

Abstract
Enterobacter sakazakii(ES) is an emerging pathogen that causes sepsis, meningitis, and necrotizing enterocolitis in neonates. Very limited information is available regarding the pathogenesis of these diseases and the specific virulence factors of ES. Here, we demonstrate, for the first time using a newborn rat model, that outer membrane protein A (OmpA) expression is important for the onset of meningitis by ES. Orally administered OmpA+ ES traverses the intestinal barrier, multiplies in blood, and subsequently penetrates the blood-brain barrier. OmpA+ ES were present in high numbers in the brains of infected animals along with associated neutrophil infiltration, hemorrhage, and gliosis. In contrast, OmpA− ES could not bind to the intestinal epithelial cells in vitro and in vivo efficiently. The bound OmpA+ ES also caused apoptosis of enterocytes in the intestinal segments of infected animals; OmpA− ES did not. Furthermore, OmpA− ES is very susceptible to blood and serum killing whereas OmpA+ ES is resistant. Of note, 100% mortality rates were observed in OmpA+ ES infected newborn rats while OmpA− ES infected rats survived without any pathological manifestations. The inability of OmpA− ES to cause disease was restored by complementation with the ompA gene. These results suggest that OmpA expression in ES is necessary for the colonization of the gastrointestinal tract and for subsequent survival in blood to cause meningitis.
Keywords: Enterobacter sakazakii, Outer membrane protein A, meningitis, rat brain, survival
Enterobacter sakazakii (ES) is an emerging opportunistic human pathogen and the etiological agent of life-threatening bacterial infections.1–3 ES infections continue to be rare but are found more commonly among neonates and infants than adults. This pathogen has a propensity to cause bacteremia, meningitis and necrotizing enterocolitis.4 Mortality and morbidity rates due to ES meningitis are high, and are often associated with serious sequelae including brain abscess and impaired sight and hearing.5–7 Furthermore, virtually all patients recovering from central nervous system infections suffer from subsequent mental and/or physical developmental delay, which may have devastating long-term ramifications for the patient. The majority of adults with ES infections had underlying diseases identified and notably 4 out of 8 adults had malignancies. The increase in incidence of neonatal infections caused by ES has stimulated a renewed research interest in this pathogen.8–10 ES has been categorized as “severe hazard for restricted populations, life threatening or substantial chronic sequelae or long duration” by the International Commission for Microbiological Specification for Foods.11
Currently, very little information is available on the virulence factors of ES and its pathogenic mechanisms. Some strains of ES produce an enterotoxin that is lethal in suckling mice and produces cytopathic effects in cell lines.12 Outbreaks of sepsis, meningitis and necrotizing enterocolitis in newborns are attributed to ES contamination of powdered infant formula, indicating that the most probable route of ES entry is oral. Therefore, the most likely primary site of ES colonization is the intestinal tract. The presence of LPS in infant formula has also been implicated in increasing compromise of intestinal barrier integrity.13 After colonizing the intestine, ES should gain access to bloodstream, survive the host defense mechanisms before entering the central nervous system. Therefore, it would be expected that ES possesses some armory for the successful establishment of the disease. In mammalian tissue culture, the organism can attach to host cells and survive internally in macrophages.14 However, the contribution of specific bacterial virulence factors involved in these processes is unknown. Recently, outer membrane protein A (OmpA) of ES has been demonstrated to play an important role in attachment and invasion of Caco-2 cells.15 In contrast, studies from our lab demonstrated that ES binds to rat intestinal epithelial cells (IEC-6 cells) with no significant invasion.16 The interaction of ES with IEC-6 cells and enterocytes in animal model of NEC also induces apoptosis, indicating that the bacteria disseminates through the compromised gut barrier. Our studies recently demonstrated that OmpA expression in ES contributes to the invasion of human brain microvascular endothelial cell (HBMEC).17 Of note, the invasion of ES in intestinal epithelial cells requires microfilaments whereas microtubules are necessary for the invasion of HBMEC.17 However, the role of OmpA in the pathogenesis of ES meningitis in vivo has yet to determined. Townsend et al demonstrated that ES causes meningitis in newborn rats leading to brain abscess formation.18 In these studies, the bacteria have been given via intracisternal route, which is not a physiological route of infection for ES, and thereby did not explore the bacterial interaction with intestine and blood.
In this study, for the first time, we demonstrate that OmpA expression is critical for the onset of meningitis in newborn rats by employing oral administration of isogenic OmpA+ ES or OmpA− ES. The attenuated virulence of OmpA− ES mutant is attributed to their inability to bind to intestinal cells and resist serum bactericidal activity.
Materials and Methods
Bacterial strains, cells, and reagents
Enterobacter sakazakii (51329) and rat intestinal epithelial cells IEC-6 (used between passages 20–26) were obtained from ATCC (Manassas, VA). IEC-6 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 1 U/ml insulin, 100 U/ml penicillin G, and 100 U/L streptomycin. Rodent formula (Esbilac) was obtained from PetAg (Hampshire, IL). All general chemicals were procured from Sigma (St Louis, MO) unless otherwise stated.
Generation of OmpA− ES and complementation with ompA gene
ES 51329 was grown in LB or Tryptic Soy Broth medium without any antibiotics. ES was transformed with the plasmid pUC13 containing the gfp gene. Transformants were selected by ampicillin (100 μg/ml) and assessed for GFP expression by viewing under ultraviolet light. The ompA-deletion mutant of ES was constructed by replacing ompA with a kanamycin (Km) cassette. Briefly, a spontaneous rifampicin-resistant mutant was isolated and named ES51R. A 1.77-kb DNA containing ompA was amplified from ES with primers: 5′-GTGAGCTCCGGGCTAAAAATTCACTCAA (containing a SacI site), and 5′-CAGGTACCATCGTGCAGCTGATTGA (containing a KpnI site). The DNA was cloned into pEP185.2 at the same sites, and the internal 876-bp NruI-BglII fragment was replaced with a 1.2-kb Km cassette from pUC-4K (Pharmacia). The recombinant plasmid was transferred from E. coli to ES51R by conjugation and double-crossover mutants were selected. The deletion of ompA in ES51R was verified by PCR with the above primers. To restore the OmpA expression in the OmpA− ES, the plasmid pEP185.2 containing the ES ompA gene was transferred into the mutant and selecting for chlroamphenicol resistant colonies. Expression of OmpA in the complemented strain, pOmpA+ ES, and OmpA− ES was confirmed by Western blot using rabbit antiserum developed against E. coli OmpA, which also recognizes the OmpA of ES.17
Binding assays using IEC-6 cells
ES (107 CFU/ml) were added to confluent monolayers of IEC-6 cells separately at a bacteria to cell ratio of 100:1 and incubated for 2–6 h.16 The cells were washed with HBSS medium and were lysed with 0.1% Triton-X 100 for 8 min. Serial dilutions were made from the lysates, plated on blood agar, and incubated at 37°C overnight. The number of bacterial colonies was recorded and the percent binding calculated based on the inoculum size.
Newborn rat model of meningitis
The animal studies were approved by the Institutional Animal Care and Use Committee of CHLA and followed National Institutes of Health guidelines for the performance of animal experiments. Timed pregnant Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA) were obtained and gave birth in our vivarium after a 21-day gestation period. Litters averaged 12 pups were kept in an opaque, polypropylene cage under a small animal isolator. Two-day old rat pups were randomly divided into various groups and infected orally with 104 CFU of OmpA+, OmpA− or pOmpA+ ES in 20 μl of saline with feeding tube to ensure proper administration of dose (L-CATH, BD Infusion therapy systems, Sandy, Utah). Control mice received saline through the same route. 12 rat pups were used for each strain as well as for control group. The infected newborn rats were separated from dams and fed orally with infant formula and given water for every 6 h. At defined time intervals (6, 12, 24, 48, 72, 96 and 120 h) the impact of bacterial feeding on the disease severity of the animals was determined in the following manner. Animals were turned on their backs and the length of time needed for the animals to turn to their normal upright position was tested and scored by numbers: 5 (normal), 4 (turns upright in less than 5 s), 3 (turns upright in less than 30 s), 2 (does not turn upright) and 1 (coma /death).19 Blood was collected from the tail or facial vein at different time periods of post-infection and plated on ampicillin LB agar plates to ensure bacteremia and success of infection. CSF samples were obtained aseptically under anesthesia and directly inoculated into broth containing antibiotics. Growth of ES in LB broth from CSF samples was considered positive for meningitis. The rats were then euthanized at 48 hours or earlier with CO2 after infection in case of OmpA+ and pOmpA+ ES, and at 120 h in case of OmpA− ES infection. Brain, liver, spleen, lung, kidney and intestine were aseptically removed and homogenized in sterile PBS. Bacterial counts in the blood as well as in the homogenates of other organs were determined by plating ten-fold serial dilutions on ampicillin LB agar plates. Cytokine levels present in the brains were calculated after correcting for blood contamination using the blood concentration and a conservative estimate of the rat cerebral blood volume.20
Histopathology
Brain and intestine samples were fixed in 10% buffered formalin, routinely processed and embedded in paraffin. 4–5 μm sections were cut in on Leica microtome and stained with hematoxylin and eosin (H & E) or used for immunohistochemistry. Intestine sections were graded microscopically by a pathologist blinded to groups, from grade 1 (normal) to grade 4 (severe), based on pathological manifestations including submucosal edema, villus core edema, epithelial sloughing/obliteration, neutrophil infiltration, intestinal perforation, and necrosis.16 Pictures were taken with Zeiss Axiovert Microscope connected to a JVC 3-chip color video camera and read by the pathologist in a blinded fashion. Furthermore, cryosections were also prepared and stained with DAPI.16 Immunofluorescent microscopy was used to assess the association of GFP-ES with the intestinal epithelial layer. Apoptosis of intestine sections was assessed after staining with ApopTag Red in situ apoptosis detection kit (Chemicon, Temecula, CA).
Immunohistochemistry
Slides were deparaffinized prior to immunohistochemical staining with a 5-min wash in 100% xylene. Samples were treated for 10 min with 70% ethanol followed by 5-min treatments each with 95% ethanol, 100% ethanol, and distilled water. Sections were microwaved at high temperature in antigen unmasking solution for 2–3 min (Vector Laboratories, Burlingame, CA) and then allowed to cool for antigen retrieval. Endogenous peroxidases were quenched by incubation with 7.5% hydrogen peroxidase in water for 5 min. Samples were incubated with PBST (0.1 M PBS plus 0.01% Triton X-100) containing 5% normal donkey serum to block non-specific binding sites followed by incubation with rabbit anti-OmpA antibodies (1:1000 dilution) or pre-immune rabbit serum (negative control) for 30 min. Samples were then rinsed with PBST containing 1% normal donkey serum, incubated with biotin labeled goat anti-rabbit antibodies (1:500 dilution) for 30 min, and then again rinsed with PBST. The sections were further incubated with streptavadin-peroxidase and the color was developed with diaminobenzidine containing hydrogen peroxide, which yields a brown reaction product. Counterstaining was performed using Mayer’s hematoxylin. Slides were digitized using a Zeiss Axiovert Microscope connected to a JVC 3-chip color video camera and read by a pathologist blinded to samples.
Assessment of neutrophil response
Infiltration of neutrophils into tissues was quantified using a myeloperoxidase (MPO) assay.21 Tissues were homogenized in 2 ml of 50 mM potassium phosphate, pH 6.0 with 5% hexadecyltrimethylammonium bromide and 5 mM EDTA. Homogenates were sonicated and centrifuged. Supernatants were mixed at the ratio of 1:15 with assay buffer and read at 490 nm. MPO units were calculated as the change in absorbance over time and expressed as U/g tissue.
Determination of lactate dehydrogenase (LDH), malionaldehyde (MDA) and gluthathione (GSH)
LDH levels were determined in blood as well as in brain and intestine using QuantiChrom LDH kit from BioAssay systems (Hayward, CA). MDA levels were determined in brain and intestine following the method of Wills et al.22 Briefly, tissue supernatant was added to equal amount of Tris HCl (0.1M, pH 7.4) and incubated at 37°C for 2 h. After incubation, trichloroacetic acid was added and centrifuged at 700 × g for 10 min. Supernatant was mixed with equal volume of thiobarbituric acid (0.67% w/v) and kept in boiling water bath for 10 min. After cooling, volume was made to 3 ml with double distilled water and absorbance was taken at 532 nm. Amount of MDA formed was expressed in nanomoles per milligram protein (n moles/mg) using an extinction coefficient of 1.56 × 105 M− 1 cm− 1. GSH content was determined following the method of Tuğtepe et al.23 Briefly, after centrifugation at 2000 × g for 10 min, 0.5 ml of supernatant was added to 2 ml of 0.3 mol/l Na2HPO4·2H2O solution and 0.2 ml solution of dithiobisnitrobenzoate (0.4 mg/ml 1% sodium citrate). The absorbance at 412 nm was measured immediately after mixing. GSH levels were calculated using an extinction coefficient of 1.36 × 105 M− 1 cm− 1 and the results were expressed in nmoles/mg.
Bactericidal assays
Susceptibility of ES strains to serum killing was carried out using normal neonatal rat serum at a final concentration of 40% diluted in gelatin Veronal buffer (GVB).24 Bacteria (106 CFU) were suspended in serum and incubated at 37°C for various time points. The bacteria were then plated on blood agar in serial dilutions for quantitative determination. Heat inactivated serum was obtained by incubating serum in a water bath for 30 min at 56°C. To perform bactericidal assays using whole blood, 120 μl of 100 mM CaCl2 in the presence of heparin (2 U/ml) was added to 10 ml of citrated blood and aliquots of the blood were incubated with bacteria for varying periods. The experiments were repeated for at least five times in triplicates.
Cytokines determination
The levels of TNF-α, IL-1β, IL-6, IL-10 and macrophage inflammatory protein-2 (MIP-2) were measured in blood, brain and intestine homogenates of rat pups infected with ES using ELISA kits from Biosource (Carlsbad, CA) according to the manufacturer’s instructions.
Statistical analysis
ANOVA, Fischer test, Wilcoxon signed rank test, Chi-square test, and Student’s t-test were applied for statistical analysis of data. P values <0.05 were considered statistically significant. Box plots were generated using STATA software version 10.
Results
OmpA expression is critical for the onset of meningitis in newborn rats by ES
The invasion of ES in HBMEC, an in vitro model of the blood-brain barrier, depends on the expression of OmpA.17 Therefore, to determine the role of OmpA in the virulence of ES in vivo, newborn rats on the second day of life were orally infected with 104 or 105 CFU of either OmpA+ or OmpA− ES. The onset of infection and development of meningitis was tracked by scoring the disease severity in animals at different post-infection time intervals, through quantitative determination of bacteremia, and the occurrence of meningitis. Infection with 104 CFU of OmpA+ ES induced a steady increase in the disease severity of the animals, which ended in a moribund state at 48 h post-infection (Table 1). Infection with 105 CFU of OmpA+ ES induced the disease severity within 16 h (data not shown). In contrast, infection with either 104 or 105 CFU of OmpA− ES caused only slightly reduced physical activity in the animals that recovered completely at 96 h. Bacterial counts in the blood of these animals revealed that OmpA+ ES was detectable as early as 6 h post-infection in case of 104 CFU; in contrast, OmpA− ES were absent at this time point. At subsequent times, the titer of the OmpA+ ES increased showing peak log10 CFU of 9.31 ml−1 at 48 h post-infection (Table 2). In contrast, the titer of OmpA− ES in blood was significantly lower (P<0.001, by Chi-square test) compared to OmpA+ ES showing peak log10 CFU of 3.25 ml−1 at 48 h post-infection and then reduced to below detection levels by 96 h. Of note, 100% of animals infected with OmpA+ ES were positive for meningitis while none of the animals fed with OmpA− ES developed positive cultures. To further substantiate the role of OmpA in the onset of meningitis, newborn rats were also fed with pOmpA+ ES generated by transforming OmpA− ES with a plasmid containing ompA gene17. The pOmpA+ ES expresses similar levels of OmpA as that of wild type ES. As shown in Tables 1 and 2, the complemented strain reverted from non-invasive to invasive phenotype and induced the onset of meningitis similar to that of OmpA+ ES. Mortality in the group inoculated with 104 CFU of OmpA+ or pOmpA+ ES was 100% by 48 h post-infection and occurred at 16 h in the groups inoculated with 105 CFU (Fig. 1). Animals were sacrificed for ethical reasons if the activity score is <2. No mortality was observed in animals infected with OmpA− ES at any inoculum size. For subsequent experiments, 104 CFU of ES was chosen as an appropriate dose. Serum LDH levels, a marker of tissue damage, were determined in these animals, which were significantly increased following infection with OmpA+ ES. LDH levels ranged from 500 to 3100 IU/L in OmpA+ infected rats between 6 and 48 h post-infection, whereas it varied from 220 to 600 IU/L in OmpA− ES fed rats (data not shown).
Table 1.
Activity scoresa of animals infected orally with 104 CFU/ml of ES at different post-infection times.
| Bacterial Strain | Activity score over timeb
|
||||||
|---|---|---|---|---|---|---|---|
| 6h | 12h | 24h | 48h | 72h | 96h | 120h | |
| OmpA+ | 5 ± 0 | 4 ± 0 | .7 ± 0.33 | 1.5 ± 0.22 | |||
| OmpA− | 5 ± 0 | 5 ± 0 | 4.8 ± 0.15 | 4.5 ± 0.13 | 4.9 ± 0 | 5 ± 0 | 5 ± 0 |
| pOmpA+ | 5 ± 0 | 4 ± 0 | 3.8 ± 0.30 | 1.3 ± 0.20 | |||
Animals were turned on their back and scored as: 5 = normal, 4 = turns upright <5 s, 3 = turns upright <30 s, 2 = does not turn upright, 1 = coma/death.
Values are mean ± SE (n = 12 animals per group)
Table 2.
Occurrence of meningitis in newborn rats infected with ES strains
| Bacterial strain | n | Bacteremia* (log10 CFU/ml of blood) | Number of animals with positive CSF culture (%) |
|---|---|---|---|
| Omp A+ ES | 12 | 9.31 ± 0.41 | 12 (100%) |
| OmpA− ES | 12 | 3.25 ± 0.16 | 0** (0%) |
| pOmpA+ ES | 12 | 9.20 ± 0.38 | 12 (100%) |
Counts after 48 h post-infection
p<0.001, compared to OmpA+ or pOmpA+ by Chi-square test
Values are mean ± SE
Figure 1. Survival curves of newborn rats infected with ES strains.
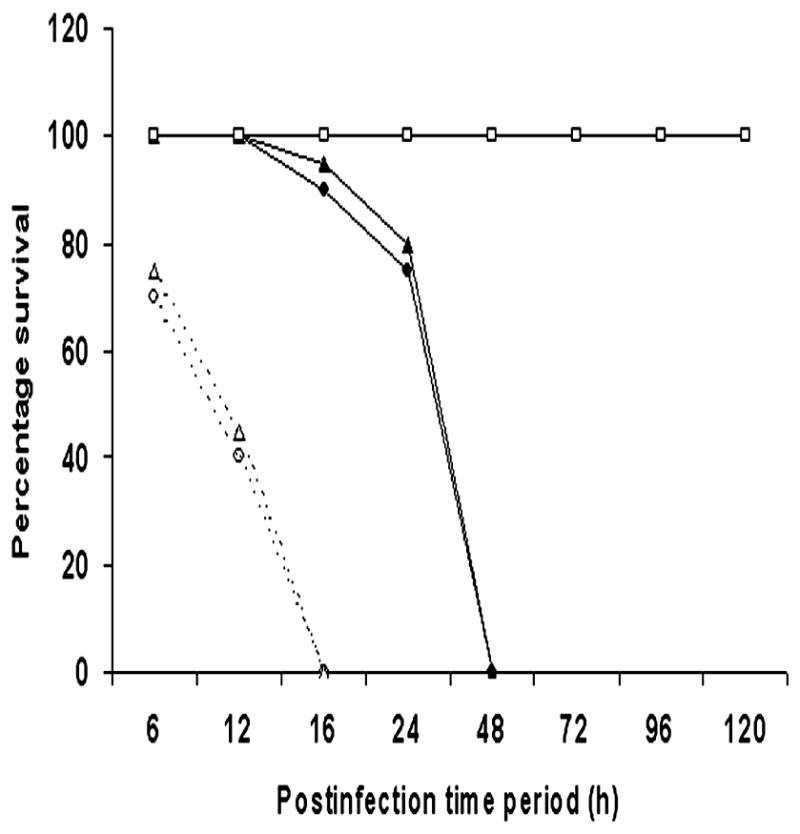
Newborn rats at day 2 were orally infected with 104 (black lines) or 105 (dotted lines) CFU of OmpA+, OmpA−, or pOmpA+ ES. Results are expressed as percentage of survival over time with death either spontaneous or by killing for ethical reasons (severe disease with activity score <2). n=12 rats/group.
OmpA+ ES penetrates through intestinal- and blood-brain barrier efficiently to induce neutrophil influx and cause tissue damage
Since OmpA+ ES and not OmpA− ES induced meningitis in newborn rats, we speculated that OmpA− ES is unable to cross either intestine or blood-brain barrier efficiently. Therefore, first we examined the bacterial load in various organs of infected newborn rats. Brain and intestine samples of rats infected with OmpA+ ES showed a log10 CFU of approximately 3.00 even at 6 h post-infection, which increased to a log CFU of 8.00 by 48 h (Fig. 2). In contrast, the bacterial counts in brains of rats infected with OmpA− ES were not detectable any time point although revealed a four-fold lower number of bacteria in the intestine when compared to OmpA+ ES. The ability of OmpA− ES to infect brain and intestine was restored following complementation with ompA gene where the bacterial titer was comparable to that of wild type ES infected rats. Studies from our lab with other meningitis causing bacteria have demonstrated that a certain level (105 to 106 CFU/ml) of bacteremia is required for the onset of meningitis in newborn rats.25 To reach the required bacteremia levels, bacteria should multiply in blood or other tissues to evade host defense mechanisms. Since the number of OmpA− ES present in the blood was significantly lower, we also examined the colonization of bacteria in liver, spleen, lungs, and kidneys following infection with ES. No detectable bacterial counts were observed in either of the organs of rat pups infected with OmpA− ES whereas OmpA+ ES were observed as early as 6 h post-infection (Fig. 2). Detectable numbers of OmpA− ES were observed after 24 h post infection and were cleared from these tissues by 120 h post-infection, suggesting that the bacterium is killed either in blood or in tissues. Correction of the OmpA mutation correlated with an increase in virulence comparable to that of the parent strain indicated by high bacterial titer in all these tissues. Further, myeloperoxidase activity (MPO) in brain and intestine samples was measured as an index of neutrophil influx. MPO is an enzyme specific for azurophil granules and represents 5% of the total cellular protein of neutrophils.26 Figure 3A shows the MPO activity in the brain and intestine of rats after infection with 104 CFU of either the OmpA+ or OmpA− ES. Higher levels of MPO activity were observed at 48 h following infection with OmpA+ ES or pOmpA+ ES, indicating neutrophil recruitment in both brain and intestine. In contrast, MPO activity was significantly lower in OmpA− ES infected rats even further reduced after 120 h post-infection.
Figure 2. Bacterial colonization in the tissues of newborn rats infected with ES.

Newborn rat pups were orally infected with various strains of ES and different tissues were harvested at indicated time intervals. Equal weights of tissues were homogenized and 20 μl of homogenates were plated on antibiotic containing agar. The number of bacteria grown overnight after incubation at 37°C were enumerated and expressed as log10 cfu/g tissue ± SD *P<0.001 by Student’s t test.
Figure 3. Neutrophil and tissue damage markers in the brains and intestines of newborn rats infected with various strains of ES.

Tissues from newborn rats infected orally with 104 CFU of ES strains were analyzed at 48 h post-infection for MPO (A), MDA (B), LDH (C), and GSH (D). The animals analyzed here were same as those used in Fig. 1. Control group animals were treated with saline only. The data represent mean ± SE from three independent measurements carried out in triplicate. The increase or decrease of the markers observed with OmpA+ or pOmpA+ ES is statistically significant compared to OmpA− ES, *P<0.001 by Student’s t test.
Malondialdehyde (MDA) is the end product of the peroxidation of cell membrane lipids that occurs during the septic cascade; lipid peroxidation (LPO) is triggered by a variety of pro-inflammatory mediators.27 In addition, peroxynitrite, a reactive nitrogen species, is a major cause of tissue damage induced during inflammation. To determine whether LPO occurred during ES infection, brain and intestine MDA levels were evaluated. Rat pups infected with OmpA+ ES showed significantly higher levels of MDA when compared to control group (P<0.001, by Student’s t test), indicating peroxidation of lipid membranes (Fig. 3B). In addition, increase in LDH levels was also observed in OmpA+ ES infected rat pups indicating tissue damage (Fig. 3C). No difference in MDA and LDH levels was observed in brain and intestine of OmpA− ES infected animals. Glutathione (GSH) is the major antioxidant produced by the cell, protecting it from free radicals, which plays a crucial role in maintaining a normal balance between oxidation and anti-oxidation. Determination of GSH levels in the brain and intestine samples revealed that GSH content was significantly depleted in OmpA+ and pOmpA+ ES infected rat pups, suggesting that infection-triggered oxidative reactions contribute to tissue damage in meningitis (Fig. 3D). However, GSH content in OmpA− ES infected rat pups was comparable to control, indicating no tissue damage.
ES infection is associated with gliosis, apoptosis, hemorrhage and neutrophil infiltration in the brain
Since biochemical markers indicated significant neutrophil infiltration and tissue damage in the brains of OmpA+ ES infected animals, we sought to characterize the pathology of the brain by histology. First, the presence of GFP-ES in the brain tissue of rat pups was confirmed by immunohistochemistry. Staining with rabbit antiserum against OmpA showed that OmpA+ ES were present in different areas of the brain including the cortical region, white matter, and meninges of infected rat pups (Fig. 4D–F). Fluorescence microscopy also revealed that OmpA+ ES were distributed in clusters in several parts of the brain, whereas no bacteria were demonstrable in brain tissue of OmpA− ES infected rat pups as observed by the presence of GFP expressing bacteria (Fig. 4G–H). Histopathological examination of the brain tissue revealed acute inflammation involving leptomeninges and spreading along penetrating vessels into the cerebral cortex and white matter compared to control brain sections (Fig. 5a–j). Pooling of neutrophils was more prominent at the base of the brain, cisternal areas, and choroidal fissures. The cerebral cortex exhibited reactive gliosis, which was more prominent in the molecular layer. Spotty neuronal necrosis and scattered apoptotic bodies were seen in the both cortex and hippocampal dentate gyrus. Areas of severe inflammation and focal hemorrhage were noted in the choroid plexus, basal thalamus, and enthorrinal cortex. OmpA− ES rats, in contrast, had normal cerebral architecture without detectable inflammation or hemorrhage by gross appearance or microscopy (P<0.001, by Wilcoxon signed rank test, n=12 animals per strain)(Fig. 5k–o). Comparative analysis of the brain using various sections showed a significant difference between rats infected with OmpA+ ES and OmpA− ES with severe pathology in the former group. Complementation of OmpA− ES with ompA gene restored its ability to cause damage in brain of infected rats (Fig. 5p–t). Thus, histopathology clearly shows an association between the occurrence of brain damage and the presence of OmpA+ ES. Also, inflammatory and hemorrhagic lesions with reactive gliosis in the brain coincided with the presence of bacteria in these lesions.
Figure 4. Newborn rat brain sections showing the presence of OmpA+ ES.

Newborn rats at day 2 were orally infected with saline (A–C) or OmpA+ ES (D–F). Rat brains were harvested after 48 h, processed as described in materials and methods section, and stained with rabbit antiserum to OmpA. Counter staining was performed with Mayer’s hematoxylin. Arrows indicate the clusters of bacteria. Cryosections of brains from different animals treated similarly were viewed under a fluorescent microscope to visualize the GFP expressing bacteria (green) and the nuclei (blue)(H–J).
Figure 5. Histopathological examination of newborn rat brains infected with ES strains.

Coronal brain sections from newborn rats infected with various strains of ES were stained with H & E. Normal morphology of various regions in the brains from control uninfected animals treated with saline was seen (Panels a–e). Severe pathology was demonstrable in the brains of newborn rats infected with OmpA+ and pOmpA+ ES (Panels f–j and p–t). The leptomeninges exhibit increased cellularity composed of polymorphonuclear cells and mononuclear cells (black arrow) whereas the blood vessels are congested with plump endothelial cells (yellow arrow; f and p). In addition, hemorrhage (black arrow) and shrunken and eosinophilic neurons (yellow arrow) were detected in cortex (Panels g and q). Apoptosis (arrow) and inflammation (arrowhead) in Ammon’s horn alveus in hippocampus and hemorrhage in choroid plexus (black arrow) were also observed (Panels h, r, i, and s). Gliosis and apoptosis in white matter showing scattered cells with shrunken hyperchromatic nucleus and perinuclear halo were significantly visible (white arrows, panels t and j). In contrast, brains of newborn rats infected with OmpA− ES showed no pathological alterations and were comparable to brains of control uninfected group (k–o).
Inefficient binding of OmpA− ES to intestine is responsible for the absence of meningitis
Based on the bacterial numbers in intestine and blood samples of rat pups infected with OmpA− ES, we theorize that OmpA− ES could not efficiently attach to enterocytes and induce morphological changes as that of wild type ES. Hematoxylin and eosin stained sections of intestine segments of newborn rats infected with OmpA+ ES revealed suppurative inflammation along with apoptosis, intraepithelial neutrophil infiltration, and loss of normal villi architecture (Fig. 6Ab–d). The lumen of the small intestine was dilated and occupied by intestinal contents, cellular debris, and focal bacterial overgrowth. Different lesions were seen involving the intestinal mucosa; in some areas there were reactive epithelial changes and an increased number of intraepithelial inflammatory cells. Other areas showed epithelial degeneration and necrosis at the top of the villi, increased intraepithelial inflammatory cells and degenerative changes at the base of the crypts (Fig. 6Ac). Focally there were areas of mucosal ulceration, extensive transmural suppurative inflammation, and reactive stromal changes. In contrast, the mucosal villi morphology and intestinal wall were intact with no inflammation in intestine of OmpA− ES infected rat pups (Fig. 6Ae and f). Of note, the cryostat sections of intestine obtained from animals infected with OmpA+ ES showed the binding of significant numbers of bacteria to intestinal villi and induced apoptosis, which is in agreement with our previous data (Fig. 6Ag). In contrast, OmpA− ES bound in very low numbers and induced very minimal apoptosis of enterocytes (Fig. 6Ah). The number of apoptotic cells is similar to that of the cells observed in control animals (data not shown). Disease severity scored by a pathologist blinded to groups indicated that both OmpA+ and pOmpA+ ES induced significant damage to intestine (P<0.001 by Chi square test) when compared to OmpA− ES or control (Fig. 6B). However, lower binding capacity of OmpA− ES could be due to its inability to survive well during the passage along gastrointestinal tract. Therefore, the binding of OmpA− ES was also examined using rat intestinal epithelial cells, IEC-6, in tissue culture model. The binding of OmpA+ ES to IEC-6 cells was ten-fold greater than the binding of OmpA− ES (P<0.001 by Student’s t test, Fig. 6C). The average number of bacteria bound per enterocyte varied from 27 to 401 in case of OmpA+ ES between 2 to 6 h post-infection whereas it varied from 2 to 26 for OmpA− ES. Complemented pOmpA+ ES restored the binding ability of ES to IEC-6 cells, indicating that OmpA expression is critical for binding to intestinal epithelial cells.
Figure 6. Pathology of intestinal segments of OmpA+ ES infected rat pups.

(A) Paraffin embedded sections of intestine segments obtained from the infected rat pups with various strains of ES were stained with H & E (a–f). Panel “a” shows intact intestinal wall with normal mucosa and uniform villi. Panel “b” shows small intestine with dilated lumen containing debris and focal bacterial overgrowth (black arrow) along with flattening of villi and an increased number of intraepithelial lymphocytes (yellow arrow) in newborn rat pup infected with OmpA+ ES. In addition, epithelial degenerative changes were observable in mucosa (yellow arrow) along with vacuolation and necrosis in villi apical portion (black arrow, panel “c”). Focal ulceration with suppurative inflammation and reactive stromal changes were also observable (panel “d”, arrow). Panels “e” and “f” show small intestine with intact muscular layers and mucosa similar to control uninfected group in OmpA− ES infected rat pups. Cryosections of the intestines from the same animals were stained with ApoTag apoptosis kit and DAPI, and viewed under fluorescence microscope for bacteria (green) and for apoptotic cells (red) (panels “g” and “h”). (B) The pathology scores by a pathologist blinded to treatments are graphed and revealed significant damage in intestinal sections obtained from rats infected with OmpA+ and pOmpA+ ES compared to control or OmpA− ES, *P<0.001 by Student’ t test. (C) Confluent monolayers of IEC-6 cells were used for ES invasion assays as described in materials and methods section. The results are expressed as average number of bacteria bound per enterocyte and the data represent mean ± SD from three independent experiments performed in triplicate. The adherence of OmpA+ and pOmpA+ ES was significantly greater when compared to OmpA− ES (*P<0.001 by Student’s t test).
Infection with ES induces pro-inflammatory response in newborn rats
To gain a better understanding of the immune responses to OmpA+ and OmpA− ES, cytokines, TNF-α, IL-1β, IL-6, IL-10 and MIP-2 were measured in blood samples taken throughout the course of infection. Results showed that the systemic cytokine and chemokine production in newborn rats after infection with OmpA+ ES or pOmpA+ ES is gradual over time. All of the cytokines except IL-10 showed highest levels of production by 48 h post-infection (Fig. 7A), which may indicate an important early participation of phagocytic cells during the onset of the inflammatory response against ES. In contrast, OmpA− ES could not induce the production of these cytokines until 48 h post-infection although the levels were 3-fold lower than the levels induced by OmpA+ ES (Fig. 7B). The amounts of cytokines dropped to undetectable levels by 120 h, which corroborate with the number of bacteria present in the blood. An exception to this was the concentration of IL-10, which continued to show high levels of expression between 96 and 120 h in the animals infected with the OmpA− ES. Following infection with OmpA+ ES, the development of meningitis was associated with increase in production of TNF-α, IL-1β and IL-6 observable at 6 h in the brain and intestine, which remain elevated till 48 h post-infection (Fig. 8). The levels of these pro-inflammatory cytokines correlated with the severity of pathological lesions in the brain and intestine. In addition, MIP-2 (a CXC chemokine in rats) levels correlated with neutrophil recruitment in the brain and intestine as assessed by MPO assay. However, the production of IL-10 in OmpA+ ES infected animals was slightly greater than the control animals. No increase in production of pro-inflammatory cytokines or MIP-2 was observable in rats infected with OmpA− ES, but increased upregulation of IL-10 was observed. Complemented pOmpA+ ES restored the ability to induce upregulation of pro-inflammatory cytokines as well as MIP-2 and downregulation of anti-inflammatory cytokine IL-10, comparable to OmpA+ ES.
Figure 7. Infection of rat pups with OmpA+ ES induces the production of cytokines and chemokines in blood.

Serum samples obtained at different time points from OmpA+ (A), or OmpA− ES (B) infected newborn rats were analyzed for the production of various cytokines and chemokines using ELISA. The data represent mean ±SE values from 12 different animals and is representative of three independent measurements carried out in triplicate.
Figure 8. Cytokine levels in brains and intestines of newborn rats infected with ES.

Brains and intestines were harvested from newborn rats infected with various ES strains after 48 h and homogenized. TNF-α, IL-1β, IL-6, MIP-2 and IL-10 levels were determined from a known amount of sample by ELISA. The data represent mean ± SD values. *P<0.001 by Student’s t test, compared with OmpA− E. coli.
OmpA expression is required for ES survival in neonatal rat serum
Although a small number of OmpA− ES crossed the intestinal barrier, subsequent multiplication in the blood was not observed, suggesting that OmpA expression may also be important for the survival in the blood. E. coli K1, another meningitis causing bacterium, survives host defenses by hiding in macrophages and avoids serum bactericidal activity, for which OmpA expression is critical.24, 28 Therefore, we also examined the survival of OmpA− ES in neonatal rat blood and serum along with OmpA+ ES. Whole blood assays revealed that 60% of OmpA+ and pOmpA+ ES survived up to 1 h (Fig. 9A). In contrast, OmpA− ES could not survive efficiently and were completely killed by 1 h post-incubation. Similarly, more than 60% of OmpA+ ES was relatively resistant to the bactericidal activity of neonatal rat serum after incubation for 1 h. Lack of expression of OmpA, however, renders the bacteria susceptible to serum killing as 100% OmpA− ES were killed within 1 h post infection (Fig. 9B). Correction of the mutation in OmpA − ES restored the serum resistance phenotype equivalent to that of the parent strain. However, OmpA− ES were not killed at any time point when incubated in heat-inactivated serum, indicating that intact complement is pivotal for serum bactericidal activity. Taken together, these data suggest that serum bactericidal activity is mainly responsible for killing of OmpA− ES in blood although the phagocytic cells play some role. In addition, these results suggest that despite the presence of small number of OmpA− ES in blood after 48 h post-infection, the bacteria could not reach threshold levels of bacteremia due to their inefficiency to resist serum bactericidal activity.
Figure 9. OmpA expression is critical for ES survival in blood and serum.

Bactericidal assays using neonatal rat blood (A) and serum (B) were performed with 106 CFU of ES as described in materials and methods section. The experiments were carried out using five different blood and serum samples and expressed as percent survival considering the bacterial numbers immediately after adding to the samples as 100%. Dotted line indicates survival of ES in heat-inactivated serum. The data represent mean ± SD and is representative of five independent experiments carried out in triplicate.
Discussion
ES is an opportunistic pathogen causing invasive infections (meningitis, sepsis, necrotizing enterocolitis) with high death rates (40–80%), primarily in newborns. 29–31 In contrast to the high number of cases in newborns, infants and children, there are only a few reported cases of ES infections in adults, generally in subjects with pre-existing conditions such as neoplasms, and just one osteomyelitis of the foot.4 Existing information regarding the virulence potential of ES is mostly based on epidemiological studies. This approach, however, can only identify association between certain serogroups, bacterial virulence traits, and their role in disease process. However, no information is available regarding the pathogenesis of meningitis due to ES. We have been utilizing a newborn rat model of hematogenous meningitis for the last one and half a decade by intra-cardiac injection of the bacteria, which is similar to human disease, to understand the pathogenic mechanisms of E. coli K1 meningitis.32–34 However, ES infections usually occur due to the feeding of contaminated formula to especially pre-mature babies. Therefore, in the present study, an attempt was made to establish meningitis model in newborn rat pups by employing oral inoculation with ES, and subsequently giving them normal infant formula and water while keeping away from the mother. In this model, our studies demonstrated that feeding with 104 CFU of ES once at day 2 was sufficient to cause meningitis within 24 h of post-infection.
The unusual conservation of OmpA-like proteins across gram-negative species suggests that they serve an important cellular function.35, 36 Despite its sequence similarity with other gram-negative bacteria, the function of OmpA in vivo as a potential virulence determinant of ES has not been investigated. Our recent studies revealed that OmpA expression in ES is critical for the invasion of HBMEC, an in vitro model of the blood-brain barrier.37 In the present investigation, we demonstrated for the first time, the contribution of OmpA expression in ES induced meningitis in the newborn rat model. Deletion of OmpA expression correlated with attenuated virulence of ES as indicated by reduced bacterial load in blood as well as in the brain, intestine, spleen, liver, kidneys and lungs. Mortality rate was high in rat pups infected with OmpA+ ES compared to OmpA− ES, indicating that OmpA+ ES had a selective advantage in vivo over the OmpA− ES. Correction of the mutation in OmpA− ES restored the virulence equivalent to that of OmpA+ ES, confirming that the observed effects were due to loss of OmpA expression. Pathological examination of brain and intestine samples of infected newborn rat pups provided additional evidence in support of role for OmpA in vivo. OmpA+ ES caused severe pathological manifestations in the brains of newborn rats including hemorrhage, cortical damage, gliosis, and inflammation whereas OmpA− ES failed to cause such changes. Severe inflammation can lead to pleocytosis of the CSF, which is considered as an important hallmark of meningitis. CXC chemokines like MIP-2 in rats have been implicated in the development of neutrophilic pleocytosis in CSF during bacterial meningitis38. MPO assay revealed neutrophil recruitment in the brain and intestine of rat pups infected with OmpA+ ES that correlated with increase in MIP-2 levels, which is one of the human IL-8 homologues in the rat and has also been implied in transepithelial neutrophil migration.39
During infection, in response to bacterial colonization in tissues, there is oxidative stress leading to lipid peroxidation and tissue damage. In corroboration with pathological observations, MDA levels were significantly elevated in the brain and intestine following infection with OmpA+ ES as compared to control group while GSH content was depleted. Caksen et al also demonstrated an increase in MDA levels and decrease in GSH content in serum of children having meningitis.40 Similarly, high levels of LDH in CSF have been associated with bacterial meningitis.41, 42 In agreement with these findings, elevated levels of LDH were detectable in serum, brain, and intestine of rat pups infected with OmpA+ ES compared to the control group and OmpA− ES infected newborn rats. Multiplication of bacteria within the central nervous system compartment triggers a host response with an overshooting inflammatory reaction, which leads to brain parenchyma damage. The inflammatory response is due in large part to the activity of cytokines, which play an important role in deciding the ultimate outcome of an infection during meningitis. TNF-α, IL-1β, and IL-6 are major early response cytokines that trigger, often in synergy, a cascade of inflammatory mediators including other cytokines, arachidonic acid metabolites, chemokines, and reactive nitrogen and oxygen intermediates.43 High levels of IL1-β, TNF-α and IL-6 have been reported in serum, CSF, and brain of individuals suffering from meningitis. Consistent with the information from humans, OmpA+ ES infection was able to induce high levels of proinflammatory cytokines and very low levels of anti-inflammatory cytokines in the blood and brain as well as in the intestine of newborn rats. These findings clearly demonstrate that OmpA+ ES could cause meningitis in newborns, which may be very similar to human disease.
The establishment of disease depends upon microbe’s ability to adhere to host surfaces, such as the intestinal epithelial layer and to grow in the intestinal wall to subsequently enter the blood stream. Our studies revealed that the binding of OmpA+ ES and complemented pOmpA+ ES was significantly higher to IEC-6 cells than the binding of OmpA− ES. Of note, no invasion of OmpA+ ES was observed in IEC-6 cells. The findings of Nair and Venkitanarayan who observed that OmpA− ES was significantly attenuated in its ability to invade INT-407 cells when compared to OmpA+ ES were in contrast to ours and could be due to the differences in the methods utilized for invasion assays or different cell types used. Previous studies demonstrated that ES induces apoptosis in intestinal epithelial cells; this effect however was not observed following infection with OmpA− ES (unpublished results). This could be due to inefficient binding of OmpA− ES to intestinal enterocytes, suggesting that OmpA also contributes to the colonization of ES in the gastrointestinal tract. Alternatively, OmpA− ES may not survive in gastrointestinal tract or in the intestinal wall efficiently, thus only a small number of bacteria were attached to the enterocytes. In agreement with in vitro data, the binding of OmpA− ES to the intestine of infected newborn rats was also significantly reduced, indicating that OmpA may be playing a role in the adherence to intestinal epithelial cells or for the survival in the intestinal tissue. Furthermore, OmpA− ES could not induce morphological changes to the intestine and apoptosis of enterocytes as OmpA+ ES did. Therefore, OmpA mediated binding of ES to respective cognate receptor on enterocytes would induce apoptotic signals in the cells. Instead, OmpA interaction with receptor causes the bacteria to secrete virulence factors that would induce apoptosis. Several pathogenic bacteria possess such secretion systems to alter the host cell function45, 46 and it remains to be determined whether ES utilizes similar mechanisms in its pathogenesis. Despite the inability of ES to invade IEC-6 cells in vitro, the bacterium may gain access into intestinal wall through the compromised gut barrier due to the apoptosis of enterocytes in animals.
Many pathogenic microorganisms avoid host defense mechanisms and successfully survive in the host. Serum resistance is a crucial factor for the development of meningitis and disseminated infection by Neisseria meningitides.47 Furthermore, local inflammation at the site of infection exposes the bacteria to moderate concentrations of serum components. In earlier studies from our laboratory we observed that Escherichia coli K1 binds C4 binding protein (C4bp) and evades serum killing.48, 28 Therefore, we speculated that the observed differences in virulence of OmpA+ and OmpA− ES might be due to differential killing of the bacteria by blood or serum components. To test this hypothesis, blood and serum killing of OmpA+ and OmpA− ES was examined at different time intervals. Similar to that of E. coli K1, OmpA expression is important for ES resistance to blood and serum killing. The increased resistance of OmpA+ ES to normal rat blood and serum may account for its greater virulence in vivo. In conclusion, our findings demonstrated an important role for OmpA in the ability of ES to cause meningitis. The neonatal rat model holds promise for understanding the biology and pathogenesis of ES induced meningitis. The results of the present study may help in developing effective preventive approach against meningitis caused by ES based on mapping OmpA epitopes that interact with host tissues.
Acknowledgments
Supported by NIH grant AI40567
We thank Barbara Driscoll for critical reading of the manuscript. We also thank Fred Dorey for helping in statistical analysis.
Abbreviations
- ES
Enterobacter sakazakii
- HBMEC
human brain microvascular endothelial cells
- OmpA
Outer membrane protein A
References
- 1.Giovannini M, Verduci E, Ghisleni D, et al. An Emerging Problem in Paediatric Nutrition Enterobacter sakazakii. J Int Med Res. 2008;36:394–399. doi: 10.1177/147323000803600303. [DOI] [PubMed] [Google Scholar]
- 2.Drudy D, Mullane NR, Yuinn T, et al. Enterobacter sakazakii: An emerging pathogen in powdered infant formula. Clin Infect Dis. 2006;42:996–1002. doi: 10.1086/501019. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Enterobacter sakazakii infections associated with the use of powdered infant formula—Tennessee, 2001. MMWR Morb Mortal Wkly Rep. 2002;51:297–300. [PubMed] [Google Scholar]
- 4.Lai KK. Enterobacter sakazakii infections among neonates, infants, children, and adults: case reports and a review of the literature. Medicine (Baltimore) 2001;80:113–122. doi: 10.1097/00005792-200103000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Adamson DH, Rodgers JR. Enterobacter sakazakii meningitis and sepsis. Clin Microbiol Newslett. 1981;3:19–20. [Google Scholar]
- 6.Kleiman MB, Allen SD, Neal P, et al. Meningoencephalitis and compartmentalization of the cerebral ventricles caused by Enterobacter sakazakii. J Clin Microbiol. 1981;14:352–354. doi: 10.1128/jcm.14.3.352-354.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muytjens HL, Zanen HC, Sonderkamp HJ, et al. Analysis of eight cases of neonatal meningitis and sepsis due to Enterobacter sakazakii. J Clin Microbiol. 1983;18:115–120. doi: 10.1128/jcm.18.1.115-120.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nazarowec-White M, Farber JM. Enterobacter sakazakii: a review. Int J Food Microbiol. 1997;34:103–113. doi: 10.1016/s0168-1605(96)01172-5. [DOI] [PubMed] [Google Scholar]
- 9.van Acker J, De Smet F, Muyldermans G, et al. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in 14 powdered milk formula. J Clin Microbiol. 2001;39:293–297. doi: 10.1128/JCM.39.1.293-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowen AB, Braden CR. Invasive Enterobacter sakazakii disease in infants. Emerg Infect Dis. 2006;12:1185–1186. doi: 10.3201/eid1208.051509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anonymous. Microorganisms in food 7. Microbiological testing in food safety management. Kluwer Academic/Plenum Publishers; 2002. International Commission on Microbiological Specifications for Foods (ICMSF) [Google Scholar]
- 12.Townsend S, Hurrell E, Forsythe S. Virulence studies of Enterobacter sakazakii isolates associated with a neonatal intensive care unit outbreak. BMC Microbiol. 2008;8:64–73. doi: 10.1186/1471-2180-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Townsend S, Caubilla Barron J, Loc-Carrillo C, et al. The presence of endotoxin in powdered infant formula milk and the influence of endotoxin and Enterobacter sakazakii on bacterial translocation in the infant rat. Food Microbiol. 2007;24:67–74. doi: 10.1016/j.fm.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Pagotto FJ, Nazarowec-White M, Bidawid S, et al. Enterobacter sakazakii: infectivity and enterotoxin production in vitro and in vivo. J Food Prot. 2003;66:370–375. doi: 10.4315/0362-028x-66.3.370. [DOI] [PubMed] [Google Scholar]
- 15.Kim KP, Loessner MJ. Enterobacter sakazakii invasion in human intestinal Caco-2 cells requires the host cell cytoskeleton and is enhanced by disruption of tight junction. Infect Immun. 2008;76:562–570. doi: 10.1128/IAI.00937-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter CA, Singamsetty VK, Chokshi NK, et al. Enterobacter sakazakii enhances epithelial cell injury and apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis. 2008;198:586–593. doi: 10.1086/590186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singamsetty VK, Wang Y, Shimada H, et al. Outer membrane protein A expression in Enterobacter sakazakii is required to induce microtubule condensation in human brain microvascular endothelial cells for invasion. Microb Pathog. 2008;45:181–191. doi: 10.1016/j.micpath.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Townsend SM, Hurrell E, Gonzlaez-Gomez I, et al. Enterobacter sakazakii invades brain capillary endothelial cells, persists in human macrophages influencing cytokine secretion and induces severe brain pathology in the neonatal rat. Microbiology. 2007;153:3538–3547. doi: 10.1099/mic.0.2007/009316-0. [DOI] [PubMed] [Google Scholar]
- 19.Gehre F, Leib SL, Grandgirard D, et al. Essential role of choline for Pneumococcal virulence in an experimental model of meningitis. J Intern Med. 2008;264:143–154. doi: 10.1111/j.1365-2796.2008.01930.x. [DOI] [PubMed] [Google Scholar]
- 20.Doran KS, Engelson EJ, Khosravi A, et al. Blood-brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J Clin Invest. 2005;115:2499–507. doi: 10.1172/JCI23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mittal R, Chhibber S, Sharma S, et al. Macrophage inflammatory protein-2, neutrophil recruitment and bacterial persistence in an experimental mouse model of urinary tract infection. Microbes Infect. 2004;6:1326–1332. doi: 10.1016/j.micinf.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Wills ED. Mechanism of lipid peroxide formation in animal tissues. Biochem J. 1965;99:667–676. doi: 10.1042/bj0990667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuğtepe H, Sener G, Cetinel S, et al. Oxidative renal damage in pyelonephritic rats is ameliorated by montelukast, a selective leukotriene CysLT1 receptor antagonist. Eur J Pharmacol. 2007;557:69–75. doi: 10.1016/j.ejphar.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Wooster DG, Maruvada R, Blom AM, et al. Logarithmic phase Escherichia coli K1 efficiently avoids serum killing by promoting C4bp-mediated C3b and C4b degradation. Immunology. 2006;117:482–493. doi: 10.1111/j.1365-2567.2006.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prasadarao NV, Wass CA, Kim KS. Endothelial cell GlcNAc beta 1–4GlcNAc epitopes for outer membrane protein A enhance traversal of Escherichia coli across the blood-brain barrier. Infect Immun. 1996;64:154–160. doi: 10.1128/iai.64.1.154-160.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cherubini A, Ruggiero C, Polidori MC, et al. Potential markers of oxidative stress in stroke. Free Radic Biol Med. 2005;39:841–852. doi: 10.1016/j.freeradbiomed.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 28.Maruvada R, Blom AM, Prasadarao NV. Effects of complement regulators bound to Escherichia coli K1 and Group B Streptococcus on the interaction with host cells. Immunology. 2008;124:265–276. doi: 10.1111/j.1365-2567.2007.02764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bociaga-Jasik M, Garlicki A, Kalinowska-Nowak A, et al. The role of cytokines in bacterial meningitis. Przegl Lek. 2001;58:1055–1058. [PubMed] [Google Scholar]
- 30.Bar-Oz B, Preminger A, Peleg O, et al. Enterobacter sakazakii infection in the newborn. Acta Paediatr. 2001;90:356–358. [PubMed] [Google Scholar]
- 31.Iversen C, Forsythe SJ. Risk profile of Enterobacter sakazakii, an emergent pathogen associated with infant milk formula. Trends Food Science Technol. 2003;14:443–454. [Google Scholar]
- 32.Huang SH, Wass C, Fu Q, et al. Escherichia coli invasion of brain microvascular endothelial cells in vitro and in vivo: molecular cloning and characterization of invasion gene ibe10. Infect Immun. 1995;63:4470–4475. doi: 10.1128/iai.63.11.4470-4475.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Wen ZG, Kim KS. Role of S fimbriae in Escherichia coli K1 binding to brain microvascular endothelial cells in vitro and penetration into the central nervous system in vivo. Microb Pathog. 2004;7:287–293. doi: 10.1016/j.micpath.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Sukumaran SK, Shimada H, Prasadarao NV. Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun. 2003;71:5951–5961. doi: 10.1128/IAI.71.10.5951-5961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munson RS, Jr, Brooks GF. Outer-membrane antigens of gram-negative pathogens: summary of session. Rev Infect Dis. 1988;10:S317–318. doi: 10.1093/cid/10.supplement_2.s317. [DOI] [PubMed] [Google Scholar]
- 36.Smith SG, Mahon V, Lambert MA, et al. A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol Lett. 2007;273:1–11. doi: 10.1111/j.1574-6968.2007.00778.x. [DOI] [PubMed] [Google Scholar]
- 37.Prasadarao NV, Wass CA, Weiser JN, et al. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun. 1996;64:146–153. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diab A, Abdalla H, Li HL, et al. Neutralization of macrophage inflammatory protein 2 (MIP-2) and MIP-1alpha attenuates neutrophil recruitment in the central nervous system during experimental bacterial meningitis. Infect Immun. 1999;67:2590–2601. doi: 10.1128/iai.67.5.2590-2601.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zwijnenburg PJ, Polfliet MM, Florquin S, et al. CXC-chemokines KC and macrophage inflammatory protein-2 (MIP-2) synergistically induce leukocyte recruitment to the central nervous system in rats. Immunol Lett. 2003;85:1–4. doi: 10.1016/s0165-2478(02)00200-6. [DOI] [PubMed] [Google Scholar]
- 40.Caksen H, Cemek M, Dede S, et al. Brief clinical study: Lipid peroxidation and antioxidant status in children with acute purulent meningitis and encephalitis. Int J Neurosci. 2004;114:105–111. doi: 10.1080/00207450490249383. [DOI] [PubMed] [Google Scholar]
- 41.Lutsar I, Haldre S, Topman M, et al. Enzymatic changes in the cerebrospinal fluid in patients with infections of the central nervous system. Acta Paediatr. 1994;83:1146–1150. doi: 10.1111/j.1651-2227.1994.tb18268.x. [DOI] [PubMed] [Google Scholar]
- 42.Kepa L, Oczko-Grzesik B, Błedowski D. Evaluation of cerebrospinal fluid and plasma lactate dehydrogenase activity in patients with purulent, bacterial meningoencephalitis. Przegl Epidemiol. 2006;60:291–298. [PubMed] [Google Scholar]
- 43.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 44.Mohan Nair MK, Venkitanarayan K. Role of bacterial OmpA and host cytoskeleton in the invasion of human intestinal epithelial cells by Enterobacter sakazakii. Pediatr Res. 2007;62:664–669. doi: 10.1203/PDR.0b013e3181587864. [DOI] [PubMed] [Google Scholar]
- 45.Stavrinides J, McCann HC, Guttman DS. Host-pathogen interplay and the evolution of bacterial effectors. Cell Microbiol. 2008;10:285–292. doi: 10.1111/j.1462-5822.2007.01078.x. [DOI] [PubMed] [Google Scholar]
- 46.Coburn B, Sekirov I, Finlay BB. Type III secretion systems and disease. Clin Microbiol Rev. 2007;20:535–549. doi: 10.1128/CMR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vogel U, Frosch M. Mechanisms of Neisserial serum resistance. Mol Microbiol. 1999;32:1133–1139. doi: 10.1046/j.1365-2958.1999.01469.x. [DOI] [PubMed] [Google Scholar]
- 48.Prasadarao NV, Blom AM, Villoutreix BO, et al. A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J Immunol. 2002;169:6352–6360. doi: 10.4049/jimmunol.169.11.6352. [DOI] [PubMed] [Google Scholar]