. Mol. Pathol. 53:255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takahashi Y, Lallemand-Breitenbach V, Zhu J, de The H. 2004. PML nuclear bodies and apoptosis. Oncogene 23:2819–2824 [DOI] [PubMed] [Google Scholar]
- 37. Trotman LC, et al. 2006. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uemura Y, et al. 1994. A unique morphology of Epstein-Barr virus-related early gastric carcinoma. Cancer Epidemiol. Biomarkers Prev. 3:607–611 [PubMed] [Google Scholar]
- 39. Valentine R, et al. 2010. Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol. Cancer 9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Beek J, et al. 2004. EBV-positive gastric adenocarcinomas: a distinct clinicopathologic entity with a low frequency of lymph node involvement. J. Clin. Oncol. 22:664–670 [DOI] [PubMed] [Google Scholar]
- 41. van Beek J, et al. 2002. A rapid and reliable enzyme immunoassay PCR-based screening method to identify EBV-carrying gastric carcinomas. Mod. Pathol. 15:870–877 [DOI] [PubMed] [Google Scholar]
- 42. van Beek J, et al. 2006. Morphological evidence of an activated cytotoxic T-cell infiltrate in EBV-positive gastric carcinoma preventing lymph node metastases. Am. J. Surg. Pathol. 30:59–65 [DOI] [PubMed] [Google Scholar]
- 43. Wang ZG, et al. 1998. Role of PML in cell growth and the retinoic acid pathway. Science 279:1547–1551 [DOI] [PubMed] [Google Scholar]
- 44. Wei J, et al. 2010. Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139:1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wilson JB, Bell JL, Levine AJ. 1996. Expression of Epstein-Barr virus nuclar antigen-1 induces B cell neoplasia in transgenic mice. EMBO 15:3117–3126 [PMC free article] [PubMed] [Google Scholar]
- 46. Wu H, Kapoor P, Frappier L. 2002. Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein-Barr nuclear antigen 1. J. Virol. 76:2480–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yin Q, Flemington EK. 2006. siRNAs against the Epstein Barr virus latency replication factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells. Virology. 346:385–393 [DOI] [PubMed] [Google Scholar]
- 48. Zhong S, et al. 1999. A role for PML and the nuclear body in genomic stability. Oncogene 18:7941–7947 [DOI] [PubMed] [Google Scholar]