

Abstract
Polyethylenimines are cationic polymers with potential as delivery vectors in gene therapy and with proven antimicrobial activity. However, the antiviral activity of polyethylenimines has not been addressed in detail thus far. We have studied the inhibitory effects of a linear 25-kDa polyethylenimine on infections with human papillomaviruses and human cytomegaloviruses. Preincubation of cells with polyethylenimine blocked primary attachment of both viruses to cells, resulting in a significant reduction of infection. In addition, the dissemination of human cytomegalovirus in culture cells was efficiently reduced by recurrent administration of polyethylenimine. Polyethylenimine concentrations required for inhibition of human papillomavirus and cytomegalovirus did not cause any cytotoxic effects. Polyethylenimines and their derivatives may thus be attractive molecules for the development of antiviral microbicides.
INTRODUCTION
Polyethylenimines (PEIs) are cationic polymers with a broad molecular weight range. They are formed as either linear or branched molecules. Given their polycationic nature, PEIs have the capacity to condense with DNA, resulting in PEI-DNA complexes, and to mediate gene transfer into mammalian cells in vitro and in vivo (7). Cellular uptake of PEI-DNA complexes is dependent on heparan sulfate proteoglycans (HSPGs), which act as the interaction factor for PEI on the cell surface (27, 39, 44, 45). PEIs range among the most potent transfection agents and hence constitute an interesting alternative to viral vectors for gene therapy (2, 10, 19). Like other cationic polymers, PEIs display considerable antimicrobial activity. Synergistic antibacterial effects of PEI and antibiotics have been shown (32), and PEI has proven to be a valuable conjugate for photodynamic therapy of localized infections with Gram-positive and -negative bacterial, yeast, and fungal pathogens (21, 54). Furthermore, derivatives of PEI, which are effective in rupturing bacterial cell membranes, have also been suggested for antimicrobial coating of devices and surfaces (38). Although the effects of PEIs against bacteria have been well studied, the antiviral activities of these molecules have been only poorly analyzed thus far.
Human papillomaviruses (HPVs) are small, nonenveloped DNA viruses that comprise a family of more than 100 different types (4). After infection of epithelial cells, the low-risk HPV types cause benign epithelial warts on skin and mucosa. High-risk HPV types, including HPV type 16 (HPV16), HPV18, and HPV31, are associated with anogenital malignancies and are etiologically linked to the development of cervical cancer (16). Recent studies have elucidated that HSPGs constitute the primary attachment factors during infection, as shown for the majority of HPVs analyzed to date (24, 30). These molecules thus constitute promising targets for antiviral therapy.
Human cytomegalovirus (HCMV) is an enveloped DNA virus that belongs to the herpesvirus family. It is a ubiquitous agent that causes severe diseases predominantly in individuals with impaired or immature immune defense functions (13). HCMV also uses heparan sulfate proteoglycans for the primary tethering step in the course of infection; these viruses thus bind to a broad range of cells before internalization (11, 12, 31). Cell culture systems are available to study the molecular mechanisms of permissive HCMV infection in vitro.
In this study, we addressed the question of whether PEI is able to inhibit infections with HPV and HCMV. We found that PEI efficiently inhibited both viruses by blocking the primary binding of HPV and HCMV virions to target cells. In addition, PEI inhibited dissemination of an HCMV infection in cell culture. Taken together, these results indicate that PEIs and their derivatives may have microbicide potential as prophylactic and therapeutic antiviral agents.
MATERIALS AND METHODS
Cell culture.
The human cervix adenocarcinoma cell line HeLa and the Cos7 cell line (CV-1 in origin, carrying simian virus 40; African green monkey kidney fibroblast cells) were obtained from the German Resource Centre for Biological Material (DSMZ, Braunschweig, Germany). HaCaT cells (human adult low-calcium high-temperature, nonvirally transformed keratinocytes) were obtained from Cell Lines Services (CLS), Eppelheim, Germany. The human embryonic kidney cell line 293TT was kindly provided by Chris Buck, Bethesda, MD (8). Cells were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 1% GlutaMAX I (Invitrogen), 1% modified Eagle's medium nonessential amino acids, and antibiotics. Primary human foreskin fibroblasts (HFFs) were grown in minimal essential medium (MEM) supplemented with 5% FCS, 2 mM l-glutamine, 50 mg/liter gentamicin, and 0.5 ng/ml basic fibroblast growth factor. Chinese hamster ovary K1 (CHO-K1) cells and their glycosaminoglycan (GAG)-deficient derivative pgsA-745 (17), kindly provided by Martin Sapp, Shreveport, LA, were grown in DMEM/F-12 medium plus GlutaMAX supplemented with 10% FCS.
HPV pseudovirions and HCMV strains.
HPV pseudovirions were prepared as previously described (8). Briefly, expression plasmids carrying codon-optimized HPV L1 and L2 cDNA were cotransfected with a pcDNA3.1/luciferase reporter plasmid into 293TT cells (48). At 48 h posttransfection, the cells were processed by lysis and nuclease digestion. The pseudovirions were purified from the cell lysates by Optiprep gradient centrifugation. Codon-optimized L1 and L2 expression vectors pUF3/hu16L1 and pUF3/hu16L2 for HPV16, pe18L1bhb and pe18L2bhb for HPV18, and p31L1h and p31L2h for HPV31 were kindly provided by Martin Müller, Heidelberg, Germany, Chris Buck, Bethesda, MD, and Tadahito Kanda, Tokyo, Japan, respectively (34, 35, 43). Quantification of pcDNA3.1/luciferase-positive pseudovirions was performed by quantitative PCR in an AB 7300 reverse transcription-PCR system with luciferase forward primer 5′-CGCGGAGGAGTTGTGTTTGT-3′, luciferase reverse primer 5′-TTTTCTTGCGTCGAGTTTTCC-3′, and luciferase TaqMan probe 5′-AAGTACCGAAAGGTCTTAC-3′. For this, the marker plasmid was extracted from pseudovirions using a high pure viral nucleic acid kit (Roche) according to the manufacturer's instructions.
HCMV strains used were RV-ΔUS2-11_GFP (18), expressing green fluorescent protein (GFP) as a marker of infection, and RV-BADwt (55). In RV-ΔUS2-11, an expression cassette for GFP had been inserted into the genome of strain Ad169, thereby deleting a genomic region encoding immune evasion genes. Deletion of these genes has no impact on replication efficiency in fibroblast cell culture. The RV-BADwt strain is a derivative of the Ad169 strain of HCMV containing a functional US2-11 region and no GFP expression cassette. Virus stocks were generated as previously described (6). Infectivity in a given stock was determined by counting immediate-early 1 (IE1) protein-positive cells at 48 h postinfection (p.i.). For this, cells were stained for the IE1 protein with monoclonal antibody (MAb) p63-27 (1, 6). Multiplicity of infection (MOI) was defined as the number of IE1-inducing units per cell.
Infection assays.
For HPV infections, 100 luciferase vector-positive pseudovirions per cell were generally applied, unless stated otherwise. Infection efficiencies of HPV pseudovirions were assessed by quantification of luciferase expression at 24 h p.i. For this, cells were lysed with cell culture lysis reagent (Promega) and relative luciferase activity was measured with the luciferase assay system (Promega) according to the manufacturer's instructions, using a Tristar LB 941 luminometer (Berthold Technologies, Bad Wildbad, Germany).
HCMV infections (MOI, 1) with the RV-ΔUS2-11_GFP strain were quantified by measuring relative GFP amounts. For this, cells were lysed with cell culture lysis reagent (Promega) and GFP was quantified, using a Tristar LB 941 fluorimeter with a 485/535 filter set. RV-BADwt infections were quantified by counting IE1-positive cells stained with MAb p63-27 at 48 h p.i. (1, 6).
Binding assays.
Cells were preincubated with PEI for 30 min or left untreated (control) and were then infected for 2 h with HPV pseudovirions (500 per cell) or HCMV RV-BADwt (MOI, 5). Subsequently, cells were washed five times with phosphate-buffered saline (PBS) and then collected in SDS sample buffer for Western blotting. Cell-bound HPV16 particles were stained with anti-L1 antibody 312F (33). HCMV virions were stained with a MAb against the tegument protein pp65 (p65-33). β-Actin was stained as input control, using a MAb (Sigma). For attachment assays with prebound HPV pseudovirions, HeLa cells were infected for 1 or 2 h, cells were washed with PBS, medium with or without PEI was added to the cells for 1 h, and cells were again washed with PBS and processed for Western blotting with anti-L1 antibody.
HCMV in vitro dissemination assay.
HFF cells were seeded on 6-well plates (105 per well) and were subsequently infected with 300 IE1-inducing units per well. At 10 h p.i., cells were extensively washed with PBS. Fresh medium without or with PEI (13 or 16 nM) was added to the cells. Additional 13 or 16 nM PEI was supplemented to the respective wells in 24-h intervals. Cells were incubated for 10 days. Viral dissemination was documented by fluorescence microscopy and quantified by measuring the relative GFP amount as described above.
Polyethylenimine.
Linear polyethylenimine (molecular weight, 25,000; Polysciences, Inc.) was solubilized in Milli-Q water by addition of HCl and by ultrasound application for 10 min at room temperature in a Bandelin Sonorex Super RK 510H apparatus. The concentration was set to 13 nM PEI (corresponding to 7.5 mM nitrogen residues), and the solution was sterilized by filtration through a 0.2-μm-pore-size filter and was then stored at 4°C.
Cell viability and cytotoxicity.
Cell lysates from infection assays were additionally used to determine lactate dehydrogenase (LDH) activity with Cytotoxicity Detection KitPLUS (Roche) as a measure for relative cell number and viability. Potential cytotoxicity/cytolysis of PEI was quantified by measuring LDH activity released from damaged cells into culture medium with the Cytotoxicity Detection KitPLUS (Roche). In addition, a Cell Proliferation Kit XTT (AppliChem) was used to quantitate cell proliferation and viability according to the manufacturer's instructions. The same culture conditions (cell densities and culture times) were used for infection and viability/cytotoxicity assays.
Fluorescence microscopy.
Images were acquired using a Zeiss Axiovert 200 M microscope equipped with a Plan-Apochromat 1006 objective (numerical aperture, 1.4), a Zeiss Axiocam digital camera, and Axiovision software (version 4.6).
Statistical analysis.
The effective concentration for 50% inhibition of infection (EC50) and 50% cell cytotoxic concentration (CC50) were calculated with the 4PL model equation of the ReaderFit software (MiraiBio Group) and used to deduce the specificity index (SI; CC50/EC50).
RESULTS
Inhibitory effect of PEI on HPV16 infection.
In a first set of experiments, the effect of PEI on HPV16 infection was tested. HeLa cells were preincubated with different concentrations of PEI for 30 min and were then infected with HPV16 pseudovirions. After 24 h, infection rates were quantified by measuring luciferase activity in infected cells. The infection efficiency of HPV16 was significantly reduced by PEI in a concentration-dependent manner (Fig. 1A). A concentration of 52 nM PEI was already sufficient to inhibit HPV16 infection by more than 99%. To exclude the possibility that the inhibition of HPV16 infection was caused by toxic effects of the compound, a lactate dehydrogenase (LDH)-based cytotoxicity assay of cell culture supernatant and a 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT) proliferation/viability assay were conducted in HeLa cells incubated with increasing concentrations of PEI for 24 h (Fig. 1B; see Fig. S1 in the supplemental material). Concentrations of up to 416 nM PEI did not affect HeLa cells. Cytotoxic effects were first detected at 832 nM. This concentration was much higher than the concentration of PEI required for the blocking of HPV16 infection. These experiments showed that PEI efficiently blocks HPV16 infection at nontoxic concentrations. A specificity index (SI) of 212 was deduced from the calculated EC50 (4.04 nM) and CC50 (856.7 nM).
Fig 1.

Analysis of the inhibitory effects of PEI on HPV16 infection and determination of the cytotoxic concentration of PEI on HeLa cells. (A) HeLa cells were preincubated with the indicated concentrations of PEI for 30 min and were subsequently infected with HPV16 pseudovirions. Infection rates were assessed at 24 h postinfection by measuring luciferase activity in infected cells. The infection rate of the control (0 nM PEI) was set to 100%. Bars represent four individual experiments ± SD. (B) HeLa cells were incubated with the indicated concentrations of PEI for 24 h. The LDH activity released from damaged cells into the culture supernatant was quantified as a measure for cytotoxicity. Data represent the mean of 3 individual experiments ± SD. OD, optical density.
Dependence of HPV inhibition on infectious dose, HPV type, and cell lines.
To validate the inhibitory effects of PEI on HPV infection, we first tested the impact of increasing numbers of HPV16 pseudovirions. Given the results in the infection inhibition and cytotoxicity assay with dilution series of PEI in HeLa cells, these and subsequent analyses with HPV were carried out with 52 nM PEI. PEI-treated or untreated HeLa cells were infected with different amounts of HPV16 pseudovirions for 24 h, and infection rates were quantified by measuring luciferase activity. Infection reduction after a 30-min preincubation with PEI remained constant over a range of 100 to 1,000 HPV16 pseudovirions per cell (Fig. 2A). In a second experiment, the effect of PEI on infection efficiency of different HPV types was analyzed in different cell lines. PEI at 52 nM inhibited the infection of the predominant high-risk HPV types, 16, 18, and 31, by more than 99% in HeLa, HaCaT, Cos7, and 293TT cells (Fig. 2B). At the given concentration, no cytotoxic effect of PEI could be detected in any of the cell lines used (data not shown). These results showed that PEI was effective to prevent infection of different cell types by different types of HPV over a wide range of infectivity.
Fig 2.

Dependence of HPV inhibition by PEI on pseudovirion concentrations and on cell types. (A) Effect of PEI on different HPV16 titers. HeLa cells were preincubated with 52 nM PEI for 30 min and were then infected for 24 h with the indicated amounts of luciferase-positive HPV16 particles per cell. Infection inhibition through PEI was determined by correlating results from infected samples to mock-infected controls. Bars represent the mean of three individual experiments ± SD. (B) Inhibition of HPV infection in different cell lines. The indicated cell lines were preincubated with 52 nM PEI for 30 min and were then infected with HPV16, -18, or -31 pseudovirions for 24 h. Infection inhibition through PEI was determined by correlating results from infected samples to mock-infected controls. Bars represent the mean of three individual experiments ± SD.
PEI blocks primary attachment of HPV16 to cells.
Primary attachment to target cells is mediated through binding to heparan sulfate proteoglycans for the majority of HPV types studied so far (46). It is generally accepted that HSPGs are also involved in the uptake of PEI-DNA polyplexes, suggesting that PEI directly interacts with HSPGs (27, 39, 44, 45). Hence, it was conceivable that HPVs and PEI compete for the same primary attachment sites on cells and that inhibition of HPV infection by PEI was on the level of primary attachment. To analyze this, HeLa cells were preincubated with PEI. Binding of HPV16 pseudovirions to these cells was analyzed by Western blotting and compared to that of untreated controls (Fig. 3A). While nearly 100% of HPV16 pseudovirions attached to untreated HeLa cells at 2 h p.i., binding of pseudovirions was almost completely blocked by PEI pretreatment. These results showed that PEI inhibits the initial attachment of HPV virions to the cell surface. The relevance of HSPGs for PEI-mediated inhibition of HPV infection was confirmed by infection assays in CHO-K1 cells and the GAG-negative derivative cell line pgsA-745. Infection of CHO-K1 cells, expressing HSPGs, was efficiently blocked by PEI (Fig. 3B). Although pgsA-745 cells do not express HSPGs, they were permissive to HPV16 infection to a minor extent (1.2% ± 0.1; data not shown) compared to parental CHO-K1 cells, which is consistent with previous reports (9, 50). However, this residual infection of pgsA-745 cells could no longer be efficiently inhibited by PEI (Fig. 3B), suggesting that infection inhibition by PEI involves HSPGs.
Fig 3.

Inhibition of HPV16 attachment by PEI. (A) HeLa cells were infected with HPV16 pseudovirions for 2 h (+PEI, HeLa cells preincubated with PEI; −PEI, untreated HeLa control cells). Subsequently, cells were washed extensively with PBS to remove unbound virions. Attached virions were detected by Western blotting, using a MAb directed against the HPV16 capsid protein L1. β-Actin was probed as a loading control. (B) Infection of GAG-deficient (pgsA-745) and parental CHO-K1 cells in absence or presence of PEI (52 nM). Infection was scored at 24 h p.i. Control infections (without PEI) were set to 100%. Bars represent the mean of 4 individual experiments ± SD.
Pre- and postinfection inhibition by PEI.
To determine the time frame of inhibitory activity, 52 nM PEI was added to HeLa cells at different time points before or after infection with HPV16 pseudovirions. Infection efficiency was measured at 24 h p.i. by quantifying luciferase in infected cells. PEI significantly reduced infection when administered more than 24 h before and up to 4 h after addition of pseudovirions (Fig. 4A). These results suggest that PEI not only inhibited the binding process of HPV16 virions but also might interfere with postbinding events. The inhibition of infection by PEI preincubation was likely due to a block of HPV binding to the primary HSPG receptors, which was shown before (Fig. 3A). Inhibition by PEI treatment after infection might result from competition for the primary receptors, detachment of pseudovirions from HSPGs by PEI, inhibition of postbinding processing of viral particles, or deviation of viral particles into a noninfectious endocytotic pathway. To further elucidate postinfection inhibition mechanisms of PEI, binding assays were performed. Pseudovirions preattached to HeLa cells for 1 or 2 h were efficiently removed by incubation with PEI for 1 h (Fig. 4B). Hence, PEI administered after infection competes HPV out before penetration occurs. Whether further mechanisms account for the postinfection inhibition by PEI remains to be clarified.
Fig 4.

Impact of the time point of PEI application on HPV16 infection. (A) HeLa cells were treated with PEI (52 nM) at the indicated times before and after infection with HPV16 pseudovirions. Infection was scored at 24 h postinfection. Control infections without PEI were set to 100%. Bars represent the mean of three individual experiments ± SD. (B) Effect of PEI on prebound pseudovirions. Cells were infected for 1 or 2 h and then incubated or not with PEI (52 nM) for 1 h. Cell-bound virions were detected by Western blotting, using an anti-L1 antibody. β-Actin was probed as a loading control.
Inhibition of HCMV infection by PEI.
We have shown that PEI inhibited HPV infection by blocking primary binding of the viral particles to HSPG receptors on target cells. To investigate whether this inhibition was specific for papillomaviruses or if this was a more general mechanism of how polyethylenimines interfered with viral infections, we tested the impact of PEI on HCMV. This enveloped herpesvirus also uses HSPGs as primary attachment molecules (11, 12). Primary HFF culture cells were permissively infected with the GFP-expressing mutant RV-ΔUS2-11_GFP. PEI was added to the culture medium at different concentrations at the time of infection. At 24 h, expression of GFP was measured as a correlate of infection (Fig. 5A). The experiment showed that HCMV infection was significantly inhibited by PEI in a concentration-dependent manner. Infection inhibition was also observed with the HCMV parental strain RV-BADwt on a per cell basis (see Fig. S2 in the supplemental material). To determine possible toxic effects of PEI on HFF cells, LDH release and XTT proliferation/viability were quantified (Fig. 5B; see Fig. S1 in the supplemental material). Although PEI was toxic to the HFFs at lower concentrations than those for HeLa cells, the window between inhibitory and toxic concentrations was still considerably broad (Fig. 5B). The SI of 32.4 was calculated from the EC50 (5.73 nM) and CC50 (185.5 nM).
Fig 5.

Inhibitory effects of PEI on HCMV infection and cytotoxic effects of PEI on HFF cells. (A) Inhibition of HCMV infection by PEI. HCMV RV-ΔUS2-11_GFP particles (MOI, 1) and PEI were coadministered to HFF cells. Infection rates were determined after 24 h through quantification of GFP expressed by infected cells. Control infection (0 nM PEI) was set to 100%. Bars represent the mean of three individual experiments ± SD. (B) Toxicity of PEI on HFF cells. Cells were incubated with the indicated concentrations of PEI for 48 h. LDH activity in the culture supernatant was determined to be a measure for toxicity. Bars represent the mean of three individual experiments ± SD. (C) Inhibition of HCMV attachment by PEI. HFF cells were infected at an MOI of 5 with the HCMV strain RV-BADwt for 2 h (+PEI, HFFs preincubated with PEI; −PEI, untreated HFF controls). Subsequently, cells were washed extensively with PBS to remove unbound virions. Attached virions were detected by Western blotting, using a MAb directed against the HCMV tegument protein pp65. β-Actin was probed as a loading control.
As for HPV16, the effect of PEI on HCMV cell adsorption was tested in a virus binding assay. After preincubation with PEI and subsequent infection, cells were incubated for 2 h. Cells were then washed to remove unbound virions and processed for Western blotting, using a MAb directed against the viral tegument protein pp65. Again, PEI efficiently inhibited cell-virus interaction and subsequent infection (Fig. 5C). Taken together these results suggest that PEI also inhibited HCMV infection at a nontoxic concentration of the compound and that this inhibition was on the level of primary attachment of virus particles to HSPGs.
Inhibition of HCMV in vitro dissemination.
Infection of HFFs by HCMV leads to permissive viral replication. Consequently, the impact of PEI on dissemination of this virus in cell culture, in contrast to HPV, was accessible to analysis. Cells were infected with the GFP-expressing HCMV strain RV-ΔUS2-11_GFP for 10 h. After that, cells were washed extensively with PBS and 13 or 16 nM PEI was added. Additional PEI was supplemented every 24 h until quantitative evaluation of viral replication. At day 10 postinfection, GFP fluorescence in cell lysates was quantified as a relative measure of total viral load (Fig. 6A). In cells treated with PEI, GFP fluorescence as a correlate for viral multiplication was reduced by 85 to 90% compared to untreated control. This indicated that, following a first round of viral replication, HCMV spread to uninfected cells in the culture was inhibited by PEI.
Fig 6.
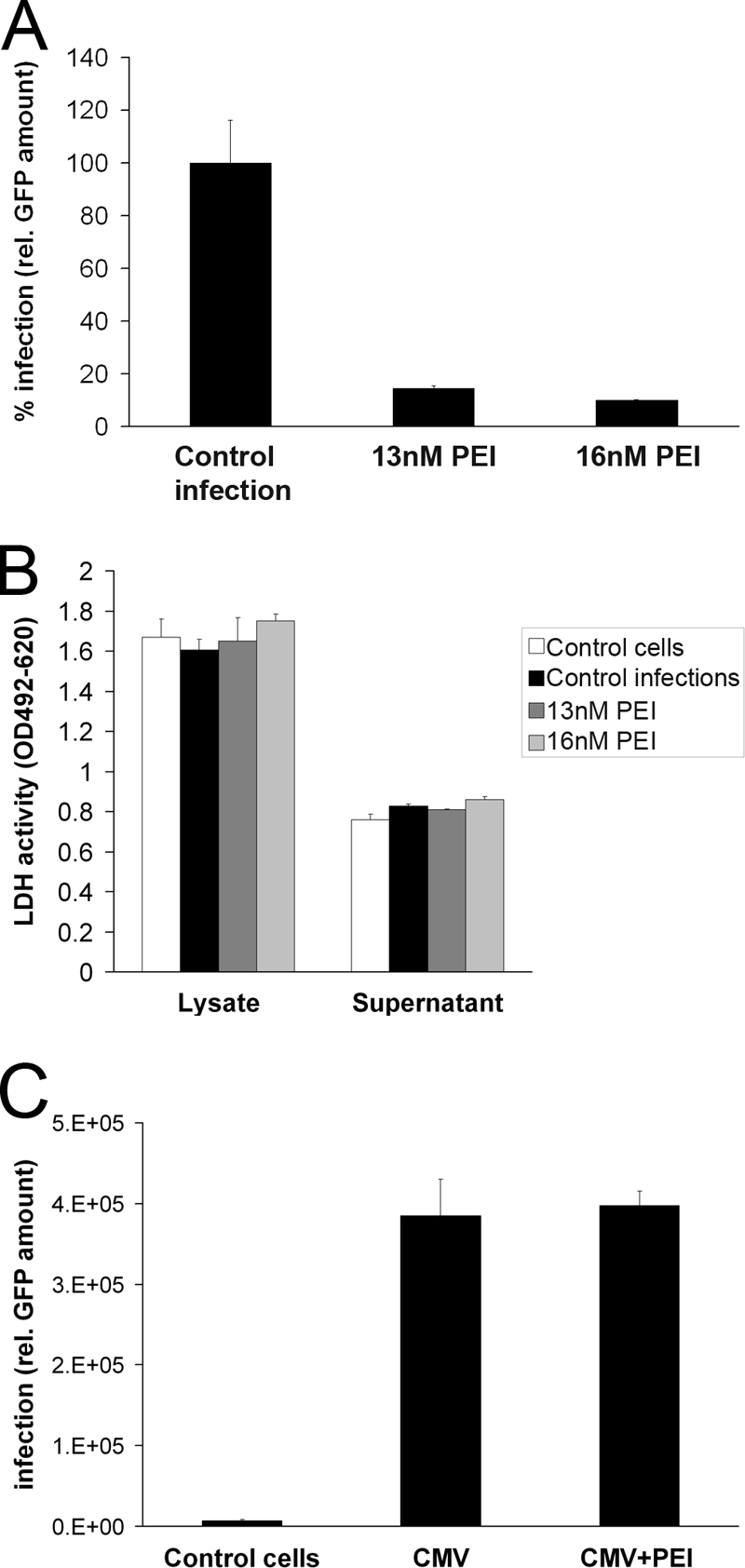
Impact of PEI on infection and spread of HCMV in permissive HFFs. (A) Inhibition of HCMV dissemination in HFF cultures by PEI. Cells were infected with RV-ΔUS2-11_GFP (MOI, 0.003). At 10 h p.i., 13 or 16 nM PEI was added to the culture medium. Additional PEI was supplemented in 24-h intervals. Total viral load was quantified 10 days after infection by measuring GFP. Bars represent the mean of three individual experiments ± SD. Control infections (no PEI) were set to 100%. (B) Determination of PEI toxicity on HFFs after 10 days of recurrent administration. LDH activity in the supernatants and lysates of the HFF cells of the infection assay (A) was determined at 10 days of culture. Bars represent the mean of three individual experiments ± SD. (C) Impact of PEI (10 h p.i.) on initial HCMV infection in HFF cultures. Cells were infected with HCMV RV-ΔUS2-11_GFP (MOI, 0.03) or mock infected. At 10 h after infection, cells were washed with PBS. PEI at 16 nM was added to some of the cultures. At 48 h postinfection, GFP expression was measured as a means for determination of primary viral infection. Bars represent the mean of three individual experiments ± SD.
To control that this reduction was not a result of cytotoxic side effects of 10 days of PEI administration, LDH in the supernatants and lysates of the infected HFF cells was quantified (Fig. 6B). The analysis showed that even prolonged application of PEI had no significant cytotoxic effects and that PEI did not affect cell viability.
To test if the impairment of HCMV multiplication in PEI-treated cultures was caused by an undefined block of viral replication after initial infection, HFFs were infected with RV-ΔUS2-11_GFP, treated with PEI at 10 h p.i., and analyzed for GFP expression at 48 h p.i. Untreated, infected cells were taken as a control (Fig. 6C). GFP expression was comparable between PEI-treated and control cells, indicating that the permissive replication program of HCMV was not affected by PEI, once the virus had entered the HFFs. To finally investigate the impact of PEI on cell-cell spread and on cell-free HCMV spread, RV-ΔUS2-11_GFP-infected cultures were treated with PEI at 10 h p.i. and monitored for plaque formation and spread, using GFP expression as a marker. Some cultures were left untreated for control. Initial inspection of the cultures showed GFP expression in single cells of the monolayer in PEI-treated and untreated HFFs at 48 h of infection (data not shown). In untreated cultures, the virus subsequently spread to finally infect all cells of the monolayer at 10 days of infection (Fig. 7). In PEI-treated cells, in contrast, small GFP-positive foci developed over time, but no spread in the culture was detectable. These results suggested that cell-cell spread was occurring in the presence of PEI but that infection by cell-free virus was blocked by the compound.
Fig 7.

Impact of PEI on HCMV cell-cell spread and cell-free virus spread. Cells were infected with HCMV RV-ΔUS2-11_GFP (MOI, 0.003). Cells were either left untreated or treated with PEI at 10 h after infection; additional PEI was supplemented in 24-h intervals. At 10 days postinfection, viral spread was monitored by fluorescence microscopy. In untreated cells (−PEI), nearly 100% of cells were infected (light cells). In PEI-treated cells (+PEI), only clusters of infected cells could be observed, suggesting that in the presence of PEI, viral dissemination occurs exclusively by cell-cell spread.
Together these results showed that PEI was effective in inhibiting HCMV infection of culture cells and that this was likely due to interference with primary attachment of viral particles to cell surface HSPGs.
DISCUSSION
In this study, the effect of polyethylenimine, a cationic polymer, on the infectivity of two different viruses, the nonenveloped HPV and the enveloped HCMV, was investigated. Infections with both viruses were significantly reduced when cells were preincubated with PEI or when PEI was administered to the cells at the time point of infection. For HPV16, we demonstrated that already 1.6 nM PEI inhibits infection significantly, by more than 20%. A concentration of 52 nM PEI reduced HPV16 infectivity below 1% compared to control infections. Interestingly, the inhibitory potential of 52 nM PEI seems to be independent of the amount of virus used for infection. Indeed, an equally efficient inhibition of more than 99% was observed in assays with increasing amounts of HPV16 pseudovirions per HeLa cell. This is in accordance with the proposed mode of action that PEI inhibits HPV infection by blocking primary receptors of HPV on target cells. Supporting this model, we could show in binding assays that viral particles were unable to bind to cells pretreated with 52 nM PEI. At the given concentration of PEI, no primary receptors for HPV particles are accessible on the surface of target cells, making infection inhibition equally efficient independent of virus amounts. In addition, we could show that low residual infection in a GAG-deficient CHO-K1 cell line (pgsA-745) is no longer inhibited by PEI, while infection in the parental CHO-K1 cells is still efficiently blocked by PEI. This is further evidence for the importance of HSPGs in PEI-mediated infection inhibition. A similar mode of action has been shown for another inhibitor of HPV infection, the N,N′-bisheteryl derivative of dispirotripiperazine, DSTP27 (50). This heparan sulfate-binding drug is a competitive inhibitor of HPV infections which blocks the primary attachment receptors of HPVs, the heparan sulfate proteoglycans. The majority of the human papillomaviruses studied so far bind to HSPGs as primary receptors on the cell surface. Subsequently, after a conformational change of the viral capsid, the virus is transferred to a secondary receptor for internalization (46, 53). Similar to PEI, the infection inhibition by DSTP27 is independent of the amount of HPV particles. At a given concentration, both molecules block all primary receptors for HPV16, which is then unable to attach to target cells.
It is generally accepted that polycations, including PEIs, bind to HSPGs (27, 39, 44, 45). In mammals, syndecans and glypicans constitute the predominant cell surface HSPGs (5, 20). Recently, it has been shown that PEI-DNA complexes bind to syndecans 1 and 2, inducing clustering of these syndecans as an initial step of internalization (42). Depending on the type of syndecan, endocytosis of PEI-DNA complexes follows different kinetics. While endocytosis of PEI with syndecan 1 occurs within minutes, PEI bound to syndecan 2 remains on the cell surface for hours. It is therefore conceivable that PEI inhibits binding of HPV to target cells by two mechanisms. One would be mediated by an interaction of PEI with HSPGs, thereby blocking primary attachment sites of HPVs on the cell surface. A second potential block would be by inducing a rapid internalization of syndecan 1, which was attributed to have a putative role as a primary receptor protein in natural HPV infection of keratinocytes (51).
HCMV infection was blocked by the administration of PEI at the time of infection. Like HPV, HCMV initiates infection by binding to HSPGs. This interaction is thought to play a crucial role in the initial stages of entry by enhancing the engagement of the viral particle with secondary receptors (11, 12). As shown in the binding assay, PEI appears to inhibit primary attachment of HCMV to target cells in binding assays, and hence, the mechanism of HCMV inhibition by PEI is probably the same as for HPV. Interestingly, HCMV infection can, like HPV infection, also be inhibited by DSTP27 (47). As engagement with HSPGs is a conserved mechanism in entry pathways of herpesviruses, it may be hypothesized that PEI also inhibits other members of this virus family.
Carrageenan, a sulfated polysaccharide extracted from red algae, has also been shown to inhibit HPV (9) and HCMV (3) adsorption and infection; however, inhibition is by a mechanism different than that for PEI. In contrast to PEI, carrageenans have a polyanionic nature, implicating an interaction with positively charged regions of viruses, which in consequence interferes with electrostatic interactions between viruses and HSPGs (23). Consistent with this, for HPV it has been shown that virus dose influences carrageenan inhibitory effects (9), in contrast to PEI. Virucidal activity of PEI and derivates by direct interaction with viruses is also known. N,N-Dodecyl,methyl-PEI inactivates human and avian influenza viruses (25, 26), probably by rupturing the viral envelope, and preincubation of HIV-1 with PEI significantly inhibits virus-cell binding, although it enhances infection when added postinfection by increasing membrane fluidity (41). This problem might be avoided with the latest engineered PEI molecules (see below).
PEI inhibited HPV16 significantly when administered 24 h before and up to 4 h after infection. This inhibitory effect was lost when the compound was added at 8 h after infection. Postinfection inhibition by PEI can be attributed to competition for primary HSPG receptors, as we observed in binding assays that PEI induces detachment of pseudovirions prebound to cells. Whether further mechanisms, e.g., feeding into noninfectious endocytosis pathways analogous to the pathway for DSTP27 (50), account for PEI-mediated postinfection inhibition remains to be clarified. Also, as the pseudovirus and cell culture system may not fully represent the naturally occurring HPV infection, future studies should analyze the effect of PEI on authentic HPV in keratinocyte infections (37).
In contrast to HPV, HCMV can be propagated in HFFs, and thus, these cells are a versatile system to study the effects of antiviral compounds with respect to their effects during HCMV replication. Low-multiplicity infection and subsequent application of PEI resulted in marked impairment of viral multiplication, measured at 10 days of infection. This could not be ascribed to a defect in viral gene expression, as this appeared to be unaffected by PEI. Rather, secondary rounds during multiple-cycle infections were likely blocked by the compound, as spread of infection in the monolayer was clearly suppressed. HCMV spreads in HFF culture either by the release of infectious virus into the supernatant or by cell-to-cell transmission. The former results in far distant infection events which depend on the interaction of the virus with HSPGs for primary attachment. Consequently, PEI inhibited the homogenous spread of HCMV in HFF cultures. In contrast, cell-to-cell transmission leads to the formation of foci, which expand over time (52). PEI did not appear to have an impact on cell-to-cell spread of HCMV in HFF culture.
It has been suggested that cell-to-cell spread of HCMV between endothelial cells and leukocytes occurs by the transfer of infectious virus via microfusion events of neighboring membranes (22). Cell-cell fusion has also been suggested to be a mode of transmission of HCMV between HFFs (15).
Transfer of HCMV via cell fusion events would not likely require the interaction of viral particles with HSPGs, thus explaining the lack of inhibition of focus formation by PEI. Others, however, have recently shown that focal transmission between endothelial cells could be blocked by the addition of virus-neutralizing antibodies (49). Neutralization by antibodies would require the release of infectious particles from infected cells. Interestingly, neutralizing antibodies can block supernatant-driven spread but not focus formation in HCMV-infected HFF cultures (15, 49). Cell type-specific differences in focal spread of HCMV were also described before by Jiang and colleagues (29). The underlying molecular mechanisms explaining the differences in focal spread between HFFs and endothelial cells are incompletely understood at this stage. In any way, detailed analysis of the impact of PEI on HCMV infection in different cell types will be required to further elucidate the potential of the compound to prevent local viral spread within organ tissues.
The concentrations and doses of PEI necessary for the inhibition of HPV and HCMV had no cytotoxic effect on any of the cells used. As a matter of fact, cytotoxic concentrations of PEI were significantly higher than the concentrations needed for inhibition of virus-cell binding and subsequent infection. Even the prolonged recurrent application of PEI did not impair viability of HFF cells. As PEI is one of the most promising nonviral delivery vectors for gene therapy (14, 40), much effort has been invested in the redesign and synthesis of different PEI molecules and derivatives in order to improve the safety and tolerability of the compound. As a consequence, a broad range of modified PEI-derived molecules with different molecular weights, some of which are even biodegradable, was developed (28, 36). These modifications will likely allow testing of PEI-derived molecules in clinical studies. Considering this, our findings render PEI and its derivatives attractive candidates as antiviral microbicides, either on their own or in combination with other compounds. Further studies are warranted to test optimized PEI derivatives for their microbicide potential and to investigate these compounds for antiviral efficiency in animal models.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by SFB grants of the Deutsche Forschungsgemeinschaft (DFG) (SFB490/D2 to L.F. and C.L.; SFB490/E7 to B.P. and K.B.).
We thank Thomas Shenk, Princeton, NJ, and Gabi Hahn, Ingolstadt, Germany, for the donation of HCMV strains and William Britt, Birmingham, AL, for monoclonal antibodies. We thank Chris Buck, Bethesda, MD, Martin Müller, Heidelberg, Germany, and Tadahito Kanda, Tokyo, Japan, for providing HPV codon-optimized expression plasmids. We thank Bettina Frenzel for technical support and Jens Stieler and Sabine Reyda for technical advice.
Footnotes
Published ahead of print 3 October 2011
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Andreoni M, Faircloth M, Vugler L, Britt WJ. 1989. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 23:157–167 [DOI] [PubMed] [Google Scholar]
- 2. Azzam T, Domb AJ. 2004. Current developments in gene transfection agents. Curr. Drug Deliv. 1:165–193 [DOI] [PubMed] [Google Scholar]
- 3. Baba M, Snoeck R, Pauwels R, de Clercq E. 1988. Sulfated polysaccharides are potent and selective inhibitors of various enveloped viruses, including herpes simplex virus, cytomegalovirus, vesicular stomatitis virus, and human immunodeficiency virus. Antimicrob. Agents Chemother. 32:1742–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bernard HU, et al. 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernfield M, et al. 1992. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 8:365–393 [DOI] [PubMed] [Google Scholar]
- 6. Besold K, Wills M, Plachter B. 2009. Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 391:5–19 [DOI] [PubMed] [Google Scholar]
- 7. Boussif O, et al. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78:751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buck CB, et al. 2006. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coll JL, et al. 1999. In vivo delivery to tumors of DNA complexed with linear polyethylenimine. Hum. Gene Ther. 10:1659–1666 [DOI] [PubMed] [Google Scholar]
- 11. Compton T, Feire A. 2007. Early events in human cytomegalovirus infection. InArvin A, et al. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 12. Compton T, Nowlin DM, Cooper NR. 1993. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 193:834–841 [DOI] [PubMed] [Google Scholar]
- 13. Crumpacker CS, Zhang JL. 2010. Cytomegalovirus, p 1971–1987 InMandell GL, Bennett JE, Dolin R. (ed), Principles and practice of infectious diseases, 7th ed Churchill Livingstone Elsevier, Philadelphia, PA [Google Scholar]
- 14. Demeneix B, Behr JP. 2005. Polyethylenimine (PEI). Adv. Genet. 53:217–230 [PubMed] [Google Scholar]
- 15. Digel M, Sampaio KL, Jahn G, Sinzger C. 2006. Evidence for direct transfer of cytoplasmic material from infected to uninfected cells during cell-associated spread of human cytomegalovirus. J. Clin. Virol. 37:10–20 [DOI] [PubMed] [Google Scholar]
- 16. Doorbar J. 2006. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond.) 110:525–541 [DOI] [PubMed] [Google Scholar]
- 17. Esko JD, Stewart TE, Taylor WH. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 82:3197–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falk CS, et al. 2002. NK cell activity during human cytomegalovirus infection is dominated by US2-11-mediated HLA class I down-regulation. J. Immunol. 169:3257–3266 [DOI] [PubMed] [Google Scholar]
- 19. Ferrari S, et al. 1997. ExGen 500 is an efficient vector for gene delivery to lung epithelial cells in vitro and in vivo. Gene Ther. 4:1100–1106 [DOI] [PubMed] [Google Scholar]
- 20. Fransson LA. 2003. Glypicans. Int. J. Biochem. Cell Biol. 35:125–129 [DOI] [PubMed] [Google Scholar]
- 21. Fuchs BB, Tegos GP, Hamblin MR, Mylonakis E. 2007. Susceptibility of Cryptococcus neoformans to photodynamic inactivation is associated with cell wall integrity. Antimicrob. Agents Chemother. 51:2929–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gerna G, et al. 2000. Human cytomegalovirus replicates abortively in polymorphonuclear leukocytes after transfer from infected endothelial cells via transient microfusion events. J. Virol. 74:5629–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghosh T, et al. 2009. Focus on antivirally active sulfated polysaccharides: from structure-activity analysis to clinical evaluation. Glycobiology 19:2–15 [DOI] [PubMed] [Google Scholar]
- 24. Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haldar J, An D, Alvarez de Cienfuegos L, Chen J, Klibanov AM. 2006. Polymeric coatings that inactivate both influenza virus and pathogenic bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:17667–17671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haldar J, Chen J, Tumpey TM, Gubareva LV, Klibanov AM. 2008. Hydrophobic polycationic coatings inactivate wild-type and zanamivir- and/or oseltamivir-resistant human and avian influenza viruses. Biotechnol. Lett. 30:475–479 [DOI] [PubMed] [Google Scholar]
- 27. Hess GT, Humphries WH, IV, Fay NC, Payne CK. 2007. Cellular binding, motion, and internalization of synthetic gene delivery polymers. Biochim. Biophys. Acta 1773:1583–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jere D, et al. 2009. Degradable polyethylenimines as DNA and small interfering RNA carriers. Expert Opin. Drug Deliv. 6:827–834 [DOI] [PubMed] [Google Scholar]
- 29. Jiang XJ, et al. 2008. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J. Virol. 82:2802–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joyce JG, et al. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 274:5810–5822 [DOI] [PubMed] [Google Scholar]
- 31. Kari B, Gehrz R. 1993. Structure, composition and heparin binding properties of a human cytomegalovirus glycoprotein complex designated gC-II. J. Gen. Virol. 74(Pt 2):255–264 [DOI] [PubMed] [Google Scholar]
- 32. Khalil H, Chen T, Riffon R, Wang R, Wang Z. 2008. Synergy between polyethylenimine and different families of antibiotics against a resistant clinical isolate of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52:1635–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Knappe M, et al. 2007. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J. Biol. Chem. 282:27913–27922 [DOI] [PubMed] [Google Scholar]
- 34. Kondo K, et al. 2007. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology 358:266–272 [DOI] [PubMed] [Google Scholar]
- 35. Leder C, Kleinschmidt JA, Wiethe C, Muller M. 2001. Enhancement of capsid gene expression: preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J. Virol. 75:9201–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lv H, Zhang S, Wang B, Cui S, Yan J. 2006. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 114:100–109 [DOI] [PubMed] [Google Scholar]
- 37. McLaughlin-Drubin ME, Christensen ND, Meyers C. 2004. Propagation, infection, and neutralization of authentic HPV16 virus. Virology 322:213–219 [DOI] [PubMed] [Google Scholar]
- 38. Milovic NM, Wang J, Lewis K, Klibanov AM. 2005. Immobilized N-alkylated polyethylenimine avidly kills bacteria by rupturing cell membranes with no resistance developed. Biotechnol. Bioeng. 90:715–722 [DOI] [PubMed] [Google Scholar]
- 39. Mislick KA, Baldeschwieler JD. 1996. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. U. S. A. 93:12349–12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Neu M, Fischer D, Kissel T. 2005. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J. Gene Med. 7:992–1009 [DOI] [PubMed] [Google Scholar]
- 41. Owada T, et al. 1998. Enhancement of human immunodeficiency virus type 1 (HIV-1) infection via increased membrane fluidity by a cationic polymer. Microbiol. Immunol. 42:97–107 [DOI] [PubMed] [Google Scholar]
- 42. Paris S, Burlacu A, Durocher Y. 2008. Opposing roles of syndecan-1 and syndecan-2 in polyethyleneimine-mediated gene delivery. J. Biol. Chem. 283:7697–7704 [DOI] [PubMed] [Google Scholar]
- 43. Pastrana DV, et al. 2004. Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology 321:205–216 [DOI] [PubMed] [Google Scholar]
- 44. Payne CK, Jones SA, Chen C, Zhuang X. 2007. Internalization and trafficking of cell surface proteoglycans and proteoglycan-binding ligands. Traffic 8:389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruponen M, Yla-Herttuala S, Urtti A. 1999. Interactions of polymeric and liposomal gene delivery systems with extracellular glycosaminoglycans: physicochemical and transfection studies. Biochim. Biophys. Acta 1415:331–341 [DOI] [PubMed] [Google Scholar]
- 46. Sapp M, Bienkowska-Haba M. 2009. Viral entry mechanisms: human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J. 276:7206–7216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmidtke M, et al. 2003. Binding of a N,N′-bisheteryl derivative of dispirotripiperazine to heparan sulfate residues on the cell surface specifically prevents infection of viruses from different families. Virology 311:134–143 [DOI] [PubMed] [Google Scholar]
- 48. Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell. Microbiol. 13:32–46 [DOI] [PubMed] [Google Scholar]
- 49. Scrivano L, Sinzger C, Nitschko H, Koszinowski UH, Adler B. 2011. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 7:e1001256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Selinka HC, et al. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J. Virol. 81:10970–10980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shafti-Keramat S, et al. 2003. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 77:13125–13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sinzger C, Knapp J, Plachter B, Schmidt K, Jahn G. 1997. Quantification of replication of clinical cytomegalovirus isolates in cultured endothelial cells and fibroblasts by a focus expansion assay. J. Virol. Methods 63:103–112 [DOI] [PubMed] [Google Scholar]
- 53. Spoden G, et al. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16—involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tegos GP, et al. 2006. Protease-stable polycationic photosensitizer conjugates between polyethyleneimine and chlorin(e6) for broad-spectrum antimicrobial photoinactivation. Antimicrob. Agents Chemother. 50:1402–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.