

Abstract
The first total synthesis of the marine toxin (−)-gymnodimine (1) has been accomplished in a convergent manner. A highly diastereo- and enantioselective exo-Diels–Alder reaction catalyzed by a bis-oxazoline Cu(II) catalyst enabled rapid assembly of the spirocyclic core of gymnodimine. The preparation of the tetrahydrofuran fragment utilized a chiral auxiliary based anti-aldol reaction. Two major fragments, spirolactam 56 and tetrahydrofuran 55, were then coupled through an efficient Nozaki–Hiyama–Kishi reaction. An unconventional, ambient temperature t-BuLi-initiated intramolecular Barbier reaction of alkyl iodide 64 was employed to form the macrocycle. A late stage vinylogous Mukaiyama aldol addition of a silyloxyfuran to a complex cyclohexanone 83 appended the butenolide and a few additional steps provided (−)-gymnodimine (1). A diastereomer of the natural product was also synthesized, C4-epi-gymnodimine (90), derived from the vinylogous Mukaiyama aldol addition.
Introduction
Among the various bioactive marine natural products that have been isolated and studied, marine toxins occupy a very unique, albeit notorious, position. Often produced by microalgae, especially dinoflagellates, marine toxins are the culprits of numerous cases of massive fish kills, marine wildlife death, and even human intoxication due to consumption of contaminated seafood.1 Despite the negative societal and economical impacts brought by these incidents, many of these naturally-occurring toxins including tetrodotoxin,2a domoic acid,2b and okadaic acid2c, 2d have served as extremely valuable tools for probing biological or pharmacological phenomena, primarily in the area of the human nervous system.2 For this reason, studies of marine toxins continue to be the subject of extensive research worldwide in a number of disciplines from chemistry to cell biology.
One such emerging class of marine toxins is the spirocyclic imine containing natural products,3 with representative examples including gymnodimine (1, Figure 1), spirolides (2),4 pinnatoxins (3),5 pteriatoxins,6 and spiro-prorocentrimine.7 Gymnodimine (1) has a special place in this family of spirocyclic imine toxins as it possesses a 6,6-spirocyclic imine, an elaborate butenolide appendage, and is also one of the first members isolated. Yasumoto and coworkers first isolated gymnodimine in 1995 from oysters Tiostrea chilensis collected off the coast of New Zealand and the gross structure was initially assigned based on extensive NMR studies.8 Subsequently, Munro, Blunt and coworkers established the relative and the absolute configuration of gymnodimine through X-ray crystallographic analysis of a N-acylated derivative of an imine reduced gymnodimine.9 More recently, two new analogs, gymnodimines B (4) and C (5), were isolated that include an apparent allylic oxidation with olefin transposition.10
Figure 1.
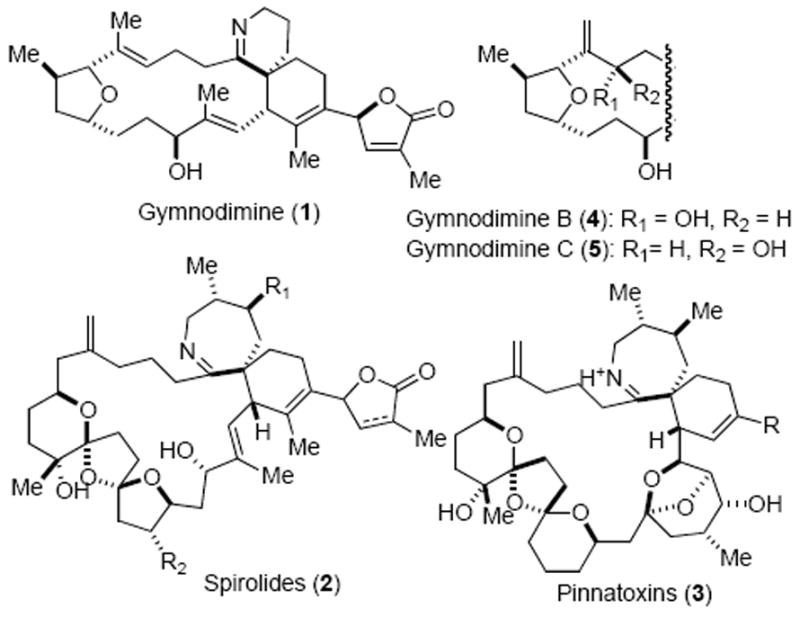
Structures of Representative Spirocyclic-imine-containing Marine Natural Products
Following its initial isolation off the coasts of New Zealand, gymnodimine has also been identified in waters surrounding Tunisia, South Africa, Europe and North American coasts likely due in part to transport via ballast water by the shipping industry.11 The worldwide occurance of gymnodimine and other marine toxins in ocean waters poses serious concerns over the potential impact to seafood safety. Indeed, gymnodimine has been associated with several incidents of neurotoxic shellfish poisoning in New Zealand.12 Gymnodimine exhibited high toxicity (LD50: 96 μg/kg by injection; 755 μg/kg by oral administration), however the toxicity was significantly reduced when administered with food.13 More recent studies demonstrated that gymnodimine, along with 13-desmethyl spirolide C and pinnatoxin A, exerts its toxic effects via binding to nicotinic acetylcholine receptors with picomolar affinities with no sign of apparent reversibility in short time frames.14 The toxicity of gymnodimine has been attributed, at least in part, to the imine functionality as its reduction leads to a non-toxic derivative.9 This hypothesis was further supported by the loss of toxicity when the imine functionality of the related toxin, spirolide B, was reduced to an amine, or in the case of spirolides E and F which bear a hydrolyzed imine ring.4b Based on recent X-ray crystal structures of gymnodimine and 13-desmethyl spirolide C bound to acetylcholine binding proteins, the origin of the pharmacological effect of the spirocyclic imine appears to be a key hydrogen bond between the protonated cyclic imine and a tryptophan residue that positions these natural toxins within the center of the binding pocket.14
This family of spirocyclic-imine-containing marine toxins has spurred intense synthetic efforts that have culminated in total or formal syntheses of the pinnatoxins by the groups of Kishi, Hirama, and Zakarian15, 14b and pteriatoxins by Kishi.16 Synthetic studies toward the spirolides have also been described.17 However, the total synthesis of gymnodimine remained elusive prior to our communication in 2009.18 The challenging structural elements of gymnodimine include a trisubstituted tetrahydrofuran within a 16-membered carbocyclic macrocycle, an azaspiro[5.5]undecadiene moiety, and a labile chiral butenolide. The total synthesis of gymnodimine and analogs will enable more detailed SAR studies of spirocyclic imine marine toxins. In addition, efficient syntheses of various fragments of gymnodimine would facilitate the development of an efficient enzyme-linked immunosorbent assay (ELISA) for detection of gymnodimine and congeners. Herein, we describe the full details of our synthetic endeavours toward this class of marine toxins, which has led to the first total synthesis of (−)-gymnodimine and (+)-C4-epi-gymnodimine.19
Results and Discussions
Retrosynthetic Analysis
Mindful of the known sensitivity of the butenolide moiety of gymnodimine to both basic and even neutral conditions,20 we decided to delay the introduction of this functionality until the late stages of the synthesis (Scheme 1). This would have the additional benefit of maximizing convergency and avoiding unnecessary adjustments of the oxidation state of the butenolide. From a retrosynthetic perspective, we initially projected a late-stage coupling to assemble the C9–C10 bond to close the 16-membered macrocycle. Among numerous possibilities considered, we were attracted to the Nozaki–Hiyama–Kishi (NHK) coupling due to its well-established functional group compatibility21 and its prior use in macrocylizations.22 While some degree of stereocontrol at the C10 carbinol center might be expected during this intramolecular NHK coupling, due to minimization of eclipsing interactions of the intermediate vinyl chromium species,22 neither the stereoselectivity nor the relative stereochemical outcome of this macrocyclization reaction could be predicted initially. In the worst-case scenario, an inversion process would be required to correct the stereochemistry of a C10 diastereomer.23 Further disconnection at the C20–C21 bond would lead to two precursors of similar complexity, namely tetrahydrofuran 6 and spirolactam 7. In the forward sense, these two fragments would be joined at C20/C21 through a nucleophilic addition of the tetrahydrofuran fragment to the δ-lactam carbonyl, with a number of possible strategies envisioned. At the outset of our studies, we recognized the challenges associated with this strategic bond disconnection given the poorly electrophilic and sterically congested nature of the C21 lactam carbonyl bearing an α-quaternary carbon. As described below, the eventual realization of such a disconnection became a primary hallmark of our synthesis.
Scheme 1.
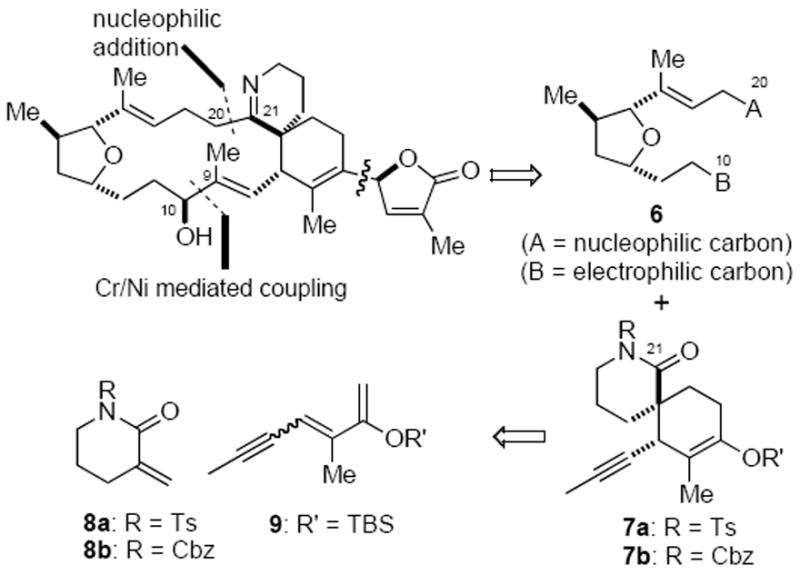
Initial Synthetic Plan for Gymnodimine
A Diels–Alder reaction appeared well suited for the preparation of spirolactam 7, leading back to α-methylene lactam 8 and diene 9 as precursors. The utility of a catalytic, enantioselective Diels–Alder process was attractive given that it had rarely been used in natural product total synthesis at the time we initiated our studies.24
Synthesis of the Tetrahydrofuran Fragment 6
In contemplating a synthetic plan towards the tetrahydrofuran fragment 6, two approaches were considered and evaluated (Scheme 2). In the first approach (‘approach a’, Scheme 2), the bicyclic ketone 10, bearing two of the three stereogenic centers necessary for the formation of fragment 6, was recognized as a good starting point.25 Conceivably, the methyl group at C15 could be introduced in a diastereoselective fashion taking advantage of the bicyclic nature of 10, while the olefin in 10 would serve as a latent ketoaldehyde 11 to ultimately reveal a cis-substituted tetrahydrofuran bearing the C13 and C16 stereocenters. Adding to the appeal of this approach was the ease of preparing known ketone 10 and its availability in high enantiomeric excess through an oxazaborolidine-catalyzed enantioselective Diels–Alder reaction of furan with α-bromoacrolein.26 Alternatively, use of acyclic stereocontrol to access tetrahydrofuran 6 would initially involve an anti-aldol reaction between carboximide 1427 and aldehyde 1528 (‘approach b’, Scheme 2) followed by a diastereoselective allylation of furanose 12a.
Scheme 2.
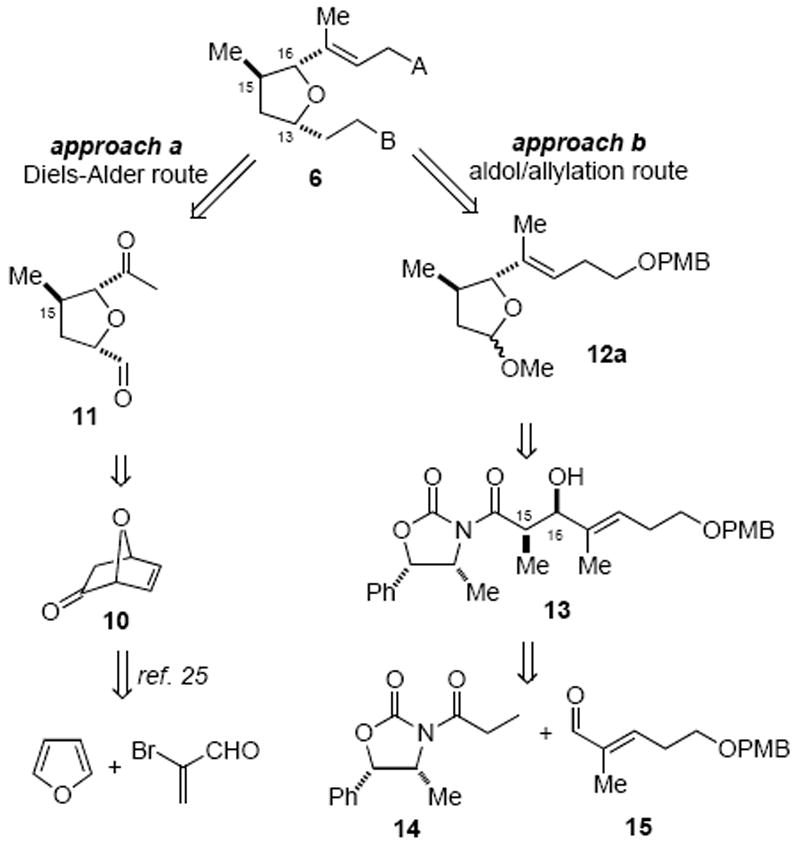
Strategies for the Synthesis of Tetrahydrofuran 6
Our initial explorations of the Diels–Alder strategy toward tetrahydrofuran 6 began with the known ketone 16, readily prepared in racemic form from furan and α-acetoxyacrylonitrile in three steps (Scheme 3).29 While methylation of the enolate derived from ketone 16 was initially complicated by incomplete conversion and self-condensation, considerable experimentation revealed conditions (LiHMDS, DMPU, MeI) to generate methyl ketone 17 in a reproducible manner with high diastereoselectivity and acceptable yield. The exo stereochemistry of the methyl group in ketone 17 was judged from the absence of the vicinal coupling between Ha and Hb (dihedral angle ~80 °). Reduction of the ketone 17 with NaBH4 delivered a 4:1 mixture of epimeric alcohols with the endo product 18 isolated in 72% yield. Oxidative elimination of the phenylseleno group afforded the vinyl bromide 19a. However, our extensive efforts to substitute the bromine for a methyl group were unsuccessful in addition to attempted deoxygenation of the thiocarbonate 19b. Therefore, we abandoned this approach and resorted to the alternative approach involving initial acyclic stereocontrol to set the stereochemistry of the tetrahydrofuran 6.
Scheme 3.
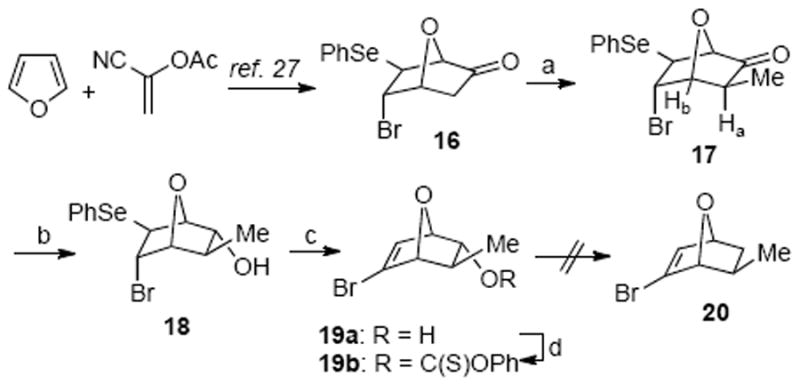
Attempted Synthesis of the Tetrahydrofuran Fragment via a Diels–Alder Approacha
a Reagents and conditions: (a) LiHMDS, MeI, DMPU, THF, −78 → 22 °C, 63%; (b) NaBH4, MeOH/THF, 72%, 0 °C; (c) H2O2, 62%; (d) PhOC(S)Cl, py., CH2Cl2, 93%.
The successful synthesis of tetrahydrofuran 6 commenced with an anti-aldol reaction according to the protocol developed by Heathcock,30 which set the C15/C16 stereogenic centers (Scheme 4). While a mixture of three diastereomers was obtained, the major diastereomer 13 could be isolated in 66% yield and possessed the desired anti stereochemistry. The relative stereochemistry was initially deduced from coupling constant analysis (i.e. H15 and H16, J = 8.7~9.3 Hz)28 and was subsequently confirmed by X-ray crystallographic analysis (Scheme 4, inset).31a Among various protocols examined to cleave the auxiliary, sodium methoxide proved to be the most efficient in delivering the corresponding methyl ester (80% yield). Following standard transformations, the ester was converted to the methyl enol ether 21 in good overall yield and cyclization with acidic methanol gave methoxy furanose 12a. Treatment of the latter with allyl trimethylsilane in the presence of BF3·OEt2 provided the allylated furan 22 in moderate diastereoselectivity (dr, 4:1), a result consistent with Woerpel’s “inside attack” model.32 Extensive screening of reaction parameters including Lewis acid, solvent, and silanes failed to provide any noticeable improvement in diastereoselectivity. Treatment of acetoxyfuranose 12b, derived from hydrolysis followed by acetylation of furanose 12a, with chiral allylating reagents (e.g. Brown’s Ipc2BAll reagent) in attempts to achieve a double diastereoselective reaction only led to marginal enhancement of the diastereoselectivity (dr, 5:1) and low yield (10%).33 Thus, the inseparable mixture of allyl diastereomers 22 was carried forward and a site selective hydroboration followed by oxidation and silylation provided the tetrahydrofuran fragment 23.
Scheme 4.
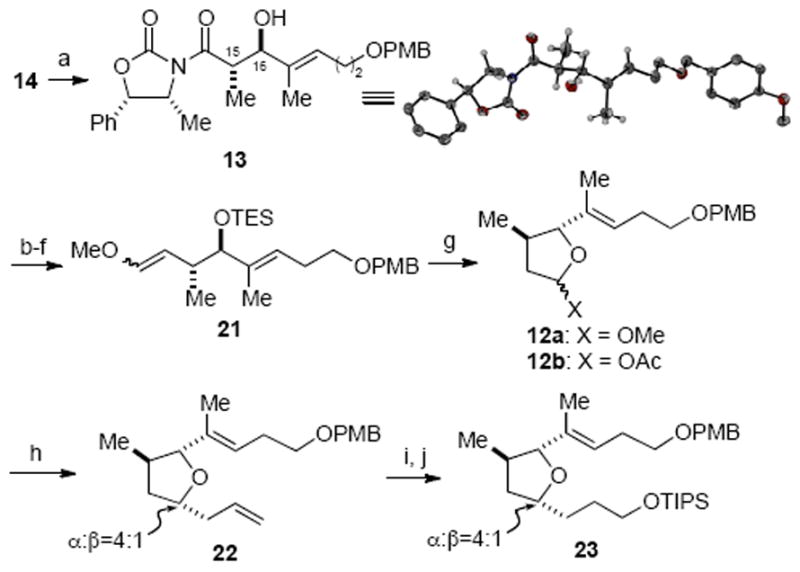
Synthesis of Tetrahydrofuran Fragment 23 via an anti-Aldol Reactiona
aReagents and conditions: (a) n-Bu2BOTf (2.0 equiv), i-Pr2NEt, Et2O, then 15, −78 °C, 68%; (b) NaOMe, MeOH, 80%; (c) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C, 90%; (d) DIBAL, CH2Cl2, −78 °C, 93%; (e) (COCl)2, DMSO, Et3N, CH2Cl2, −78 → 22 °C, 99%; (f) Ph3PCH2(OMe)Cl, KOtBu, THF, 97%; (g) p-TSA, MeOH, 86%; (h) X = OMe, allyl trimethylsilane, BF3·OEt2, PhMe/CH2Cl2 (1 : 1), −78 °C, 71%, α : β = 4 : 1; (i) 9-BBN, THF; H2O2, NaOH, 72%; (j) TIPSOTf, 2,6-lutidine, CH2Cl2, 99% (inset: ORTEP representation of X-ray crystal structure of 13).
Synthesis of the Spirolactam Fragment 7a
We initially studied the Diels–Alder reaction toward racemic spirolactam to determine the reactivity of the proposed diene and dienophile. Thus, silyloxy diene (Z)-24 was prepared from 2,4-hexadiyne in a highly stereoselective fashion featuring a (Z)-selective hydrotelluration reaction (Scheme 5).19b Diene (Z)-24 underwent a smooth cycloaddition reaction with N-tosyl lactam 8a in the presence of Et2AlCl to provide the spirolactam (±)-7a in good yield (67%) and excellent diastereoselectivity (dr > 95 : 5). The stereochemical outcome of the Diels–Alder reaction was stereoconvergent with respect to the diene geometry as the isomeric (E)-diene 24 provided the same adduct 7a in comparable yield under identical conditions (Et2AlCl, −30 °C) suggestive of a stepwise mechanism for this Diels–Alder process.19b
Scheme 5.
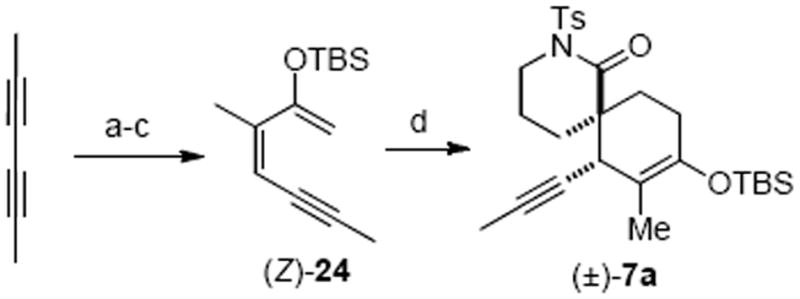
Diastereoselective Synthesis of Spirolactam (±)-7aa
aReagents and conditions: (a) Bu2Te2, NaBH4, EtOH, reflux, 77%; (b) n-BuLi, N-methyl-N-methoxyacetamide, THF, −78 °C, 90%; (c) NaHMDS, TBSOTf, THF, −78 °C, 82%; (d) 8a, Et2AlCl, CH2Cl2, −30 °C, 67%, dr > 95 : 5.
Our initial extrapolation of the asymmetric Diels–Alder variant with lactam 8 and (Z)-diene 24 uniformly met with failure providing no cycloadduct with a variety of chiral catalysts and δ-lactam N-activating groups but rather only desilylation of the diene. Reasoning that the olefin geometry may be responsible for the lack of reactivity of the diene under these conditions, due to interactions of the catalyst and (Z)-diene 24, we pursued a diastereoselective synthesis of (E)-diene 24. Use of radical hydrostannylation or Pd-catalyzed hydrostannylation was unsatisfactory, providing the undesired geometrical isomer 25b and regioisomer 25c as the major products, respectively (Table 1, entries 1 and 2). Following extensive optimization, we ultimately found that stannylcupration of 2,4-hexadiyne at low temperature gave (E)-vinyl stannane 25a in good regio- and diastereoselectivity (Table 1, entry 3). Adding methanol as a proton source gave a slightly improved yield but the regio- and diastereoselectivity were diminished (entry 4).34 The vinyl stannane 25a was then converted to silyloxy diene (E)-24 following metallation/acylation and silylation (Scheme 6). We were pleased to find that in the presence of the [Cu(box)](SbF6)2 Lewis acid developed by Evans,35 diene (E)-24 participated in a highly efficient Diels–Alder reaction with the bidentate N-Cbz lactam 8b, providing spirolactam 7b in 85% yield and high diastereoselectivity.36 After disclosing our initial results,19i we found that the efficiency of the Diels–Alder reaction could be further improved by adapting an improved procedure for the preparation of the catalyst37 and by omitting molecular sieves. Under these conditions, the catalyst loading could be decreased to 10 mol% without detriment to yield or enantioselectivity.
Table 1.
Synthesis of (E)-Vinyl Stannane 25a from 2,4-Hexadiyne
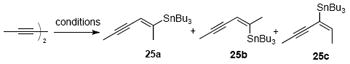 | |||
|---|---|---|---|
| entry | conditions | % yielda | ratiob (25a:25b:25c) |
| 1 | n-Bu3SnH, AIBN, PhMe, 80 °C | 90 | 1:1.4:0 |
| 2 | n-Bu3SnH, PdCl2(PPh3)2, THF | 90 | 0:0:1 |
| 3 | (n-Bu3Sn)2CuCNLi2, THF, −78 °C | 67c | 7:0:1 |
| 4 | (n-Bu3Sn)2CuCNLi2, MeOH, THF, −78 °C | 86 | 5:2:1 |
Yields refer to isolated, purified yields of the mixture of isomers.
Ratios were determined by 1H NMR (300 MHz) of the crude reaction mixtures.
Yield refers to the major isomer 25a.
Scheme 6.
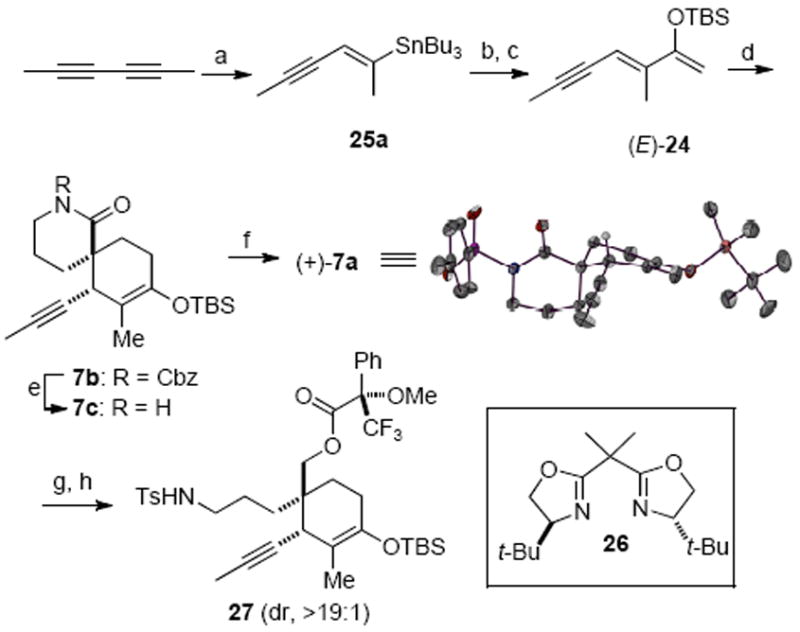
Synthesis of Spirolactam (+)-7aa
aReagents and conditions: (a) (n-Bu3Sn)2CuCNLi2, THF, −78 °C, 67%; (b) n-BuLi, N-methyl-N-methoxyacetamide, THF, −78 °C, 77%; (c) Et3N, TBSOTf, CH2Cl2, −78 °C, 90%; (d) 26 (11 mol%), AgSbF6 (20 mol%), CuCl2 (10 mol%), 8b, CH2Cl2, 85%, exo : endo > 95 : 5, 95% ee (major diast.); (e) n-BuLi, THF, −78 °C, 82%; (f) KHMDS, TsCl, THF, 80%; (g) LiBH4, THF, 91%; (h) (S)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetic acid, DCC, DMAP, CH2Cl2, 84% (inset: ORTEP representation of X-ray crystal structure of (+)-7a).
Determination of the enantiomeric excess of the Diels–Alder adduct, N-Cbz lactam (+)-7a, initially met with some difficulty by chiral HPLC or GC since neither method fully resolved enantiomers. However, the chiral shift reagent Eu(hfc)3 provided a good initial estimate of the enantiomeric excess following deprotection. Subsequently, Mosher ester analysis of the alcohol derived from reduction of lactam (+)-7a with LiBH4 by 19F NMR gave a more accurate measure of enantiopurity (95% ee).
In model studies toward fragment coupling, we found that addition of n-BuLi led to clean deprotection of the Cbz group of lactam 7b with no addition to the lactam carbonyl. Furthermore, activation with a tosyl group proved to be necessary for efficient addition to this hindered δ-lactam. Thus this two-step process gave a crystalline N-Ts lactam 7a readied for coupling with an appropriate nucleophilic coupling partner (vide infra). The N-Ts lactam (+)-7a enabled determination of absolute configuration through single-crystal X-ray crystallographic analysis (S as heavy atom) confirming correlation to the gymnodimine spirocyclic core (Scheme 6, inset).29b
Fragment Coupling
With both gymnodimine fragments, tetrahydrofuran 23 and spirolactam 7a, available in multi-gram quantities and in high enantiopurity, we next focused on identifying appropriate strategies for their coupling. Among the various approaches we attempted, a few selected strategies are described below (Scheme 7).
Scheme 7.
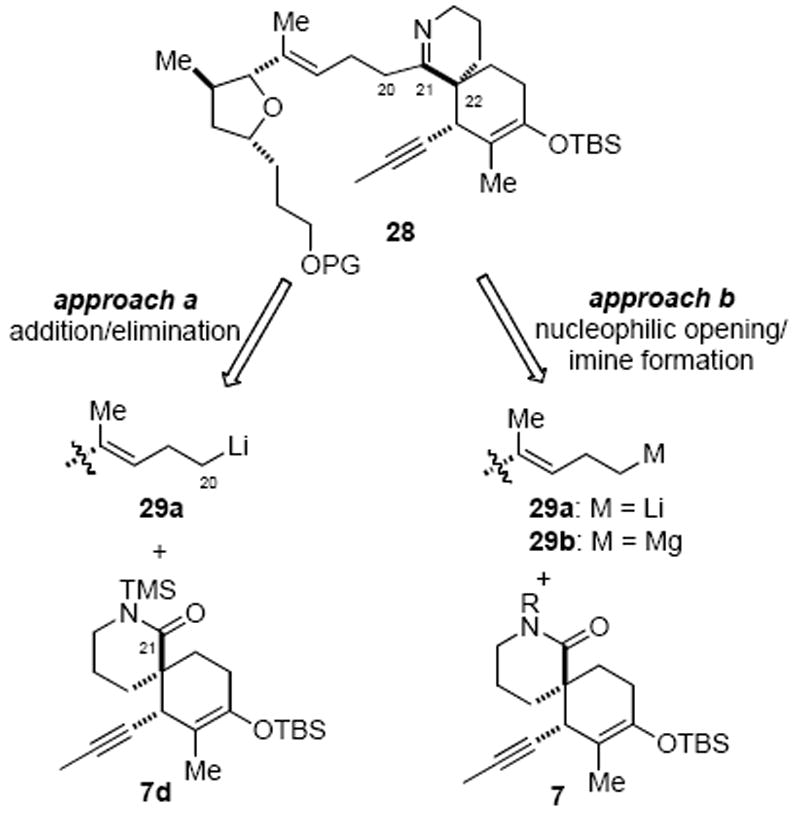
Strategies Explored for Fragment Coupling
Our initial efforts focused on nucleophilic additions to a N-(trimethylsilyl)lactam such as 7d (Scheme 7, approach a) based on a report by Hua demonstrating that nucleophilic additions of alkyllithium species to N-(trimethylsily)lactams 30 selectively provided cyclic ketimines 31 in good to excellent yields through a presumed Peterson-like olefination process (Scheme 8a).38 We modified Hua’s protocol and developed a single-pot variant involving in situ N-silylation of the hindered α,α-dimethyl δ-lactam 32a followed by addition of an alkyllithium. This procedure directly provided the corresponding cyclic ketimines without the need to isolate the labile N-trimethylsilyl lactam intermediates (Scheme 8b).19c However, all attempts to apply this protocol to the more advanced substrate 7c met with failure when the in situ generated N-(trimethylsilyl)lactam 7d was treated with (4-methyl-3-pentenyl)lithium species (Scheme 8c). Recovery of only starting material 7c from these attempts underscored the severe steric issues that any successful coupling protocol would have to overcome.
Scheme 8.
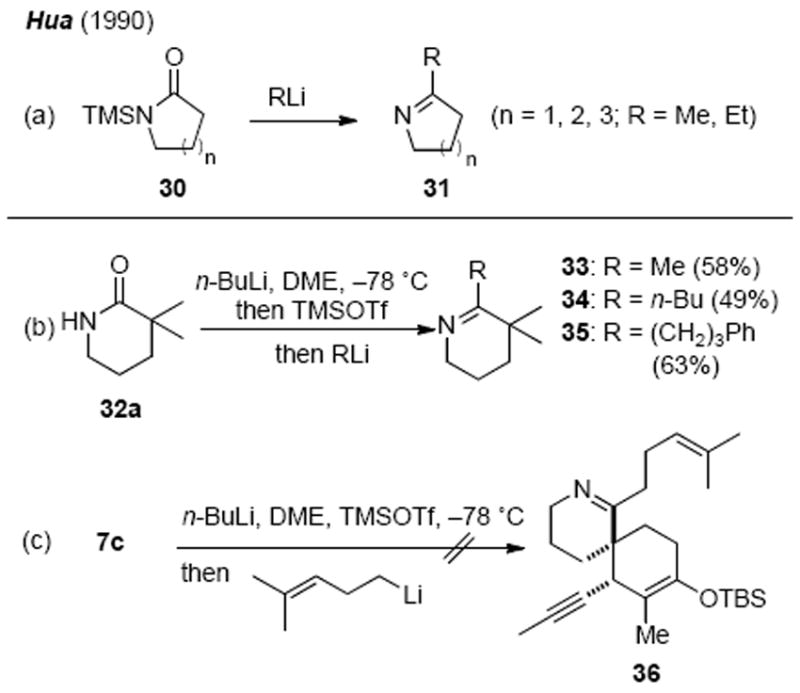
Attempted Imine Synthesis via a Modified Hua Protocol
The aforementioned failed strategy led us to focus on a potentially more challenging intermolecular monoaddition of an alkylmetal species to an electrophilic N-tosyl lactam to deliver an intermediate amino ketone that could be recyclized to an imine (Scheme 7, approach b). We anticipated that the steric issues, which thwarted our previous attempts of additions to this δ-lactam, would slow double addition to the intermediate ketone. In model studies, treatment of N-Ts lactam 32b with only a slight excess of n-BuLi (1.05 equiv) gave only the double addition product, alcohol 38a (90%, Scheme 10a). However, ketone 37b became the major product (68%) when the nucleophilicity of the alkylmetal species was attenuated by use of a Grignard reagent. In this case, use of excess Grignard reagent was required and this would not be ideal with more elaborate alkyl Grignards (i.e. the tetrahydrofuran fragment 29). This reaction also gave significant quantities of the tertiary alcohol 38b. An alternative strategy involved mono-reduction of N-Ts lactam 32b to an intermediate carbinolamine, which was readily accomplished with diisobutylaluminum hydride, and subsequent NaH/alkyllithium addition to provide the amino alcohol 39 in good overall yield (Scheme 10b). However, the required redox corrections made this route less than ideal.
Scheme 10.
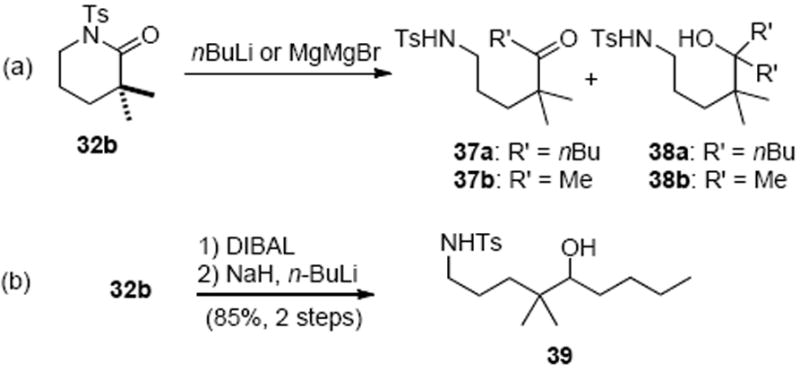
Direct Alkylmetal Addition to Model δ-Lactams
To our surprise, the more functionalized substrate N-Ts lactam 7a underwent efficient nucleophilic opening with MeMgBr to provide the desired amino ketone 40a in good yield without double addition that plagued our model substrates (Scheme 11). Furthermore, more reactive alkyllithium species such as MeLi, n-BuLi and even 4-methyl-3-pentenyl lithium smoothly added to N-Ts lactam 7a at low temperature (−78 °C) with no double addition detected. Related examples of controlled ring opening reactions of N-protected lactams have been reported; however in these cases an N-alkoxycarbonyl group assisted in stabilizing the tetrahedral intermediate in a manner similar to a Weinreb amide.39 This contrasts to the present case of a N-tosyl lactam 7a bearing an adjacent quaternary carbon where steric effects likely play a key role in preventing double addition.
Scheme 11.
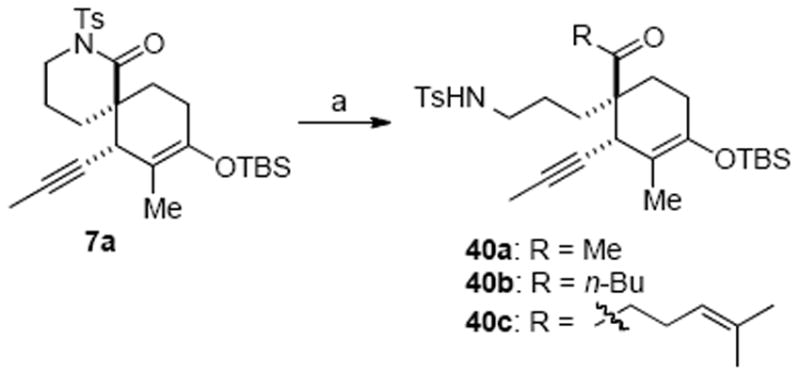
Addition of Alkyllithium Species to N-Ts Lactam 7aa
a Reagents and conditions: (a) for 40a and 40b: RLi, Et2O, −78 °C, 98% (40a), 68% (40b); for 40c: 5-bromo-2-methyl-2-pentene, t-BuLi, TMEDA, Et2O/THF, −20 °C, 63%.
Extension of these findings to the synthesis of gymnodimine required a suitably functionalized tetrahydrofuran precursor. The PMB group of tetrahydrofuran fragment 23 was cleaved under dissolving metal conditions and the resulting alcohol 41a was converted to the homoallylic iodide 42 through the intermediacy of mesylate 41b (Scheme 12). Initial experimentation under the aforementioned conditions, involving treatment of N-Ts lactam 7a with the alkyllithium derived from alkyl iodide 42 by halogen-metal exchange with t-BuLi, provided the desired amino ketone 43, albeit in low yield. Modifying the order of addition by addition of t-BuLi to a mixture of alkyl iodide 42 and N-Ts lactam 7a at low temperature dramatically improved the yield of this intermolecular addition to 92%. The success of this Barbier-type addition laid the foundation for subsequent development of the intramolecular Barbier macrocyclization protocol that proved to be the key for our successful total synthesis of gymnodimine (vide infra).
Scheme 12.
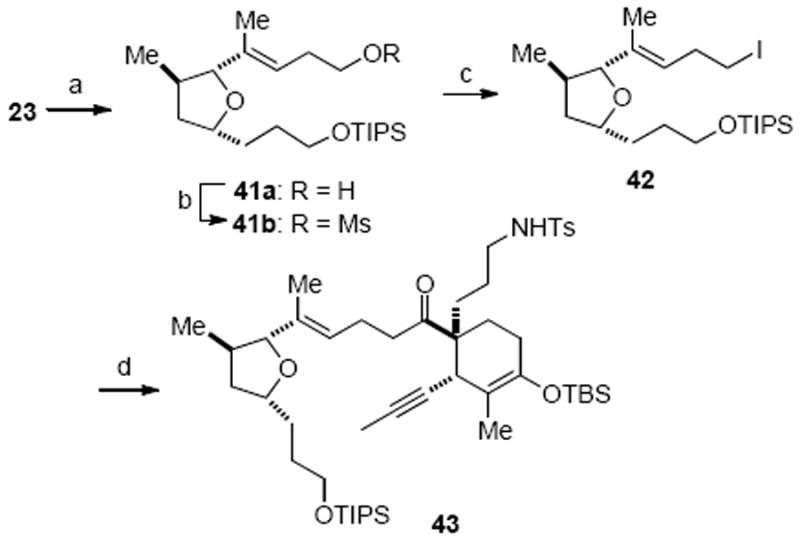
Fragment Coupling of 42 and 7a via a Barbier Reactiona
a Reagents and conditions: (a) Nao, NH3(l), THF, −78 °C, 90%; (b) MsCl, Et3N, CH2Cl2, 92%; (c) n-Bu4NI, THF, 66 °C, 91%; (d) 42 + 7a, Et2O, −78 °C, then t-BuLi, 92%.
Attempted Macrocyclization via a NHK Coupling
In preparation for the projected Cr/Ni mediated macrocyclization, acetylene 43 was subjected to a Pd-catalyzed hydrostannylation reaction under Guibé’s conditions to provide 44a in 39% yield (81% yield, based on recovered starting material) (Scheme 13).40 After tin-iodine exchange, the two silyl groups were removed with HF·pyridine to provide hydroxy ketone 45a. Oxidation with Dess–Martin periodinane provided aldehyde 46, setting the stage for the crucial intramolecular NHK macrocyclization. We were disappointed to find that aldehyde 46 failed to undergo the planned macrocyclization under a variety of conditions. The major adduct isolated from these reactions following aqueous work-up was the corresponding deiodinated substrate. Macrocyclization was also studied following treatment of vinyl iodide 46 with SmI2 or t-BuLi; however, this only led to reduction of the aldehyde and deiodination, respectively.
Scheme 13.
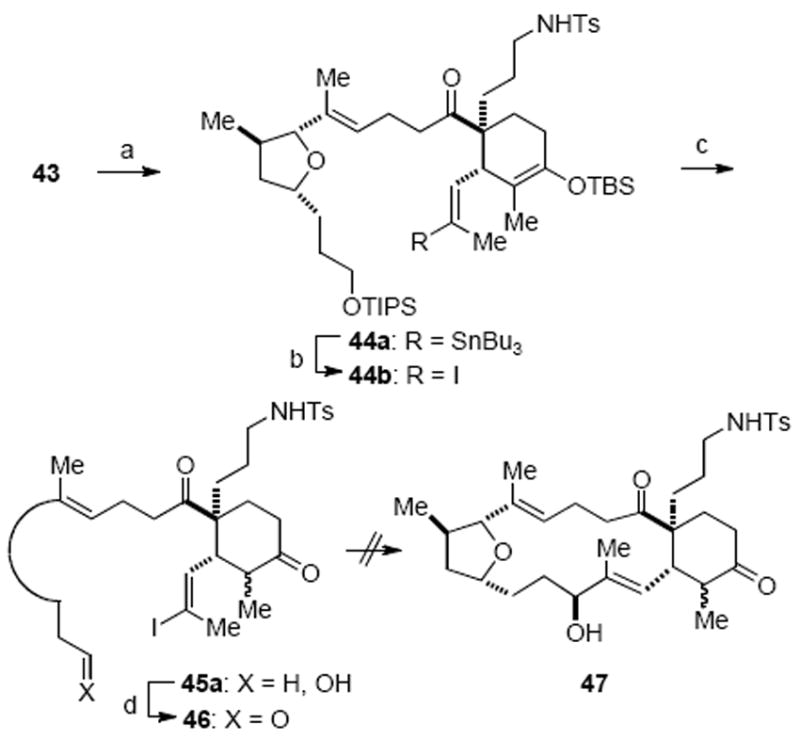
Attempted Macrocyclization of Aldehyde 46 via a Nozaki–Hiyama–Kishi Processa
a Reagents and conditions: (a) n-Bu3SnH, (Ph3P)2PdCl2, THF, 25 °C, 39%, 52% recovered 43; (b) I2, CH2Cl2, −78 °C; cyclohexene, 64%; (c) HF·Py, CH3CN, 83%; (d) Dess–Martin periodinane, CH2Cl2, 90%.
Even though successful NHK couplings of aldehydes in the presence of ketones have been documented,21 we nevertheless considered whether the failure of 46 to undergo a NHK macrocyclization was due to the acidity of the sulfonamide or ketone functionality.41 To test this hypothesis, the triethylsilyl ether 48 was prepared in a similar fashion to TIPS ether 44b, allowing for a selective removal of the triethylsilyl group with PPTS without cleavage of the sensitive silylenol ether moiety (Scheme 14). The alcohol 49 was then oxidized to aldehyde 50; however, this substrate also proved to be unsuitable for macrocyclization and only deiodination was again observed (not shown).
Scheme 14.
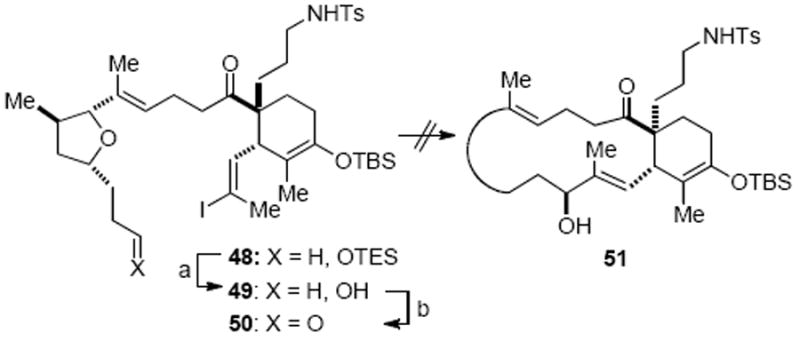
Attempted NHK Macrocyclization of Iodoaldehyde 50a
a Reagents and conditions: (a) PPTS, MeOH/CH2Cl2, 85%; (b) Dess–Martin periodinane, NaHCO3, CH2Cl2, 90%.
We postulated that the reluctance of substrates 46 and 50 to undergo an intramolecular NHK process might be due to unfavorable conformations that preclude macrocyclization. Therefore, we set out to ascertain the effect of a pre-installed spirocyclic imine moiety, which we expected would dramatically alter conformational preferences, might have on macrocyclization. Following our previously developed protocol,42 N-Ts spirolactam 45a was deprotected by conversion to the trifluoroacetamide 45b upon treatment with (CF3CO)2O in the presence of Et3N, which also led to trifluoroacetylation of the primary alcohol, and then direct reduction with SmI2 (Scheme 15).43 The trifluoroacetate ester underwent hydrolysis during work-up of the latter reaction to give alcohol 45b and cleavage of the trifluoroacetamide with concomitant cyclization to the imine 52 was realized upon treatment with ammonium hydroxide in warm methanol. Dess–Martin oxidation provided aldehyde 53 for the proposed macrocyclization reaction. Disappointingly, aldehyde 53 was once again resistant to macrocyclization under several NHK reaction conditions.
Scheme 15.
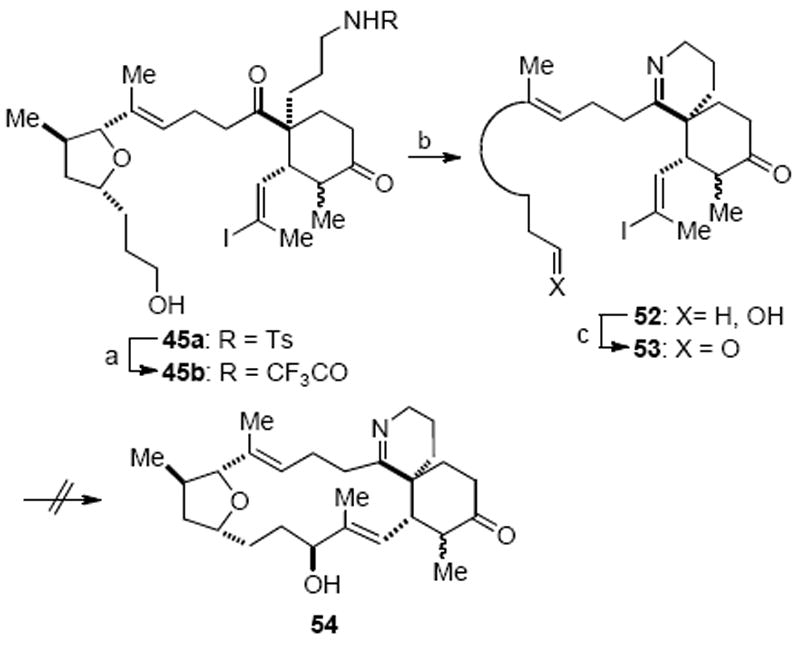
Attempted NHK Macrocyclization of Imine Substrate 53a
a Reagents and conditions: (a) (CF3CO)2O, Et3N, CH2Cl2; SmI2, 87%; (b) NH4OH, MeOH, 80 °C, 81%; (c) Dess–Martin periodinane, CH2Cl2, 83%.
Macrocyclization via an Intramolecular Barbier Reaction
The inability to form the gymnodimine macrocycle through various NHK macrocyclization strategies led us to consider reversing the order of coupling to an intermolecular NHK coupling followed by an intramolecular Barbier macrocyclization (Scheme 16). In this plan, the two fragments, choroaldehyde 55 and spirolactam 56, would be coupled through an intermolecular NHK coupling. While low diastereoselectivity would be expected in this latter process due to the absence of nearby stereocontrol elements, the novelty of the approach involving a Barbier-type macrocyclization coupled with our success of an intermolecular Barbier reaction to couple fragments 42 and 7a (vide supra) spurred our interest in this strategy. While low diastereoselectivity of the NHK process could be overcome through an oxidation/reduction sequence, a more serious challenge was the proposed late stage intramolecular Barbier reaction on a highly functionalized intermediate. To the best of our knowledge, such a Barbier-type macrocyclization had not been previously documented.44
Scheme 16.
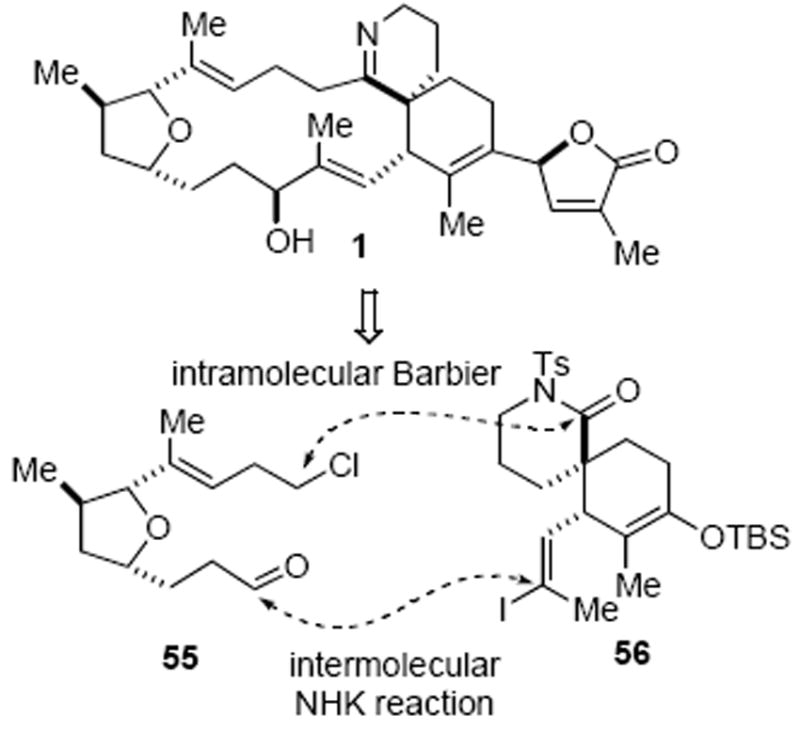
Revised Strategy Based on a Barbier-type Macrocyclization
The two fragments 42 and 7a were easily modified to incorporate the functional groups required for the proposed intramolecular Barbier strategy. Functionalization of the internal acetylene of 7a was realized through standard Pd-catalyzed hydrostannylation conditions to initially provide vinyl stannane 57 in only 25% yield along with recovered starting material (Scheme 17). Screening of other hydrometallation protocols, including hydrozirconation,45 hydroboration,46 and hydroalumination,47 either resulted in recovery of starting material or reduction of the N-Ts lactam moiety. The steric congestion around this internal alkyne due to the cyclohexyl ring and adjacent quaternary carbon posed a considerable challenge to this otherwise standard transformation. Extensive studies were undertaken to improve the efficiency of the hydrostannylation reaction of alkyne 7a. While reaction temperature did not have a notable effect on the conversion (23% conversion at 70 °C), a significant solvent effect was observed with a mixed solvent of THF/hexanes being optimal as described by Semmelhack and Lee.48 Careful analysis of the reaction mixture identified significant amounts of alkene 58, a byproduct that was not derived from protodestannylation of vinyl stannane 57 during stannylation or purification.49 It is known that in Pd-catalyzed hydrostannylation of acetylenes, a principal competitive pathway is the dimerization of tributyltin hydride to provide bis(tributyltin) and molecular hydrogen.40a We suspected alkene 58 was formed from hydrogenation of alkyne 7a in the presence of minute amounts of precipitated Pd(0). Therefore, we postulated that by keeping the concentration of n-Bu3SnH low throughout the reaction, the competing hydrogenation pathway could be suppressed. Indeed, when tributyltin hydride was added slowly to the reaction via syringe pump, the vinyl stannane 57 could be obtained reproducibly in 85% yield with minimal amounts of alkene 58. The subsequent iodine-tin exchange was executed at low temperature in order to minimize competing α-iodination of the reactive silyl enol ether. A neutral workup and use of cyclohexene to quench excess iodine allowed the sensitive vinyl iodide 56 to be isolated in good yield, which was immediately used in the subsequent intermolecular NHK process.
Scheme 17.
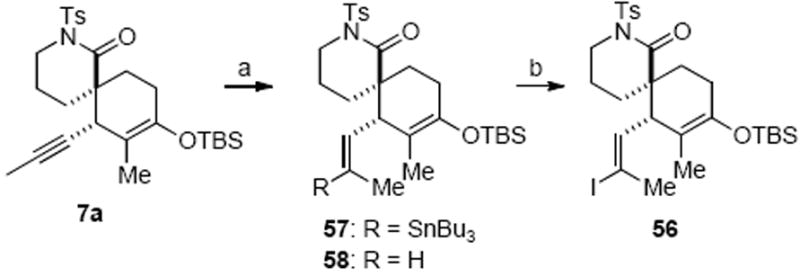
Synthesis of Vinyl Iodide 56a
a Reagents and conditions: (a) PdCl2(PPh3)2, n-BuSnH (syringe pump addition), THF/hexanes (1:6), 85%; (b) I2, CH2Cl2, −78 °C, then cyclohexene, 76% (8% recovered starting material).
The other required fragment for the proposed NHK coupling was prepared from PMB ether 22 (Scheme 18). Cleavage of the PMB group under dissolving metal conditions afforded the primary alcohol 59a, which was then converted to alkyl chloride 59b in 85% yield. A site selective hydroboration followed by Dess–Martin oxidation yielded aldehyde 55 for the projected NHK reaction. A smooth coupling took place between aldehyde 55 and iodide 56 in the presence of excess CrCl2 doped with catalytic amounts of NiCl2, providing readily separable diastereomeric allylic alcohols 61a and 61b in excellent yield but with essentially no diastereoselectivity as anticipated (dr, 1:1.3; 500 MHz 1H NMR). The minor diastereomer 61a was readily converted to the desired diastereomer 61b through an oxidation/asymmetric reduction sequence using the Corey–Itsuno protocol.50 While modified Mosher ester analysis51 did not provide unambiguous assignment of the absolute stereochemistry of the C10 stereocenter of these alcohols, the mechanistic model proposed for the Corey–Itsuno reduction50b was employed to initially assign relative stereochemistry. The stereochemistry of the major diastereomer 61b was subsequently confirmed by X-ray crystallographic analysis of an advanced intermediate (vide infra). The allylic alcohol 61b was then protected as a silyl ether and the minor C13 diastereomer, resulting from previous allylation of the furanose (cf. Scheme 4), could be separated at this stage. A Finkelstein reaction provided the alkyl iodide 64 in high yield, thus setting the stage for the key Barbier-type macrocyclization.
Scheme 18.
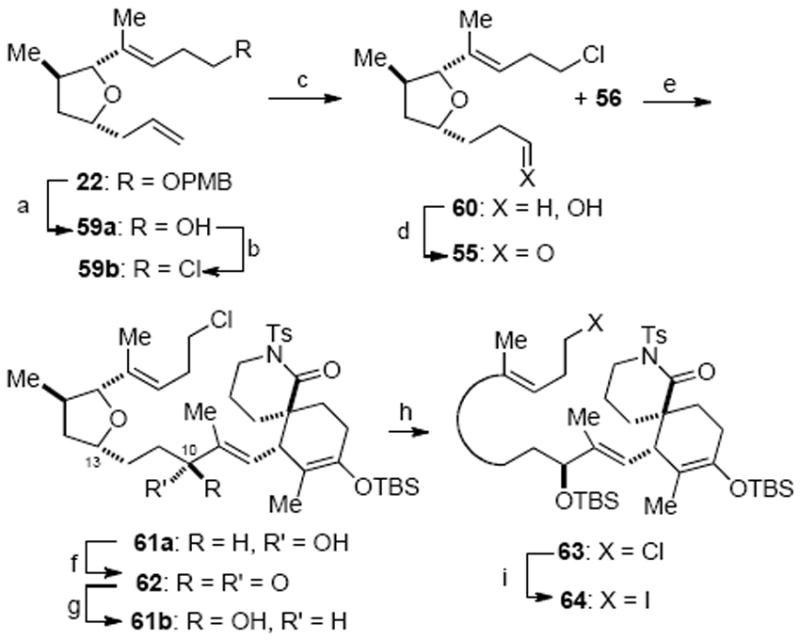
Fragment Coupling through a NHK reactiona
a Reagents and conditions: (a) Na, NH3(l), THF, −78 °C, 92%; (b) PPh3, CCl4, DMF, 65 °C, 85%; (c) 9-BBN, THF; NaOH, H2O2, 98%; (d) Dess–Martin periodinane, CH2Cl2, NaHCO3, 71%; (e) CrCl2/NiCl2, DMF/THF (1 : 1), 97% (β-OH:α-OH, 1.3:1); (f) Dess–Martin periodinane, NaHCO3, CH2Cl2, 88%; (g) (R)-Me-CBS, catecholborane, CH2Cl2, 0 °C, 81% (dr, 6:1); (h) TBSOTf, Et3N, CH2Cl2, −78 °C, 86%; (i) NaI, acetone, 65 °C, 99%.
Our initial macrocyclization attempts involving halogen-metal exchange with iodide 64 and t-BuLi, under conditions previously developed for the intermolecular process (−78 °C, Et2O), only provided trace amounts of the desired product 65 (Scheme 19). At least two competitive reaction pathways could be discerned after careful analysis of the reaction mixture. One was the unproductive quenching of the formed alkyllithium species either during the reaction or following quenching providing deiodinated adduct 66. Surprisingly, t-butyl ketone 67 derived from t-BuLi addition to the highly hindered N-Ts lactam was isolated and this material had also undergone deiodination. To enable macrocyclization, we reasoned that the rate of intermolecular opening by t-BuLi had to be suppressed by rapid consumption of this reagent while at the same time enabling the intramolecular process. Screening of solvents (Et2O, THF, and Trapp solvent52) and additives (TMEDA, HMPA) failed to provide any improvement. We briefly explored the possibility of forming a different alkylmetal species by varying metallation reagents. Attempted formation of a Grignard reagent by treating iodide 64 with i-PrMgCl led to only addition to the N-Ts lactam, whereas SmI2 cleaved the N-Ts group.
Scheme 19.
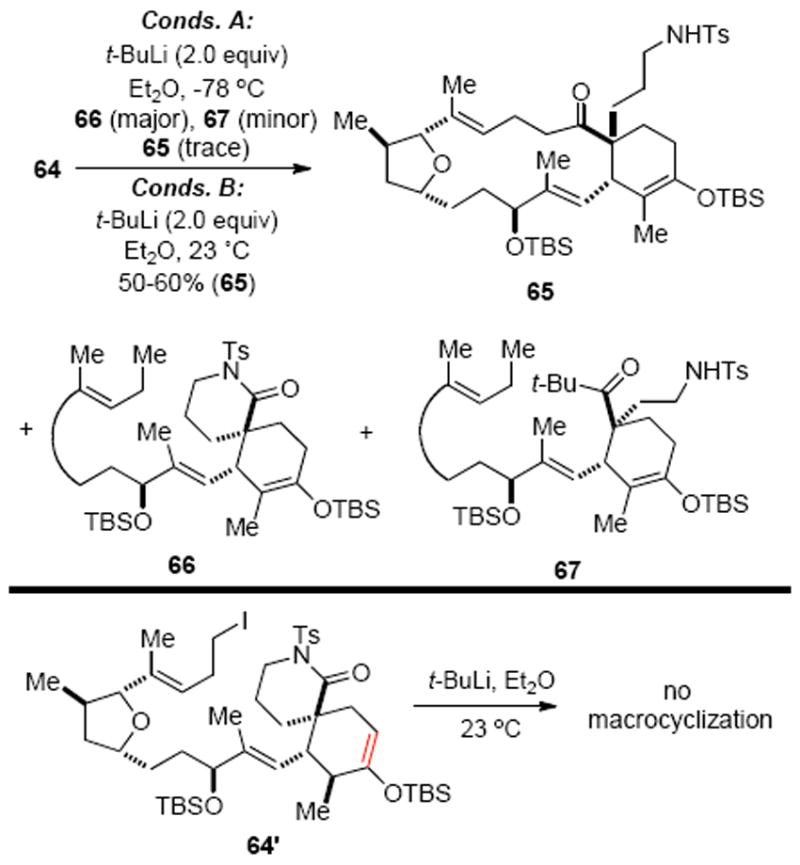
Successful and Unsucessful Barbier-Type Macrocyclizations
Ultimately, we varied reaction temperature reasoning that conformational effects were again preventing macrocyclization and this proved to be the defining parameter for successful macrocyclization. By conducting the reaction at 0 °C, the desired product 65 was obtained in 25% yield. Further optimization provided macrocycle 65 in 50–60% yield when t-BuLi was added to the iodide in Et2O at ambient temperature (23 °C), with 66 and 67 constituting the minor components of the reaction mixture! The exact reason for the dramatic effect of temperature on the outcome of this reaction is not clear; however, one possibility is that a dramatic change in conformational equilibria occurs at higher temperatures, which may facilitate the desired macrocyclization by enabling access to a productive conformer. The iodine-lithium exchange is likely the fastest step;53 however the fact that the desired macrocyclization is faster than both intermolecular addition of the second equivalent of t-BuLi to the N-Ts lactam and elimination of t-BuI by t-BuLi is intriguing and may be partially rationalized by virtue of the fact that each of these processes have different temperature profiles. Similarly, the observation that a closely related substrate, olefin regioisomer 64’, failed to undergo macrocyclization under similar conditions might reflect the importance of substrate conformation in this Barbier cyclization reaction.
Introduction of the Butenolide and Completion of the Synthesis of (−)-Gymnodimine
Having prepared the macrocyclic structure of gymnodimine, the remaining challenge was to introduce the butenolide moiety. Due to the fragile nature of this fragment,20 we planned to keep manipulations following introduction of this moiety to a minimum. Towards this goal, we considered various strategies among which two approaches were of particular interest. One utilized a Stille coupling between the triflate 68 and vinyl stannane 69 (Scheme 20, approach a).54 A selenolactonization of the derived alkene followed by oxidation and elimination would provide the butenolide.55 The other option involved use of a rarely studied vinylogous Mukaiyama aldol reaction with a ketone, which rapidly assembles the butenolide in a single operation (Scheme 20, approach b).56 A subsequent dehydration reaction would then provide the penultimate intermediate 72.
Scheme 20.
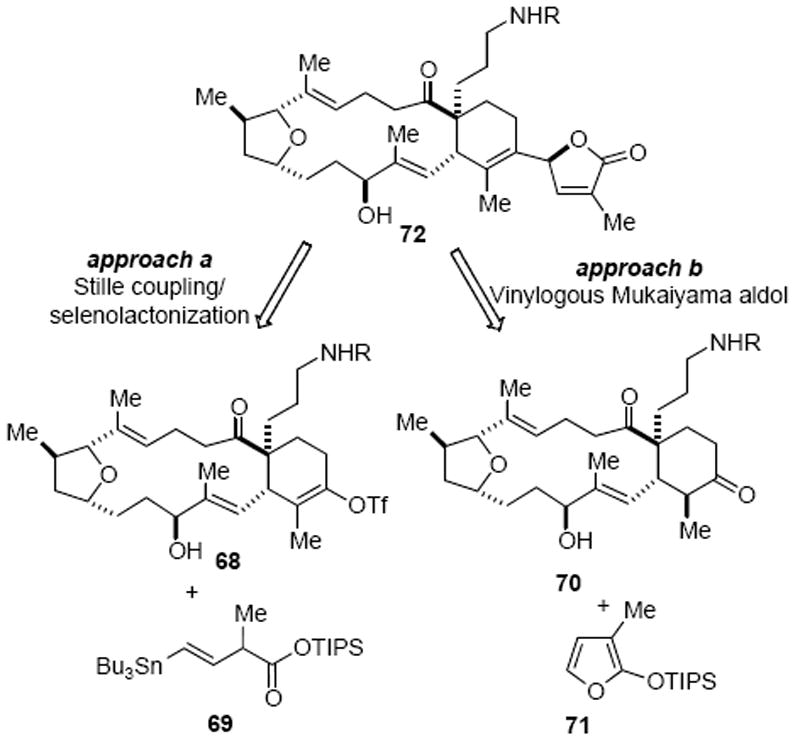
Strategies for Introduction of the Butenolide Moiety
Our initial effort to execute the proposed cross-coupling strategy was conducted on the model substrate spirolactam 7a (Scheme 21). Attempted preparation of vinyl triflate 73, directly from the silyl enol ether precursor 7a under modified conditions reported by Corey, were unsuccessful.57 We then resorted to a two-step procedure involving ketone 74 prepared from acidic deprotection of silyl enol ether 7a. Standard triflation conditions only provided the undesired regioisomer, vinyl triflate 75. In parallel with extensive studies to obtain the required regioisomeric triflate, the subsequent proposed coupling was explored to incorporate the butenolide. A Stille coupling reaction between triflate 75 and vinyl stannane 6958 under Farina’s conditions provided the diene 76 in good yield.59 After cleavage of the silyl group, the carboxylic acid 77 underwent a smooth diastereoselective selenolactonization reaction in the presence of in situ generated phenylselenyl chloride (PhSeSePh, SO2Cl2) to provide the selenide 78 as a mixture of two diastereomers. Treatment of 78 with H2O2 initiated an oxidation/elimination sequence providing butenolide 79 as a mixture of two epimers (epimeric at C4). While this approach was eventually abandoned due to the inability to identify an efficient protocol for the formation of the requisite vinyl triflate 73, this efficient sequence nevertheless provides a concise strategy for butenolide incorporation onto highly functionalized substrates that may find use in other contexts.
Scheme 21.
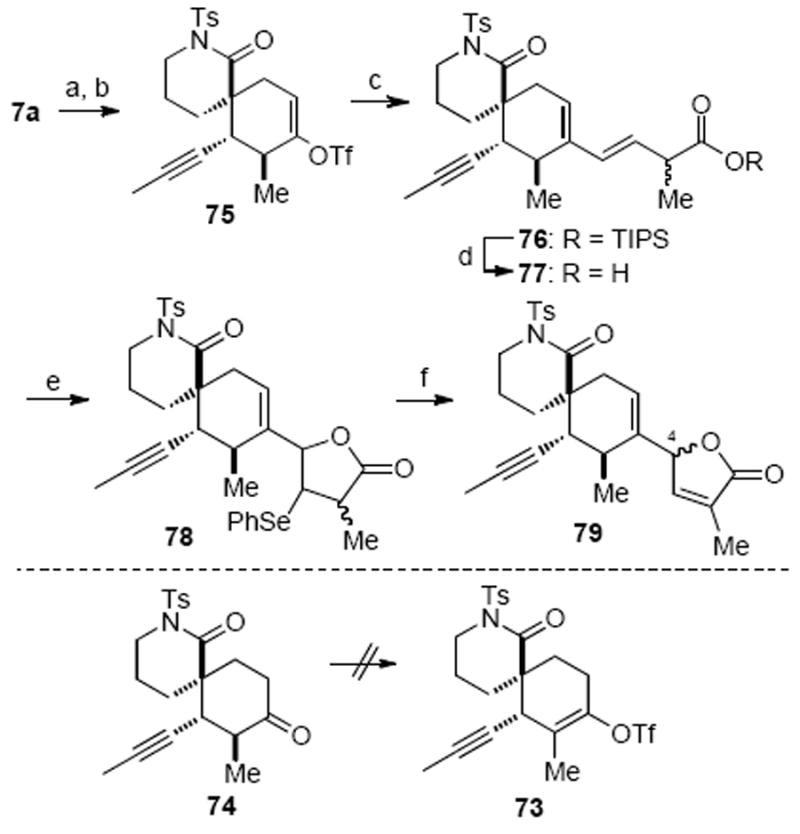
Appending the Butenolide through a Stille Coupling Reactiona
a Reagents and conditions: (a) p-TsOH, CH2Cl2/MeOH, 88%; (b) LiHMDS, PhNTf2, THF, 52%; (c) 69, Pd2(dba)3·CHCl3, Ph3As, THF, 84%; (d) TBAF, THF, −78 °C, 84%; (e) PhSeSePh, SO2Cl2, CH2Cl2, 60%; (f) H2O2, CH2Cl2, 90%. (TBAF = tetrabutylammonium fluoride)
A vinylogous Mukaiyama aldol reaction of a cyclic ketone was next explored for butenolide incorporation, further motivated by the fact that only a few examples of this process have been reported.60 This strategy was not without risks given the potential functional group incompatibility issues since the expected low reactivity of ketone substrates in a Mukaiyama aldol reaction would necessitate the use of strong Lewis or Brönsted acids. Furthermore, the stereochemical outcome of the reaction was unclear. Preliminary studies of the vinylogous Mukaiyama aldol reaction between simple α-alkyl cyclohexanones and several silyloxy furans demonstrated that these reactions could effectively be promoted by TiCl4 to provide the desired adducts in good yield and moderate to high diastereoselectivities.61 More importantly, studies with the advanced model substrate 80 revealed some key factors for success of this reaction including: 1) use of the more robust triisopropylsilyl version of the siloxyfuran 71 as the butenolide precursor, 2) the requirement of having the allylic hydroxyl group unprotected in order to prevent elimination to a conjugated diene, and 3) the requirement of very short reaction times following brief exposure of a mixture of the silyloxyfuran and ketone to excess TiCl4 (Scheme 22).
Scheme 22.
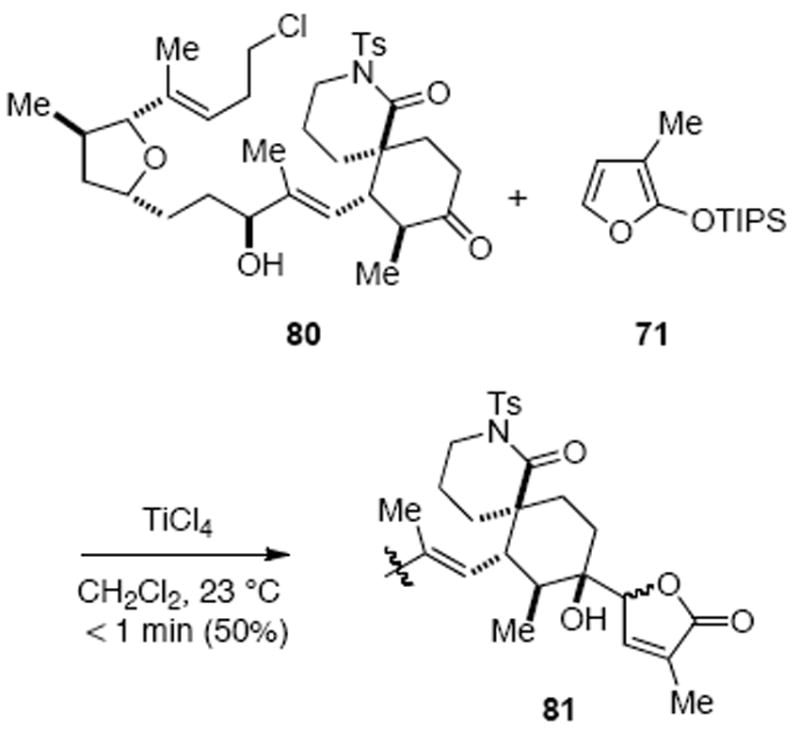
Butenolide Addition via a Vinylogous Mukaiyama Aldol Reaction: Advanced Model Substrate
The substrate for the vinylogous Mukaiyama aldol reaction, ketone 83, was prepared in a few straightforward transformations from silylenol ether 65 (Scheme 23). First, the robust N-tosyl group was replaced with a more readily cleaved trifluoroacetamide employing our previously reported conditions.42 Both silyl groups in 82 were then removed under acidic conditions furnishing a crystalline hydroxy ketone 83 that enabled determination of relative stereochemistry of this advanced intermediate by X-ray crystallographic analysis,31c confirming the earlier assignment of the C10 allylic alcohol 61b (Scheme 23, inset). Brief exposure (<1 min) of a mixture of ketone 83 and silyloxyfuran 71 to TiCl4 led to a smooth addition reaction to provide lactones 84 as a 1.1:1 mixture of two diastereomers (epimeric at C4) in 61% yield. After silyl protection of the C10 allylic hydroxyl group, the two epimeric alcohols 85a and 85b were readily separated. Comparison of 13C NMR data for hydroxyketones 85a and 85b with that of related adducts from our previous model studies, whose structures were secured through X-ray crystallographic analysis,61 enabled preliminary assignment of the relative stereochemistry of butenolides 85a/85b.58 It was found that the undesired silyl ether 85b could be equilibrated to a 1:2 mixture of 85a and 85b upon treatment with DBU at ambient temperature, improving material throughput.62 Definitive confirmation that hydroxy ketone 85a possessed the correct C4 relative stereochemistry would ultimately come from subsequent conversion to gymnodimine (vide infra).
Scheme 23.
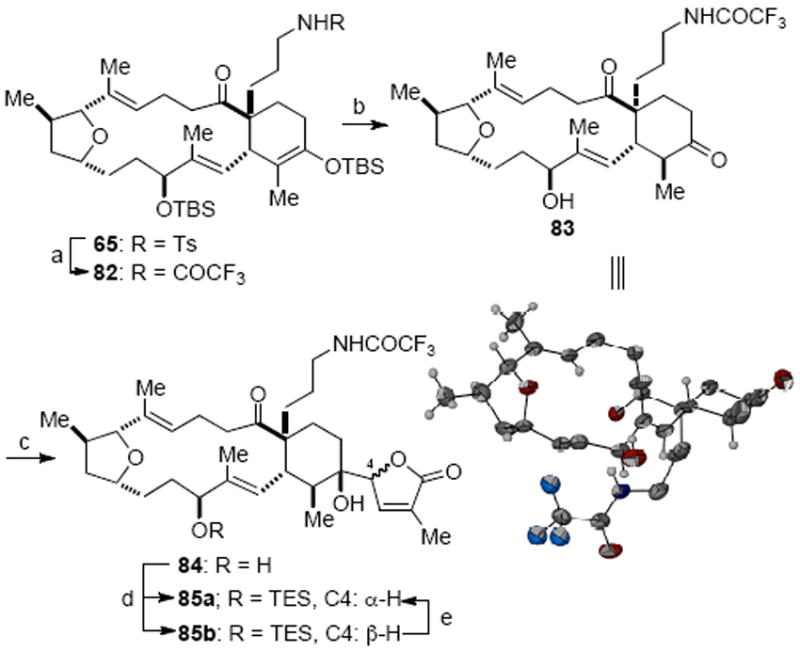
Vinylogous Mukaiyama Aldol Addition to Append Butenolidea
a Reagents and conditions: (a) (CF3CO)2O, Et3N, CH2Cl2; SmI2, 73%; (b) p-TsOH, THF/CH2Cl2/MeOH, 84%; (c) 71, TiCl4, CH2Cl2, 23 °C, 1 min, dr = 1.1 : 1, 61%; (d) TESCl, imidazole, DMAP, CH2Cl2, 76%; (e) DBU, CH2Cl2, 85a : 85b = 1 : 2, 60% (inset: ORTEP representation of X-ray structure of 83; DMAP = 4-dimethylamino pyridine; DBU = diazobicycloundecane).
The tertiary alcohol 85a underwent dehydration under standard conditions (Et3N, SOCl2) to deliver olefin 86 as the major regioisomer (3:1, Scheme 24). Conditions for cleavage of the trifluoroacetamide group required mild basic conditions to avoid cleavage of the butenolide moiety. Unfortunately, attempts to cleave the trifluoroacetamide under a variety of basic conditions (LiOH/THF/H2O, NaHCO3/MeOH, K2CO3/MeOH, NH3·H2O/MeOH) only led to decomposition of this sensitive substrate, in good agreement with Miles’ finding.20 Acidic hydrolysis (2M HCl/MeOH, 44 °C)63 also proved too harsh for this highly functionalized substrate. An undesirable but required activation of the trifluoroacetamide group was required and a t-butoxycarbonyl group was chosen given that this could then be removed by mild acidic conditions. Hence, treatment of the trifluoroacetamide 86 with (Boc)2O and brief exposure to hydrazine cleaved the trifluoroacetamide and provided N-Boc amine 87.64 Acid hydrolysis of N-Boc amine 87 with CF3CO2H cleaved the N-Boc group and concomitant desilylation of the triethylsilyl group to provide the corresponding primary amine salt. Upon standing at ambient temperature (23 °C) under high vacuum overnight, the free amine underwent a smooth cyclocondensation to provide gymnodimine as a white solid. The spectral data of synthetic gymnodimine was in full agreement with that reported previously for the natural product.8,9 Furthermore, the trifluoroacetate salt of gymnodimine was prepared and its optical rotation ([α]22D −14.1 (c 0.06, MeOH)) was in good agreement with that reported ([α]25D −10.4 (c 0.13, MeOH)).
Scheme 24.
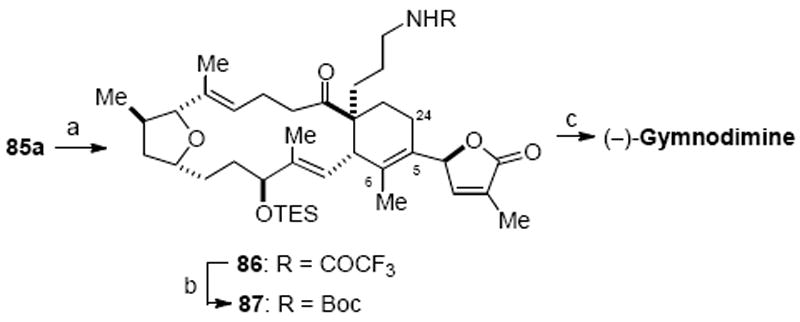
Synthesis of Gymnodiminea
a Reagents and conditions: (a) Et3N, SOCl2, CH2Cl2, −78 °C, 82%, Δ5,6 :Δ5,24 = 3 : 1; (b) Et3N, (Boc)2O, DMAP, CH2Cl2; hydrazine, 99%; (c) TFA, CH2Cl2; high vacuum overnight, 68%. (DMAP = 4-dimethylamino pyridine, TFA = trifluoroacetic acid; Boc = t-butoxy carbonyl)
Synthesis of C4-epi-Gymnodimine
In an analogous fashion to the natural product, C4-epi-gymnodimine (90) was prepared starting from the C4-epimer 85b (Scheme 25). For this diastereomer, the elimination step leading to 88 was non-regioselective and provided an approximately equal mixture of two regioisomers. After separation of the regioisomers, C4-epi-gymnodimine (90) was isolated as a white solid in two additional steps. Comparison of 1H and 13C NMR of C4-epi-gymnodimine with that of gymnodimine provided evidence that they are epimeric at C4, further supporting the previous stereochemical assignment of alcohols 85a and 85b.
Scheme 25.
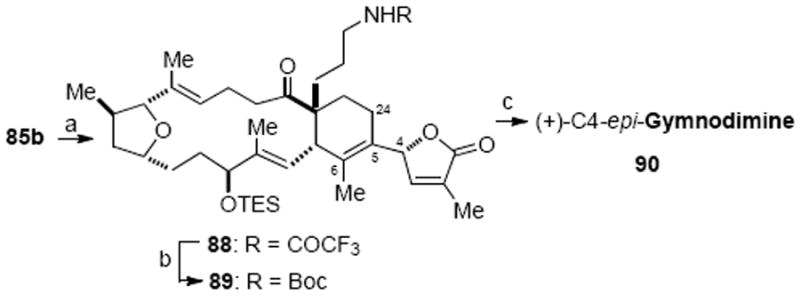
Synthesis of C4-epi-Gymnodiminea
a Reagents and conditions: (a) Et3N, SOCl2, CH2Cl2, 0 °C, 50%, Δ5,6 : Δ5,24 = 1 : 1.3; (b) Et3N, (Boc)2O, DMAP, CH2Cl2; hydrazine, 87%; (c) TFA, CH2Cl2; high vacuum overnight, 58%. (DMAP = 4-dimethyalmino pyridine, TFA = trifluoroacetic acid)
Conclusions
The first total synthesis of the marine toxin (−)-gymnodimine was achieved featuring a convergent coupling between two principal fragments, tetrahydrofuran 55 and spirolactam 56, prepared in multi-gram quantity and high enantiomeric excess through an anti-aldol reaction and a catalytic, asymmetric Diels–Alder reaction, respectively. Several strategies were studied for fragment coupling, macrocyclization, formation of the cyclic imine, and butenolide annulation. Ultimately, a successful macrocyclization was achieved employing an unprecedented intramolecular Barbier reaction proceeding at ambient temperature (23 °C) with t-BuLi and a highly advanced alkyl iodide intermediate. To the best of our knowledge, this represents the first application of a Barbier-type reaction to form a macrocycle. A late stage vinylogous Mukaiyama aldol reaction of an α-alkyl ketone is also noteworthy as it constitutes a highly efficient route to attach the labile butenolide moiety of gymnodimine. The application of this vinylogous Mukaiyama aldol reaction may prove useful for the synthesis of other butenolide-containing natural products such as the spirolides, which are of topical interest.65 The synthetic approach toward gymnodimine described herein is robust and has provided valuable analogs (e.g. C4-epi-gymnodimine) to enable more detailed SAR studies of these marine toxins, known to interact with nicotinic acetylcholine receptors. The described synthesis has allowed preparation of potential gymnodimine immunogens as promising candidates for the development of a congener-independent ELISA that could prove useful for monitoring gymnodimine and congeners in the oceans. Collaborative efforts in both these directions are underway and results emanating from these studies will be reported in due course.
Supplementary Material
Acknowledgments
We are grateful for generous support from the National Institutes of Health (GM52964) and the Welch Foundation (A-1280). We thank Dr. Joseph Reibenspies and Dr. Nattamai Bhuvanesh (TAMU) for X-ray structure analyses and Prof. John W. Blunt and Prof. Murray H. G. Munro for providing characterization data for natural gymnodimine. We thank Prof. John L. Wood (Colorado State University) for valuable support and Prof. Brian Connell (Texas A&M University) for helpful discussions. We thank Mrs. Ju Yang and Mr. Stephen T. Cohn for their contributions to initial synthetic studies toward gymnodimine and Ms. Supakarn Chamni, Mr. Gang Liu, and Dr. Yong Ahn for assistance with the synthesis of starting materials.
Footnotes
Supporting Information Available. Complete experimental and characterization details for all new compounds reported. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Anderson DM. Sci Am. 1994;271:62–68. doi: 10.1038/scientificamerican0894-62. [DOI] [PubMed] [Google Scholar]; (b) Van Dolah FM. Environ Health Perspect Suppl. 2000;108:133–141. doi: 10.1289/ehp.00108s1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narahashi T. J Toxicol Toxin Rev. 2001;20:67–84. and references therein. MacGeer EG, Olney JW, MacGeer PL. Kainic Acid as a Tool in Neurogiology. Raven; New York: 1978. . Tachibana K, Scheuer PJ, Tsukitani Y, Kikuchi H, Engen DV, Clardy J, Gopichand Y, Schmitz FJ. J Am Chem Soc. 1981;103:2469–2471.. Haystead TAJ, Sim ATR, Carling D, Honnor RC, Tsukitani Y, Cohen P, Hardie DG. Nature. 1989;337:78–81. doi: 10.1038/337078a0.. Bhakuni DS, Rawat DS. Bioactive Marine Natural Products. Springer, Anamaya Publishers; New York, New Delhi: 2005.
- 3.For a review on imine-containing natural products, see: Kita M, Uemura D. Chem Lett. 2005;34:454–459.
- 4.(a) Hu T, Curtis J, Oshima Y, Quilliain M, Walter J, Wright W, Wright J. J Chem Soc Chem Commum. 1995:2159–2161. [Google Scholar]; (b) Hu T, Curtis JM, Walter J, Wright JLC. Tetrahedron Lett. 1996;37:7671–7674. [Google Scholar]; (c) Hu T, Burton IW, Cembella AD, Curtis JM, Quilliam MA, Walter JA, Wright JLC. J Nat Prod. 2001;64:308–312. doi: 10.1021/np000416q. [DOI] [PubMed] [Google Scholar]; (d) Falk M, Burton IW, Hu T, Walter JA, Wright JLC. Tetrahedron. 2001;57:8659–8665. [Google Scholar]
- 5.(a) Uemura D, Chou T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chen H. J Am Chem Soc. 1995;117:1155–1156. [Google Scholar]; (b) Chou T, Osamu K, Uemura D. Tetrahedron Lett. 1996;37:4023–4026. [Google Scholar]; (c) Chou T, Haino T, Kuramoto M, Uemura D. Tetrahedron Lett. 1996;37:4027–4030. [Google Scholar]; (d) Takada N, Umemura N, Suenaga K, Chou T, Nagatsu A, Haino T, Yamada K, Uemura D. Tetrahedron Lett. 2001;42:3491–3494. [Google Scholar]
- 6.Takada N, Umemura N, Suenaga K, Uemura D. Tetrahedron Lett. 2001;42:3495–3497. [Google Scholar]
- 7.Lu C-K, Lee G-H, Huang R, Chou H-N. Tetrahedron Lett. 2001;42:1713–1716. [Google Scholar]
- 8.Seki T, Satake M, Mackenzie L, Kaspar H, Yasumoto T. Tetrahedron Lett. 1995;36:7093–7096. [Google Scholar]
- 9.Stewart M, Blunt JW, Munro MHG, Robinson WT, Hannah DJ. Tetrahedron Lett. 1997;38:4889–4890. [Google Scholar]
- 10.(a) Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J Agric Food Chem. 2000;48:1373–1376. doi: 10.1021/jf991031k. [DOI] [PubMed] [Google Scholar]; (b) Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J Agric Food Chem. 2003;51:4838–4840. doi: 10.1021/jf030101r. [DOI] [PubMed] [Google Scholar]
- 11.(a) Biré R, Krys S, Frémy JM, Dragacci S, Stirling D, Kharrat R. J Nat Toxins. 2002;11:269–275. [PubMed] [Google Scholar]; (b) Krock B, Pitcher GC, Ntuli J, Cembella AD. Afr J Mar Sci. 2009;31:113–118. [Google Scholar]
- 12.Stirling JD. New Zealand J Mar Freshwater Res. 2001;35:851–857. [Google Scholar]
- 13.Munday R, Towers NR, Mackenzie L, Beuzenberg V, Holland PT, Miles CO. Toxicon. 2004;44:173–178. doi: 10.1016/j.toxicon.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bourne Y, Radić Z, Aráoz R, Talley TT, Benoit E, Servent D, Taylor P, Molgó J, Marchot P. Proc Nat Acad Sci. 2010;107:6076–6081. doi: 10.1073/pnas.0912372107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Araoz R, Servent D, Molgó J, Iorga BI, Fruchart-Gaillard C, Benoit E, Gu Z, Stivala C, Zakarian A. J Am Chem Soc. 2011;133:10499–10511. doi: 10.1021/ja201254c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCauley JA, Nagasawa K, Lander PA, Mischke SG, Semones MA, Kishi Y. J Am Chem Soc. 1998;120:7647–7648.. Sakamoto S, Sakazaki H, Hagiwara K, Kamada K, Ishii K, Noda T, Inoue M, Hirama M. Angew Chem Int Ed. 2004;43:6505–6510. doi: 10.1002/anie.200461802.. Stivala CE, Zakarian A. J Am Chem Soc. 2008;130:3774–3775. doi: 10.1021/ja800435j.. Nakamura S, Kikuchi F, Hashimoto S. Angew Chem Int Ed. 2008;47:7091–7094. doi: 10.1002/anie.200802729.. For a review of synthetic efforts towards pinnatoxins, see: Beaumont S, Ilardi EA, Tappin NDC, Zakarian A. Eur J Org Chem. 2010:5743–5765. doi: 10.1002/ejoc.201000842.
- 16.(a) Matsuura F, Peters R, Anada M, Harried SS, Hao J, Kishi Y. J Am Chem Soc. 2006;128:7463–7465. doi: 10.1021/ja0618954. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hao J, Matsuura F, Kishi Y, Kita M, Uemura D, Asai N, Iwashita T. J Am Chem Soc. 2006;128:7742–7743. doi: 10.1021/ja061893j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Furkert DP, Brimble MA. Org Lett. 2002;4:3655–3658. doi: 10.1021/ol026605c. [DOI] [PubMed] [Google Scholar]; (b) Ishihara J, Ishizaka T, Suzuki T, Hatakeyama S. Tetrahedron Lett. 2004;45:7855–7858. [Google Scholar]; (c) Meilert K, Brimble MA. Org Lett. 2005;7:3497–3500. doi: 10.1021/ol051260u. [DOI] [PubMed] [Google Scholar]; (d) Meilert K, Brimble MA. Org Biomol Chem. 2006;4:2184–2192. doi: 10.1039/b604334h. [DOI] [PubMed] [Google Scholar]; (e) Stivala CE, Zakarian A. Org Lett. 2009;11:839–842. doi: 10.1021/ol8027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong K, Romo D, Lee C. Angew Chem Int Ed. 2009;48:7402–7405. doi: 10.1002/anie.200903432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For synthetic studies towards gymnodimine, see: Ishihara J, Miyakawa J, Tsujimoto T, Murai A. Synlett. 1997:1417–1419.. Yang J, Cohn ST, Romo D. Org Lett. 2000;2:763–766. doi: 10.1021/ol005510c.. Ahn Y, Cardenas GI, Yang Y, Romo D. Org Lett. 2001;3:751–754. doi: 10.1021/ol0155081.. Tsujimoto T, Ishihara J, Horie M, Murai A. Synlett. 2002:399–402.. White JD, Wang G, Quaranta L. Org Lett. 2003;5:4109–4112. doi: 10.1021/ol030101c.. White JD, Wang G, Quaranta L. Org Lett. 2003;5:4983–4986. doi: 10.1021/ol035939e.. Brimble MA, Trzoss M. Tetrahedron. 2004;60:5613–5622.. Johannes JW, Wenglowsky S, Kishi Y. Org Lett. 2005;7:3997–4000. doi: 10.1021/ol051553n.. Kong K, Moussa Z, Romo D. Org Lett. 2005;7:5127–5130. doi: 10.1021/ol051840r.. White JD, Quaranta L, Wang G. J Org Chem. 2007;72:1717–1728. doi: 10.1021/jo062396o.
- 20.Miles CO, Hawkes AD, MacKenzie AL, Munday R, Towers NR, Prinsep MR. Marine Biotoxin Science Workshop. Vol. 99. New Zealand: 1999. pp. 94–98. [Google Scholar]
- 21.Takai K, Tagashira M, Kuroda T, Oshima K, Utimoto K, Nozaki H. J Am Chem Soc. 1986;108:6048–6049. doi: 10.1021/ja00279a068.. Jin H, Uenishi J, Christ WJ, Kishi Y. J Am Chem Soc. 1986;108:5644–5645.. For an excellent review, see: Fürstner A. Chem Rev. 1999;99:991–1045. doi: 10.1021/cr9703360.
- 22.For example see: Macmillan DWC, Overman LE, Pennington LD. J Am Chem Soc. 2001;123:9033–9044. doi: 10.1021/ja016351a.
- 23.Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]
- 24.Evans DA, Johnson JS. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Springer-Verlag; Berlin: 2000. [Google Scholar]
- 25.Taken in part from the M.S. thesis of Ms. Ju Yang, Texas A&M University. 1999. [Google Scholar]
- 26.Corey EJ, Loh T-P. Tetrahedron Lett. 1993;34:3979–3982. [Google Scholar]
- 27.Evans DA, Mathre DJ, Scott WL. J Org Chem. 1985;50:1830–1835. [Google Scholar]
- 28.Hirama M, Noda T, Takeishi S, Itô S. Bull Chem Soc Jpn. 1988;61:2645–2646. [Google Scholar]
- 29.(a) Warm A, Vogel P. J Org Chem. 1986;51:5348–5353. [Google Scholar]; (b) Arjona O, Dominguez C, Pradilla R, Mallo A, Manzano C, Plumet J. J Org Chem. 1989;54:5883–5887. [Google Scholar]
- 30.Hansen MM, Danda H, Heathcock CH. J Org Chem. 1990;55:173–181. [Google Scholar]
- 31.(a) CCDC-801790 contains the supplementary crystallographic data for compound 13. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (b) CCDC-801791 contains the supplementary crystallographic data for compound (+)-7a. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (c) CCDC-655682 contains the supplementary crystallographic data for compound 83. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 32.Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J Am Chem Soc. 1999;121:12208–12209. [Google Scholar]
- 33.Jadhav PK, Bhat KS, Perumal PT, Brown HC. J Org Chem. 1986;51:432–439. [Google Scholar]
- 34.(a) Piers E, Morton HE. J Org Chem. 1980;45:4263–4264. [Google Scholar]; (b) Singer RD, Hutzinger MW, Oeschlager AC. J Org Chem. 1991;56:4933–4938. [Google Scholar]; (c) Cummins CH, Gordon EJ. Tetrahedron Lett. 1994;35:8133–8136. [Google Scholar]; (d) Betzer J-F, Ardisson J, Lallemand J-Y, Pancrazi A. Tetrahedron Lett. 1997;38:2279–2282. [Google Scholar]
- 35.(a) Evans DA, Miller SJ, Lectka T, Matt PV. J Am Chem Soc. 1999;121:7559–7573. [Google Scholar]; (b) Evans DA, Barnes DM, Johnson JS, Lectka T, Matt PV, Miller SJ, Murry JA, Norcross RD, Shaughnessy EA, Campos KR. J Am Chem Soc. 1999;121:7582–7594. [Google Scholar]
- 36.While our work was in progress, Murai and coworkers disclosed a similar catalytic, asymmetric Diels–Alder reaction of α-methylene caprolactam derivatives, see: Ishihara J, Horie M, Shimada Y, Tojo S, Murai A. Synlett. 2002:404–406.
- 37.Evans DA, Peterson GS, Johnson JS, Barnes DM, Campos KR, Woerpel KA. J Org Chem. 1998;63:4541–4544. [Google Scholar]
- 38.Hua DH, Miao SW, Bharathi SN, Katsuhira T, Bravo AA. J Org Chem. 1990;55:3682–3684. [Google Scholar]
- 39.(a) Ohta T, Hosoi A, Kimura T, Nozoe S. Chem Lett. 1987:2091–2094. [Google Scholar]; (b) Giovannini A, Savoia D, Umani-Ronchi A. J Org Chem. 1989;54:228–234. [Google Scholar]
- 40.Zhang HX, Guibé F, Balavoine G. J Org Chem. 1990;55:1857–1867.. For a review, see: Smith ND, Mancuso J, Lautens M. Chem Rev. 2000;100:3257–3282. doi: 10.1021/cr9902695.
- 41.The sulfonamide N–H proton is more acidic compared to the cyclohexanone α-proton. Although temporary masking of the sulfonamide with trifluoroactamide would eliminate this acidic proton, the resulting N-acyl, N-sulfonyl intermediate was deemed too labile to test this hypothesis.
- 42.Moussa Z, Romo D. Synlett. 2006:3294–3298. [Google Scholar]
- 43.For another recently developed protocol for removal of a tosyl group from primary sulfonamides, see: Ankner T, Hilmersson G. Org Lett. 2009;11:503–506. doi: 10.1021/ol802243d.
- 44.For use of Barbier reactions to form medium-sized rings on simple substrates, see: Cooke MP, Jr, Houpis IN. Tetrahedron Lett. 1985;26:4987–4990.. Souchet M, Clark RD. Synlett. 1990:151–152.
- 45.Hart DW, Blackburn TF, Schwartz J. J Am Chem Soc. 1975;97:679–680. [Google Scholar]
- 46.(a) Brown HC, Gupta SK. J Am Chem Soc. 1972;94:4370–4371. [Google Scholar]; (b) Brown HC, Hamaoka T, Ravindran N. J Am Chem Soc. 1973;95:5786–5788. [Google Scholar]
- 47.Zweifel G, Whitney CC. J Am Chem Soc. 1967;89:2753–2754. [Google Scholar]
- 48.(a) Semmelhack MF, Hooley RJ. Tetrahedron Lett. 2003;44:5737–5739. [Google Scholar]; (b) Yuan Y, Men H, Lee C. J Am Chem Soc. 2004;126:14720–14721. doi: 10.1021/ja0447154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Similar difficulties were previously noted by Pancrazi and coworkers during hydrostannylation of hindered alkynes, however no possible explanations were suggested, see: Betzer J-F, Delaloge F, Muller B, Pancrazi A, Prunet J. J Org Chem. 1997;62:7768–7780.
- 50.(a) Hirao A, Itsuno S, Nakahama S, Yamazaki N. J Chem Soc Chem Commun. 1981:315–317. [Google Scholar]; (b) Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551–5553. [Google Scholar]
- 51.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 52.Köbrich G, Trapp H. Chem Ber. 1966;99:680–688. [Google Scholar]
- 53.Bailey WF, Punzalan ER. J Org Chem. 1990;55:5404–5406.. Negishi E, Swanson DR, Rousset CJ. J Org Chem. 1990;55:5406–5409.. Iodine-lithium exchange is known to be an extremely fast process proceeding faster than deprotonation since this transformation can be performed in the presence of MeOH in some cases, see: Bailey WF, Patricia JJ, Nurmi TT, Wang W. Tetrahedron Lett. 1986;27:1861–1864.. Clayden J. Organolithiums: Selectivity for Synthesis. Pergamon; Oxford, UK: 2002.
- 54.Stille JK. Angew Chem Int Ed Eng. 1986;25:508–523. [Google Scholar]
- 55.Campos MM, Petragnani N. Chem Ber. 1960;93:317.. Nicolaou KC, Seitz SP, Sipio WJ, Blount JF. J Am Chem Soc. 1979;101:3884–3893.. For a recent mechanistic study of selenolactonization reactions, see: Denmark SE, Edwards MG. J Org Chem. 2006;71:7293–7306. doi: 10.1021/jo0610457.
- 56.(a) Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 2000;100:1929–1972. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Heemstra JR, Beutner GL. Angew Chem Int Ed. 2005;44:4682–4698. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]; (c) Kalesse M. Top Curr Chem. 2005;244:43–76. [Google Scholar]
- 57.Mi Y, Schreiber JV, Corey EJ. J Am Chem Soc. 2002;124:11290–11291. doi: 10.1021/ja027373f. [DOI] [PubMed] [Google Scholar]
- 58.See Supporting Information for further details.
- 59.Farina V, Krishnan B. J Am Chem Soc. 1991;113:9585–9595. [Google Scholar]
- 60.(a) Asaoka M, Yanagida N, Ishibashi K, Takei H. Tetrahedron Lett. 1981;22:4269–4270. [Google Scholar]; (b) Ollevier T, Bouchard J-E, Desyroy V. J Org Chem. 2008;73:331–334. doi: 10.1021/jo702085p. [DOI] [PubMed] [Google Scholar]; (c) Yanai H, Yoshino Y, Takahashi A, Taguchi T. J Org Chem. 2010;75:5375–5378. doi: 10.1021/jo100915e. [DOI] [PubMed] [Google Scholar]
- 61.Kong K, Romo D. Org Lett. 2006;8:2909–2912. doi: 10.1021/ol060534q. [DOI] [PubMed] [Google Scholar]
- 62.Prolonged treatment of 85b with DBU triggered a retro-vinylogous Mukaiyama aldol reaction leading to the formation of a ketone.
- 63.King SB, Ganem B. J Am Chem Soc. 1994;116:562–570. [Google Scholar]
- 64.Burk MJ, Allen JG. J Org Chem. 1997;62:7054–7057. [Google Scholar]
- 65.For some lead references on the synthesis of the spirolides, see: Ishihara J, Ishizaka T, Suzuki T, Hatakeyama S. Tetrahedron Lett. 2004;42:7855–7858.. Stivala CE, Zakarian A. Org Lett. 2009;11:839–842. doi: 10.1021/ol8027797.. Guéret SM, Furkert DP, Brimble MA. Org Lett. 2010;12:5226–5229. doi: 10.1021/ol102525w.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.