

Abstract
Although survival-promoting effects of insulin-like growth factor-1 (IGF-1) during neurogenesis are well characterized, mitogenic effects remain less well substantiated. Here, we characterize cell cycle regulators and signaling pathways underlying IGF-1 effects on embryonic cortical precursor proliferation in vitro and in vivo. In vitro, IGF-1 stimulated cell cycle progression and increased cell number without promoting cell survival. IGF-1 induced rapid increases in cyclin D1 and D3 protein levels at 4 h and cyclin E at 8 h. Moreover, p27KIP1 and p57KIP2 expression were reduced, suggesting downregulation of negative regulators contributes to mitogenesis. Furthermore, the phosphatidylinositol 3-kinase (PI3K)/Akt pathway specifically underlies IGF-1 activity, because blocking this pathway, but not MEK (mitogen-activated protein kinase kinase)/ERK (extracellular signal-regulated kinase), prevented mitogenesis. To determine whether mechanisms defined in culture relate to corticogenesis in vivo, we performed transuterine intracerebroventricular injections. Whereas blockade of endogenous factor with anti-IGF-1 antibody decreased DNA synthesis, IGF-1 injection stimulated DNA synthesis and increased the number of S-phase cells in the ventricular zone. IGF-1 treatment increased phospho-Akt fourfold at 30 min, cyclins D1 and E by 6 h, and decreased p27KIP1 and p57KIP2 expression. Moreover, blockade of the PI3K/Akt pathway in vivo decreased DNA synthesis and cyclin E, increased p27KIP1 and p57KIP2 expression, and prevented IGF-1-induced cyclin E mRNA upregulation. Finally, IGF-1 injection in embryos increased postnatal day 10 brain DNA content by 28%, suggesting a role for IGF-1 in brain growth control. These results demonstrate a mitogenic role for IGF-1 that tightly controls both positive and negative cell cycle regulators, and indicate that the PI3K/Akt pathway mediates IGF-1 mitogenic signaling during corticogenesis.
Keywords: corticogenesis, IGF-1, p27KIP1, p57KIP2, PI3K/Akt, proliferation
Introduction
Cerebral cortex function depends on cellular networks including excitatory glutamatergic projection neurons and inhibitory interneurons. Glutamatergic neurons are generated from dividing progenitors of the neocortical ventricular zone (Tan et al., 1998; Rakic, 2002). Precise regulation of progenitor cell cycle exit is an important requisite because it determines cell number and laminar fate (McConnell, 1988; Caviness et al., 1995).
Extrinsic signals directly influence progression from G1 into S phase of cortical precursors (Cunningham and Roussel, 2001). Current models suggest that D cyclins are primary targets of mitogenic signals (Sherr and Roberts, 1999). On stimulation, D cyclins bind and activate cyclin-dependent kinase (CDK) proteins CDK4/6 in early G1. Cyclin E, a transcriptional target dependent on cyclin D/CDK4/6 activation, in turn activates cyclin E/CDK2 complex during late G1, and is required for S-phase entry. CDK activity is also regulated through inhibitory interactions with CDK inhibitors (CKIs) of the CIP/KIP family, including p21CIP1, p27KIP1, and p57KIP2, which represent additional targets of extracellular cues (Nourse et al., 1994; Li and DiCicco-Bloom, 2004). Progression into S phase is determined by the balance between promitogenic cyclins and antimitogenic CKIs. For example, antimitogenic PACAP (pituitary adenylate cyclase-activating polypeptide) reduces S-phase commitment of cortical precursors by upregulating p57KIP2 (Carey et al., 2002), whereas mitogenic basic fibroblastic growth factor (bFGF) stimulates cell cycle reentry by increasing cyclins D1 and E, and downregulating p27KIP1 (Li and DiCicco-Bloom, 2004). Accordingly, bFGF injection into rat embryo cerebral ventricles dramatically increases neuron production and brain size (Vaccarino et al., 1999). Therefore, defining how extracellular signals regulate cell cycle machinery provides insight into emergence of neocortical cytoarchitecture.
Insulin-like growth factor-1 (IGF-1) is another signal important for nervous system development (Carson et al., 1993). Numerous studies in vitro support IGF-1 roles in proliferation (DiCicco-Bloom and Black, 1988; D'Ercole et al., 1996; Aberg et al., 2003), survival (Aizenman and de Vellis, 1987; Wilkins et al., 2001), and differentiation (Hsieh et al., 2004; McCurdy et al., 2005) of neuronal and glial progenitors. Acting through type 1 receptor (Adams et al., 2000), IGF-1 activates phosphatidylinositol 3-kinase (PI3K)/Akt and mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathways (Clemmons and Maile, 2003), which can mediate proliferation and/or survival (Liang and Slingerland, 2003; Manning and Cantley, 2007).
IGF-1 appears critical for controlling brain growth and cell number (Beck et al., 1995; Cheng et al., 1998). Transgenic mice overexpressing IGF-1 during development display increases in cortical volume and cell number (Popken et al., 2004). Increased neurogenesis involved reducing G1 phase and recruiting additional cells into cycle, suggesting a mitogenic role for IGF-1 (Hodge et al., 2004). However, transduction and cell cycle pathways mediating mitogenic stimulation and requirements for IGF-1 during corticogenesis in vivo remain undefined. Because compensatory mechanisms may accompany genetic manipulations, we performed acute transuterine intracerebroventricular injections into embryos in addition to culture studies. We report that IGF-1 is a mitogen for cortical precursors during embryonic development, acting through upregulation of G1 cyclins and downregulation of CKIs p27KIP1 and p57KIP2, without altering cell survival. Furthermore, we demonstrate that IGF-1 mitogenic signaling is mediated by the PI3K/Akt pathway. These studies suggest that IGF-1 signaling may influence cellular output during corticogenesis.
Materials and Methods
Reagents.
Human recombinant IGF-1 was purchased from Cell Sciences. bFGF was obtained from Scios. DMEM, F12 medium, penicillin, glutamine, and streptomycin were from Invitrogen. Transferrin, PI3K inhibitor [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002)], and MEK inhibitors [2-amino-3-methoxyflavone (PD98059); 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U0126)] were obtained from Calbiochem. Putrescine, progesterone, selenium, glucose, bovine serum albumin (BSA), bromodeoxyuridine (BrdU), propidium iodide (PI), fluorescein diacetate (FDA), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), poly-d-lysine, and protease inhibitors were purchased from Sigma-Aldrich. Monoclonal BrdU antibody was from BD Biosciences. Phosphohistone H3 (Ser10) polyclonal antibody was from Millipore. Cleaved caspase-3 polyclonal antibody (Asp175), monoclonal phospho-Akt (Ser473), polyclonal Akt, polyclonal phospho-ERK (Thr202/Tyr204), polyclonal ERK, polyclonal phospho-GSK-3β (Ser9), and rabbit monoclonal GSK-3β antibodies were from Cell Signaling. Cyclin D1 (SC-20044), cyclin D3 (SC-182), p27KIP1 (SC-528), and p57KIP2 (SC-8298) antibodies were obtained from Santa Cruz. Cyclin E polyclonal antibody was from Abcam. β-Actin monoclonal antibody was from Millipore Bioscience Research Reagents. Alexa Fluor 488 and 594 secondary antibodies were purchased from Invitrogen. Mouse recombinant IGF-1, monoclonal antibody specifically neutralizing the bioactivity of mouse IGF-1, and anti-mouse IgG were obtained from R&D Systems.
Cortical precursor cell culture.
Time-mated pregnant Sprague Dawley rats were obtained from Hilltop Laboratory Animals. The day of plug was considered as embryonic day 0.5 (E0.5) and the day of birth as postnatal day 0 (P0). On E14.5, skin, skull, and meninges were removed from embryos. The dorsolateral cortex was dissected and mechanically dissociated using a fire-polished glass pipette. Cortical precursors were then plated at 1750 cells/mm2 on poly-d-lysine (5 μg/ml)-coated 60 mm dishes (5 million cells/dish), 35 mm dishes (1.7 millions cells/dish), or 24-multiwell plates (313,000 cells/well), in defined medium, as previously described (Lu and DiCicco-Bloom, 1997). Culture medium was composed of a 50:50 (v/v) mixture of DMEM and F12 containing penicillin (50 U/ml) and streptomycin (50 μg/ml) and supplemented with transferrin (100 μg/ml), putrescine (100 μm), progesterone (20 nm), selenium (30 nm), glutamine (2 mm), glucose (6 mg/ml), and BSA (10 mg/ml). Cultures were maintained in an incubator at 37°C with 5% CO2. In most experiments, human IGF-1 was added into culture medium at plating. For the analysis of ERK and Akt phosphorylation by Western blotting, cells were preincubated for 1 h before IGF-1 treatment. For inhibitor experiments, cells were plated for 1 h, and then PI3K inhibitor (LY294002) or MEK inhibitor (PD98059 or U0126) was added 30 min before IGF-1 treatment and maintained throughout the subsequent incubation period.
Anti-IGF-1 neutralizing experiments in vitro.
Analysis of the neutralizing activity of anti-IGF-1 antibody to mouse IGF-1 was performed on rat E14.5 cortical precursors in vitro in defined medium in 24-multiwell plates. Mouse IGF-1 (3 ng/ml) was incubated with anti-IGF-1 (10 μg/ml) or anti-IgG (10 μg/ml; negative control) under agitation in 1 ml of medium for 2 h at 37°C. To determine specificity of the anti-IGF-1 antibody, bFGF (10 ng/ml) was incubated with anti-IGF-1 or anti-IgG (10 μg/ml) in the same conditions. After the preincubation period, the mixtures or growth factors alone were added into cultures, and [3H]thymidine incorporation assay was performed at 24 h as described below.
Intracerebroventricular injections.
Pregnant rats and pregnant mice (C57BL/6; Hilltop Laboratory Animals) were anesthetized with a mixture of ketamine (100 mg/kg body weight; Ketaset; Fort Dodge Animal Health) and xylazine (10 mg/kg body weight; Xyla-ject; Phoenix Pharmaceuticals). Then, the uterine horns were exposed by laparotomy and embryos were transilluminated to visualize the cerebral cortex. All injected solutions contained 0.05% fast green and were manually microinjected (3 μl) into the left lateral ventricle through the uterine wall using a Hamilton syringe (33/1 inch gauge). Injection success was indicated by spread of the dye from left to right lateral ventricle. At the indicated time, embryos were collected and dissected in ice-cold PBS. For IGF-1 injection experiments, PBS alone (control) and PBS solution containing IGF-1 (30 or 100 ng per embryo of the human recombinant form) were injected into E15.5–E16.5 rat embryos. For LY294002 injection experiments, control solution (DMSO vehicle) and LY294002 solution (184 ng per embryo; 3 μl of 200 μm solution) were injected into E16.5 rat embryos. For IGF-1 neutralizing experiments, anti-IgG (control) or anti-mouse IGF-1 antibody was injected into E14.5–E16.5 mouse embryos (0.5–2 μg per embryo, using 0.5 μg/μl antibody solution prepared in PBS). Because the concentration of IGF-1 in embryonic CSF was unknown, we performed neutralizing experiments with a fourfold range of anti-IGF-1 and anti-IgG antibodies. Because there was no difference in effects among the doses, we combined all the experiments.
[3H]Thymidine incorporation assays.
Incorporation of [3H]thymidine in vitro was used as a marker of DNA synthesis (Lu and DiCicco-Bloom, 1997). Cells plated in 24-well plates were incubated with 1 μCi/ml [3H]thymidine (GE Healthcare) for 2 h before culture termination. Subsequently, cells were collected with a semiautomatic harvester (Skatron), and incorporation was assessed by liquid scintillation spectroscopy.
To assess DNA synthesis in cerebral cortex of injected embryos, we used a percentage [3H]thymidine incorporation assay (Tao et al., 1996; Wagner et al., 1999; Suh et al., 2001). After removing meninges, cortices were dissected from embryos and stored at −80°C until use. Tissues were homogenized in 300 μl of distillated water and equal volume aliquots were taken to determine total isotope uptake in tissue homogenate and isotope incorporated into DNA. DNA was precipitated with 10% trichloroacetic acid (TCA), sedimented by centrifugation, and washed twice with 10% TCA. The final pellet and the aliquot of homogenate were dissolved and counted by liquid scintillation spectroscopy. The percentage incorporation is calculated as the ratio of radiolabel incorporated into DNA to total tissue uptake.
DNA quantification.
Brains were removed from P10 animals and dissected from the rostral extent of the cerebral hemispheres (excluding the olfactory bulbs) to their caudal extent (corresponding to the back of the occipital cortex) and homogenized in ice-cold distilled water. DNA was purified and quantified using a DNA dye-binding assay (Burton, 1956; Cheng et al., 2001). Briefly, DNA was extracted from homogenates by precipitation with 10% TCA and washed with ethanol. Then, the pellet containing the DNA was treated with 1N KOH and sedimented using 5% TCA. After solubilization in 5% TCA at 90°C, the supernatant containing the DNA was used for the dye-binding assay. Experimental samples and DNA standards (calf thymus DNA; Sigma-Aldrich) were mixed with diphenylamine reagent and incubated at 37°C overnight. Optical densities were measured at 600 nm.
Immunocytochemistry.
Cortical precursor cells were cultured in 35 mm dishes. At the end of incubation, culture medium was removed, cells were fixed in 4% paraformaldehyde (PFA) for 20 min, and then washed 5 min twice with PBS before immunolabeling. Embryonic brains were fixed by immersion in 4% PFA for 2 h. Cells or tissue sections were incubated overnight at room temperature in primary antibody diluted in PBS containing 0.3% Triton X-100 (PBS-T), 1% BSA, 10% dry milk, and 0.01% sodium azide. Labeling was revealed with a secondary antibody conjugated to Alexa Fluor 488 or 594 (1:500) prepared in the same buffer for 1 h at room temperature (Mairet-Coello et al., 2005).
Incorporation of BrdU.
Cells were plated in 35 mm dishes and BrdU (10 μm final) was added 2 h before culture termination. At the end of culture, medium was removed, and cells were fixed and then treated with 2N HCl for 30 min, rinsed twice with PBS, incubated with monoclonal anti-BrdU (1:100) for 1 h, followed by anti-mouse secondary antibody. The BrdU labeling index (LI), defined as the proportion of total cells incorporating BrdU into the nucleus, was determined by counting BrdU-immunolabeled cells over total cells under phase (4000–5000 cells), using 10× objective from 10 randomly selected fields in each of three dishes per group (Li and DiCicco-Bloom, 2004).
To determine whether cells that entered S phase were able to progress completely through the cell cycle in vitro, a cohort of cells was marked during a 2 h BrdU pulse at 22 h of culture, and then the absolute numbers of BrdU-labeled cells were compared between 36 and 24 h of culture. If cells that entered S phase during the 2 h BrdU pulse were able to divide, then the absolute number of BrdU-labeled cells is expected to increase from 24 to 36 h. BrdU (10 μm final) was added into culture medium at 22 h in control and IGF-1 cultures and then removed at 24 h by washing cells twice with PBS. A first group of control and IGF-1 cultures was fixed and processed for BrdU immunolabeling at 24 h. In a parallel group, culture medium was replaced for the following 12 h of culture. Then, cells were fixed at 36 h and processed for BrdU immunolabeling. The absolute number of BrdU-labeled cells per field was determined by counting as above.
To estimate the BrdU LI in vivo, BrdU was injected subcutaneously into the pregnant dam (100 μg/g body weight) 1 h before the killing. Brains from intracerebroventricularly injected embryos were removed, fixed by immersion in 4% PFA overnight at 4°C, and then processed for paraffin embedding. Brains were cut serially into 6 μm coronal sections on a microtome (Leica), and then mounted on Superfrost Plus slides (VWR). Before BrdU immunolabeling, sections were rinsed 5 min in PBS, treated with trypsin solution (0.1% trypsin in 0.1 m Tris buffer, 0.1% CaCl2, pH 7.5) for 20 min at 37°C, and then with 2N HCl for 30 min. Then sections were incubated with anti-BrdU antibody (1:100 in PBS-T) overnight at room temperature. Signal was revealed by incubating with secondary antibody conjugated to Alexa Fluor 488 (1:500) for 1 h at room temperature. Finally, sections were counterstained with PI. To determine the BrdU labeling index, analysis was performed in the dorsolateral cortical area on three nonadjacent coronal sections per brain. BrdU-positive nuclei and total nuclei were scored within a 100 μm width sector based on the ventricular surface and extending to the ventricular zone/intermediate zone boundary (Suh et al., 2001).
Assessment of precursor mitosis.
The relative number of cells in M phase was estimated using phosphohistone H3 antibody (1:200) as a marker. Phosphohistone H3-labeled cells were counted in three 1 cm rows in three dishes per group.
Cell death analysis.
At the indicated times, 15 μg/ml PI, a red fluorescent DNA marker whose uptake reveals plasma membrane damage, was added directly to living cultures in 35 mm dishes and incubated at 37°C for 10 min. A green fluorescent staining of living cells was obtained by adding 15 μg/ml FDA at the same time (Vaudry et al., 2003). The percentage of cell death was determined by counting damaged (red) and living (green) cells (1000–1500 cells), using 40× objective from 10 randomly selected fields in each of three dishes per group. For double staining of nuclei with PI and DAPI, cells were washed with PBS after the 10 min incubation with PI (staining damaged cells only), fixed with 4% PFA, permeabilized with PBS-T, and DAPI (15 μg/ml) was added for 10 min to visualize nuclear morphology. The percentage of cell death was determined by counting damaged cells (PI) over total cells (observed under phase microscopy), using 40× objective from 10 randomly selected fields in each of three dishes per group (1000–1500 cells). Alternatively, percentage of cell death was assessed by visualizing morphology of nuclei with DAPI. Nuclei were considered normal when glowing bright and homogenously. Apoptotic nuclei were identified by the condensed chromatin gathering at the periphery of the nuclear envelope or a total fragmented morphology of nuclear bodies.
Apoptosis analysis.
Cells plated in 35 mm dishes were processed for cleaved (Asp175) caspase-3 (1:200) immunocytochemistry. The apoptotic labeling index, defined as the proportion of total cells immunolabeled for activated caspase-3, was determined in culture by counting cells (1000–1500 cells), using 40× objective from 10 randomly selected fields in each of three dishes per group. For cleaved caspase-3 immunohistochemistry of brain tissues, sections were incubated in boiling 0.01 m citrate buffer, pH 6, for 5 min before immunohistochemical procedure.
Protein extract preparation and Western blot analysis.
Cells cultured in 60 mm dishes were washed twice with PBS, pH 7.4, detached using a rubber policeman in PBS, and then pelleted and resuspended in lysis buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm EDTA, 2 mm EGTA, 1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 0.5% NP-40, 1% Triton X-100, and a mix of protease inhibitors consisting of 10 μg/ml leupeptin, 10 μg/ml aprotinin, 20 μg/ml trypsin inhibitor, 50 mm NaF, 1 mm PMSF, 0.5 μm microcystin, and 1 mm o-vanadate. For in vivo experiments, both the right and left cortices were dissected after removing the meninges, pooled, and then were homogenized in the same lysis buffer. Lysates were sonicated twice 30 s and then centrifuged at 20,000 × g for 20 min (Carey et al., 2002). The supernatants were assayed for protein content using the Bio-Rad Protein Assay (Bio-Rad).
The specificity of the p57KIP2 polyclonal antibody used for the present study was assessed on brain homogenates obtained from E14.5 wild-type (positive control) and p57KIP2 gene knock-out (negative control) mouse embryos. Mouse embryos were obtained by mating heterozygote mice (p57KIP2+/−; a gift from Dr. S. Elledge, Harvard Medical School, Boston, MA) (Zhang et al., 1997), and their genotype was determined by standard PCR.
Equal amounts of proteins (30–80 μg/lane) obtained from cell cultures or tissues were loaded on 10 or 12% SDS-polyacrylamide gels. The separated proteins were electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad). The membranes were blocked with blocking buffer containing 5% fat-free dry milk in Tris-buffered saline solution and Tween 20 (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.05% Tween 20) and incubated overnight at 4°C with different primary antibodies diluted in blocking buffer. Incubations with HRP-conjugated secondary antibodies (1:1000) were performed for 1 h at room temperature and visualization was performed by chemiluminescence (ECL; GE Healthcare). Dilutions of primary antibodies were as follows: anti-phospho-Akt (1:1000), anti-Akt (1:1000), anti-phospho-GSK-3β (1:1000), anti-GSK-3β (1:1000), anti-phospho-ERK (1:1000), anti-ERK (1:1000), anti-cyclin D1 (1:500), anti-cyclin D3 (1:1000), anti-p27KIP1 (1:400), anti-p57KIP2 (1:250), anti-cyclin E (1:400), anti-β-actin (1:5000), and anti-cleaved caspase 3 (1:1000). Autoradiographic films were analyzed to quantify signals using the Bio-Rad Gel Doc 2000 with Quantity One, version 4.2.1, software (Bio-Rad). To control for loading, blots were stripped and reanalyzed for β-actin.
Quantitative reverse transcription-PCR.
Total RNA were extracted from cell cultures prepared in 35 mm dishes or from tissue using the RNeasy Mini Kit (QIAGEN). Contaminating DNA was removed by DNase I treatment, and 2 μg of total RNA was reverse transcribed with SuperScript II reverse transcriptase (Invitrogen). Quantitative reverse transcription-PCR (Q-RT-PCR) was performed on cDNA using the PCR Master Mix (Applied Biosystems), which contains buffer and preset concentrations of MgCl2, dNTPs, and SYBR Green, in the presence of forward and reverse primers for the gene of interest (900 nm each), or forward and reverse primers for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (200 nm each) used as reference. Reactions were performed using the ABI Prism 7000 Sequence Detection system (Applied Biosystems). The relative differences between groups were calculated based on the values for the gene of interest normalized over the values of the GAPDH gene. The primers used were as follows: p27KIP1 forward primer, 5′-CCTTCGACGCCAGACGTAAA-3′, and p27KIP1 reverse primer, 5′-AGCAGTGATGTATCTAATAAACAAGGAATT-3′; p57KIP2 forward primer, 5′-ACCCCGCGCAAACGT-3′, and p57KIP2 reverse primer, 5′-AGATGCCCAGCAAGTTCTCTCT-3′; cyclin D1 forward primer, 5′-GGCCCAGCAGAACATCGAT-3′, and cyclin D1 reverse primer, 5′-GACCAGCTTCTTCCTCCACTTC-3′; cyclin D3 forward primer, 5′-TCTCTGCCCAGTGACCATCA-3′, and cyclin D3 reverse primer, 5′-GGGCCCAAGACGTTTGG-3′; cyclin E forward primer, 5′-AGCCCCCTGACCATTGTG-3′, and cyclin E reverse primer, 5′-TCGTTGACGTAGGCCACTTG-3′. Primers for rodent GAPDH were obtained from Applied Biosystems (proprietary sequences).
Acquisition and processing of microscope images.
Cell cultures and tissue sections were examined with fluorescence microscope (Axiovert 200M; Carl Zeiss MicroImaging) coupled to an Apotome module, under control of AxioVision software (Carl Zeiss MicroImaging). Depending on the cell culture experiments, observations and images were made under phase or fluorescence using 10, 20, or 40 NeoFluar objectives. For the analyses of cleaved caspase-3 immunostaining and BrdU LI on tissue sections, acquisitions were made using 20 or 40 AxioPhot objectives with Apotome. Figure production was made using Adobe Photoshop software, minimally altering captured images.
Statistical analysis.
Data are expressed as means or percentages ± SEM. Statistical analyses were performed using Student's t test for two-group comparison and ANOVA followed by Dunnett's posttest for multiple-group comparison, using GraphPad Instat 3.05 software (GraphPad Software).
Results
IGF-1 stimulates proliferation of embryonic cerebral cortical precursors in vitro
To characterize IGF-1 effects on cerebral cortical precursors, we cultured cortical cells from E14.5 rat embryos in a serum-free defined medium lacking insulin (Carey et al., 2002; Li and DiCicco-Bloom, 2004). We examined cultures at early incubation times to minimize possible trophic activity that might be elicited by the growth factor, as previously studied (Lu et al., 1996). At 24 h of incubation, IGF-1 elicited a concentration-dependent increase in DNA synthesis, measured by [3H]thymidine incorporation assay (Fig. 1 A). A significant increase in DNA synthesis was observed at a minimal concentration of 0.1 ng/ml, and a plateau twofold increase was obtained at 10 ng/ml, a concentration used for additional studies. At this concentration, the factor produced an increase in DNA synthesis of 27% as early as 8 h and 82% at 24 h (data not shown) (p < 0.05 and p < 0.01, respectively). To further define IGF-1 mechanisms, we added BrdU (10 μm) into culture medium 2 h before 24 h culture termination, and then performed BrdU nuclear labeling to assess cells that entered S phase. IGF-1 treatment increased the BrdU labeling index by twofold (Fig. 1 B), indicating that increased DNA synthesis detected by thymidine incorporation reflected increased G1/S-phase progression. Moreover, the factor stimulated mitosis, because there was a 39% increase in the number of cells exhibiting M-phase marker phosphohistone H3 at 24 h (Fig. 1 C).
Figure 1.
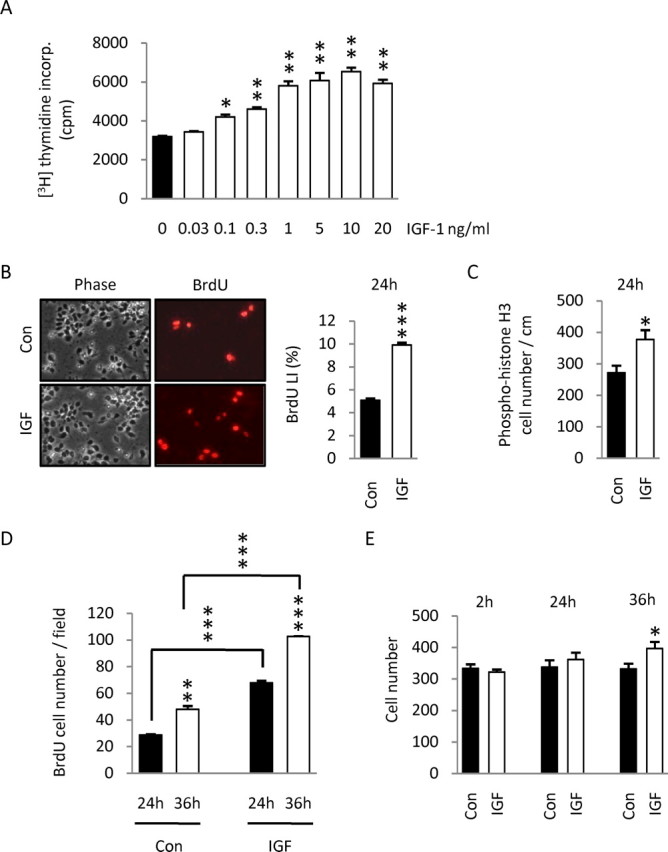
Effects of IGF-1 on DNA synthesis, cell cycle progression, and cortical precursor cell number in vitro. A, DNA synthesis was measured in E14.5 rat cortical precursors using [3H]thymidine incorporation assay. Addition of IGF-1 (0.03–20 ng/ml) to media elicited a dose-dependent increase in DNA synthesis at 24 h. Data are representative of one of three experiments, four wells per group. Statistical analysis was performed with ANOVA followed by Dunnett's posttest. cpm, Absolute counts per minute per well. B, BrdU (10 μm) was added 2 h before culture termination at 24 h. BrdU-labeled cells and total cells (observed under phase microscopy) were counted in 10 randomly selected fields. IGF-1 (10 ng/ml) increased the BrdU labeling index by twofold, indicating the factor stimulated G1/S progression. Data are representative of three experiments, three dishes per group per experiment. C, IGF-1 also increased the number of cells labeled for the M-phase marker phosphohistone H3 by 39%. Phosphohistone H3-labeled cells were counted in three 1 cm rows in three dishes per group. D, Proliferating cells were labeled during a 2 h BrdU (10 μm) pulse at 22 h, and the absolute number of BrdU-labeled cells was counted at 24 and 36 h. At 24 h, the number of BrdU-positive cells increased by twofold after IGF-1 exposure. At 36 h, the number of BrdU-labeled cells increased in both groups with twice as many in the IGF-1 group, indicating that cells that entered S phase between 22 and 24 h progressed through the cell cycle and divided in both groups. E, Quantification of cell number at 2, 24, and 36 h in control and IGF-1-treated cultures. The y-axis corresponds to the mean number of cells counted in 10 different fields per 35 mm dish. Whereas no difference in cell number among groups was observed between 2 and 24 h, a 19% increase was detected in IGF-1-treated cultures at 36 h. Note that no change in cell number was detected in control group at any time analyzed. Data are from three experiments. Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To determine whether cells that entered S phase after IGF-1 exposure were able to progress completely through the cell cycle, precursors were labeled with BrdU during a 2 h pulse at 22 h of culture, and then the number of BrdU-positive cells was counted at 24 and 36 h. At 24 h, the absolute number of BrdU-labeled cells was twofold higher in the IGF-1-exposed group compared with control (Fig. 1 D), paralleling the increased BrdU LI (Fig. 1 B). At 36 h, the number of BrdU-positive cells increased in both the control and IGF-1 groups, by 66 and 51%, respectively, with twice as many labeled precursors in the IGF-1 group. These results suggest that (1) IGF-1 increased the number of cells entering S phase at 24 h and (2) these cells completed cell cycle progression and divided over the following 12 h. Finally, we counted total cell number over the 36 h incubation period to detect evidence of proliferation. Whereas no change was observed between 2 and 24 h in the IGF-1 group, a 19% increase in cell number was detected at 36 h (Fig. 1 E). In contrast, there was no change in cell number in the control group over the same period. Together, these results suggest that IGF-1 primarily serves as a mitogen of cortical precursors in this culture model.
We next determined whether IGF-1 effects on DNA synthesis were attributable to a possible increase in precursor survival. Because IGF-1 treatment did not alter total cell number at 2 and 24 h (Fig. 1 E), it is not likely that the factor promoted survival in our model, although cell production balanced by cell death cannot be excluded. To assess this possibility, we analyzed cell survival at 2, 8, and 24 h, using PI/FDA staining (Fig. 2 A) to discriminate dead and living cells. There were no differences in cell survival among groups at these times. In addition, we estimated apoptosis at 8 and 24 h using immunolabeling for activated caspase-3 (Fig. 2 B). No differences in apoptotic cell numbers were observed among groups at each of these times. Together, these data suggest that IGF-1 exhibits no trophic activity during this culture incubation period, but rather stimulates DNA synthesis of cortical precursors by increasing entry into the mitotic cycle and cell division, resulting in cell proliferation.
Figure 2.

Effects of IGF-1 on cortical precursor cell survival and apoptosis. Cells incubated in 35 mm dishes were counted in 10 randomly selected fields, in three dishes per group. A, PI (red signal) and FDA (green signal) were used to distinguish dead cells (arrows) and living cells at 2, 8, and 24 h. Percentage cell survival was determined as the ratio of FDA-stained cells over total cells stained for FDA and PI. This value was then normalized to the value of the control group at 2 h, which was set at 100%. No significant difference was observed at each time among groups. Data are expressed as percentage ± SEM of 2 h control value (p > 0.05). B, Apoptotic cells immunolabeled for activated caspase-3 were counted and are expressed as percentage of total cell number observed under phase microscopy. Whereas the percentage of activated caspase-3-labeled cells increased from 5% at 8 h to 13% at 24 h among groups, no difference between IGF-1 and control groups was observed at each time analyzed (p > 0.05). Data are from three experiments, three dishes per group per experiment. Values shown represent the mean ± SEM.
IGF-1 rapidly increases the expression of G1 cyclins in vitro
To identify mechanisms involved in G1/S progression after IGF-1 treatment, we analyzed positive cyclin regulators of the cell cycle by Western blotting. D cyclins are known to be induced in response to mitogenic signals, and according to current models, D cyclin upregulation precedes that of cyclin E. We therefore analyzed D cyclins at an early time point: cyclin D1 protein levels were increased by 30% (Fig. 3 A) and cyclin D3 by 45% (Fig. 3 B), as early as 4 h of culture, indicating that IGF-1 rapidly regulates two different D cyclins, results consistent with traditional cell cycle models (Sherr and Roberts, 1999). We also measured cyclin D1 and D3 transcript levels using Q-RT-PCR, because cyclin D expression can be regulated at the transcriptional and/or posttranslational level (Liang and Slingerland, 2003). We found no changes in either cyclin mRNA level at 4 h (Fig. 3 A,B), suggesting that posttranslational mechanisms are probably involved in early increases of cyclin D1 and D3 protein levels after IGF-1 treatment.
Figure 3.
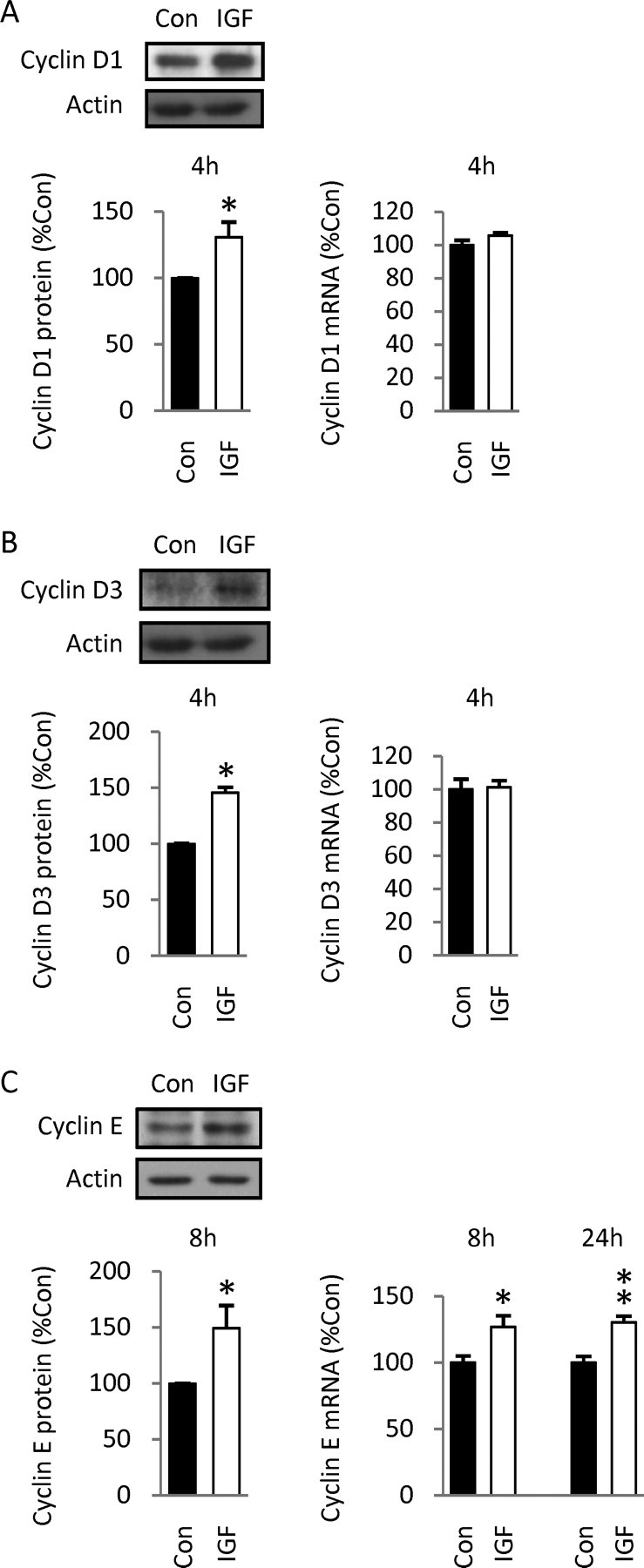
Effects of IGF-1 on positive regulators of the cell cycle. A, Western blot showing that IGF-1 increased cyclin D1 protein levels by 30% as early as 4 h. Data are from five experiments. However, no changes in cyclin D1 mRNA levels, measured by Q-RT-PCR, were observed at 4 h. Data are representative of three experiments, three dishes per group. B, IGF-1 also increased cyclin D3 protein levels, by 45% at 4 h (4 experiments), but no changes in cyclin D3 mRNA levels were detected (data are representative of 3 experiments). C, Cyclin E protein levels increased by 49% at 8 h of culture, after IGF-1 exposure. Data are from six experiments. In addition, IGF-1 increased cyclin E mRNA levels by 25% at 8 and 24 h. Data are representative of three experiments. Autoradiographic signals were quantified by densitometric analysis. Levels of protein of interest were normalized to β-actin. Levels of cyclin mRNA were normalized to GAPDH. Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01.
Next, we measured the level of cyclin E at 8 h because DNA synthesis was already increased at this time. IGF-1 treatment induced a 49% increase in cyclin E protein levels (Fig. 3 C), suggesting that enhanced S-phase entry depends on this positive regulator. We also analyzed cyclin E gene expression by Q-RT-PCR because it is known to be transcriptionally regulated as a consequence of increased D cyclin-dependent kinase activation (Sherr and Roberts, 1999): cyclin E mRNA levels were increased by 25% at 8 and 24 h after IGF-1 treatment (Fig. 3 C), indicating that expression of the cyclin E gene is positively regulated by the growth factor.
p27KIP1 and p57KIP2 are downregulated in response to IGF-1 treatment in vitro
Cell cycle progression is also subject to negative regulation by CKIs of the CIP/KIP family that bind and inhibit cyclin E/CDK2 complexes, thereby preventing commitment to S phase. We first focused our analyses on cell cycle inhibitor p27KIP1, because it is apparently the most widespread and abundant CIP/KIP family member expressed in cortex during embryonic development (van Lookeren Campagne and Gill, 1998; Nguyen et al., 2006). IGF-1 treatment decreased p27KIP1 protein levels by 25% at 8 h and by 70% at 12 h (Fig. 4 A). The inhibitory protein levels remained reduced even at 24 h of incubation, exhibiting a 40% decrease. p27KIP1 is known to be downregulated by posttranslational mechanisms involving the proteosomal pathway (Liang and Slingerland, 2003). However, recent evidence suggests that p27KIP1 is also regulated at the transcriptional level (Chassot et al., 2007; Itoh et al., 2007). Using Q-RT-PCR in our model, we measured p27KIP1 mRNA levels at 12 and 24 h and detected a 20% reduction after IGF-1 treatment at both times (Fig. 4 A), suggesting that transcriptional regulation is also involved.
Figure 4.

Effects of IGF-1 on negative regulators of the cell cycle. p27KIP1 and p57KIP2 protein levels were analyzed by Western blotting, and mRNA levels by Q-RT-PCR, at different times of culture. A, p27KIP1 protein levels were decreased by 25% at 8 h, 70% at 12 h, and 40% at 24 h in IGF-1-treated cultures. Data are from three to four experiments. A 20% reduction in p27KIP1 mRNA levels was measured at 12 and 24 h after IGF-1 treatment. Data are representative of three experiments. B, IGF-1 treatment elicited a 17% decrease in p57KIP2 protein levels at 12 h (3 experiments), and a 20% decrease in mRNA levels at 12 and 24 h (data are representative of 3 experiments). p57KIP2-specific signal (arrows) was determined by comparison to signal obtained from brain homogenates of E14.5 wild-type (p57+/+; positive control) and p57KIP2 gene knock-out (p57−/−; negative control) mouse embryos. Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01.
We next analyzed the expression of p57KIP2 gene. Previous studies suggested that p57KIP2 protein is expressed in a small subset of cortical neurons during embryonic development (Nguyen et al., 2006), and more recent work from our laboratory suggests that the protein is expressed throughout the embryonic brain (Ye et al., 2007, 2008). In the present study, detection of p57KIP2 protein by Western blotting from cortical precursor cultures was difficult because most commercially available antibodies revealed nonspecific signals. To overcome this issue, protein extracts from cultures were analyzed together with brain homogenates from E14.5 wild-type (positive control) and p57KIP2−/− (negative control) mouse embryos. The specificity of the p57KIP2 signal from rat cultures was determined by comparing band sizes to positive and negative mouse controls. IGF-1 treatment produced a small decrease in p57KIP2 protein levels (from 8 to 17%) at 8, 12, and 24 h (data not shown), but only the decrease at 12 h (17%) was statistically significant (Fig. 4 B). We also used Q-RT-PCR to analyze changes in p57KIP2 expression and found that IGF-1 treatment elicited a 20% decrease in p57KIP2 mRNA levels at 12 and 24 h (Fig. 4 B). In aggregate, these results suggest that, in addition to stimulating promitogenic cyclins, IGF-1 also induces downregulation of cell cycle inhibitors, p27KIP1 and p57KIP2, to promote S-phase entry of cortical precursors.
IGF-1 activates the PI3K/Akt and MEK/ERK signaling pathways in vitro
IGF-1 biological actions are usually mediated by the PI3K/Akt and/or MEK/ERK signaling pathways. Although both pathways are known to enhance G1/S progression in several cellular models, their role in IGF-1 mitogenic effects on cortical precursors has not been defined. To address this issue, cortical precursors were preincubated in defined medium for 1 h, stimulated with IGF-1 for different times, and then phosphorylation of Akt and ERKs was detected by Western blotting. As expected, IGF-1 provoked a robust increase in Akt and ERK phosphorylation at 30 and 10 min, respectively (Fig. 5 A,B). ERK2 (p42) was preferentially phosphorylated by IGF-1 treatment, whereas levels of phospho-ERK1 (p44) were barely detectable in either group (Fig. 5 B). We also analyzed phosphorylation of GSK-3β, the downstream target of phospho-Akt, because it is involved in cyclin D1 stabilization (Diehl et al., 1998). IGF-1 induced a marked increase in GSK-3β phosphorylation by 30 min (Fig. 5 C). These results indicate that both the PI3K/Akt and MEK/ERK pathways are activated after IGF-1 treatment and that they are both potentially involved in IGF-1 mitogenic effects on cortical precursors.
Figure 5.

IGF-1 activates the PI3K/Akt and MEK/ERK signaling pathways. A, B, IGF-1 increases the phosphorylation of Akt at 30 min (A) and ERK at 10 min (B). IGF-1 treatment preferentially induced phosphorylation of ERK2 (p42), whereas phospho-ERK1 (p44) signal was barely detectable. C, IGF-1 treatment increased the phosphorylation of GSK-3β at 30 min. Data are from three experiments. Values shown represent the mean ± SEM. *p < 0.001.
Inhibition of the PI3K/Akt but not MEK/ERK pathway abrogates IGF-1 mitogenic effects
To determine whether the PI3K/Akt and/or the MEK/ERK transduction pathways are required for IGF-1 mitogenic action, we used the PI3K inhibitor LY294002 or the MEK inhibitors PD98059 and U0126, and assessed DNA synthesis. Cortical precursors were preincubated for 1 h in defined medium, and then pretreated with inhibitors for 30 min before IGF-1 exposure. We assayed [3H]thymidine incorporation at an early incubation time, 12 h, because prolonged exposure to metabolic inhibitors could result in increased cell death. In the absence of inhibitor, IGF-1 elicited a twofold increase in [3H]thymidine incorporation (Fig. 6 A). In contrast, in the presence of PI3K inhibitor, IGF-1 stimulation was markedly reduced (Fig. 6 A), suggesting that this pathway contributes to mitogenic stimulation. The inhibitor alone had no effect on ongoing DNA synthesis. To assess the effectiveness of the PI3K/Akt inhibitor, we examined Akt phosphorylation at 30 min and found major pathway blockade (Fig. 6 B). Because the decrease in [3H]thymidine incorporation may potentially reflect an increase in cell death, we analyzed cell survival in the same experimental conditions by counting living cells and dead cells based on phase microscopy combined with PI, or nuclear morphology using DAPI staining (Fig. 6 C). Neither IGF-1 or LY294002 alone, nor IGF-1 plus LY294002, altered cell survival, indicating that the PI3K inhibitor indeed prevented IGF-1 stimulatory effects through cell cycle mechanisms.
Figure 6.

Blockade of PI3K/Akt signaling, but not the MEK/ERK pathway, inhibits IGF-1 stimulatory effects on DNA synthesis of cortical precursors. A, Cells were preincubated with PI3K inhibitor LY294002 (LY) for 30 min before IGF-1 addition, and DNA synthesis was assayed at 12 h. IGF-1 stimulation of DNA synthesis was markedly reduced by 10 μm LY294002. Data are representative of three experiments. B, Western blot showing that IGF-1-induced Akt phosphorylation was blocked by the PI3K inhibitor LY294002 (10 μm), at 30 min of incubation. C, Cell survival at 12 h was estimated by counting living and dead cells in 10 randomly selected fields using phase microscopy combined with PI (staining dead cells), or using DAPI staining to assess living and dead cells based on nuclear morphology. No differences were observed among groups (p > 0.05). Data are from three experiments. D–F, Cortical precursors were preincubated with different concentrations of MEK inhibitor PD98059 (D) or U0126 (F) for 30 min before the addition of IGF-1 for 12 h. Blockade of the MEK/ERK pathway with either drug resulted in an inhibitor dose-dependent decrease in DNA synthesis in both control and IGF-1-treated groups. Data are representative of three experiments. The Western blot in E shows that IGF-1-induced ERK phosphorylation is blocked by MEK inhibitor PD98059 (PD) (50 μm), at 10 min of culture. Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01.
In contrast to PI3K/Akt inhibition, addition of MEK inhibitor PD98059 up to 50 μm had no effect on IGF-1 stimulation of [3H]thymidine incorporation at 12 h (Fig. 6 D), whereas stimulation of ERK2 phosphorylation was blocked (Fig. 6 E). At 70 and 100 μm, the drug induced a dose-dependent decrease in [3H]thymidine incorporation of the same magnitude in both control and IGF-1-treated groups, suggesting that it induced cell death. Increased cell death at these concentrations was indeed apparent when observing cells under phase microscopy (data not shown). Similar results were obtained using another MEK inhibitor, U0126 (Fig. 6 F). Together, these data suggest that the PI3K/Akt pathway, but not the MEK/ERK pathway, is likely involved in rapid IGF-1 mitogenic action on embryonic cortical precursors.
Effects of intracerebroventricular IGF-1 microinjections on cortical precursor DNA synthesis, S-phase entry, and survival in vivo
Although the foregoing studies define roles of IGF-1 and the PI3K/Akt pathway in cortical precursor cell cycle in culture, their relevance to normal development in vivo remains unexplored. To determine whether IGF-1 stimulates cortical precursor proliferation in the developing embryo, we administered IGF-1 by transuterine intracerebroventricular injection into E16.5 rat brains, as performed previously (Suh et al., 2001), and analyzed effects on second messengers and cell cycle regulators in dorsolateral cerebral cortex tissue. We first assessed DNA synthesis after IGF-1 injection (30 ng per embryo) using the percentage [3H]thymidine incorporation assay (Suh et al., 2001) using four to five pregnant dams per time point. IGF-1 injection resulted in a 12% increase in [3H]thymidine incorporation at 6 h and a 26% increase at 12 h (Fig. 7 A).
Figure 7.

Effects of IGF-1 intracerebroventricular injections on DNA synthesis, S-phase entry, and apoptosis in the cerebral cortex of developing embryos. IGF-1 (30 ng) was injected into the lateral ventricles of E16.5 rat embryos. A, DNA synthesis was assessed in the cerebral cortex at 6 and 12 h after IGF-1 injection, using percentage [3H]thymidine incorporation assay. [3H]Thymidine was injected subcutaneously into pregnant dams 1 h before the killing. IGF-1 elicited a 12% increase in [3H]thymidine incorporation at 6 h (PBS-treated animals, n = 29; IGF-1-treated animals, n = 30; 5 pregnant dams) and 26% increase at 12 h (PBS-treated animals, n = 20; IGF-1-treated animals, n = 17; 4 pregnant dams) compared with the control group. B, C, Analysis of apoptosis 12 h after injection using activated caspase-3 as a marker. B, Rare cleaved caspase-3-labeled cells (arrows), exhibiting cytoplasmic signal, were revealed in the proliferating and postmitotic cortical zones of both PBS- and IGF-1-injected embryos. C, Western blotting analyses performed on the cortices of injected embryos revealed two bands (17 and 19 kDa) corresponding to the large fragments of activated caspase-3. No difference in protein levels assessed by densitometry was observed between control and IGF-1 treated animals (p > 0.05). Data are representative of three experiments, four animals per group. D, Analysis of cells in S-phase using BrdU immunohistochemistry. BrdU was injected into pregnant dams 1 h before the killing. BrdU-labeled cells were counted over total cells in the VZ of the dorsolateral cortex. IGF-1 intracerebroventricular injection elicited a 26% increase in the BrdU LI at 12 h. Data are from four animals per group. LV, Lateral ventricle. Values shown represent the mean ± SEM. *p < 0.01; **p < 0.001.
To determine whether increased DNA synthesis was possibly attributable to enhanced survival elicited by IGF-1, we analyzed activation of caspase-3 at 12 h. Very few cleaved caspase-3-immunolabeled cells were detected in either control and IGF-1-injected animals (Fig. 7 B). Labeled cells were sparsely distributed in the ventricular zone (VZ) and postmitotic compartments of the developing cerebral cortex, and were notable at the injection site. Because cleaved caspase-3-immunolabeled cells were very rare in both groups, we assessed activated caspase-3 protein levels in the entire cerebral cortex by Western blotting. No changes were observed between control and IGF-1-injected embryos (Fig. 7 C), indicating that IGF-1 treatment did not alter survival at 12 h.
To identify and quantify cells responsive to IGF-1 treatment, we assessed the number of cells in S phase in the cortical VZ of injected animals using BrdU immunohistochemistry at 12 h. IGF-1 injection elicited a 26% increase in the proportion of BrdU-labeled cells that increased from 38% in control to 48% in IGF-1-treated embryos (Fig. 7 D), a result that is identical with the change in percentage thymidine incorporation (Fig. 7 A). Collectively, these results suggest that IGF-1 rapidly elicited G1/S progression of cortical precursors in vivo and parallels the direct mitogenic action of the factor defined in precursor cultures.
DNA synthesis in cortical precursors is inhibited by neutralizing antibody to IGF-1 in vitro and in vivo
Although exogenous IGF-1 stimulates precursor mitosis, the role of endogenous IGF-1 in cortical proliferation in vivo remains unexplored. To address this question, we injected mice with a specific monoclonal antibody that neutralizes mouse IGF-1 biological activities, because neutralizing antibodies against rat IGF-1 were not available. The effects of anti-IGF-1 were compared with a monoclonal anti-IgG antibody (control) exhibiting the same isotype. Before examining the effects of the antibodies in vivo, their biological activities were first defined in vitro at 24 h on E14.5 rat cortical precursor cultures, using the [3H]thymidine incorporation assay to assess DNA synthesis. Whereas mouse IGF-1 elicited a 70% increase in DNA synthesis, the effect was completely blocked in the presence of anti-IGF-1 but not with anti-IgG antibody (Fig. 8 A), indicating that anti-IGF-1 antibody effectively neutralizes IGF-1 activity. Furthermore, to examine specificity of neutralizing activity, cortical cultures were exposed to bFGF. bFGF increased DNA synthesis by fourfold as previously described (Li and DiCicco-Bloom, 2004), an effect that was not altered by either anti-IGF-1 or anti-IgG antibody (Fig. 8 A), indicating that the neutralizing activity of anti-IGF-1 antibody is specific for IGF-1.
Figure 8.

Effects of anti-IGF-1 antibody on [3H]thymidine incorporation in rat cortical precursor cultures and in mouse embryonic cortex in vivo. A, The neutralizing activity of anti-IGF-1 antibody on IGF-1-induced mitosis was defined in E14.5 rat cortical precursor cultures. Mouse IGF-1 (3 ng/ml) or bFGF (10 ng/ml) were preincubated alone or in the presence of anti-IGF-1 (10 μg/ml) or anti-IgG (10 μg/ml) antibody under agitation for 2 h in 1 ml of culture medium. Then, growth factors or mixtures were applied to cultures, and [3H]thymidine incorporation was measured at 24 h. Mouse IGF-1 increased DNA synthesis by 70%, an effect that was abolished when the growth factor was preincubated with anti-IGF-1, but not with anti-IgG antibody. In the presence of bFGF, [3H]thymidine incorporation increased by fourfold, and the effect was not altered when preincubated with either anti-IGF-1 or anti-IgG antibody. Antibodies alone had no effects on [3H]thymidine incorporation in vitro. B, After intracerebroventricular injection of antibodies into E14.5–E16.5 mouse embryos, [3H]thymidine incorporation was assayed in the cortices at 4 h. IGF-1 neutralizing antibody (anti-IGF-1; 0.5, 1, or 2 μg using a 0.5 μg/μl solution) reduced DNA synthesis by 28% in the cortex of E14.5–E16.5 mice, compared with control (anti-IgG) (anti-IgG-treated animals, n = 17; anti-IGF-1-treated animals, n = 13; 4 pregnant dams; the data from the different doses were combined because effects were overlapping). Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
After control experiments in vitro, antibodies (0.5–2 μg using a 0.5 μg/μl solution) were injected intracerebroventricularly into four different litters consisting of E14.5–E16.5 mouse embryos, and DNA synthesis was examined in the cortices at 4 h using the percentage [3H]thymidine incorporation assay. Administration of IGF-1-neutralizing antibody reduced DNA synthesis by 28% compared with control conditions (Fig. 8 B). These experiments reveal a stimulatory role for endogenous IGF-1 in cortical cell proliferation during embryonic development in vivo.
IGF-1 activates the PI3K/Akt and MEK/ERK pathways in vivo
Based on our in vitro studies, we next defined the effects of IGF-1 on the PI3K/Akt and MEK/ERK pathways in vivo. Injection of IGF-1 induced a marked fourfold increase in Akt phosphorylation (Fig. 9 A) at 30 min, indicating that the growth factor activated the pathway. The downstream target of activated Akt, GSK-3β, was also inhibited by 30 min, as indicated by the twofold increase in GSK-3β phosphorylation (Fig. 9 B), results that also paralleled those obtained in culture. GSK-3β inhibition by IGF-1 was transient, because no changes in phosphorylation levels were detected 3 h after the injection (data not shown). Furthermore, the MEK/ERK pathway was also activated, because we detected a 40% increase in phospho-ERKs by 10 min (Fig. 9 C), an effect that was smaller than that observed in vitro. The small change in ERK phosphorylation levels may have resulted from additional time needed for cortical dissection (∼10 min). ERK activation was also transient as observed in vitro, because no differences were observed at 30 min (data not shown). These results indicate that pathways defined in cortical precursors in vitro are similarly activated by IGF-1 in the developing embryo, suggesting a major role for PI3K/Akt signaling.
Figure 9.
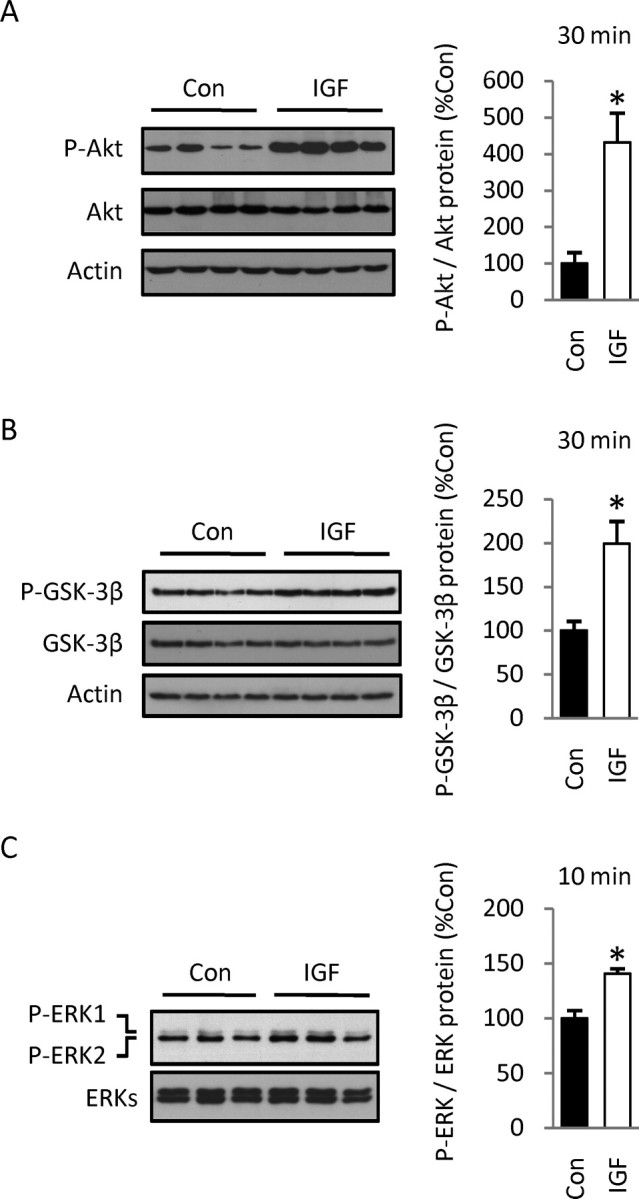
Effects of IGF-1 intracerebroventricular injections on the PI3K/Akt and MEK/ERK pathways. A, IGF-1 injections into the lateral ventricles of E16.5 rat embryos (30 ng per embryo) resulted in a fourfold increase in the phosphorylation of Akt by 30 min, revealed by Western blotting. B, The phosphorylation of GSK-3β increased by twofold at 30 min. C, IGF-1 injection induced a 40% increase in ERK phosphorylation by 10 min. Densitometric values of phosphoproteins were normalized to values of respective total proteins. Data are representative of three experiments, three to four animals per group. Values shown represent the mean ± SEM. *p < 0.01.
IGF-1 rapidly increases the expression of G1 cyclins and downregulates p27KIP1 and p57KIP2 expression in vivo
Because inhibition of GSK-3β through its phosphorylation would be expected to lead to an increase in cyclin D1 levels, we investigated changes in positive and negative cell cycle regulators in vivo. IGF-1 injection elicited a 56% increase in cyclin D1 protein levels at 6 h (Fig. 10 A), consistent with its role as a sensor of extracellular mitogens and as defined in vitro. Similar to culture studies (Fig. 3 A), no change in cyclin D1 mRNA levels was detected, suggesting the involvement of posttranslational regulatory mechanisms (Fig. 10 A). Furthermore, we detected a twofold increase in cyclin E protein levels at 6 h, and a 60% increase in cyclin E mRNA levels 6 and 12 h after IGF-1 treatment (Fig. 10 B), consistent with enhanced cell entry into S phase. However, we failed to detect any changes in p27KIP1 protein levels 6, 12, or 20 h after IGF-1 injection (data not shown). To further address this issue, we also examined mRNA levels: whereas IGF-1 stimulated cyclin E expression, the factor negatively regulated p27KIP1 and p57KIP2 transcript levels by 20–25% (Fig. 10 C). Together, these results suggest that IGF-1 promotes S-phase commitment of cortical precursors in vivo by regulating positive and negative components of the cell cycle machinery in opposing manner through mechanisms involving the PI3K/Akt signaling pathway.
Figure 10.
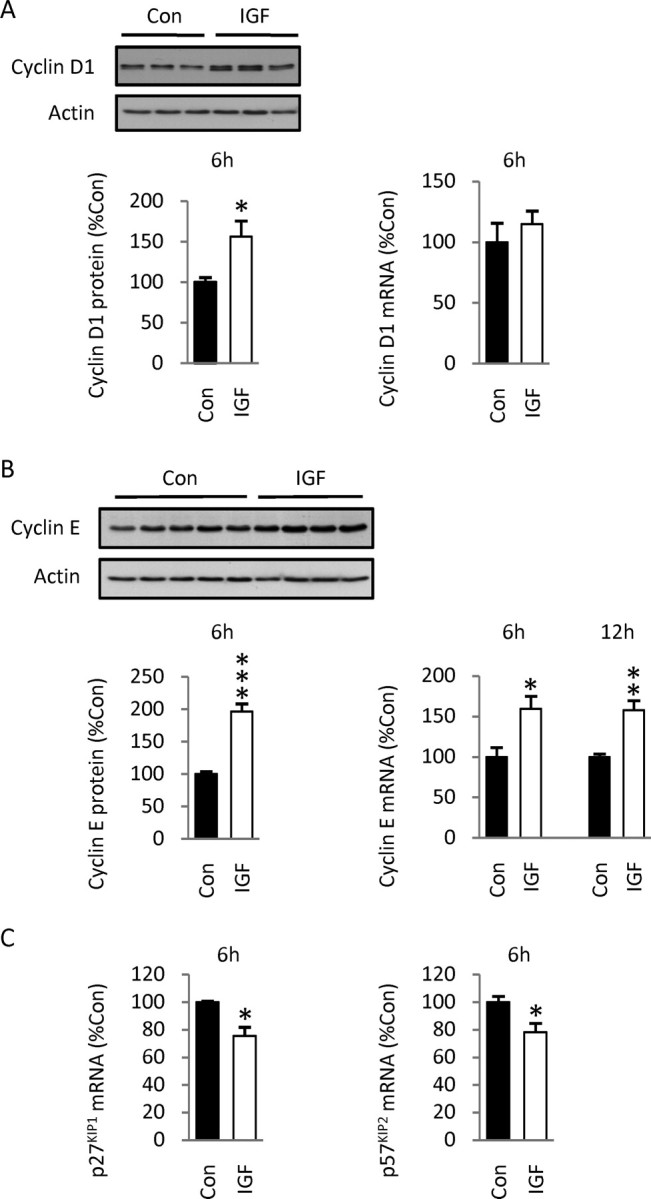
Effects of IGF-1 intracerebroventricular injections on the expression of cyclins D1 and E, and the CKIs p27KIP1 and p57KIP2. A, IGF-1 injection (30 ng per embryo) produced a 56% increase in cyclin D1 protein level at 6 h. Values represent the mean of seven embryos per group obtained from two pregnant dams. However, no changes in mRNA levels were observed using Q-RT-PCR. B, Injection of IGF-1 elicited a twofold increase in cyclin E protein levels at 6 h, and a 60% increase in cyclin E transcript at 6 and 12 h. C, IGF-1 injection decreased p27KIP1 and p57KIP2 mRNA levels by 25 and 22%, respectively, in the same samples at 6 h. Data are representative of three experiments, four to five animals per group. Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Role of the PI3K/Akt pathway in cortical precursor mitosis in vivo
The foregoing data indicating robust IGF-1 stimulation of Akt phosphorylation in vivo (Fig. 9 A) suggest that the pathway may be involved in mitogenic regulation. To begin exploring the role of PI3K/Akt in mitogenic extracellular signals in vivo, including IGF-1, we performed transuterine intracerebroventricular injections of the PI3K inhibitor LY294002 and assessed DNA synthesis and levels of cyclin E, p27KIP1, and p57KIP2 mRNA in the cortices at 6 h. We defined the effective dose of inhibitor in vivo by assessing the effects of different doses on Akt phosphorylation 30 min after injection. We found that 184 ng (3 μl of 200 μm solution) of LY294002 efficiently reduced ongoing phosphorylation of Akt (Fig. 11 A). At this dose, the drug decreased DNA synthesis by 13% (Fig. 11 B) and elicited a 23% decrease in cyclin E mRNA at 6 h (Fig. 11 C), suggesting that active PI3K/Akt signaling plays a stimulatory role in cell cycle progression during cortical development. Conversely, inhibition of the PI3K/Akt pathway elicited a 36 and 27% increase in p27KIP1 and p57KIP2 mRNA levels, respectively (Fig. 11 D), 6 h after injection, suggesting that the PI3K/Akt pathway negatively regulates these CKIs to promote proliferation during cortical development.
Figure 11.
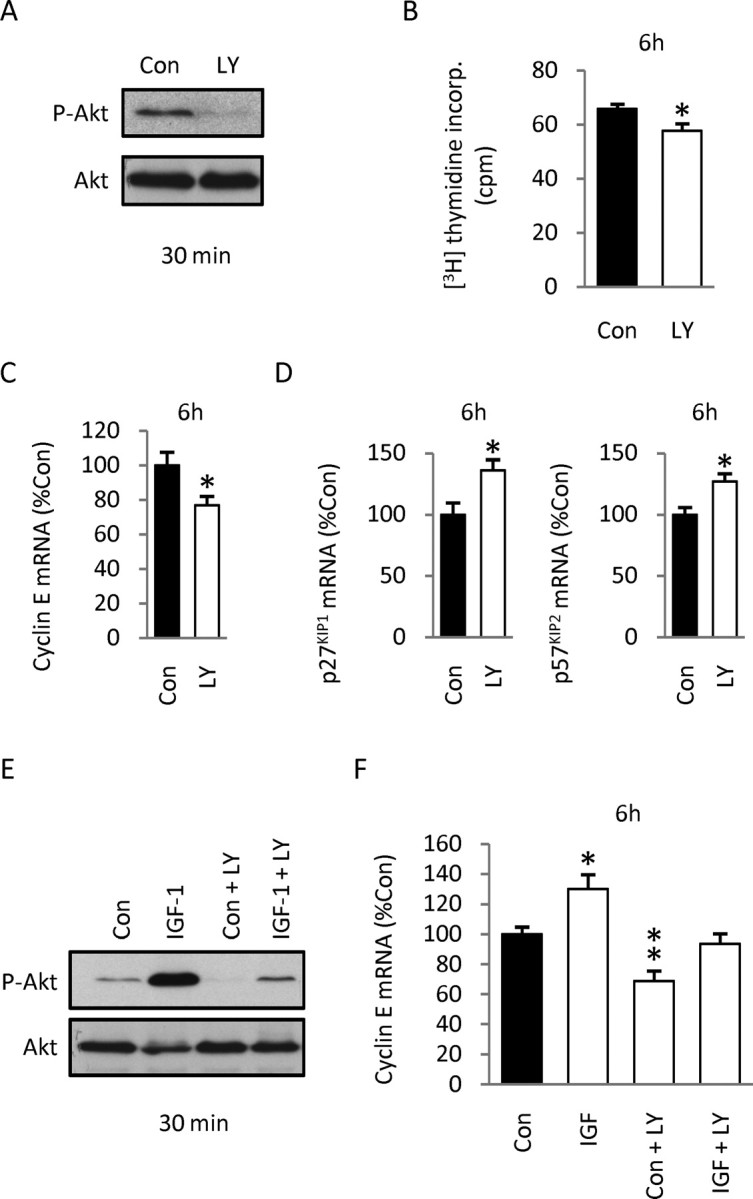
Effects of blocking PI3K/Akt signaling on DNA synthesis, expression of cell cycle regulators, and IGF-1 mitogenesis in the cortex of developing embryos. A, Injection of the PI3K/Akt inhibitor LY294002 (LY) (184 ng per embryo; 3 μl of 200 μm solution) into E16.5 rat embryos efficiently decreased ongoing Akt phosphorylation at 30 min. B, C, Intracerebroventricular injection of LY294002 elicited a 13% decrease in DNA synthesis at 6 h (data are representative of 3 experiments, 4 animals per group) (B), and 23% reduction in cyclin E mRNA levels at 6 h (data are from 3 experiments, 4 animals per group) (C). D, Conversely, injection of LY294002 increased p27KIP1 and p57KIP2 mRNA levels by 36 and 27%, respectively, at 6 h. Data are from three experiments, four animals per group. E, Western blot showing that intracerebroventricular injection of LY294002 30 min before IGF-1 injection blocked IGF-1-induced phosphorylation of Akt at 30 min. F, Whereas IGF-1 injection increased cyclin E mRNA levels by 30% at 6 h, preinjection of LY294002 prevented IGF-1 stimulatory effect on cyclin E expression (Con group, n = 4; IGF group, n = 3; LY294002 group, n = 4; LY294002 plus IGF-1 group, n = 4; data are from 3 experiments). Values shown represent the mean ± SEM. *p < 0.05; **p < 0.01.
To assess the role of the PI3K/Akt pathway directly in IGF-1 mitogenic stimulation, we performed sequential transuterine intracerebroventricular treatments consisting of an injection of the PI3K inhibitor LY294002 (184 ng per embryo; or vehicle) followed 20–30 min by an injection of IGF-1 (30 ng; or vehicle) into the same lateral ventricle. We first defined the efficiency of the inhibitor on Akt phosphorylation by Western blotting at 30 min: whereas injection of inhibitor alone decreased ongoing levels of phospho-Akt, LY294002 reduced IGF-1-induced Akt phosphorylation to levels comparable with controls (Fig. 11 E). As an index of cell cycle progression, we measured cyclin E mRNA levels by Q-RT-PCR from cortical tissues 6 h after IGF-1 injection. Whereas IGF-1 elicited a 30% increase in cyclin E mRNA levels in the absence of the inhibitor, the stimulatory effect of the growth factor was abrogated when the inhibitor was injected beforehand (Fig. 11 F). Also, cyclin E mRNA level decreased when the inhibitor was injected alone (Fig. 11 C,F), consistent with the activity of endogenous IGF-1 in vivo as shown above (Fig. 8 B). Together, these results demonstrate that the PI3K/Akt pathway participates in mediating IGF-1 stimulatory effects on the G1/S transition of cortical precursors during forebrain development.
Effects of intracerebroventricular IGF-1 microinjections on brain growth in vivo
Although our foregoing studies characterize the mitogenic role of IGF-1 in embryonic cortical precursors, the relationship to brain growth remains unknown. To begin addressing the effects of IGF-1 mitogenic activity on forebrain development in vivo, we injected the growth factor (100 ng) into the lateral ventricles of E15.5 rat brains and analyzed overall brain growth at P10. Gross examination of the brains of IGF-1-injected animals did not reveal any particular morphological features compared with controls (data not shown). To evaluate possible differences in total cell number, we assessed total DNA content, a marker for total cell number (Burton, 1956; Cheng et al., 2001), in the forebrain (PBS-treated animals, n = 15; IGF-1-treated animals, n = 25; two pregnant dams per group). Interestingly, the forebrains of IGF-1-injected animals exhibited a marked 28% increase in total DNA content (PBS-treated animals, 559 ± 19 μg; IGF-1-treated animals, 715 ± 31 μg; p = 0.0009). These data indicate that a single injection of IGF-1 into cerebral ventricles during embryonic development may increase the total number of cells in the brain after birth, suggesting the factor stimulates precursor proliferation in the CNS, potentially through mitogenic and/or trophic mechanisms of action.
Discussion
Our observations indicate that IGF-1 has a direct mitogenic rather than a trophic effect, in cortical precursors in vitro and in vivo, and thus serves a proliferative role during corticogenesis. IGF-1 mitogenic stimulation involves rapid, sequential, and opposite regulation of positive and negative cell cycle regulators, which occurs through transcriptional and posttranslational mechanisms. Furthermore, the PI3K/Akt pathway, but not the MEK/ERK cascade, mediates IGF-1 mitogenic signaling to downstream cell cycle regulators.
IGF-1 stimulates mitosis of embryonic cortical precursors
We report that IGF-1 serves primarily as a mitogen for cortical precursors, as indicated by stimulation of cell cycle machinery and proliferation in vitro. IGF-1 increased DNA synthesis as early as 8 h, the number of precursors in S and M phases at 24 h and the absolute number of cells at 36 h. This effect may reflect either a reduction of cell cycle length and/or promotion of cycle reentry, causing precursors to undergo additional rounds of division (Tarui et al., 2005). A mitogenic role for IGF-1 has been reported only rarely in culture models, including embryonic sympathetic neuroblasts (DiCicco-Bloom and Black, 1988) and adult hippocampal progenitors (Aberg et al., 2003). IGF-1 is better known to act in association with other mitogens to enhance proliferation (Arsenijevic et al., 2001; Jiang et al., 2001). These observations suggest proliferative effects of IGF-1 are cell type specific.
Paralleling culture results, IGF-1 injections into embryos rapidly stimulated G1/S transition of precursors, increasing DNA synthesis and BrdU labeling. Conversely, neutralizing endogenous IGF-1 activity reduced DNA synthesis, demonstrating the mitogenic role of IGF-1 during normal corticogenesis. Importantly, a single injection of IGF-1 into embryos increased DNA content in P10 brain, supporting a role in cell production through regulation of precursor proliferation. These findings mirror genetic models. Mice overexpressing IGF-1 exhibited increased brain size and neuron/glia numbers in adult cortex because of reduced G1-phase length and increased precursor cell cycle reentry (Hodge et al., 2004). Conversely, IGF-1 gene disruption led to reduced brain size and cell number (Beck et al., 1995). Similar to IGF-1, bFGF dramatically stimulates cortical proliferation after embryonic injection, increasing either glia or neuron numbers in adult cortex as a function of embryo age. However, contrary to IGF-1 (Hodge et al., 2004), bFGF did not affect precursor cell cycle kinetics but rather increased the pool of proliferating precursors (Vaccarino et al., 1999). Consistent with mitogenic activity, mice lacking bFGF gene or expressing dominant-negative receptors displayed fewer cortical neurons and glia (Vaccarino et al., 1999; Shin et al., 2004). These observations suggest that growth factors, by acting on precursor proliferation, affect cellular output and consequently modify the composition of adult cerebral cortex.
Although IGF-1 also promotes survival of proliferating precursors from E10 mouse forebrain and midbrain (Drago et al., 1991), we found no evidence of trophic activity in E14.5 rat cortical precursors, a discrepancy likely attributable to both precursor stage and species, and our use of high cell density that enhances survival. Without factors, Drago et al. reported mostly nonviable cells at 24 h, contrasting with sustained survival in our model.
IGF-1 promotes sequential activation of G1 cyclins
Current cell cycle models describe sequential activation of G1 regulators, leading to increased cyclin E/CDK2 activity, which is indispensable for G1/S transition (Sherr and Roberts, 1999), a model supported by our studies. In vitro, IGF-1 produced early and sequential increases in G1 cyclins, including cyclin D1/D3 proteins (but not mRNAs) at 4 h, followed by cyclin E mRNA and protein at 8 h. Cyclin increases at 8 h were associated with a 27% increase in DNA synthesis, which peaked approximately twofold by 24 h. Similar rapid activations occurred in vivo, with cyclin D1 and E increases already present at 6 h. In contrast, although we previously found a similar cyclin sequence after bFGF, the temporal schedule was significantly delayed: cyclin D1 (but not D3) increased only at 8 h, whereas cyclin E required 20 h (Li and DiCicco-Bloom, 2004).
The existence of different growth factor temporal profiles implies they activate distinct signaling cascades. For example, cyclin D1 can be regulated transcriptionally, as defined in cortical precursors after bFGF (Li and DiCicco-Bloom, 2004), likely involving MEK/ERK signaling and cyclin D1 promoter activation (Lavoie et al., 1996). Alternatively, cyclin D1 can be regulated by promoting nuclear accumulation and stabilization through Akt phosphorylation-induced inhibition of GSK-3β (Diehl et al., 1998). In our study, IGF-1 indeed increased GSK-3β phosphorylation and cyclin D1 protein levels without changes in mRNA, suggesting IGF-1 activates G1 progression by stabilizing cyclin D1 through the PI3K/Akt/GSK-3β pathway, as shown in oligodendrocyte progenitors (Frederick et al., 2007).
Although IGF-1 and bFGF are both cortical mitogens, it is unknown whether they act on the same or different subpopulations. Whereas we find the combined factors exhibit additive effects on precursor proliferation (our unpublished observations), others demonstrate synergistic activity (Arsenijevic et al., 2001; Frederick and Wood, 2004). The current and previous studies (Li and DiCicco-Bloom, 2004) suggest IGF-1 and bFGF stimulate cortical proliferation through different signaling cascades, PI3K/Akt and MEK/ERK, respectively. The two mitogens may thus regulate distinct precursor populations that may produce different cell types in developing cerebral cortex.
p27KIP1 and p57KIP2 are downregulated by IGF-1 mitogenic signaling
Although IGF-1 sequential activation of cyclins may underlie G1/S progression, the small changes in cyclin protein levels suggested other mechanisms may contribute, such as negative regulation of cyclin/CDK by CKIs (Sherr and Roberts, 1999). We found that p27KIP1 and p57KIP2 both contribute to IGF-1 regulation of G1/S, although limited p57KIP2 protein changes suggest a minor role. By decreasing CKI expression, IGF-1 likely relieves inhibitory constraint on cyclin E/CDK2 activity. Mitogen downregulation of CKIs has been reported previously in cortical (Li and DiCicco-Bloom, 2004; Itoh et al., 2007) and oligodendrocyte progenitors (Frederick and Wood, 2004).
The decrease in p27KIP1 protein levels coincided with increases in cyclin E, suggesting downregulation may depend on cyclin E/CDK2 activity. Negative regulation of CKI expression involves the ubiquitin-dependent proteasome pathway, which requires cyclin E/CDK2 phosphorylation of the CKIs (Kamura et al., 2003; Leibovitch et al., 2003). In this pathway, the PI3K/Akt cascade regulates p27KIP1 degradation by upregulating Skp2, a key component of the SCFSKP2 ubiquitin ligase complex (Liang and Slingerland, 2003). Mitogens also negatively regulate CKI expression through transcription, as reported in cortical precursors (Itoh et al., 2007). Our results support this mechanism because IGF-1 reduced p27KIP1 and p57KIP2 mRNA levels rapidly in culture, and in vivo. IGF-1 downregulation of p27KIP1 promoter activity may involve phosphorylation-induced inhibition of Forkhead transcription factor FoxO1 by PI3K/Akt signaling, as shown in muscle (Machida et al., 2003). Thus, IGF-1 may control the G1/S transition via posttranslational mechanisms that depend on cell cycle machinery (cyclin/CDK complexes) as well as via second messenger-regulated transcriptional pathways that are independent of cycle machinery.
The PI3K/Akt pathway mediates IGF-1 mitogenic signaling to downstream cell cycle regulators
We show that IGF-1 activates PI3K/Akt signaling in cortical precursors, and suggest that the pathway directly regulates expression of positive and negative cell cycle regulators. PI3K/Akt activation by IGF-1 increased cyclin E and decreased p27KIP1 and p57KIP2 expression. Conversely, pharmacological inhibition reversed effects on regulator expression, suggesting the pathway contributes to the dynamic system controlling the cell cycle in cortical precursors. Although PI3K/Akt signaling is best known for mediating cell survival, accumulating evidence supports additional roles in regulating G1/S progression (Liang and Slingerland, 2003; Manning and Cantley, 2007), stem cell self-renewal, and brain cell number and composition (Easton et al., 2005; Tschopp et al., 2005). In vivo, we found that, at E16.5, (1) endogenous levels of Akt phosphorylation are relatively low, and (2) inhibition of PI3K/Akt decreased DNA synthesis by 12%. These observations indicate PI3K/Akt signaling may play a minor role in proliferation or, alternatively, stimulates a subset of cortical progenitors. Interestingly, recent studies suggest important roles for PI3K/Akt in an embryonic stem cell subset that exhibits enhanced Akt levels and displays pleiotropic responses to growth factor activation in survival, proliferation, and self-renewal (Sinor and Lillien, 2004). Furthermore, we show that PI3K/Akt signaling mediates IGF-1 mitogenic stimulation in cortical precursors, whereas MEK/ERK has a minimal role, at least in vitro. These results contrast with IGF-1-induced proliferation of adult hippocampal stem cells, which requires both PI3K/Akt and MEK/ERK pathways in vitro (Aberg et al., 2003), suggesting IGF-1 differential activation of mitogenic signaling pathways is cell type or stage dependent.
In conclusion, IGF-1 signaling influences precursor proliferation during corticogenesis, an effect mediated by the PI3K/Akt pathway and driven by rapid activation of G1 cyclins and downregulation of CKIs. These studies, based on acute regulation of IGF-1 and signaling pathways in utero, directly address mechanistic concerns raised by using mutant mice regarding cell selection and molecular compensation that can accompany genetic manipulations present since conception. As observed for bFGF, IGF-1 may play a role in controlling the output from the proliferative population. Additional experiments will need to address whether different growth factors, IGF-1 and bFGF, (1) differentially regulate the production of layer-specific neuronal cell types, (2) bias cortical precursors to a neuronal versus glial fate, and (3) affect cellular differentiation during corticogenesis in vivo.
Footnotes
This work was supported by National Institutes of Health Grant HD23315. We thank Dr. Akiva Marcus for technical advice on intracerebroventricular injections and Corinna Panlilio for technical support.
References
- Aberg et al., 2003.Aberg MA, Aberg ND, Palmer TD, Alborn AM, Carlsson-Skwirut C, Bang P, Rosengren LE, Olsson T, Gage FH, Eriksson PS. IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol Cell Neurosci. 2003;24:23–40. doi: 10.1016/s1044-7431(03)00082-4. [DOI] [PubMed] [Google Scholar]
- Adams et al., 2000.Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenman and de Vellis, 1987.Aizenman Y, de Vellis J. Brain neurons develop in a serum and glial free environment: effects of transferrin, insulin, insulin-like growth factor-I and thyroid hormone on neuronal survival, growth and differentiation. Brain Res. 1987;406:32–42. doi: 10.1016/0006-8993(87)90766-9. [DOI] [PubMed] [Google Scholar]
- Arsenijevic et al., 2001.Arsenijevic Y, Weiss S, Schneider B, Aebischer P. Insulin-like growth factor-I is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J Neurosci. 2001;21:7194–7202. doi: 10.1523/JNEUROSCI.21-18-07194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck et al., 1995.Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Burton, 1956.Burton K. A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric estimation of deoxyribonucleic acid. Biochem J. 1956;62:315–323. doi: 10.1042/bj0620315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey et al., 2002.Carey RG, Li B, DiCicco-Bloom E. Pituitary adenylate cyclase activating polypeptide anti-mitogenic signaling in cerebral cortical progenitors is regulated by p57Kip2-dependent CDK2 activity. J Neurosci. 2002;22:1583–1591. doi: 10.1523/JNEUROSCI.22-05-01583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson et al., 1993.Carson MJ, Behringer RR, Brinster RL, McMorris FA. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-o. [DOI] [PubMed] [Google Scholar]
- Caviness et al., 1995.Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- Chassot et al., 2007.Chassot AA, Turchi L, Virolle T, Fitsialos G, Batoz M, Deckert M, Dulic V, Meneguzzi G, Busca R, Ponzio G. Id3 is a novel regulator of p27kip1 mRNA in early G1 phase and is required for cell-cycle progression. Oncogene. 2007;26:5772–5783. doi: 10.1038/sj.onc.1210386. [DOI] [PubMed] [Google Scholar]
- Cheng et al., 1998.Cheng CM, Joncas G, Reinhardt RR, Farrer R, Quarles R, Janssen J, McDonald MP, Crawley JN, Powell-Braxton L, Bondy CA. Biochemical and morphometric analyses show that myelination in the insulin-like growth factor 1 null brain is proportionate to its neuronal composition. J Neurosci. 1998;18:5673–5681. doi: 10.1523/JNEUROSCI.18-15-05673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng et al., 2001.Cheng Y, Tao Y, Black IB, DiCicco-Bloom E. A single peripheral injection of basic fibroblast growth factor (bFGF) stimulates granule cell production and increases cerebellar growth in newborn rats. J Neurobiol. 2001;46:220–229. doi: 10.1002/1097-4695(20010215)46:3<220::aid-neu1004>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Clemmons and Maile, 2003.Clemmons DR, Maile LA. Minireview: integral membrane proteins that function coordinately with the insulin-like growth factor I receptor to regulate intracellular signaling. Endocrinology. 2003;144:1664–1670. doi: 10.1210/en.2002-221102. [DOI] [PubMed] [Google Scholar]
- Cunningham and Roussel, 2001.Cunningham JJ, Roussel MF. Cyclin-dependent kinase inhibitors in the development of the central nervous system. Cell Growth Differ. 2001;12:387–396. [PubMed] [Google Scholar]
- D'Ercole et al., 1996.D'Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- DiCicco-Bloom and Black, 1988.DiCicco-Bloom E, Black IB. Insulin growth factors regulate the mitotic cycle in cultured rat sympathetic neuroblasts. Proc Natl Acad Sci U S A. 1988;85:4066–4070. doi: 10.1073/pnas.85.11.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl et al., 1998.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago et al., 1991.Drago J, Murphy M, Carroll SM, Harvey RP, Bartlett PF. Fibroblast growth factor-mediated proliferation of central nervous system precursors depends on endogenous production of insulin-like growth factor I. Proc Natl Acad Sci U S A. 1991;88:2199–2203. doi: 10.1073/pnas.88.6.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton et al., 2005.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick and Wood, 2004.Frederick TJ, Wood TL. IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol Cell Neurosci. 2004;25:480–492. doi: 10.1016/j.mcn.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Frederick et al., 2007.Frederick TJ, Min J, Altieri SC, Mitchell NE, Wood TL. Synergistic induction of cyclin D1 in oligodendrocyte progenitor cells by IGF-I and FGF-2 requires differential stimulation of multiple signaling pathways. Glia. 2007;55:1011–1022. doi: 10.1002/glia.20520. [DOI] [PubMed] [Google Scholar]
- Hodge et al., 2004.Hodge RD, D'Ercole AJ, O'Kusky JR. Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J Neurosci. 2004;24:10201–10210. doi: 10.1523/JNEUROSCI.3246-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh et al., 2004.Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Nakashima K, Gage FH. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol. 2004;164:111–122. doi: 10.1083/jcb.200308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh et al., 2007.Itoh Y, Masuyama N, Nakayama K, Nakayama KI, Gotoh Y. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J Biol Chem. 2007;282:390–396. doi: 10.1074/jbc.M609944200. [DOI] [PubMed] [Google Scholar]
- Jiang et al., 2001.Jiang F, Frederick TJ, Wood TL. IGF-I synergizes with FGF-2 to stimulate oligodendrocyte progenitor entry into the cell cycle. Dev Biol. 2001;232:414–423. doi: 10.1006/dbio.2001.0208. [DOI] [PubMed] [Google Scholar]
- Kamura et al., 2003.Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, Hatakeyama S, Nakayama K, Nakayama KI. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A. 2003;100:10231–10236. doi: 10.1073/pnas.1831009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie et al., 1996.Lavoie JN, L'Allemain G, Brunet A, Müller R, Pouysségur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Leibovitch et al., 2003.Leibovitch MP, Kannengiesser C, Leibovitch SA. Signal-induced ubiquitination of p57Kip2 is independent of the C-terminal consensus Cdk phosphorylation site. FEBS Lett. 2003;543:125–128. doi: 10.1016/s0014-5793(03)00425-3. [DOI] [PubMed] [Google Scholar]
- Li and DiCicco-Bloom, 2004.Li B, DiCicco-Bloom E. Basic fibroblast growth factor exhibits dual and rapid regulation of cyclin D1 and p27 to stimulate proliferation of rat cerebral cortical precursors. Dev Neurosci. 2004;26:197–207. doi: 10.1159/000082137. [DOI] [PubMed] [Google Scholar]
- Liang and Slingerland, 2003.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- Lu and DiCicco-Bloom, 1997.Lu N, DiCicco-Bloom E. Pituitary adenylate cyclase-activating polypeptide is an autocrine inhibitor of mitosis in cultured cortical precursor cells. Proc Natl Acad Sci U S A. 1997;94:3357–3362. doi: 10.1073/pnas.94.7.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu et al., 1996.Lu N, Black IB, DiCicco-Bloom E. A paradigm for distinguishing the roles of mitogenesis and trophism in neuronal precursor proliferation. Brain Res Dev Brain Res. 1996;94:31–36. doi: 10.1016/0165-3806(96)00050-8. [DOI] [PubMed] [Google Scholar]
- Machida et al., 2003.Machida S, Spangenburg EE, Booth FW. Forkhead transcription factor FoxO1 transduces insulin-like growth factor's signal to p27Kip1 in primary skeletal muscle satellite cells. J Cell Physiol. 2003;196:523–531. doi: 10.1002/jcp.10339. [DOI] [PubMed] [Google Scholar]
- Mairet-Coello et al., 2005.Mairet-Coello G, Tury A, Fellmann D, Risold PY, Griffond B. Ontogenesis of the sulfhydryl oxidase QSOX expression in rat brain. J Comp Neurol. 2005;484:403–417. doi: 10.1002/cne.20411. [DOI] [PubMed] [Google Scholar]
- Manning and Cantley, 2007.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell, 1988.McConnell SK. Development and decision-making in the mammalian cerebral cortex. Brain Res. 1988;472:1–23. doi: 10.1016/0165-0173(88)90002-1. [DOI] [PubMed] [Google Scholar]
- McCurdy et al., 2005.McCurdy RD, Féron F, McGrath JJ, Mackay-Sim A. Regulation of adult olfactory neurogenesis by insulin-like growth factor-I. Eur J Neurosci. 2005;22:1581–1588. doi: 10.1111/j.1460-9568.2005.04355.x. [DOI] [PubMed] [Google Scholar]
- Nguyen et al., 2006.Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM, Guillemot F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourse et al., 1994.Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, Massague J, Crabtree GR, Roberts JM. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- Popken et al., 2004.Popken GJ, Hodge RD, Ye P, Zhang J, Ng W, O'Kusky JR, D'Ercole AJ. In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur J Neurosci. 2004;19:2056–2068. doi: 10.1111/j.0953-816X.2004.03320.x. [DOI] [PubMed] [Google Scholar]
- Rakic, 2002.Rakic P. Neurogenesis in adult primate neocortex: an evaluation of the evidence. Nat Rev Neurosci. 2002;3:65–71. doi: 10.1038/nrn700. [DOI] [PubMed] [Google Scholar]
- Sherr and Roberts, 1999.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Shin et al., 2004.Shin DM, Korada S, Raballo R, Shashikant CS, Simeone A, Taylor JR, Vaccarino F. Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J Neurosci. 2004;24:2247–2258. doi: 10.1523/JNEUROSCI.5285-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor and Lillien, 2004.Sinor AD, Lillien L. Akt-1 expression level regulates CNS precursors. J Neurosci. 2004;24:8531–8541. doi: 10.1523/JNEUROSCI.1470-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh et al., 2001.Suh J, Lu N, Nicot A, Tatsuno I, DiCicco-Bloom E. PACAP is an anti-mitogenic signal in developing cerebral cortex. Nat Neurosci. 2001;4:123–124. doi: 10.1038/83936. [DOI] [PubMed] [Google Scholar]
- Tan et al., 1998.Tan SS, Kalloniatis M, Sturm K, Tam PP, Reese BE, Faulkner-Jones B. Separate progenitors for radial and tangential cell dispersion during development of the cerebral neocortex. Neuron. 1998;21:295–304. doi: 10.1016/s0896-6273(00)80539-5. [DOI] [PubMed] [Google Scholar]
- Tao et al., 1996.Tao Y, Black IB, DiCicco-Bloom E. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF) J Comp Neurol. 1996;376:653–663. doi: 10.1002/(SICI)1096-9861(19961223)376:4<653::AID-CNE11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Tarui et al., 2005.Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS. Overexpression of p27 Kip 1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex. 2005;15:1343–1355. doi: 10.1093/cercor/bhi017. [DOI] [PubMed] [Google Scholar]
- Tschopp et al., 2005.Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- Vaccarino et al., 1999.Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne and Gill, 1998.van Lookeren Campagne M, Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene Bax. J Comp Neurol. 1998;397:181–198. doi: 10.1002/(sici)1096-9861(19980727)397:2<181::aid-cne3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Vaudry et al., 2003.Vaudry D, Falluel-Morel A, Basille M, Pamantung TF, Fontaine M, Fournier A, Vaudry H, Gonzalez BJ. Pituitary adenylate cyclase-activating polypeptide prevents C2-ceramide-induced apoptosis of cerebellar granule cells. J Neurosci Res. 2003;72:303–316. doi: 10.1002/jnr.10530. [DOI] [PubMed] [Google Scholar]
- Wagner et al., 1999.Wagner JP, Black IB, DiCicco-Bloom E. Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999;19:6006–6016. doi: 10.1523/JNEUROSCI.19-14-06006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins et al., 2001.Wilkins A, Chandran S, Compston A. A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia. 2001;36:48–57. doi: 10.1002/glia.1094. [DOI] [PubMed] [Google Scholar]
- Ye et al., 2007.Ye W, Mairet-Coello G, DiCicco-Bloom E. DNAse I pre-treatment markedly enhances detection of nuclear cyclin-dependent kinase inhibitor p57Kip2 and BrdU double immunostaining in embryonic rat brain. Histochem Cell Biol. 2007;127:195–203. doi: 10.1007/s00418-006-0238-6. [DOI] [PubMed] [Google Scholar]
- Ye et al., 2008.Ye W, Mairet-Coello G, Pasoreck E, Dicicco-Bloom E. Patterns of p57Kip2 expression in embryonic rat brain suggest roles in progenitor cell cycle exit and neuronal differentiation. Dev Neurobiol. 2008;69:1–21. doi: 10.1002/dneu.20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 1997.Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]