

Abstract
Human coronaviruses (HCoV) are recognized respiratory pathogens. Some HCoV strains, including HCoV-OC43, can invade the central nervous system, where they infect neurons, with unclear consequences. We have previously reported that HCoV-OC43 infection of human neurons activates the unfolded-protein response and caspase-3 and induces cell death and that the viral spike (S) glycoprotein is involved in the process. We now report on underlying mechanisms associated with the induction of programmed cell death (PCD) after infection by the reference HCoV-OC43 virus (rOC/ATCC) and a more neurovirulent and cytotoxic HCoV-OC43 variant harboring two point mutations in the S glycoprotein (rOC/US183-241). Even though caspase-3 and caspase-9 were both activated after infection, the use of caspase inhibitors neither reduced nor delayed virus-induced PCD, suggesting that these proteases are not essential in the process. On the other hand, the proapoptotic proteins BAX, cytochrome c (CytC), and apoptosis-inducing factor (AIF) were relocalized toward the mitochondria, cytosol, and nucleus, respectively, after infection by both virus variants. Moreover, LA-N-5 neuronal cells treated with cyclosporine (CsA), an inhibitor of the mitochondrial permeabilization transition pore (mPTP), or knocked down for cyclophilin D (CypD) were completely protected from rOC/ATCC-induced neuronal PCD, underlining the involvement of CypD in the process. On the other hand, CsA and CypD knockdown had moderate effects on rOC/US183-241-induced PCD. In conclusion, our results are consistent with mitochondrial AIF and cyclophilin D being central in HCoV-OC43-induced PCD, while caspases appear not to be essential.
INTRODUCTION
Human coronaviruses (HCoV) are enveloped positive-stranded single-stranded RNA viruses. They are recognized respiratory pathogens (70) with neurotropic and neuroinvasive properties (4, 11, 43, 66). We reported previously that the OC43 strain of HCoV (HCoV-OC43) could infect primary cultures of human and murine central nervous system (CNS) cells (11, 41), as well as infect and persist in human neural cell lines (5) and human brains (4). We also demonstrated that neurons are the main target of infection in murine CNS (41), as well as in cocultures of human NT2 neuronal cells and primary astrocytes (M. Desforges and P. J. Talbot, unpublished data). Furthermore, HCoV-OC43 induced a chronic encephalitis in susceptible mice (41) and was associated with acute disseminated encephalomyelitis in a human case (77). Considering that murine hepatitis virus (MHV), the murine counterpart of HCoV-OC43, induces a neurological disease in mice (14), we hypothesized that HCoV-OC43 might be associated with some human neurological diseases of unknown etiology. Recently, we reported that HCoV-OC43 induces the unfolded-protein response (UPR) in infected human neurons, while inducing significant neuronal death (27). Moreover, we showed that caspase-3 was activated upon HCoV-OC43 infection of human neurons (27). However, the molecular cell death pathways involved remain to be defined.
One of the major cell death-associated complexes is the mitochondrial permeability transition pore (mPTP), which has been linked to several neurodegenerative diseases, such as experimental autoimmune encephalomyelitis (30) and amyotrophic lateral sclerosis (53). Following cellular stress, such as accumulation of reactive oxygen species (ROS) and high Ca2+ levels, the mPTP opens and allows the release of proapoptotic factors such as cytochrome c (CytC) and apoptosis-inducing factor (AIF) (48). CytC is known to participate in the formation of the apoptosome, leading to the cascade of caspase activation associated with apoptotic programmed cell death (PCD) (8), while AIF translocates to the nucleus and promotes high-molecular-weight DNA fragmentation and chromatin condensation (67), which is considered a hallmark of caspase-independent apoptosis-like PCD (20, 44, 67, 78). Indeed, evidence is accumulating regarding the role of AIF in neuronal death in both chronic and acute neurodegeneration (3, 46, 74). Characterization of the mPTP is being intensely pursued, and one of its major components is cyclophilin D (CypD), which is a member of the cyclophilin family possessing peptidyl-prolyl cis-trans isomerase activity (29, 68). CypD is localized at the inner mitochondrial membrane and is known to be responsible for modulation of mPTP in various types of cell death (6, 62). However, it is not clear which cellular protein(s) specifically interacts with CypD to promote mPTP formation. Numerous putative partners have been identified, such as the adenine nucleotide transporter (38), the voltage-dependent anion channel (21), and BAX (79), and this is the subject of an intense debate (7, 22, 32, 36, 45). There are some apparent discrepancies in the literature regarding the type of cell death regulated by CypD and mPTP. Overexpression of CypD has been associated with enhanced necrosis but not apoptosis in mouse embryonic fibroblasts (57) and the neuronal cell line B50 (50). Conversely, CypD−/− cells were protected from death induced by focal cerebral ischemia (62), thapsigargin, and oxidative stress but not staurosporine or tumor necrosis factor alpha (TNF-α) (6, 57). This indicates that CypD can regulate various types of cell death in a specific cell type- and stimulus-dependent manner. In fact, it is clear that CypD knockout or inhibition by cyclosporine (CsA) (37) prevents cell death from numerous injuries or apoptotic insults, such as axonal degeneration (9), motoneurons axotomy (73), oxidative stress in cultured cerebellar granule neurons (71), amyloid-β-induced neuronal apoptosis in cultured neurons (25), experimental autoimmune encephalomyelitis-induced axonal injury (30), and excitotoxic neuronal death (61).
Considering that the roles of mPTP, AIF and CypD in neurodegenerative diseases are recognized and currently being characterized, particularly in neurons (46, 54, 62, 74), we sought to determine whether these factors were involved in human neuronal cell death induced by HCoV-OC43, which is associated with the viral S glycoprotein (27). Consequently, we characterized the type of cell death induced by HCoV-OC43 in human neurons, using the LA-N-5 model (40, 59), and evaluated the contribution of caspases, CypD, and different cellular proteins associated with different forms of PCD related to the mPTP. We also investigated the importance of the N-terminal portion of S (a putative receptor-binding domain) in PCD induction by comparing wild-type HCoV-OC43 (rOC/ATCC) to a previously reported variant (rOC/US183-241) which harbors two point mutations within the spike S glycoprotein (H183R and Y241H) (27, 42), is more neurovirulent (42), and induces a modified virus-induced neuropathology involving hind-limb paralysis and demyelination in susceptible mice (13).
MATERIALS AND METHODS
Cell lines, viruses, caspase inhibitors and cyclosporine treatment.
The LA-N-5 cell line (a kind gift of Stephan Ladisch, George Washington University School of Medicine) was cultured in RPMI medium supplemented with 15% (vol/vol) fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM nonessential amino acids (Gibco-Invitrogen). Cells were differentiated into human neurons as previously described (40). Briefly, cells were seeded in Cell+ petri dishes (5 × 105 cells) or in 6-well (4 × 104 cells), 24-well (5 × 103 cells), or 96-well (1.8 × 103 cells) plates (Sarstedt) in RPMI medium supplemented with 10% (vol/vol) FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM nonessential amino acids. The next day and every 2 days for 6 days, the medium was replaced with the same medium containing 10 μM all-trans retinoic acid (Sigma-Aldrich).
The recombinant wild-type reference HCoV-OC43 virus, designated rOC/ATCC, and the recombinant HCoV-OC43 variant virus containing two point mutations within the spike S glycoprotein (H183R and Y241H), designated rOC/US183-241, were generated by reverse genetics using the previously described full-length cDNA clone pBAC-OC43FL (64). Sequencing was performed in order to confirm that the two single point mutations in the S gene were the only differences between the two recombinants. The S gene of rOC/ATCC was identical to that of the HCoV-OC43 ATCC VR-759 strain obtained several years ago from the American Type Culture Collection (ATCC). The S gene of rOC/US183-241 differed from rOC/ATCC only by the two mutations inserted into the gene encoding the S glycoprotein. The rOC/ATCC designation refers to the recombinant virus identical to HCoV-OC43 ATCC VR-759 strain; rOC/US183-241 refers to the recombinant virus containing the aforementioned two point mutations within the S glycoprotein. Both viruses were propagated on the HRT-18 cell line, as previously described (55). LA-N-5 cells were infected at a multiplicity of infection (MOI) of 0.2 or mock infected and then were incubated at 37°C for 2 h, washed with phosphate-buffered saline (PBS), and incubated at 37°C with fresh RPMI medium supplemented with 2.5% (vol/vol) FBS for different periods of time. Cells and supernatants were harvested at the indicated times postinfection. For the study with inhibitors, cells were treated with 2.5 μM cyclosporine (Calbiochem), 50 μM Z-VAD-FMK (MBL), 10 μM Z-LEHD-FMK (MBL), or dimethyl sulfoxide (DMSO) and harvested at the indicated time points.
Generation of cell populations knocked down for CypD.
LA-N-5 cells knocked down for CypD expression were obtained using the Mission pLKO.1 short hairpin RNA (shRNA) expression vector (Sigma-Aldrich) packaged within lentiviral pseudoparticles. Lentiviral pseudoparticles were obtained by cotransfecting the Mission pLKO.1 shRNA vector encoding one of five different sequences for CypD silencing (TRCN0000049263, TRCN0000049264, TRCN0000049265, TRCN0000049266, and TRCN0000049267) or the empty vector and pLP1, pLP2, and pLP-VSVG vectors in HEK293T cells and were retrieved in the supernatants 72 h later. LA-N-5 cells were then transduced by lentiviral pseudoparticles, and cell populations were selected 24 h later with puromycin (2 μg/ml). Populations of CypD knockdown LA-N-5 cells were generated and maintained in regular medium supplemented with puromycin. Five different shRNA sequences were used to generate CypD knockdown cells, and only two different populations, K and M, corresponding to shRNA sequences TRCN0000049263 and TRCN0000049265, respectively, survived the selection process and were used for further experiments. A population of LA-N-5 cells transduced with the empty vector and selected with puromycin were used as reference cells in CypD knockdown studies and were named LA-N-5 empty. The expression levels of CypD in both CypD knockdown populations were measured by quantitative PCR using the Livak 2−ΔΔCT method (50a) and are presented as expression relative to that in the LA-N-5 empty population.
Immunofluorescence.
Cells were fixed with 4% (wt/vol) paraformaldehyde for 20 min at room temperature, permeabilized with methanol at −20°C for 5 min, incubated with primary rabbit polyclonal antibody against activated caspase-3 (1/50) (R&D Systems), rabbit polyclonal antibody against activated BAX (1/200) (sc-493; Santa Cruz), mouse monoclonal antibody against AIF (1/500) (a kind gift of Guido Kroemer, INSERM, France), or mouse monoclonal antibody against AIF (1/200) (sc-13116; Santa Cruz) for 1 h at room temperature, and washed three times with PBS. Cells were then incubated for 1 h at room temperature with the secondary antibodies (Molecular Probes-Invitrogen) anti-rabbit Alexa Fluor 488 or anti-mouse Alexa Fluor 488 (1/1500). Cells incubated with antibody to activated caspase-3 were then incubated with 4′,6′-diamidino-2-phenylindole (DAPI) for 5 min and washed three times with PBS prior to imaging. MitoTracker Red CMXROS (200 nM) Molecular Probes-Invitrogen) was added to viable cells and left for 15 min prior to fixation and staining with antibody directed against activated BAX or AIF.
Protein extraction and Western immunoblotting.
Cytoplasmic and nuclear proteins were extracted using the NucBuster extraction protein kit (71183-3; Novagen) according to the manufacturer's instructions. Briefly, cells were harvested, permeabilized, and centrifuged at 16,000 × g for 5 min, and cytoplasmic proteins were retrieved from the supernatant. The pellet was then washed with cold PBS, solubilized, and centrifuged at 16,000 × g for 5 min at 4°C, and nuclear proteins were retrieved from the supernatant. Mitochondrial proteins were extracted using the ProteoExtract Cytosol/Mitochondria fractionation kit (QIA88; EMD Bioscience) according to the manufacturer's instructions. Briefly, cells were harvested, washed with ice-cold PBS, centrifuged at 600 × g for 5 min at 4°C, incubated for 10 min at 4°C with Cytosol extraction buffer, homogenized using a Dounce tissue homogenizer, and centrifuged at 700 × g for 10 min at 4°C. Cytosolic proteins were retrieved from the supernatant. Mitochondrial proteins were obtained from the pellet, which was then washed with cold PBS, incubated with Mitochondria extraction buffer, and vortexed for 10 s.
Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit (Novagen) according to the manufacturer's protocol. Equal amounts of proteins were subjected to SDS-PAGE using a 10% or 4 to 12% Novex NuPage gradient gel (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) with the Bio-Rad semidry Transblot apparatus. Membranes were blocked overnight at 4°C with Tris-buffered saline (TBS) containing 1% (vol/vol) Tween 20 (TBS-T) and 5% (wt/vol) nonfat milk and then incubated with rabbit polyclonal antibody against activated BAX (1/200) (N20 sc-493; Santa Cruz), mouse monoclonal antibody against CytC (1/500) (BD Pharmingen), mouse monoclonal antibody against p84 (1/500) (AbCam), mouse monoclonal antibody against VDAC (1/500) (AbCam), or mouse monoclonal antibody against AIF (1/1,000) (sc-13116; Santa Cruz) for 1 h at room temperature. After three TBS-T washes, the membranes were incubated with anti-mouse or anti-rabbit secondary antibody coupled to horseradish peroxidase (GE Life Sciences), and detection was by chemiluminescence using the ECL kit (GE Life Sciences) with the ChemiGenius2 Syngene apparatus.
Cell viability assay.
Cell viability was monitored through the reduction of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H tetrazolium inner salt (MTS) in the presence of phenazine methosulfate (PMS), as previously described (19). Briefly, infected, mock-infected, inhibitor-treated, and DMSO-treated cells cultured in 96-well plates were incubated in the presence of 0.6 mM MTS (Promega) and 14 μM PMS (Sigma-Aldrich) at 24, 48, or 72 h postinfection and absorbance read at 492 nm every 20 min for 3 h. Viability was determined by slope regression analysis for each sample and is expressed as a relative percentage compared the slope obtained with mock-infected cells. Student's t test was performed to determine statistical significance of the differences in slopes between samples, using the SPSS software, version 16.0.
Caspase activity.
Caspase-3 and caspase-9 activities were assessed using caspase-3 and caspase-9 colorimetric assays (R&D Systems), respectively, according to the manufacturer's protocol. Briefly, 1.2 × 107 cells were harvested, lysed, and centrifuged at 10,000 × g for 10 min at 4°C, and the concentration of protein in the supernatant was assessed with a BCA protein assay kit (Novagen) according to the manufacturer's protocol. A 200-μg aliquot of protein from each infection condition was incubated with the colorimetric substrate LEHD-pNA for the caspase-9 assay or DEVD-pNA for the caspase-3 assay for 2 h at 37°C. Photometric analysis was performed at 405 nm, and background values obtained from wells without colorimetric substrate were subtracted. The fold increase of caspase activity in cells infected under different conditions was quantitated relative to mock-infected reference cells. Analysis of variance (ANOVA) tests followed by post hoc Tahame analysis were performed to determine the statistical significance of the differences in the percentages of caspase activity between samples, using the SPSS software, version 16.0.
TUNEL, permeability assay, and intracellular DNA fragmentation assay.
Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) and permeability assay were performed using an in situ cell death detection kit (Roche) according to the manufacturer's instructions. Briefly, 4 × 106 cells were harvested, incubated with 0.5 μg/ml of 7-aminoactinomycin D (7-AAD) for 5 min, washed with PBS, and fixed with 1% (wt/vol) paraformaldehyde for 20 min at room temperature. After three washes with PBS, they were then permeabilized with methanol at −20°C for 5 min, washed three times with PBS, incubated with labeling solution and enzyme solution for 1 h at 37°C, washed twice with PBS, and analyzed using a FACSCalibur cytofluorimeter. Data analysis was performed using the Cell Quest Pro software (BD Bioscience). ANOVA tests followed by post hoc Tahame analysis were performed to determine the statistical significance of the differences in the percentages of TUNEL-labeled and 7-AAD positive cells between samples, using the SPSS software, version 16.0. The intracellular DNA fragmentation assay was performed using the cell death detection enzyme-linked immunosorbent assay (ELISA) plus (Roche) according to the manufacturer's instructions. Briefly, cells in 96-well plates were centrifuged at 200 × g for 10 min at 4°C, the supernatant was removed, and the cell pellet, containing intact cells and apoptotic bodies, was lysed for 30 min at room temperature. Lysates were centrifuged at 200 × g for 10 min to separate intact genomic DNA from fragmented DNA, and supernatants, containing fragmented DNA, were subjected to sandwich ELISA in streptavidin-coated microplates with antihistone antibody coupled with biotin and anti-DNA antibody coupled to horseradish peroxidase for 2 h at room temperature. The 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS) substrate was added, and photometric analysis was performed at 405 nm with a reference wavelength of 490 nm. The fold increase of intracellular fragmented DNA in cells infected under different conditions was quantitated relative to mock-infected reference cells. ANOVA tests followed by post hoc Tahame analysis were performed to determine the statistical significance of the differences in the percentages of fragmented DNA between samples, using the SPSS software, version 16.0.
Quantitation of infectious virus titers by IPA.
An immunoperoxidase assay (IPA) was performed on HRT-18 cells as previously described (49). Briefly, the primary antibody used was monoclonal antibody 1-10C3 directed against the S protein of rOC/ATCC and rOC/US183-241. The secondary antibody was horseradish peroxidase-conjugated goat anti-mouse immunoglobulin (KPL). Immune complexes were detected by incubation with 0.025% (wt/vol) 3,3′-diaminobenzidine tetrahydrochloride (Bio-Rad) and 0.01% (vol/vol) hydrogen peroxide in PBS, and infectious virus titers were calculated by the Karber method as previously described (49).
RESULTS
Both rOC/ATCC and rOC/US183-241 induce programmed cell death in human neurons.
We previously showed that rOC/ATCC and rOC/US183-241 can induce neuronal death (27). Here we sought to identify both the underlying mechanisms and the type of cell death caused by infection of human neurons with two variants of HCoV-OC43, namely, rOC/ATCC and rOC/US183-241. We first confirmed that infection by both viruses led to a loss of viability of human neurons, starting at 48 h postinfection, and that the rOC/US183-241 mutant induced more neuronal death than rOC/ATCC, as shown by the MTS-PMS assay (Fig. 1A). Moreover, using an intracellular DNA fragmentation ELISA, we also showed that there was an increase in DNA fragmentation within infected neurons, compared to mock-infected cells, as soon as 48 h postinfection (Fig. 1B). Indeed, we confirmed that rOC/ATCC and rOC/US183-241 induced fragmentation of DNA in infected neurons, as shown by an increase in the percentage of positive TUNEL-labeled cells (Fig. 1C). The increase in the percentage of 7-AAD-positive cells started after the increase in percentage of TUNEL-labeled cells, indicating that DNA fragmentation occurred while the cell membrane was still intact in human neurons following infection by both viruses. Together, the loss of cell viability and the detection of intracellular fragmented DNA with subsequent cellular permeability alterations strongly suggests that neuronal death induced by rOC/ATCC and rOC/US183-241 is characteristic of a programmed cell death (PCD), as has recently been described by the unified criteria of the Nomenclature Committee on Cell Death (NCCD) (47). Moreover, we showed that rOC/US183-241 induced a faster and stronger PCD than rOC/ATCC in human neurons.
Fig 1.
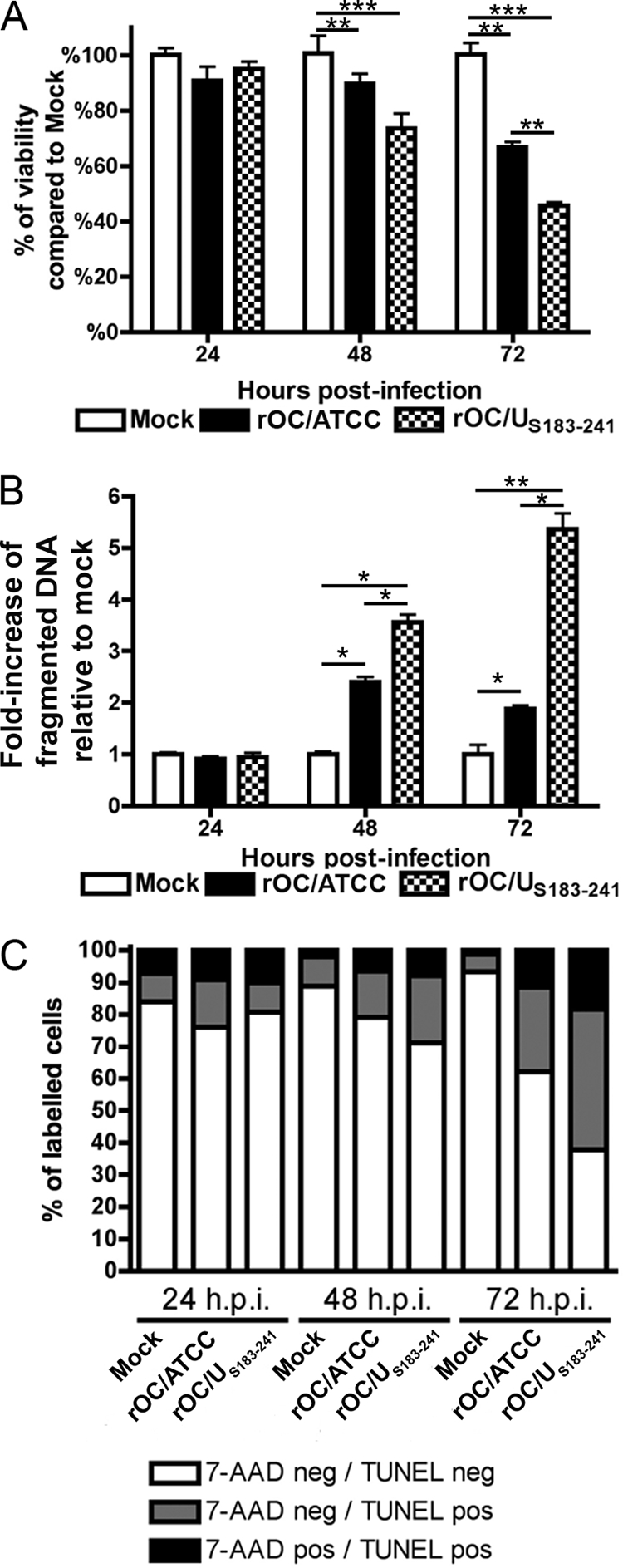
rOC/ATCC and rOC/US183-241 induce programmed cell death in human neurons. Differentiated LA-N-5 cells were infected with rOC/ATCC or rOC/US183-241 for 24, 48, or 72 h. (A) Viability of infected human neurons. Cell viability was evaluated by the MTS-PMS assay and is expressed as relative percentage compared to that of mock-infected cells. (B) Fragmentation of DNA in infected neurons. Fragmented DNA was quantified by sandwich ELISA against histone and DNA and is expressed as a relative fold increase compared to that in mock-infected cells. (C) Fluorescence-activated cell sorting (FACS) TUNEL/7-AAD labeling of infected neurons. Cells stained with 7-AAD and TUNEL labeled were analyzed by FACS. The graph represent the percentage of quadrants. Nonsignificant percentages of 7-AAD-positive/TUNEL-negative cells were omitted. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Caspases are activated following infection of human neurons by rOC/ATCC and rOC/US183-241, but virus-induced PCD is not inhibited by Z-VAD-FMK.
We previously showed that infection of human neurons by rOC/ATCC and rOC/US183-241 can lead to activation of caspase-3 (27). In order to monitor the cascade of activation of caspases in relation to neuronal death induced by both viruses, we quantitated the activities of caspase-9, the main initiator caspase, and caspase-3, the main effector caspase. We showed that both caspase-9 and caspase-3 were activated in human neurons at 48 h postinfection by at least 2-fold compared to in mock-infected cells (Fig. 2A and B). Moreover, rOC/US183-241 promoted a stronger activity of both caspases following infection, compared to rOC/ATCC. The relative reduction of activity of both caspases at 72 h postinfection is likely related to an increased protein degradation that might occur at late times in the induced PCD. Considering that both initiator and effector caspases were activated following neuronal infection by both viruses, we sought to determine whether they were essential in the induction of PCD. The pan-caspase inhibitor Z-VAD-FMK and the caspase-9 inhibitor Z-LEHD-FMK were added to infected neurons, and viability was assessed with the MTS-PMS assay. The presence of both inhibitors did not result in any change in cell viability at any time during infection (Fig. 3A and B). Indeed, there was no statistical difference between the viabilities of infected cells treated with the caspase inhibitors and their respective vehicle (DMSO)-treated infected control cells. Moreover, the difference in cell viability between infected and mock-infected cells remained even in the presence of the inhibitors (Fig. 3A and B). Immunofluorescence analysis confirmed the relative activation of caspase-3 following the infection by both viruses compared to in mock-infected cells, as well as the efficiency of Z-VAD-FMK in inhibiting caspase-3 activation, as no detectable level of activation of this caspase was observed (Fig. 3C). Taken together, our data indicate that initiator caspase-9 and effector caspase-3 were activated by rOC/ATCC and rOC/US183-241 and that their efficient inhibition did not impair or delayed neuronal death, which clearly indicates that neuronal PCD induced by both viruses is not inhibited by Z-VAD-FMK and strongly suggests that caspases are not essential factors in the process.
Fig 2.
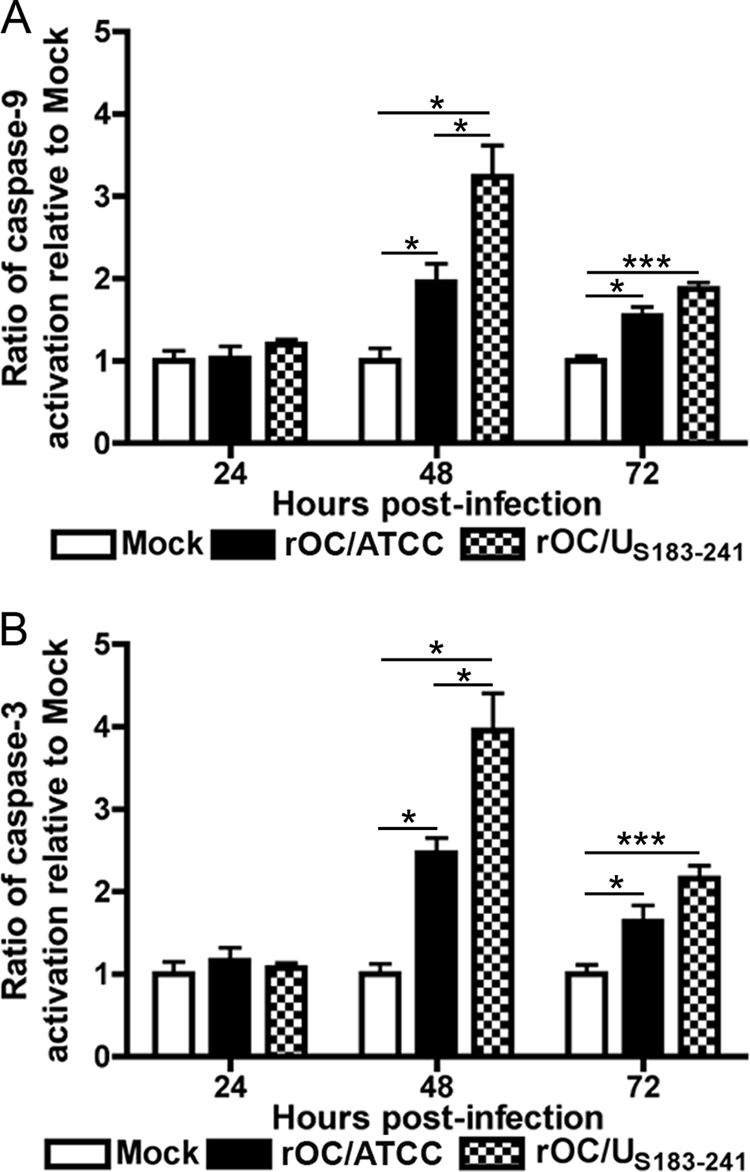
Infection by rOC/ATCC and rOC/US183-241 activates caspase-3 and -9 in human neurons. Differentiated LA-N-5 cells were infected with rOC/ATCC or rOC/US183-241 for 24, 48, or 72 h. The relative activity of caspase-9 was measured using LEHD-pNA (A), and the relative activity of caspase-3 was measured using DEVD-pNA (B); results are expressed as a relative fold increase compared to mock-infected cells. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig 3.

Caspases are activated following infection of human neurons by rOC/ATCC and rOC/US183-241, but the induced programmed cell death is not inhibited by Z-VAD-FMK. (A and B) Differentiated LA-N-5 cells were infected with rOC/ATCC or rOC/US183-241 and treated with either the pan-caspase inhibitor Z-VAD-FMK (VAD) (A) or the caspase-9 inhibitor Z-LEHD-FMK (LEHD) (B) for 24, 48, or 72 h. Viability of infected human neurons was evaluated with the MTS-PMS assay and expressed as relative percentage compared to that of mock-infected cells. (C) Immunofluorescence of active caspase-3. Vehicle-treated (left column) and Z-VAD-FMK-treated (right column) cells were incubated with anti-activated caspase-3 antibody (green). DAPI (blue) insets demonstrate equivalent cell density in the microscope fields analyzed. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Infection by rOC/ATCC and rOC/US183-241 promotes BAX, CytC, and AIF relocalization in human neurons.
The mitochondrion is thought to play a central role in PCD by releasing proapoptotic factors, such as CytC and AIF, after organelle permeabilization (38). Given that BAX is one of the major proapoptotic factors promoting permeabilization of the mitochondrial outer membrane (75), we analyzed its translocation toward the mitochondria. Making use of an antibody which is specific for the activated form of BAX, analysis by immunofluorescence confocal microscopy revealed that activated BAX was translocated to mitochondria at 48 h after infection of neuronal cells by both viruses, as shown by the colocalization of BAX with MitoTracker (Fig. 4A). These results were confirmed by Western immunoblotting analysis of mitochondrial extracts, which indicated an increased amount of BAX in this subcellular fraction (Fig. 4B). Moreover, we demonstrated that this translocation of BAX at 48 h postinfection was concurrent with the release of CytC from the mitochondria (Fig. 4B), indicating an effective permeabilization of mitochondria during the course of infection of humans neurons by both viruses. However, since caspases appear to be dispensable in rOC/ATCC- and rOC/US183-241-induced PCD, we analyzed the translocation of another proapoptotic protein, AIF, which is involved in a different type of neuronal death (3, 46, 74). Using immunofluorescence confocal microscopy, we demonstrated that AIF was effectively translocated from the mitochondria to the nucleus at 48 h postinfection, as shown by the colocalization of AIF with the nuclear marker DRAQ5 (Fig. 4C, arrows), with some residual mitochondrion-located AIF (Fig. 4C, arrowheads). Moreover, this translocation was confirmed by Western immunoblotting analysis of nuclear extracts (Fig. 4D). Altogether, our results demonstrate that in humans neurons infected by rOC/ATCC or rOC/US183-241, BAX was translocated to the mitochondria concurrently with the permeabilization of this organelle and the release of CytC and AIF.
Fig 4.

Infections by rOC/ATCC and rOC/US183-241 promote BAX, CytC, and AIF relocalization in human neurons. Differentiated LA-N-5 cells were infected with rOC/ATCC or rOC/US183-241. (A) Immunofluorescent detection of activated BAX. Cells were incubated with the MitoTracker Red CMXROS (red), fixed, and incubated with an anti-activated BAX antibody (green). Colocalization is represented by merged BAX and MitoTracker Red CMXROS signals (yellow) as indicated by white arrows. (B) Western immunoblotting of mitochondrial BAX and CytC. Mitochondrial protein fractions were subjected to Western immunoblotting analysis using antibodies directed against BAX or CytC. VDAC served as a loading control. (C) Immunofluorescent detection of AIF. Cells were incubated with the MitoTracker Red CMXROS (red), fixed, and incubated with an anti-AIF antibody (green) and DRAQ5 (blue). Nuclear colocalization is represented by merged AIF and DRAQ5 signals (turquoise) as indicated by white arrows, and residual mitochondrial colocalization is represented by merged AIF and MitoTracker (yellow) as indicated by white arrowheads. (D) Western immunoblotting of nuclear AIF. Nuclear protein fractions were subjected to Western immunoblotting analysis using antibodies directed against AIF. p84 served as a loading control.
Human neuronal PCD induced by HCoV-OC43 rOC/ATCC involves cyclophilin D.
Considering that one of the putative partners of BAX in the pore-forming unit at the mitochondria is CypD (48) and that CypD is being associated with numerous neurodegenerative diseases (9, 25, 30, 61, 73), we sought to determine whether chemical inhibition or knocked down expression of CypD could prevent or modulate rOC/ATCC- or rOC/US183-241-induced PCD in infected human neurons. In a first experimental approach, cells were treated with a chemical inhibitor of CypD, cyclosporine (CsA), to inhibit its ability to promote mPTP formation. In a second experimental approach, two stable LA-N-5 cell populations, knocked down for CypD, were generated using shRNA expression vector transduced via lentiviral pseudoparticles. To determine their suitability for CypD knockdown experiments, the selected populations were assessed for their levels of expression of CypD compared to LA-N-5 population transduced with the empty vector (LA-N-5 empty) by quantitative PCR (see Fig. 6F). The LA-N-5 CypD knockdown population K (pop K CypD-kd) expressed only 6.75% of the amount of CypD expressed by LA-N-5 empty (see Fig. 6F). Moreover, the LA-N-5 CypD knockdown population M (pop M CypD-kd) expressed 34.55% of the CypD expressed by LA-N-5 empty (see Fig. 6F). Treatment with CsA or knockdown of CypD expression completely abolished the cytopathic effect (CPE) induced by rOC/ATCC infection, as shown by phase-contrast microscopy at 48 h postinfection (Fig. 5). CsA treatment or CypD knockdown also reduced the retractions of dendrites and axons (Fig. 5, arrows), as well as cell rounding and shrinking (Fig. 5, arrowheads), while promoting the preservation of a characteristic neuronal shape (Fig. 5). It is noteworthy that pop K CypD-kd had greater protection from rOC/ATCC infection-induced CPE than pop M CypD-kd, which is in exact correlation with the level of expression of CypD in these two populations of LA-N-5 cells knocked down for CypD. However, CsA and CypD knockdown only moderately reduced CPE induced by rOC/US183-241, which induced a stronger PCD in human neurons (Fig. 5A). Using the MTS-PMS assay, we confirmed that inhibition of CypD by CsA and CypD knockdown efficiently reduced neuronal death induced by rOC/ATCC (Fig. 6A). Indeed, survival of cells treated with CsA and infected with rOC/ATCC was equal to that of mock-infected CsA-treated cells at all times tested after infection (Fig. 6A). In contrast, CypD inhibition only slightly and transiently improved neuronal survival after infection by rOC/US183-241 at 48 h postinfection, correlating with phase-contrast microscopy results (Fig. 6A). These differences correlated with the level of expression of CypD (Fig. 6F), which was significantly lower in pop K CypD-kd, and demonstrate that protection from cell death was linked to the expression level of CypD. However, CypD inhibition by CsA or knockdown of CypD only moderately and transiently improved neuronal survival after infection by rOC/US183-241 at 48 h postinfection (Fig. 6A and B), correlating with phase-contrast microscopy results showing partial protection against CPE (Fig. 5). Moreover, analysis of DNA fragmentation confirmed that CypD inhibition by CsA led to a significant reduction of fragmented DNA in neurons infected with rOC/ATCC during the course of infection (Fig. 6C), as well as to a reduction of the percentage of TUNEL- and 7-AAD-positive cells (Fig. 6E). Indeed, the pop K CypD-kd infected by rOC/ATCC also showed a significantly reduced DNA fragmentation at all times after infection, which was comparable to that of mock-infected cells, demonstrating that CypD is involved in neuronal death induced by rOC/ATCC (Fig. 6D). On the other hand, pop M CypD-kd infected by rOC/ATCC showed only a moderate reduction of DNA fragmentation at 48 and 72 h postinfection (Fig. 6D). Here again, these differences were in correlation with the relative level of expression of CypD (Fig. 6F), which was significantly lower in pop K CypD-kd, which demonstrates that protection from cell death was linked to the expression level of CypD. However, CsA treatment or knockdown of CypD only moderately delayed DNA fragmentation at 48 or 72 h after infection by the variant harboring two S point mutations (rOC/US183-241) (Fig. 6C and D)). Moreover, CsA treatment also did not significantly reduced the percentage of TUNEL- and 7-AAD-positive cells (Fig. 6E). As the replication of other viruses was shown to be impaired in the presence of CsA (23, 39), we measured the production of intra- and extracellular infectious viral particles and demonstrated that the reduced PCD induced by rOC/ATCC was not due to an impaired production of infectious virus at the concentration used (data not shown). Altogether, our data clearly demonstrate that human neuronal PCD induced by the reference wild-type rOC/ATCC is CypD dependent. However, although inhibition of CypD by CsA or genetic knockdown of CypD appeared to play a role in the rOC/US183-241-induced PCD, the outcome was only a partial inhibition of neuronal death. This suggests that rOC/US183-241-induced PCD does not rely exclusively on CypD.
Fig 6.

Human neuronal PCD induced by rOC/ATCC is cyclophilin D dependent. Wild-type LA-N-5 cells, treated with cyclosporine (CsA) or mock treated (vehicle) for 24, 48, or 72 h, and two LA-N-5 populations knocked down for CypD, pop K CypD-kd (pop K) and pop M CypD-kd (pop M) and the LA-N-5 empty vector population (LAN5 empty) were infected with rOC/ATCC or rOC/US183-241. (A and B) Cell viability of infected wild-type LA-N-5 cells treated with cyclosporine (A) and of infected LA-N-5 K and M populations knocked down for CypD (B) was evaluated with the MTS-PMS assay and is expressed as relative percentage compared to that of mock-infected cells. (C and D) Fragmentation of DNA in infected wild-type LA-N-5 cells treated with cyclosporine (C) and of infected LA-N-5 K and M populations knocked down for CypD (D) was evaluated and expressed as a relative fold increase compared to that in mock-infected cells. (E) FACS TUNEL labeling/7-AAD staining of infected wild-type LA-N-5 cells treated with cyclosporine. Cells were analyzed by FACS at 72 h postinfection. The graph represents percentages of quadrants. Nonsignificant percentages of 7-AAD-positive/TUNEL-negative cells were omitted. (F) Quantification of CypD expression in LA-N-5 knockdown populations. Expression of CypD in the two populations of LA-N-5 neurons knocked down for CypD was evaluated by quantitative PCR and is expressed as a relative percentage compared to that in the LA-N-5 empty vector population. Nonsignificant percentages of 7-AAD-positive/TUNEL-negative cells were omitted. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to corresponding noninfected LA-N-5 population). #, P < 0.05 compared to LA-N-5 empty vector infected with the same virus.
Fig 5.

Cyclosporine treatment and cyclophilin D knockdown protects human neurons from rOC/ATCC-induced programmed cell death. Wild-type LA-N-5 cells were treated with cyclosporine (CsA), and two LA-N-5 populations knocked down for CypD (pop K CypD-kd and pop M CypD-kd) were infected with rOC/ATCC or rOC/US183-241. Images represent phase-contrast microscopy pictures taken at 48 h postinfection. The control LA-N-5 empty cell population is not presented since these cells were comparable to nontreated CsA wild-type LA-N-5 cells. Arrows indicates dendrites and axons. Arrowheads indicate rounding and shrinking bodies.
Chemical inhibition of cyclophilin D alters AIF nuclear translocation in human neurons infected by rOC/ATCC and rOC/US183-241.
AIF has been associated with several neurodegenerative diseases (3, 46, 74). Here we show that it was translocated to the nucleus following infection by both viruses (Fig. 4C [arrows] and D). Moreover, AIF is thought to be mainly involved in neuronal death and caspase-independent PCD (16, 46, 74). Therefore, we analyzed AIF localization after infection of neurons. Immunofluorescence confocal microscopy showed that AIF nuclear translocation was impaired following CypD inhibition by CsA (Fig. 7A). CypD inhibition also slightly favored the retention of AIF and CytC in mitochondria (Fig. 7B) following infection by rOC/ATCC and rOC/US183-241. It is striking to observe that translocation of AIF followed the same kinetics in both rOC/ATCC- and rOC/US183-241-infected-cells, treated or not with CsA. Therefore, one might consider that the stronger PCD induced by rOC/US183-241 in human neurons could use other mechanisms that do not involve CypD or AIF. Nevertheless, taken together, our data strongly suggest that AIF is translocated to the nucleus following infection by both viruses and that the impairment of this process by inhibition of CypD with CsA significantly altered virus-induced cell death.
Fig 7.

Cyclophilin D inhibition alters AIF nuclear translocation in infected neurons. Differentiated LA-N-5 cells were infected with rOC/ATCC or rOC/US183-241 and treated with cyclosporine (CsA). (A) Detection of AIF by immunofluorescence. Cells were fixed and incubated with an anti-AIF antibody (green) and DRAQ5 (blue). Colocalization is represented by merged AIF and DRAQ5 signals (turquoise) as indicated by white arrows. (B) Western immunoblotting of mitochondrial AIF and CytC. Mitochondrial proteins fractions were subjected to Western immunoblotting analysis using antibodies directed against AIF or CytC. VDAC served as a loading control. Intensities of the bands were evaluated with ChemiGenius2 Syngene software and expressed as percentage relative to the loading control. Results are representative of two independent experiments.
DISCUSSION
Using reverse genetics with our cDNA infectious clone pBAC-OC43FL to generate new virus recombinants (64), herein we characterize the type of neuronal death induced by two viral variants, reference wild-type virus (rOC/ATCC) or mutant virus (rOC/US183-241), and identify proapoptotic factors involved in this process. The results reported here define neuronal death induced by rOC/ATCC and rOC/US183-241 as an apoptosis-like programmed cell death that is not inhibited by Z-VAD-FMK and is CypD dependent, with an involvement of AIF. Furthermore, our data confirm that the acquisition of persistence-associated mutations in the HCoV-OC43 S glycoprotein, which leads to an increased neurovirulence in mice (42), also causes enhanced neuronal cytopathic effects, which appear to involve other cell death factors. Analysis of binding studies and of recently obtained crystals of a viral S protein fragment containing the H183R and Y241H mutations suggests that these two mutated amino acids are involved in binding of S to 9-O-acetyl-sialic acid (9-O-acetyl-SA) (M. Desforges, A. Liavonchanka, A. Mansouri, C. Sharon, H. Yu, Y. Chen, X. Chen, P. J. Talbot, and J. M. Rini, unpublished data). This may represent a viral determinant of the fate of infected cells.
Careful attention must be paid when classifying and attributing a characteristic type of cell death (47). The use of recent neologisms (e.g., necropoptosis or aponecrosis) (58) to define nonclassical mechanisms with diverse heterogenous functional markers of apoptosis is bewildering. Based on the recently described NCCD guidelines (47), we found that human neurons infected by rOC/ATCC or rOC/US183-241 underwent cell death associated with modified morphological aspects and specific hallmarks of programmed cell death.
Caspases are known for their involvement in several cellular death pathways (33). We confirmed that infection of neurons by both viruses led to the release of CytC, which is known to be involved in the formation of the apoptosome (8), and to the activation of initiator caspase-9 and effector caspase-3. However, viability assays in the presence of caspase inhibitors revealed that PCD induced by rOC/ATCC and rOC/US183-241 was not inhibited by Z-VAD-FMK, since neuronal survival after infection remained the same as for controls (Fig. 3A and B). The NCCD guidelines (47) and others (15) bring caution in stating that the absence of cell survival protection by Z-VAD-FMK treatment is a consequence of caspase-independent death. It cannot be excluded that undetectable residual caspase activity does remain, even in the presence of Z-VAD-FMK in cultured neurons, or that some caspases could not be inhibited (15). However, even though a slight level of activation of caspases cannot be ruled out in our model, our immunofluorescence data (Fig. 3C) showed that caspase-3 activation in the presence of Z-VAD-FMK was below a detectable level. Therefore, we conclude that PCD induced in neurons by rOC/ATCC and rOC/US183-241 infection is not inhibited by Z-VAD-FMK, while there is a possibility of a caspase-dispensable PCD, which cannot be associated with classical apoptotic mechanisms and is therefore more characteristic of an apoptosis-like PCD (reviewed in reference 47). However, we cannot completely rule out the possibility that caspases play an accessory role during the course of neuronal death. On the other hand, caspase activation has also been reported in other cellular functions unrelated to death (34), such as neuronal plasticity (12) and long-term potentiation (35) or processing of viral proteins after infection by the coronavirus transmissible gastroenteritis virus (26) and influenza virus (80).
Since caspases appear to be nonessential factors in rOC/ATCC- and rOC/US183-241-induced apoptosis-like PCD, we sought to identify other proapoptotic factors potentially involved in this virus-induced neuronal death. AIF is one such factor, which has been shown to be involved in several neurological diseases and caspase-independent neuronal death (3, 46, 74), apparently by promoting high-molecular-weight DNA fragmentation (67). Here we demonstrate that AIF translocation from mitochondria to the nucleus occurred during the course of infection by rOC/ATCC and rOC/US183-241. Moreover, inhibition of CypD by CsA caused an increased mitochondrial retention of AIF, leading to impaired nuclear localization, which was accompanied by almost complete cell survival after infection by rOC/ATCC compared to that of mock-infected cells, and to a slightly delayed neuronal death after infection by rOC/US183-241. These results suggest that AIF is a neuronal death-related factor in rOC/ATCC infection, likely through its involvement in high-molecular-weight fragmentation of DNA. The sustained neuronal death induced by rOC/US183-241 even in the absence of nuclear localization of AIF leads us to speculate that this viral mutant might activate other death factors. For instance, poliovirus infection is known to induce competing death programs that involve canonical cytopathic effect or classic apoptosis (1). In this case, pan-caspase inhibitors promote a switch in death programs leading to cytopathic effect (1, 2, 72). Moreover, it is established that vesicular stomatitis virus (VSV) can induce apoptosis via mitochondrial and caspase-dependent pathways (24). However, VSV could also use alternative death programs, such as passive necrosis, as shown by sustained cell death even in the presence of pan-caspase inhibitors (24).
Alteration of mitochondrial membrane permeability and the release of the proapoptotic factors CytC and AIF are also hallmarks of PCD, in which CypD and BAX represent two major inducing factors, through their possible interaction leading to the formation of pores in the mitochondrial membrane (48). The translocation of BAX toward mitochondria after infection by both viruses, as shown in our study, suggests that it might be involved in inducing mitochondrial membrane permeabilization. Indeed, it was previously suggested that, when localized with the mitochondria, BAX can interact with CypD to promote mPTP formation (48). Our data showing that neurons treated with CsA or knocked down for CypD completely recover their viability following rOC/ATCC infection clearly argue for a contribution of CypD in the apoptosis-like PCD induced by rOC/ATCC in human neurons. Furthermore, the correlation between protection of neuronal viability and the level of CypD knockdown strengthens our conclusion that CypD is involved in the PCD induced by rOC/ATCC and rOC/US183-241. It is worth noting that the involvement of CypD in different types of cell death has retained much attention and remains under considerable debate, mainly regarding its role in apoptosis and necrosis. Indeed, it was reported that CypD overexpression protected the B50 neuronal cell line from apoptosis while promoting necrosis (50). On the other hand, another study showed that overexpression of the proapoptotic factor Apop-1 induced CypD-dependent apoptotic cell death in vascular smooth muscle cells (76). Others have argued that CypD is instead involved in necrotic cell death induced by Ca2+ overload or ROS in mouse embryonic fibroblasts and hepatocytes (6, 57), while CypD-null mice were nearly completely protected from mPTP-related apoptosis (54). Therefore, the apparent discrepancies between these studies investigating the role of CypD in different types of cell death might be reconciled by taking into consideration the cell type- and stimulus-specific function of this important death-regulating factor. Also, the absence of generalized use of recognized definitions established by the National Committee on Cell Death (NCCD) (47) to define or describe the type of cell death observed in a particular situation may amplify such apparent discrepancies. Nevertheless, considering the facts that DNA fragmentation occurred while the cell membrane was still intact (Fig. 1) and that the CypD knockdown cell population was protected from death, we argue that CypD functions as a pro-PCD factor in the type of apoptosis-like PCD induced by rOC/ATCC and rOC/US183-241 infection of neurons. It is striking to observe that neuronal survival following infection varies depending on whether the wild-type reference virus (rOC/ATCC) or the mutant virus (rOC/US183-241) is used. As cited above (Desforges et al., unpublished data), the H183R and Y241H mutations are within the 9-O-acetyl-sialic acid (9-O-acetyl-SA)-binding domain of the viral S protein. Indeed, X-ray crystallographic data indicate that these two residues may be involved in an optimal interaction of the S protein with SA. Furthermore, binding studies strongly suggest that these two S mutations in rOC/US183-241 increase the affinity of S for 9-O-acetyl-SA. Interestingly, other viruses, such as reovirus (18), are able to induce apoptosis in relation to binding with higher affinity to sialic acid domains on their cellular receptors (17). The 9-O-acetyl-SA can be found on different types of glycans, including the GD3 gangliosides (51). This glycolipid is found in different cell membranes, including at the cell surface and at the mitochondria (56). When acetylated, this ganglioside is beneficial to the membranes, but when the acetylated group is removed, the GD3 molecule acquires pro-PCD properties (28). Indeed, deacetylation of GD3, by either the hemagglutinin esterase (HE) protein of influenza C virus (10) or the cellular O-acetylesterase (69), is known to induce the capacity of this ganglioside to participate in the opening of the mPTP, with the involvement of BAX, and to promote the release of cell death proteins, such as CytC and AIF (52, 60, 63). Moreover, we reported previously that the rOC/US183-241 variant produced increased amounts of viral proteins and at least 10 times more infectious virus within infected human neurons and led to an increased activation of the unfolded-protein response (27). Considering this increased number of infectious virions, all harboring S proteins, in the infected cells, the apparent higher affinity of rOC/US183-241 S protein for 9-O-acetyl sialic acid, and the presence of an active HE protein in the HCoV-OC43 envelope (M. Desforges, J. Desjardins, and P. J. Talbot, unpublished data), which is known to remove the 9-O-acetyl-ester group on sialic acid (C. Zhang, personal communication), it is likely that rOC/US183-241 infection may lead to an increased cleavage of 9-O-acetyl GD3 by the larger amount of HE protein molecules present in the infected cell and produce a larger amount of the pro-PCD deacetylated GD3 gangliosides than rOC/ATCC infection. The increase of deacetylated GD3 that promotes mPTP formation could participate in a stronger insult at the mitochondria, in addition to the role of CypD, to allow the release of CytC and AIF (31, 63). Therefore, we speculate that rOC/US183-241, a more neurovirulent mutant (42), could promote increased GD3 deacetylation through its HE viral protein and induce a much stronger cellular insult, which might circumvent CypD inhibition or knockdown and promote the increased cell death observed compared to rOC/ATCC. Our results suggest that virus persistence-associated mutations in the viral S protein led to a modification of the underlying mechanisms involved in neuronal death.
In summary, the results of the study presented here strongly suggest that rOC/ATCC and rOC/US183-241 induce a neuronal death characteristic of an apoptosis-like programmed cell death, which is not inhibited by Z-VAD-FMK, accompanied by an accessory and dispensable activation of major initiator caspase-9 and effector caspase-3 and a concomitant release of CytC from mitochondria. Furthermore, we demonstrate that neuronal death induced by rOC/ATCC is CypD dependent, consistent with a proapoptotic role of CypD in this human neuronal model. In addition, our data also strongly suggest a role for AIF in rOC/ATCC-induced neuronal death. Moreover, the use of a mutant virus (rOC/US183-241) which harbors persistence-associated mutations in the viral S protein (65) and possesses an enhanced capacity to accumulate in infected cells (27) and an apparent higher affinity for 9-O-acetyl-sialic acid points toward a central role for the viral S glycoprotein, which could indirectly influence the capacity of the viral HE protein to regulate the fate of 9-O-acetylated pro-PCD molecules such as GD3 in neuronal cell death pathways. Further studies to characterize the early events of PCD leading to CypD activation and the possible involvement of the S protein of rOC/US183-241 in 9-O-acetyl-sialic acid metabolism and related mechanisms of cell death, such as GD3 deacetylation, in human neurons are ongoing.
ACKNOWLEDGMENTS
This work was supported by grant MT-9203 from the Institute of Infection and Immunity (III) of the Canadian Institutes of Health Research (CIHR) to Pierre J. Talbot, who is the holder of the Tier 1 (Senior) Canada Research Chair in Neuroimmunovirology award. Dominique J. Favreau acknowledges a doctoral studentship from the Fonds de la recherche en santé du Québec (FRSQ).
Footnotes
Published ahead of print 19 October 2011
REFERENCES
- 1. Agol VI, et al. 1998. Two types of death of poliovirus-infected cells: caspase involvement in the apoptosis but not cytopathic effect. Virology 252:343–353 [DOI] [PubMed] [Google Scholar]
- 2. Agol VI, et al. 2000. Competing death programs in poliovirus-infected cells: commitment switch in the middle of the infectious cycle. J. Virol. 74:5534–5541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alano CC, et al. 2010. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30:2967–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arbour N, Day R, Newcombe J, Talbot PJ. 2000. Neuroinvasion by human respiratory coronaviruses. J. Virol. 74:8913–8921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arbour N, et al. 1999. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J. Virol. 73:3338–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baines CP, et al. 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662 [DOI] [PubMed] [Google Scholar]
- 7. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. 2007. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9:550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baliga B, Kumar S. 2003. Apaf-1/cytochrome c apoptosome: an essential initiator of caspase activation or just a sideshow? Cell Death Differ. 10:16–18 [DOI] [PubMed] [Google Scholar]
- 9. Barrientos SA, et al. 2011. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 31:966–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Birks SM, et al. 2011. Targeting the GD3 acetylation pathway selectively induces apoptosis in glioblastoma. Neuro-oncology 13:950–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonavia A, Arbour N, Yong VW, Talbot PJ. 1997. Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J. Virol. 71:800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bravarenko NI, et al. 2006. Caspase-like activity is essential for long-term synaptic plasticity in the terrestrial snail Helix. Eur. J. Neurosci. 23:129–140 [DOI] [PubMed] [Google Scholar]
- 13. Brison E, Jacomy H, Desforges M, Talbot PJ. 2011. Glutamate excitotoxicity is involved in the induction of paralysis in mice after infection by a human coronavirus with a single point mutation in its spike protein. J. Virol. 85:12362–12375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchmeier MJ, Dalziel RG, Koolen MJ, Lampert PW. 1987. Molecular determinants of CNS virulence of MHV-4. Adv. Exp. Med. Biol. 218:287–295 [DOI] [PubMed] [Google Scholar]
- 15. Chauvier D, Ankri S, Charriaut-Marlangue C, Casimir R, Jacotot E. 2007. Broad-spectrum caspase inhibitors: from myth to reality? Cell Death Differ. 14:387–391 [DOI] [PubMed] [Google Scholar]
- 16. Cheung EC, et al. 2005. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J. Neurosci. 25:1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clarke P, Tyler KL. 2003. Reovirus-induced apoptosis: a mini-review. Apoptosis 8:141–150 [DOI] [PubMed] [Google Scholar]
- 18. Connolly JD, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 75:4029–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cory AH, Owen TC, Barltrop JA, Cory JG. 1991. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 3:207–212 [DOI] [PubMed] [Google Scholar]
- 20. Cregan SP, et al. 2002. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 158:507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crompton M, Barksby E, Johnson N, Capano M. 2002. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 84:143–152 [DOI] [PubMed] [Google Scholar]
- 22. Crompton M, Virji S, Ward JM. 1998. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 258:729–735 [DOI] [PubMed] [Google Scholar]
- 23. Damaso CR, Keller SJ. 1994. Cyclosporin A inhibits vaccinia virus replication in vitro. Arch. Virol. 134:303–319 [DOI] [PubMed] [Google Scholar]
- 24. Desforges M, et al. 2002. Matrix protein mutations contribute to inefficient induction of apoptosis leading to persistent infection of human neural cells by vesicular stomatitis virus. Virology 295:63–73 [DOI] [PubMed] [Google Scholar]
- 25. Du H, et al. 2008. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 14:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eleouet JF, et al. 2000. The viral nucleocapsid protein of transmissible gastroenteritis coronavirus (TGEV) is cleaved by caspase-6 and -7 during TGEV-induced apoptosis. J. Virol. 74:3975–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Favreau DJ, Desforges M, St Jean JR, Talbot PJ. 2009. A human coronavirus OC43 variant harboring persistence-associated mutations in the S glycoprotein differentially induces the unfolded protein response in human neurons as compared to wild-type virus. Virology 395:255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernández-Checa JC. 2003. Redox regulation and signaling lipids in mitochondrial apoptosis. Biochem. Biophys. Res. Commun. 304:471–479 [DOI] [PubMed] [Google Scholar]
- 29. Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. 1989. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337:476–478 [DOI] [PubMed] [Google Scholar]
- 30. Forte M, et al. 2007. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 104:7558–7563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. García-Ruiz C, Colell A, París R, Fernández-Checa JC. 2000. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition, cytochrome c release, and caspase activation. FASEB J. 14:847–858 [DOI] [PubMed] [Google Scholar]
- 32. Giorgio V, et al. 2010. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 1797:1113–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grutter MG. 2000. Caspases: key players in programmed cell death. Curr. Opin. Struct. Biol. 10:649–655 [DOI] [PubMed] [Google Scholar]
- 34. Gulyaeva NV. 2003. Non-apoptotic functions of caspase-3 in nervous tissue. Biochemistry Mosc. 68:1171–1180 [DOI] [PubMed] [Google Scholar]
- 35. Gulyaeva NV, Kudryashov IE, Kudryashova IV. 2003. Caspase activity is essential for long-term potentiation. J. Neurosci. Res. 73:853–864 [DOI] [PubMed] [Google Scholar]
- 36. Halestrap AP, Brenner C. 2003. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 10:1507–1525 [DOI] [PubMed] [Google Scholar]
- 37. Halestrap AP, Davidson AM. 1990. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 268:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Halestrap AP, McStay GP, Clarke SJ. 2002. The permeability transition pore complex: another view. Biochimie 84:153–166 [DOI] [PubMed] [Google Scholar]
- 39. Henry SD, et al. 2006. Mycophenolic acid inhibits hepatitis C virus replication and acts in synergy with cyclosporin A and interferon-alpha. Gastroenterology 131:1452–1462 [DOI] [PubMed] [Google Scholar]
- 40. Hill DP, Robertson KA. 1998. Differentiation of LA-N-5 neuroblastoma cells into cholinergic neurons: methods for differentiation, immunohistochemistry and reporter gene introduction. Brain Res. Protoc. 2:183–190 [DOI] [PubMed] [Google Scholar]
- 41. Jacomy H, Fragoso G, Almazan G, Mushynski WE, Talbot PJ. 2006. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/c mice. Virology 349:335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jacomy H, et al. 2010. Mutations in the spike glycoprotein of human coronavirus OC43 modulate disease in BALB/c mice from encephalitis to flaccid paralysis and demyelination. J. Neurovirol. 16:279–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jacomy H, Talbot PJ. 2003. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology 315:20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joza N, et al. 2001. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410:549–554 [DOI] [PubMed] [Google Scholar]
- 45. Kokoszka JE, et al. 2004. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427:461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krantic S, Mechawar N, Reix S, Quirion R. 2007. Apoptosis-inducing factor: a matter of neuron life and death. Prog. Neurobiol. 81:179–196 [DOI] [PubMed] [Google Scholar]
- 47. Kroemer G, et al. 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16:3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumarswamy R, Chandna S. 2009. Putative partners in Bax mediated cytochrome-c release: ANT, CypD, VDAC or none of them? Mitochondrion 9:1–8 [DOI] [PubMed] [Google Scholar]
- 49. Lambert F, Jacomy H, Marceau G, Talbot PJ. 2008. Titration of human coronaviruses, HCoV-229E and HCoV-OC43, by an indirect immunoperoxidase assay. Methods Mol. Biol. 454:93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li Y, Johnson N, Capano M, Edwards M, Crompton M. 2004. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem. J. 383:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50a. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 25:402–408 [DOI] [PubMed] [Google Scholar]
- 51. Maccioni HJ, Daniotti JL, Martina JA. 1999. Organization of ganglioside synthesis in the Golgi apparatus. Biochim. Biophys. Acta 1437:101–118 [DOI] [PubMed] [Google Scholar]
- 52. Malisan F, et al. 2002. Acetylation suppresses the proapoptotic activity of GD3 ganglioside. J. Exp. Med. 196:1535–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin LJ. 2010. The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta 1802:186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin LJ, Adams NA, Pan Y, Price A, Wong M. 2011. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J. Neurosci. 31:359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mounir S, Talbot PJ. 1992. Sequence analysis of the membrane protein gene of human coronavirus OC43 and evidence for O-glycosylation. J. Gen. Virol. 73:2731–2736 [DOI] [PubMed] [Google Scholar]
- 56. Mukherjee K, et al. 2008. O-acetylation of GD3 prevents its apoptotic effect and promotes survival of lymphoblasts in childhood acute lymphoblastic leukaemia. J. Cell. Biochem. 105:724–734 [DOI] [PubMed] [Google Scholar]
- 57. Nakagawa T, et al. 2005. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–658 [DOI] [PubMed] [Google Scholar]
- 58. Nicotera P, Melino G. 2004. Regulation of the apoptosis-necrosis switch. Oncogene 23:2757–2765 [DOI] [PubMed] [Google Scholar]
- 59. Pleasure SJ, Lee VM. 1993. NTera 2 cells: a human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J. Neurosci. Res. 35:585–602 [DOI] [PubMed] [Google Scholar]
- 60. Rippo MR, et al. 2000. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 14:2047–2054 [DOI] [PubMed] [Google Scholar]
- 61. Schinder AF, Olson EC, Spitzer NC, Montal M. 1996. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16:6125–6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schinzel AC, et al. 2005. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U. S. A. 102:12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scorrano L, Petronilli V, Di Lisa F, Bernardi P. 1999. Commitment to apoptosis by GD3 ganglioside depends on opening of the mitochondrial permeability transition pore. J. Biol. Chem. 274:22581–22585 [DOI] [PubMed] [Google Scholar]
- 64. St Jean JR, et al. 2006. Recovery of a neurovirulent human coronavirus OC43 from an infectious cDNA clone. J. Virol. 80:3670–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. St Jean JR, Desforges M, Talbot PJ. 2006. Genetic evolution of human coronavirus OC43 in neural cell culture. Adv. Exp. Med. Biol. 581:499–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. St Jean JR, et al. 2004. Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J. Virol. 78:8824–8834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Susin SA, et al. 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441–446 [DOI] [PubMed] [Google Scholar]
- 68. Takahashi N, Hayano T, Suzuki M. 1989. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 337:473–475 [DOI] [PubMed] [Google Scholar]
- 69. Takematsu H, Diaz S, Stoddart A, Zhang Y, Varki A. 1999. Lysosomal and cytosolic sialic acid 9-O-acetylesterase activities can be encoded by one gene via differential usage of a signal peptide-encoding exon at the N terminus. J. Biol. Chem. 274:25623–25631 [DOI] [PubMed] [Google Scholar]
- 70. Talbot PJ, Jacomy H, Desforges M. 2008. Pathogenesis of human coronaviruses other than severe acute respiratory syndrome coronavirus, p 313–324. In Perlman S, Gallagher T, Snijder EJ. (ed), Nidoviruses. ASM Press, Washington, DC [Google Scholar]
- 71. Teshima Y, et al. 2003. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar granule neurons from apoptosis induced by oxidative stress. Stroke 34:1796–1802 [DOI] [PubMed] [Google Scholar]
- 72. Tolskaya EA, et al. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 69:1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vanderluit JL, McPhail LT, Fernandes KJ, Kobayashi NR, Tetzlaff W. 2003. In vivo application of mitochondrial pore inhibitors blocks the induction of apoptosis in axotomized neonatal facial motoneurons. Cell Death Differ. 10:969–976 [DOI] [PubMed] [Google Scholar]
- 74. Wang H, et al. 2004. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 24:10963–10973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wei MC, et al. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yasuda O, et al. 2006. Apop-1, a novel protein inducing cyclophilin D-dependent but Bax/Bak-related channel-independent apoptosis. J. Biol. Chem. 281:23899–23907 [DOI] [PubMed] [Google Scholar]
- 77. Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H. 2004. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 113:e73–76 [DOI] [PubMed] [Google Scholar]
- 78. Yu SW, Wang H, Dawson TM, Dawson VL. 2003. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 14:303–317 [DOI] [PubMed] [Google Scholar]
- 79. Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS. 2008. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J. Biol. Chem. 283:3476–3486 [DOI] [PubMed] [Google Scholar]
- 80. Zhirnov OP, Konakova TE, Garten W, Klenk H. 1999. Caspase-dependent N-terminal cleavage of influenza virus nucleocapsid protein in infected cells. J. Virol. 73:10158–10163 [DOI] [PMC free article] [PubMed] [Google Scholar]