

Abstract
Objectives
To identify genetic factors that would be predictive of individuals who require an implantable cardioverter-defibrillator (ICD), we conducted a genome-wide association study among individuals with an ICD who experienced a life-threatening arrhythmia (LTA; cases) vs. those who did not over at least a 3-year period (controls).
Background
Most individuals that receive implantable cardioverter-defibrillators never experience a life-threatening arrhythmia. Genetic factors may help identify who is most at risk.
Methods
Patients with an ICD and extended follow-up were recruited from 34 clinical sites with the goal of oversampling those who had experienced LTA, with a cumulative 607 cases and 297 controls included in the analysis. A total of 1,006 Caucasian patients were enrolled during a time period of 13 months. Arrhythmia status of 904 patients could be confirmed and their genomic data were included in the analysis. In this cohort, there were 704 males, 200 females, and the average age was 73.3 years. We genotyped DNA samples using the Illumina Human660 W Genotyping BeadChip and tested for association between genotype at common variants and the phenotype of having an LTA.
Results and Conclusions
We did not find any associations reaching genome-wide significance, with the strongest association at chromosome 13, rs11856574 at P = 5×10−6. Loci previously implicated in phenotypes such as QT interval (measure of the time between the start of the Q wave and the end of the T wave as measured by electrocardiogram) were not found to be significantly associated with having an LTA. Although powered to detect such associations, we did not find common genetic variants of large effect associated with having a LTA in those of European descent. This indicates that common gene variants cannot be used at this time to guide ICD risk-stratification.
Trial Registration
ClinicalTrials.gov NCT00664807
Introduction
Sudden cardiac arrest death (SCD) accounts for the loss of over 300,000 individuals each year in the United States [1] and approximately 80% of those affected have underlying coronary artery disease [1]. Many studies have provided evidence that there is a genetic contribution to SCD by demonstrating family history as being a risk factor for sudden cardiac death or cardiac arrest [2], [3], [4]. In addition, multiple family studies have emphasized the importance of heritability in SCD, with relative risks of 1.5 to 2.7 in case-control studies among first-degree relatives of individuals who have died suddenly [5], [6].
Recently, several studies have demonstrated specific gene variants or genomic loci that are associated with SCD. These include variants in the cardiac ion channels KCNQ1 and SCN5A [7], nitric oxide synthase 1 adaptor protein [8], and a susceptibility locus at 21q21 for ventricular fibrillation in patients who have had acute myocardial infarction [9]. Furthermore, common variants in at least 10 genomic loci have been correlated with QT duration, a key indicator of cardiac repolarization [10], [11].
While considerable research has been directed to the identification of the genomics of life threatening arrhythmias (LTA), there has not yet been a genome-wide assessment of patients who have received an implantable cardioverter-defibrillator (ICD). ICDs are implanted in approximately 250,000 individuals in the United States annually for criteria that include diminished ejection fraction, symptomatic heart failure, and to a lesser extent, prolongation of the QRS interval or other primary arrhythmogenic cardiomyopathies. While ICDs have a success rate of more than 97% for sensing and terminating the LTA [12], they are never activated in approximately 80% of patients over the duration of their lives [13]–[15]. Accordingly, our current criteria for selecting patients for ICD therapy are rather crude, particularly when one considers up-front cost approaching $30,000 and the risk, albeit small, of infection, lead and device malfunctions, and inappropriate shocks. At the same time, many patients who could benefit from an ICD do not receive one. Therefore, there is a need for better approaches to risk stratification.
The hypothesis of the current study was that a genome-wide assessment of patients with ICDs would identify common DNA sequence variants associated with LTA and would refine ICD selection criteria. Furthermore, by better defining the population that could benefit from ICD therapy, the information might be extrapolated to identify individuals at risk in the general population who do not currently meet guidelines for primary prevention ICD therapy.
We present the results of a retrospective analysis on patients with an ICD and extended follow-up who had experienced LTA, with a cumulative 607 cases and 297 controls included in the analysis. We genotyped DNA samples using the Illumina Human660 W Genotyping BeadChip and tested for association between genotype at common variants and the phenotype of having an LTA.
Methods
Ethics Statement
The Scripps Institutional Review Board reviewed and approved the protocol entitled, MEDTRONIC GAME: Genetic Arrhythmia Markers for Early Detection, IRB #08-4985, on June 6, 2008. The protocol underwent continuing review on May 29, 2009 and was closed with a Final Report on July 22, 2009. Written informed consent was obtained from all participants involved in this study. A copy of the last approved informed consent form is included as Appendix S2.
Barbara G. Bigby, ALM, CIP, Scripps IRB Officer
Patient Population
The overall study design is shown in Figure 1. The inclusion criteria for all patients was that an ICD or cardiac resynchronization therapy with defibrillator (CRT-D) was implanted, that the patient was considered to be on optimal medical therapy by the treating physician, that coronary artery disease was present with either prior myocardial infarction, percutaneous coronary intervention or coronary artery bypass surgery, and that the patient was of European ancestry, with both parents and all 4 grandparents self-reported as Caucasian. To qualify as cases, the additional inclusion criteria were (1) age >40 years at the time of ICD implantation, and (2) at least one fully documented life threatening arrhythmia, as defined by a spontaneous ventricular arrhythmia with cycle length ≤400 ms appropriately treated by the device. Two hundred of the 607 case patients were secondary prevention patients. Although all patients in the study were expected to have a myocardial infarction (MI) or coronary artery disease (CAD), it was not required that the MI/CAD had to be diagnosed prior to the arrhythmic event date for the secondary prevention patients. To qualify as controls, patients required at least 3 years of ICD therapy with a center-verified absence of any appropriately treated life-threatening arrhythmias and no pre-implantation history of LTA or SCA (i.e. a primary-prevention ICD indication). All control patients all had ICDs for primary prevention. The control data were treated as right censoring at exam date. The demographic data from cases and controls were compared using a t-test.
Figure 1. Study design for genome-wide association study of ICD activation with LTA.

Patients were recruited at 34 sites across the United States (Appendix S1) and the local institutional review boards approved the protocol. A cohort of 500 cases and 500 controls was targeted for enrollment in keeping with the success of the genome-wide association studies that had a similar sample size in related cardiovascular phenotypes of coronary heart disease [16], atrial fibrillation [17], and QT interval [18]. All patients provided informed consent prior to enrollment.
DNA isolation
All patients had a single tube of blood drawn at the time of enrollment, which was collected centrally for subsequent DNA extraction. A 0.2 ml aliquot of whole blood from each participant was used for DNA isolation using the Qiagen QIAamp DNA Mini Kit (Qiagen, Valencia, CA; Catalog #51185) and QiaCube Robotic workstation for automated DNA purification. The typical yield was 2–10 ug DNA from 0.2 ml blood. DNA concentrations were determined using a nanodrop spectrophotometer and DNA concentrations were adjusted to 50 ng/ul.
Genotyping and Data Quality Control
Genotyping was performed using the Illumina 660 W BeadChip, following the manufacturer's instructions. Genotypes were clustered within GenomeStudio using all samples with >98% call rates. SNPs with call rates <95%, or heterozygote frequencies >65% after re-clustering were removed. Cluster separation scores were generated using Illumina's Genome Studio software. After visual inspection of the range of cluster separation scores in this dataset, thresholds for filtering were determined. Autosomal SNPs with cluster separation scores ≤0.30 and X-chromosome SNPs with cluster separation scores ≤0.38 were removed. Sample call rates were recalculated and individuals with call rates <99% were removed (median call rate = 99.989%). Genotype concordance was calculated based on 12 duplicate samples (99.998%).
Additional quality control was undertaken using the genetic analysis program PLINK [19]. Individuals were tested for gender consistency, cryptic relatedness, and ancestry. All genders determined by genotype data agreed with the clinical data. Cryptic relatedness was tested using the –genome command, which was run on SNPs that had been filtered for linkage equilibrium (N = 105,837, r2<0.5 within a window of 50 SNPs). Based on the proportion of relatedness (PI_HAT), 3 pairs of samples were duplicate or twin samples (PI_HAT ∼1) and 1 pair of siblings (PI_HAT ∼0.5) were present. In these cases, one individual from each related pair was removed. Samples were clustered by multidimensional scaling with HapMap III samples using the set of 105,837 SNPs in linkage equilibrium. There were 4 samples that clustered outside the European (CEU and TSI) ancestry groups and were removed. A total of 904 samples passed all quality control filters, consisting of 607 cases, 297 controls.
SNPs were additionally filtered for minor allele frequency (>0.01 in all samples) and Hardy-Weinberg equilibrium (P<10−6 in the controls). Finally, if SNPs were not present in dbSNP or did not map uniquely to the reference genome, they were removed (N = 645). A total of 534,690 genotyped SNPs passed all quality control filters.
Genotype Imputation
We imputed genotypes in genotyped individuals for all HapMap (phase II, release 22) SNPs using the program MaCH [20] and an r2 threshold of 0.3. The best estimate of the quantitative allele dosage was used as the predictor in association tests. The CEU HapMap phased haplotypes were used as a reference (N = 60 unrelated individuals). We estimated error rates by masking 1% of the genotypes: genome-wide, the allelic error rate was 0.017 and the genotypic error rate was 0.034.
Association
Case-control association was performed using logistic regression in PLINK. For SNPs that were directly genotyped, the genotype calls were used, while MaCH maximum likelihood dosages (0–2) were used for SNPs that were imputed. Genetic background was estimated using multidimensional scaling (MDS) and the top 10 MDS components were tested for association with the phenotype [21]. Q-Q plots and genomic inflation factors were calculated in PLINK using the –qq-plot command. The survival analysis was performed using a Cox proportional hazards model as implemented in the survival [22], [23]. For the subset of cases with length of follow-up (N = 258), the time between implant and having an adjudicated LTA was used, while for controls (N = 297), the length of follow-up was the time between implant date and exam date. Gender was included as a covariate.
Power
Power was calculated for a range of allele frequencies and genotyping relative risks based on sample size of cases and controls, minor allele frequency, and genotyping relative risk assuming an additive genetic model. Power at each genotypic relative risk was calculated for an alpha of 5×10−8 with CaTS [24].
CNV analysis
Copy number variants (CNVs) were predicted based on intensity and genotype information from CNV and SNP probes using PennCNV [25] (2010May01 version) and QuantiSNP [26] (v2.3) with default parameters. A total of 33 samples were excluded for high log R Ratio standard deviation, high waviness factor, or drifting B allele frequency. CNVs covering at least 10 probes and with a length of at least 100 kb were obtained using each method. Regions that overlapped between the methods were merged and analyses were performed using CNVs predicted by either method. Only regions that overlapped between the methods were used in the final analysis. There were 4,467 total autosomal CNV regions consisting of 2,193 deletions and 2,274 duplications. Association between cases and controls was run using the rare CNV commands in PLINK with 100,000 permutations. Three analyses were run: all CNVs, deletions (copy number = 0 or 1), and duplications (copy number = 3, 4, or 5). P-values were calculated using a max(T) permutation using the –mperm function in PLINK [19]. This test compares each observed test statistic against the maximum of all permuted statistics over all SNPs for each single replicate. Regions with permuted P-value of less than 0.05 are reported.
Results
The baseline demographics are summarized in Table 1, the electrophysiologic features at baseline in Table 2, and medications in Table 3.
Table 1. Demographics of Cases and Controls at Time of ICD Implantation.
| Patient Characteristics | Case Arm (N = 607) | Control Arm (N = 297) | p-value |
| Age at Visit | 71.1±10.0 | 77.9±5.8 | <0.0001** |
| Gender (Male) | 474 (89%) | 230 (84%) | 0.059 |
| Most Recent LVEF | 34.3±12.9 | 36.7±13.0 | 0.012* |
| LVEF Not Available | 43 (7%) | 22 (7%) | 0.891 |
| Weight (Kilograms) | 90.1±19.1 | 85.2±15.9 | <0.0001** |
| Height (Meters) | 1.76±0.09 | 1.74±0.09 | 0.010* |
| Body Mass Index | 29.1±5.4 | 28.0±4.8 | 0.006** |
| RV - MI | 44 (7%) | 23 (8%) | 0.788 |
| LVA - MI | 143 (24%) | 89 (30%) | 0.043* |
| LVP - MI | 72 (12%) | 19 (6%) | 0.010* |
| Septal MI | 37 (6%) | 27 (9%) | 0.128 |
| Unknown MI Location | 264 (43%) | 118 (40%) | 0.316 |
| No Documented MI | 100 (16%) | 55 (19%) | 0.453 |
| Approximate Age at First MI | 55.5±11.6 | 60.9±10.8 | <0.0001** |
| Family History of Sudden Cardiac Arrest or Death | 148 (37%) | 85 (43%) | 0.154 |
| Earliest Age of SCA/SCD for Parents/Siblings | 61.7±13.3 | 64.8±13.5 | 0.115 |
Table 2. Electrophysiological characteristics of cases and controls.
| Electrophysiological Characteristics | Case Arm (N = 607) | Control Arm (N = 297) | p-value |
| QRS> = 120 ms | 218 (54%) | 122 (66%) | 0.005** |
| Most Recent Intrinsic QRS Duration | 128.4±35.8 | 138.0±39.8 | 0.004** |
| QRS Duration Not Available | 203 (33%) | 113 (38%) | 0.182 |
| Atrial Fibrillation | 250 (41%) | 145 (49%) | 0.032* |
| Atrial Flutter | 59 (10%) | 30 (10%) | 0.906 |
| Atrial Tachycardia | 24 (4%) | 7 (2%) | 0.248 |
| AV Block | 49 (8%) | 53 (18%) | <0.0001** |
| AV Nodal Re-Entrant Tachycardia | 4 (1%) | 0 (0%) | 0.309 |
| AV Re-Entrant Tachycardia | 1 (0%) | 1 (0%) | 0.549 |
| Familial or Inherited Conditions with High Risk for VT | 10 (2%) | 1 (0%) | 0.113 |
| Intraventricular Conduction Delay | 14 (2%) | 13 (4%) | 0.097 |
| Left Bundle Branch Block | 67 (11%) | 50 (17%) | 0.020* |
| Right Bundle Branch Block | 38 (6%) | 23 (8%) | 0.400 |
Table 3. Baseline use of key medications.
| Medication Use | Case Arm (N = 607) | Control Arm (N = 297) | p-value |
| ACE Inhibitor Use - Current | 294 (61%) | 143 (58%) | 0.426 |
| ACE Inhibitor Use - Ever | 444 (92%) | 216 (87%) | 0.046* |
| Angiotensin Receptor Blocker Use - Current | 117 (36%) | 57 (31%) | 0.286 |
| Angiotensin Receptor Blocker Use - Ever | 174 (53%) | 85 (46%) | 0.119 |
| Aldosterone Antagonist Use - Current | 81 (27%) | 31 (17%) | 0.019* |
| Aldosterone Antagonist Use - Ever | 130 (43%) | 49 (27%) | 0.001** |
| Beta Blocker Use - Current | 467 (82%) | 229 (82%) | 1.000 |
| Beta Blocker Use - Ever | 559 (98%) | 274 (98%) | 0.800 |
| Diuretics Use - Current | 362 (74%) | 188 (75%) | 1.000 |
| Diuretics Use - Ever | 445 (92%) | 223 (88%) | 0.187 |
| Anti-Arrhythmic/Class I Use - Current | 44 (16%) | 10 (6%) | 0.002** |
| Anti-Arrhythmic/Class I Use - Ever | 81 (29%) | 19 (11%) | <0.0001** |
| Anti-Arrhythmic/Class II Use - Current | 34 (13%) | 16 (10%) | 0.283 |
| Anti-Arrhythmic/Class II Use - Ever | 56 (22%) | 16 (10%) | 0.001** |
| Anti-Arrhythmic/Class III Use - Current | 167 (42%) | 36 (18%) | <0.0001** |
| Anti-Arrhythmic/Class III Use - Ever | 259 (65%) | 68 (34%) | <0.0001** |
Cases and controls differed by age, height, weight, location of myocardial infarction, and age of first myocardial infarction (Table 1). In addition, there were 704 males and 200 females in the study. Gender specific differences were observed in mean left ventricular ejection fraction (LVEF), weight, height, smoking, and approximate age for the first MI.
The electrophysiologic characteristics at baseline also differed between the groups with respect to incidence of atrioventricular block, left bundle branch block, and a history of spontaneous ventricular arrhythmia (Table 2). For the 102 patients with qualifying tachycardias, the mean cycle length was 297 msec (S.D. = 56.5). The histogram of average ventricular cycle length for these 102 patients is shown in Figure S1. Finally, there were also some imbalances in baseline medications, particularly anti-arrhythmic drugs (Table 3).
A genome-wide association was performed between individuals that had an LTA (cases) and those that did not (controls) over the course of at least three years (Figure 2). Gender and genetic background were tested for association with case-control status, were not found to be significant, and were not included in the model. We did not detect any regions associated at P<5×10−8 and report top regions at P<1×10−5 (Table 4). The strongest association at rs11856574 (P = 5.0×10−6), located in the hypothetical gene KIAA0574 (hypothetical protein LOC23359). We detected no evidence of population stratification (genomic inflation factor = 1.0035, Figure S2). On a subset of individuals for which we had length of follow-up, we performed survival analysis GWA (Figure S3). We did not detect any associations at P<5×10−8, but do note a region at 13q14.2 (rs2854357 P = 8.2×10−7) with a consistent signal across multiple SNPs.
Figure 2. Genome-wide association results of Cases with LTA vs. Controls without a LTA.

Association −log(P-values) are plotted according to position in the genome. Points indicate genotyped (circle) or imputed (triangle) SNPs.
Table 4. Top regions associated at P<10−5.
| Top SNP | Alleles (A1/A2) | Chr | Pos | #SNPs at P<10−4 | Freq (A1) | OR (A1) | P | Nearest gene, description, relative SNP location |
| rs11856574* | G/A | 15 | 27518736 | 1/13 | 0.86 | 2.02 | 5.0×10−6 | KIAA0574, hypothetical protein, intron |
| rs482329* | C/G | 1 | 232883177 | 3/18 | 0.61 | 1.60 | 5.5×10−6 | IRF2BP2, interferon regulatory factor 2 binding protein 2, 72 kb downstream |
| rs3848198* | C/T | 15 | 78426619 | 2/4 | 0.38 | 1.81 | 9.8×10−6 | ARNT2, Aryl hydrocarbon receptor nuclear translocator 2; hypoxia associated transcription factor, 57 kb upstream |
| rs6565373* | T/C | 16 | 86817543 | 0/1 | 0.59 | 0.32 | 9.8×10−6 | BANP, BTG3 associated nuclear protein isoform a; negative regulator of p53 transcription, 149 kb downstream |
*imputed; #SNPs at P<10−4 indicates the number of SNPs at P<10−4 within +/−150 kb of the top SNP that were either directly genotyped or imputed (genotyped/imputed).
We looked specifically at P-values for 42 SNPs previously reported to be implicated in prolonged QT duration or SCD [10], [11], [17], [18], [27]–[30]. None were associated in the study dataset at Bonferroni corrected P-values (P<0.05/42 = 0.0012). Table S1 shows the P-values from directly genotyped or imputed genotypes for these 42 SNPs, indicating that these SNPs may be of limited prognostic value in identifying individuals likely to have an LTA among those that are candidates for an ICD with prior myocardial infarction.
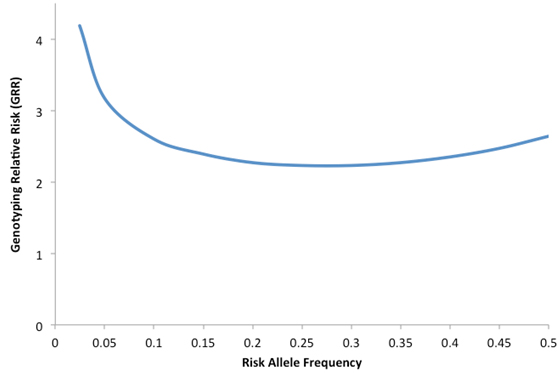
We calculated our power to detect effects at an alpha of 10−7 for the sample sizes observed here. We were well powered to detect effects of large effect size (OR>3.78) in common variants (minor allele frequency (MAF)>0.05) given our sample size (Figure S4). For very common variants (MAF>0.25), we had 80% power to detect effects of OR>2.15. Thus, it is unlikely that there is a common variant with a large effect strongly associated with LTA in individuals of European ancestry. Using genome-wide genotyping chips such as the Ilumina Human660 BeadChip, there is good coverage of common variation in European ancestry populations [31], however not all regions of the genome, in particular regions of low linkage disequilibrium, may be covered well.
We conducted a copy number variation analysis, using QuantiSNP and PennCNV to estimate copy number variation in each individual. CNV regions covering at least 10 probes and 100 kb from both methods were combined and tested for association with case/control status in PLINK. There were no regions that were associated at a multiple testing corrected P-value less than 0.05 when all CNVs were combined. However, one region on chromosome 16 (position 33,395,681–33,506,617) was associated (minimum P = 0.0097) when deletions alone were tested (Figure S5). The region is flanked by a target of p53 (TP53TG3) and a creatine transporter (SLC6A8). As this region is a duplication of a region on chromosome X [32], we tested whether having a CNV in this region was associated with gender and saw no association. These CNVs require validation using quantitative PCR and the association will require verification in a replication study.
Discussion
Although ICDs are the main therapy for preventing SCD, current criteria for patient selection remains suboptimal with only approximately 20% requiring appropriate therapy over their lifetime [13]–[15]. Through a genome-wide assessment, we were unable to identify any common sequence variants that were associated with appropriate ICD therapy for a LTA. We did detect a marginally significant deletion associated with case status, but this will require further validation and replication before it can be considered a positive finding.
Our sample size was adequate to detect common sequence variants that confer large effects with minor allele frequency of 0.25 or greater, many of which have previously been described to be genome-wide associated with prolonged QT duration or in candidate gene studies of SCD [6]–[11], [17], [18], [27]–[30]. There are several potential explanations for our findings. First, it is possible that low frequency or rare sequence variants are associated with appropriate ICD therapy. Indeed, several recent studies have demonstrated that for common clinical phenotypes, multiple rare variants with high penetrance may play an important role in accounting for heritability [33]–[35]. It should be pointed out that we screened common SNPs and copy number variants, but our methodology did not address the detection of other sequence variants such as small insertions, small deletions, or other forms of structural genomic variation. Second, there may be relatively common SNPs that confer a smaller effect (OR<2.0), but were not detected in our cohort due to inadequate statistical power or due to combining all types of LTA with rates ≤400 msec. This potential shortcoming could be overcome by combining the data from multiple like cohorts to determine whether gene variants with minor allele frequency between 5% and 25% may, at least in part, contribute to the heritability of LTA. Third, it remains possible that propensity to develop an LTA is not genetically determined, particularly in the types of patients who currently receive ICDs. Such patients have markedly reduced systolic left ventricular function. The likelihood of having appropriate ICD therapy increases over time [36], and may be related to unfavorable cardiac remodeling and adrenergic activation, representing more of a mechanical/neural substrate than one that would implicate an intrinsic abnormality of ion channels, repolarization, or electrical dispersion. This possible explanation appears to be offset to some degree by the remarkable finding of a common variant at 21q21 for ventricular fibrillation in patients with acute myocardial infarction [9], although the arrhythmogenic milieu of acute myocardial infarction differs from that of SCA in the stable patient of systolic dysfunction. Fourth, it is possible that our cohort was quite heterogeneous or suboptimal from a number of standpoints. The lack of more extended follow-up may have led to misclassification of phenotype, or the multiple paths to low ejection fraction (e.g. myocardial infarction, diffuse coronary artery disease cardiomyopathy) might represent a mosaic with inadvertent lumping of phenotypes and diminished ability to discern the genomic factors associated with any given subtype. There is some indication from the subgroup of patient with extended follow up in our study that this may represent a key issue for detection of a significant genomic association.
It is worth noting that the study cohort is representative of the demographics for patients in the United States who receive an ICD. Patients who are in the control arm were slightly older, likely because of the selection bias requiring that the individuals were event-free and followed for a minimum of three years following the device implant.
ICDs have been shown to reduce all-cause mortality by 26% and 57% reduction of SCD in randomized controlled trials [36]. Observational data from 11 case-control cohorts involving 96,951 patients has been even more impressive with a 46% reduction of all-cause mortality [36]. Yet the current selection criteria for ICD implantation are far from optimal. About 80% of patients do not require ICD activation for a life-threatening arrhythmia during their lifetime after implantation. This lack of discriminative ability is especially problematic given the cost of the devices and the array of complications, albeit at low incidence, but including implantation-related complications, lead failures, device malfunction, and inappropriate shocks.
Moreover, a large proportion of patients with SCD do not meet any current criteria for ICD therapy [1]. They have intact ejection fraction and, at present, have no identifying risk markers. Had genomic loci become evident from the present study of a cohort with ICDs, it remains possible that the findings would be informative or extrapolatable to the general population. With the negative findings here, it remains unclear whether the ICD cohort will prove to be representative of the at-large population with risk of SCD.
There have been a very large number of genome-wide association studies performed, and a subset of these have been published for over 150 complex traits [37]. The negative GWAS for LTA represents an important finding with the bias of publication of only positive results. Here we have attempted to identify genomic markers that would refine our appropriate selection of patients who might receive an ICD, above and beyond those patients with significant left-ventricular dysfunction. Our inability to demonstrate a genomic marker should not be considered an abject failure, but rather it can be viewed as the first step that lays the groundwork for more intensive efforts such as deep sequencing of candidate genes or much larger cohorts with more extended follow-up for genome-wide association. This area of medicine, exemplifying device genomics, and especially at a time of crisis in health care economics, deserves further study.
Supporting Information
Histogram of average ventricular cycle length for subjects with qualifying tachycardia event (N = 102).
(JPG)
{kind=link}
Q-Q plot of LTA association. −log(P-values) are plotted according to expected (x-axis) and observed (y-axis) values. Expected values were calculated in PLINK. Only directly genotyped SNPs were used in this plot. A line is drawn at y = x.
(JPG)
{kind=link}
Survival analysis Manhattan Plot. Genome-wide association −log(P-values) are shown for a survival analysis using individuals (258 cases, 297 controls) for which we had length of follow-up. Points indicate genotyped (circle) or imputed (triangle) SNPs.
(JPG)
{kind=link}
GWAS power. Genotypic relative risk (GRR) detected at 80% power with at alpha = 5×10−8 for 607 cases and 297 controls is plotted as a function of minor allele frequency.
(JPG)
{kind=link}
Deletions overlapping chr 16 CNV association. CNVs overlapping at least 10 probes and 100 kb were predicted using PennCNV and QuantiSNP. CNVs predicted in controls (blue) and cases (red) are plotted by location on the chromosome. SNP and CNV markers are shown as asterisks or triangles, respectively and plotted by location.
(JPG)
{kind=link}
Association at SNPs previously implicated in sudden cardiac death, prolonged QT duration, atrial fibrillation, or ventricular fibrillation.
(DOC)
Enrolling Center List.
(PDF)
IRB Approved Consent to Participate in Research Form.
(PDF)
Acknowledgments
The corresponding author had full access to all data in the study and takes responsibility for the integrity and accuracy of the data analysis.
Footnotes
Competing Interests: Funding for this study was made in part by Medtronic (OS, TN, JL, HG, RCK-consultant), Scripps Health (SSM, EJT, ENS, NV, MS, SD) and NIH/NCRR UL1RR025774 (SSM, SD, EJT). The funders' role in the study pertains to the specific contributions of the authors as listed above, including study design, data collection and analysis, decision to publish and assistance with preparation of the manuscript. This does not alter the authors' adherence to all the PLoS ONE policies on sharing data and materials.
Funding: Funding for this study was made in part by Medtronic (OS, TN, JL, HG, RCK-consultant), Scripps Health (SSM, EJT, ENS, NV, MS, SD) and NIH/NCRR UL1RR025774 (SSM, SD, EJT). The funders' role in the study pertains to the specific contributions of the authors as listed above, including study design, data collection and analysis, decision to publish and assistance with preparation of the manuscript. This does not alter the authors' adherence to all the PLoS ONE policies on sharing data and materials.
References
- 1.Sen-Chowdhry S, McKenna WJ. Sudden cardiac death in the young: the impact of inherited cardiovascular disease. Textbook of Cardiovascular Medicine. CD/online content ed. Philadelphia, PA: Lippincott Williams and Wilkins Press; 2007. [Google Scholar]
- 2.Jouven X, Desnos M, Guerot C, Ducimetiere P. Predicting sudden death in population: Paris prospective study I. Circulation. 1999;99:1978–1983. doi: 10.1161/01.cir.99.15.1978. [DOI] [PubMed] [Google Scholar]
- 3.Dekker L, Bezzina CR, Henriques JP, Tanck MW, Koch KT, et al. Familial Sudden Death Is an Important Risk Factor for Primary Ventricular Fibrillation. Circulation. 2006;114:1140–1145. doi: 10.1161/CIRCULATIONAHA.105.606145. [DOI] [PubMed] [Google Scholar]
- 4.Friedlander Y, Siscovick DS, Weinmann S, Austin MA, Psaty BM, et al. Family History as a Risk Factor for Primary Cardiac Arrest. Circulation. 1998;97:155–160. doi: 10.1161/01.cir.97.2.155. [DOI] [PubMed] [Google Scholar]
- 5.Knollmann BC, Roden DM. A genetic framework for improving arrhythmia therapy. Nature. 2008;451:929–936. doi: 10.1038/nature06799. [DOI] [PubMed] [Google Scholar]
- 6.Noseworthy PA, Newton-Cheh C. Genetic determinants of sudden cardiac death. Circulation. 2008;118:1854–1863. doi: 10.1161/CIRCULATIONAHA.108.783654. [DOI] [PubMed] [Google Scholar]
- 7.Albert CM, MacRae CA, Chasman DI, VanDenburgh M, Buring JE, et al. Common variants in cardiac ion channel genes are associated with sudden cardiac death. Circ Arrhythm Electrophysiol. 2010;3:222–229. doi: 10.1161/CIRCEP.110.944934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, et al. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. 2009;119:940–951. doi: 10.1161/CIRCULATIONAHA.108.791723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezzina CR, Pazoki R, Bardai A, Marsman RF, de Jong JS, et al. Genome-wide association study identifies a susceptibility locus at 21q21 for ventricular fibrillation in acute myocardial infarction. Nat Genet. 2010;42:688–691. doi: 10.1038/ng.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeufer A, Sanna S, Arking DE, Muller M, Gateva V, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myerburg RJ. Implantable cardioverter-defibrillators after myocardial infarction. N Engl J Med. 2008;359:2245–2253. doi: 10.1056/NEJMra0803409. [DOI] [PubMed] [Google Scholar]
- 13.Desai H, Aronow WS, Ahn C, Gandhi K, Hussain S, et al. Risk factors for appropriate cardioverter-defibrillator shocks, inappropriate cardioverter-defibrillator shocks, and time to mortality in 549 patients with heart failure. Am J Cardiol. 2010;105:1336–1338. doi: 10.1016/j.amjcard.2009.12.057. [DOI] [PubMed] [Google Scholar]
- 14.Kruger A, Aronow WS, Lai H, Desai H, Singla A, et al. Prevalence of appropriate cardioverter-defibrillator shocks in 1038 consecutive patients with implantable cardioverter-defibrillators. Am J Ther. 2009;16:323–325. doi: 10.1097/MJT.0b013e3181727a59. [DOI] [PubMed] [Google Scholar]
- 15.Otmani A, Trinquart L, Marijon E, Lavergne T, Waintraub X, et al. Rates and predictors of appropriate implantable cardioverter-defibrillator therapy delivery: results from the EVADEF cohort study. Am Heart J. 2009;158:230–237 e1. doi: 10.1016/j.ahj.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 16.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 18.Arking DE, Pfeufer A, Post W, Kao WH, Newton-Chew C, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–651. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Y, Liu Z, Ott J. Systematic removal of outliers to reduce heterogeneity in case-control association studies. Hum Hered. 2010;70:227–231. doi: 10.1159/000320422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 23.Therneau T, Lumley T. 2008. survival: Survival analysis, including penalised likelihood; S original; R package version 2.34-1.
- 24.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Optimal designs for two-stage genome-wide association studies. Genet Epidemiol. 2007;31:776–788. doi: 10.1002/gepi.20240. [DOI] [PubMed] [Google Scholar]
- 25.Wang K, Li M, Hadley D, Liu R, Glessner J, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colella S, Yau C, Taylor JM, Mirza G, Butler H, et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 28.Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–244. doi: 10.1038/ng.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 30.Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, et al. Genome-wide association study of PR interval. Nat Genet. 2010;42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eberle MA, Ng PC, Kuhn K, Zhou L, Peiffer DA, et al. Power to detect risk alleles using genome-wide tag SNP panels. PLoS Genet. 2007;3:1827–1837. doi: 10.1371/journal.pgen.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eichler EE, Lu F, Shen Y, Antonacci R, Jurecic V, et al. Duplication of a gene-rich cluster between 16p11.1 and Xq28: a novel pericentromeric-directed mechanism for paralogous genome evolution. Hum Mol Genet. 1996;5:899–912. doi: 10.1093/hmg/5.7.899. [DOI] [PubMed] [Google Scholar]
- 33.Johansen CT, Wang J, Lanktree MB, Cao H, McIntyre AD, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ezekowitz JA, Rowe BH, Dryden DM, Hooton N, Vandermeer B, et al. Systematic review: implantable cardioverter defibrillators for adults with left ventricular systolic dysfunction. Ann Intern Med. 2007;147:251–262. doi: 10.7326/0003-4819-147-4-200708210-00007. [DOI] [PubMed] [Google Scholar]
- 37.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histogram of average ventricular cycle length for subjects with qualifying tachycardia event (N = 102).
(JPG)
Q-Q plot of LTA association. −log(P-values) are plotted according to expected (x-axis) and observed (y-axis) values. Expected values were calculated in PLINK. Only directly genotyped SNPs were used in this plot. A line is drawn at y = x.
(JPG)
Survival analysis Manhattan Plot. Genome-wide association −log(P-values) are shown for a survival analysis using individuals (258 cases, 297 controls) for which we had length of follow-up. Points indicate genotyped (circle) or imputed (triangle) SNPs.
(JPG)
GWAS power. Genotypic relative risk (GRR) detected at 80% power with at alpha = 5×10−8 for 607 cases and 297 controls is plotted as a function of minor allele frequency.
(JPG)
Deletions overlapping chr 16 CNV association. CNVs overlapping at least 10 probes and 100 kb were predicted using PennCNV and QuantiSNP. CNVs predicted in controls (blue) and cases (red) are plotted by location on the chromosome. SNP and CNV markers are shown as asterisks or triangles, respectively and plotted by location.
(JPG)
Association at SNPs previously implicated in sudden cardiac death, prolonged QT duration, atrial fibrillation, or ventricular fibrillation.
(DOC)
Enrolling Center List.
(PDF)
IRB Approved Consent to Participate in Research Form.
(PDF)