

Abstract
Please cite this paper as: Ambrose et al. (2011) An integrated, multistudy analysis of the safety of Ann Arbor strain live attenuated influenza vaccine in children aged 2–17 years. Influenza and Other Respiratory Viruses 5(6), 389–397.
Background Trivalent, Ann Arbor strain, live attenuated influenza vaccine (LAIV) is approved in several countries for use in eligible children aged ≥2 years.
Objective To describe the safety of Ann Arbor strain LAIV in children aged 2–17 years.
Methods An integrated analysis of randomized, controlled trials of LAIV.
Results A total of 4245 and 10 693 children received ≥1 dose of LAIV in year 1 of 6 trivalent inactivated influenza vaccine (TIV)‐controlled and 14 placebo‐controlled studies, respectively; 3212 children were revaccinated in year 2 of 4 placebo‐controlled studies. Compared with placebo for days 0–10 post‐vaccination, LAIV recipients exhibited increased runny/stuffy nose (+7%), headache (+7%), and tiredness/decreased activity (+2%) after dose 1; and a higher rate of decreased appetite (+4%) after year 2 revaccination. Compared with TIV, only runny/stuffy nose was increased (dose 1, +12%; dose 2, +4%). Compared with initial vaccination, LAIV reactogenicity was lower after dose 2 in year 1 and revaccination in year 2. Unsolicited adverse events (AEs) increased with LAIV in some comparisons were headache, nasal congestion/rhinorrhea, rhinitis, and pyrexia; ear pain and lower respiratory illness were decreased. There was no evidence of an increase in any potential vaccine‐related serious AE in LAIV recipients. Among children aged 2–17 years and specifically aged 24–35 months, there was no evidence that lower respiratory illness or wheezing illness occurred at a higher rate in LAIV recipients.
Conclusion This analysis supports the safety of Ann Arbor strain LAIV in children aged 2–17 years and provides a consensus assessment of events expected after vaccination.
Keywords: Adverse events, Ann Arbor strain live attenuated influenza vaccine, reactogenicity events, trivalent inactivated influenza vaccine
Introduction
A trivalent, intranasal, live attenuated influenza vaccine (LAIV) manufactured by MedImmune (Gaithersburg, MD, USA) is currently approved for use in individuals aged 2–49 years in the United States, South Korea, Israel, Hong Kong, and Macau; in Canada in individuals 2–59 years of age; and in the European Union in children 2–17 years of age. The vaccine was originally derived at the University of Michigan by cold adaptation of an influenza type A strain (A/Ann Arbor/6/60 H2N2) and a type B strain (B/Ann Arbor/1/66) through serial passage at sequentially lower temperatures. 1 During this process, the Ann Arbor strains acquired multiple mutations in genes encoding internal non‐glycosylated proteins, resulting in master donor viruses with a cold‐adapted, temperature‐sensitive, and attenuated phenotype. These vaccine strains are updated annually to include A/H3N2, A/H1N1, and Type B influenza strains with hemagglutinin (HA) and neuraminidase (NA) proteins that match those of the strains selected for the specific annual formulation. A frozen formulation of trivalent Ann Arbor strain LAIV was first licensed in 2003 in the United States. A refrigerated formulation was licensed in 2007, based on the demonstration of comparable immunogenicity and safety. 2 Both formulations contain 106·5–107·5 fluorescent focus units of each of the three virus strains per dose, with no preservatives or adjuvants.
Ann Arbor strain LAIV has been used extensively in the United States, with the majority of use occurring in children, adult healthcare workers, and US military personnel. More than 39 million doses have been distributed for use between licensure in 2003 and November 2010. In three randomized studies comparing Ann Arbor strain LAIV and trivalent inactivated influenza vaccine (TIV) in children aged 6 months to 17 years, LAIV recipients had 35–53% fewer cases of culture‐confirmed influenza. 3 , 4 , 5 Additionally, LAIV has demonstrated effectiveness in children and adults against influenza strains that were antigenically mismatched to those contained in the vaccine. 6 , 7 Nasal administration has facilitated use of the vaccine in mass vaccination and alternative‐site clinics such as school‐based influenza vaccination programs. 8 , 9 , 10 , 11 , 12 , 13
The safety of Ann Arbor strain LAIV has been studied in more than 140 000 subjects in 73 completed or ongoing studies in multiple regions of the world. The safety of LAIV has been generally comparable with placebo and TIV in these studies; however, one study demonstrated that, compared with TIV, LAIV was associated with an increased rate of all‐cause hospitalization among children aged 6–11 months and an increased rate of medically attended wheezing in children aged 6–23 months. There was no increase in hospitalizations in children aged ≥12 months and no increase in medically attended wheezing in children aged ≥24 months. 3
Although the safety of Ann Arbor strain LAIV was analyzed for individual studies, an integrated analysis of safety across these studies has not been conducted. To enhance the understanding of the safety of LAIV in children for whom it is approved, an integrated safety analysis was conducted for subjects 2–17 years of age. The goals of this analysis were to describe solicited reactogenicity events, unsolicited adverse events (AEs), and serious AEs (SAEs) associated with LAIV administration.
Methods
Study design
All clinical studies conducted with the Ann Arbor strain LAIV were reviewed for inclusion, with a data cutoff date of April 2008. Studies were included if any subjects were 2–17 years of age and if a randomized control group of placebo or TIV recipients was included. Ongoing studies and studies without individual subject data were excluded. Overall, 20 studies contributed to the integrated analysis. Subject demographics summarized included age, gender, and region but not race or ethnicity, because these data were not collected by common terms across the studies.
Reactogenicity events, AEs, and SAEs
For reactogenicity events and AEs, only data from subjects who received the refrigerated formulation were analyzed to optimize relevance. However, data from studies using frozen and refrigerated LAIV were combined for SAE analyses to maximize detection of a rare SAE. Classification of AEs and SAEs was consistent with the International Conference on Harmonisation Guidelines for Good Clinical Practice. Reactogenicity events were pre‐defined AEs that were actively solicited after study product administration and included runny/stuffy nose, sore throat, cough, vomiting, headache, muscle ache, chills, decreased activity, irritability, abdominal pain, decreased appetite, and fever. Reactogenicity events may also have been collected as AEs (e.g. a reactogenicity event of runny/stuffy nose may have been reported as a rhinitis/rhinorrhea AE) in accordance with study design and investigator judgment.
Reactogenicity events and AEs were summarized through 11 days (days 0–10 post‐vaccination). Serious adverse events were summarized from day of vaccination through 42 days after the last dose, because this was the data collection period in common across most studies, and from day of vaccination to day 180 after the last dose for those studies that collected safety information for this period, which represents the longest duration of follow‐up. Adverse events and SAEs were summarized by system organ class and preferred term using MedDRA version 8.0 (MedDRA MSSO, Chantilly, VA, USA) and by investigator‐reported severity. To calculate incidence, each subject contributed only once to a category. Adverse events and SAEs because of lower respiratory illness and wheezing were analyzed as events of special interest in the entire population and specifically in children 24–35 months of age. All reported AEs considered to be related to wheezing, asthma, bronchial obstruction, or bronchospasm (including bronchiolitis) were grouped into wheezing events of special interest for analysis. A similar lower respiratory illness category included, in addition to all wheezing events of special interest, any term that referred to disease of the lower respiratory system or to respiratory difficulty.
Statistical analysis
Rate differences were calculated as the LAIV rate minus the comparator (TIV or placebo) rate. Although all analyses are descriptive in nature, Fisher’s exact P‐values were calculated for rate differences between LAIV and comparator groups for reactogenicity events, AEs, and SAEs for the purpose of screening for differences of potential significance. No adjustment was made for multiplicity, and P‐values presented should be interpreted in this context.
Results
Subject disposition and demographics
Data were available for 4245 and 10 693 subjects aged 2–17 years who received at least one dose of LAIV in the first year of six TIV‐controlled and 14 placebo‐controlled studies, respectively (1, 2). Data were available for 3212 subjects aged 2–7 years who were revaccinated with LAIV in the second year of four placebo‐controlled studies (Table 2); some subjects did not provide data for the year 1 analysis because they were aged <2 years in year 1. For the analysis of a second dose in the first year of dosing, subjects aged <9 years who received the same study vaccine at dose 1 and dose 2 were included. Because some studies randomized children to a single dose in year 1, population sizes for first and second dose analyses differed; similarly, populations for year 1 vaccination and year 2 revaccination differed because there were a limited number of 2‐year studies. In placebo‐controlled studies, more subjects received LAIV than placebo as a result of randomization ratios. In year 1, study follow‐up was completed by 95·9% of subjects, 2·0% were lost to follow‐up, and 1·6% withdrew consent; the remainder did not complete because of protocol violations, investigator decisions, or other reasons. Only five subjects (LAIV, n = 2; placebo, n = 2; TIV, n = 1) withdrew because of an AE. Study completion and discontinuation rates were similar for individuals who received LAIV in year 2 and across treatment groups in both years. Demographic data are shown in Table 2; results were similar for LAIV, TIV, and placebo recipients for each corresponding comparison group.
Table 1.
Number of subjects in safety populations from TIV‐ and placebo‐controlled clinical studies
| AE and reactogenicity event populations (refrigerated LAIV) | Year 1 | Year 2 | ||
|---|---|---|---|---|
| Dose 1 | Dose 2 | |||
| TIV‐controlled studies | ||||
| Reactogenicity event population: LAIV | 4108 | 2187 | NA | |
| Reactogenicity event population: TIV | 4118 | 2223 | NA | |
| AE population: LAIV | 4147 | 2230 | NA | |
| AE population: TIV | 4182 | 2270 | NA | |
| Placebo‐controlled studies | ||||
| Reactogenicity event population: LAIV | 3245 | 2503 | 2287 | |
| Reactogenicity event population: Placebo | 1994 | 1702 | 1248 | |
| AE population: LAIV | 3278 | 2533 | 2295 | |
| AE population: Placebo | 2026 | 1734 | 1256 | |
| SAE populations (Frozen & Refrigerated LAIV) | Year 1 | Year 2 | ||
|---|---|---|---|---|
| Days 0–42 PLD | Days 0–180 PLD | Days 0–42 PLD | Days 0–180 PLD | |
| TIV‐controlled studies | ||||
| LAIV | 4245 | 4130 | NA | NA |
| TIV | 4278 | 4163 | NA | NA |
| Placebo‐controlled studies | ||||
| LAIV | 10 693 | 2408 | 3212 | 2295 |
| Placebo | 5667 | 1546 | 1697 | 1256 |
AE, adverse event; LAIV, Ann Arbor live attenuated influenza vaccine; NA, data not available; PLD, post‐last dose (dose 1 or, if administered, dose 2); SAE, serious AE; TIV, trivalent inactivated influenza vaccine.
Table 2.
Demographics of subjects by study type and vaccine received
| SAE population | Reactogenicity event/AE population | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TIV‐controlled | Placebo‐controlled | TIV‐controlled | Placebo‐controlled | |||||||||
| Year 1 | Year 1 | Year 2 | Year 1 | Year 1 | Year 2 | |||||||
| LAIV | TIV | LAIV | Placebo | LAIV | Placebo | LAIV | TIV | LAIV | Placebo | LAIV | Placebo | |
| Number of subjects | 4245 | 4278 | 10 693 | 5677 | 3212 | 1697 | 4147 | 4182 | 3278 | 2026 | 2295 | 1256 |
| Age, year Mean (SD) | 4·9 (3·9) | 4·8 (3·8) | 5·8 (4·3) | 5·7 (4·4) | 3·0 (1·1) | 2·9 (1·0) | 4·9 (3·9) | 4·8 (3·8) | 2·4 (2·0) | 2·7 (2·7) | 2·6 (0·6) | 2·6 (0·6) |
| Range | 2–17 | 2–17 | 2–17 | 2–17 | 2–7 | 2–7 | 2–17 | 2–17 | 2–17 | 2–17 | 2–4 | 2–4 |
| Age, 24–35 month, n | 1650 | 1647 | 4117 | 2383 | 1223 | 660 | 1650 | 1647 | 3149 | 1900 | 1063 | 574 |
| Age, 36–59 month, n | 1219 | 1247 | 1636 | 790 | 1615 | 866 | 1219 | 1247 | 11 | 4 | 1232 | 682 |
| Age, 5–17 year, n | 1376 | 1384 | 4940 | 2504 | 374* | 171* | 1278 | 1288 | 118 | 122 | 0 | 0 |
| Gender, % Male | 53·8 | 55·5 | 50·0 | 50·0 | 51·1 | 49·4 | 53·9 | 55·7 | 52·3 | 51·2 | 53·0 | 49·5 |
| Region, % | ||||||||||||
| USA | 22·1 | 22·4 | 68·7 | 63·4 | 28·5 | 26·0 | 20·3 | 20·6 | 0·5 | 0·8 | 0·0 | 0·0 |
| Asia/Oceania† | 3·1 | 3·1 | 14 | 17·5 | 25·4 | 31·2 | 3·2 | 3·2 | 45·7 | 48·9 | 35·5 | 42·1 |
| Latin America | 0·0 | 0·0 | 7·8 | 6·5 | 15·7 | 9·8 | 0·0 | 0·0 | 25·0 | 17·8 | 22·0 | 13·2 |
| Africa‡ | 0·0 | 0·0 | 2·7 | 2·7 | 11·6 | 8·0 | 0·0 | 0·0 | 8·8 | 7·7 | 16·2 | 10·8 |
| Europe§ | 74·8 | 74·5 | 6·7 | 9·9 | 18·8 | 25·0 | 76·5 | 76·2 | 19·9 | 24·8 | 26·4 | 33·8 |
AE, adverse event; LAIV, live attenuated influenza vaccine; SAE, serious AE; TIV, trivalent inactivated influenza vaccine; USA, United States of America.
*Age 5–7 year.
†East Asia, Southeast Asia, South Asia, and Australia.
‡South Africa only.
§Western Europe, Eastern Europe including Scandinavia, Lebanon, and Israel.
Solicited reactogenicity events days 0–10 post‐vaccination
In all studies, reactogenicity events were common in all study groups, including placebo, after the first dose of vaccine. In TIV‐controlled studies, the only reactogenicity event that was statistically increased among LAIV recipients was runny/stuffy nose (rate differences, 11·8% after dose 1 and 4·1% after dose 2; P < 0·01 for both); runny/stuffy nose was also the most commonly reported reactogenicity event (Figure 1). For all other reactogenicity events, the rate difference was ≤1·5% points. After dose 1, the incidence of muscle aches was lower in LAIV than in TIV recipients (P = 0·04). Overall, the incidence of reactogenicity events was lower among LAIV and TIV recipients after the second dose of vaccine.
Figure 1.
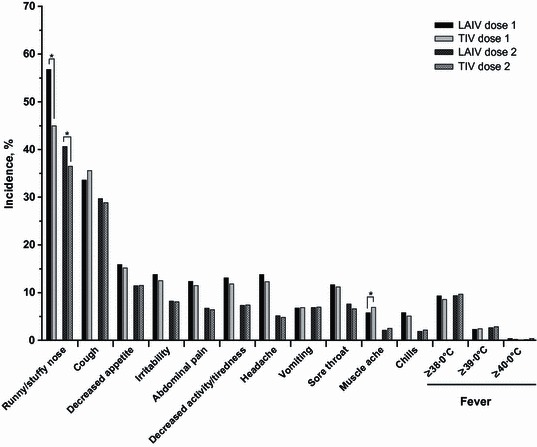
Solicited reactogenicity events days 0–10 post‐vaccination in TIV‐controlled studies. *P < 0·05, unadjusted for multiplicity. LAIV, live attenuated influenza vaccine; TIV, trivalent inactivated influenza vaccine.
In placebo‐controlled studies, the most common reactogenicity event was runny/stuffy nose, which was statistically increased in LAIV recipients only after dose 1 (rate difference, 6·8%; P < 0·01) (Figure 2). Headache (rate difference, 6·9%; P = 0·02) and tiredness/decreased activity (rate difference, 2·1%; P = 0·03) were also significantly increased among LAIV recipients after dose 1. No other reactogenicity events were significantly increased, and all had rate differences <3·0% points. After dose 2, the incidence of reactogenicity events was lower for both treatment groups compared with dose 1. The only statistically significant difference between LAIV and placebo recipients after dose 2 was a lower rate of decreased appetite among LAIV recipients (rate difference, −2·9%; P = 0·04). Upon revaccination in year 2 (Figure 3), the only statistically significant difference was a higher rate of decreased appetite among LAIV recipients (rate difference, 3·9%; P = 0·03). Data for headache, muscle ache, and chills were not collected in year 2 of any studies.
Figure 2.
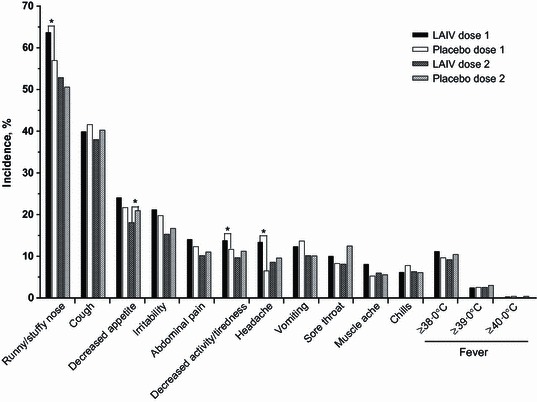
Solicited reactogenicity events days 0–10 post‐vaccination in year 1 of placebo‐controlled studies. *P < 0·05, unadjusted for multiplicity. LAIV, live attenuated influenza vaccine.
Figure 3.
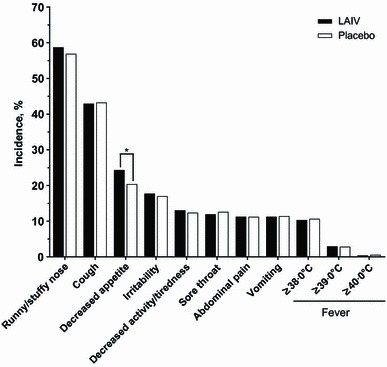
Solicited reactogenicity events days 0–10 post‐vaccination in year 2 of placebo‐controlled studies. Data for headache, muscle ache, and chills were not collected in year 2 of any studies. *P < 0·05, unadjusted for multiplicity. LAIV, live attenuated influenza vaccine.
When reactogenicity events were analyzed by number of days reported, the median number of days for runny/stuffy nose and cough for LAIV, placebo, and TIV recipients was 4–5 days. Median values were ≤3 days for decreased appetite and sore throat, ≤2 days for irritability, abdominal pain, tiredness/decreased activity, headache, muscle aches, and chills, and 1 day for elevated temperatures and vomiting. When LAIV recipients were compared with TIV or placebo recipients, the difference in the median number of days for each reactogenicity event was ≤1 day.
Unsolicited AEs days 0–10 post‐vaccination
In TIV‐controlled studies, during days 0–10 after dose 1, more LAIV recipients reported ≥1 AE (rate difference, 2·7%; P < 0·01); rates were similar for both groups after dose 2 (Table 3). Statistically significant rate differences were seen in infections (1·5%; P < 0·01), nervous system disorders (0·6%; P = 0·04), and respiratory disorders (1·5%; P < 0·01) after dose 1. The rate differences in nervous system and respiratory disorders were primarily attributable to headache (0·6%; P = 0·03) and nasal congestion and rhinorrhea (0·3%; P = 0·01 and 1·2%; P < 0·01), respectively. The infection increase could not be attributed to a specific AE, but the largest rate difference (0·4%, P = 0·10) was in rhinitis. Fewer surgical procedures were reported in LAIV recipients after dose 2 (rate difference, 0·4%; P = 0·02), which was attributed to increased reports of prophylaxis (i.e. preoperative administration of antibiotics) among TIV recipients (rate difference of 0·4%; P < 0·01). Rates of lower respiratory illness and wheezing illness were similar among LAIV and TIV recipients after dose 1 and dose 2.
Table 3.
Adverse events (AEs) during days 0–10 after LAIV and TIV administration in TIV‐controlled studies
| AEs | Year 1, dose 1 | Year 1, dose 2 | ||||
|---|---|---|---|---|---|---|
| LAIV, n (%) | TIV, n (%) | Rate difference* | LAIV, n (%) | TIV, n (%) | Rate difference* | |
| Total number of events, n | 1292 | 1103 | 528 | 519 | ||
| Subjects reporting ≥1 events | 860 (20·7) | 755 (18·1) | 2·7 | 359 (16·1) | 363 (16·0) | 0·1 |
| Events by organ class with absolute rate difference ≥0·10% | ||||||
| Infections and infestations | 329 (7·9) | 267 (6·4) | 1·5† | 192 (8·6) | 175 (7·7) | 0·9 |
| Respiratory, thoracic and mediastinal disorders | 270 (6·5) | 209 (5·0) | 1·5† | 92 (4·1) | 97 (4·3) | −0·1 |
| Nervous system disorders | 79 (1·9) | 55 (1·3) | 0·6† | 5 (0·2) | 7 (0·3) | −0·1 |
| Eye disorders | 44 (1·1) | 32 (0·8) | 0·3 | 16 (0·7) | 12 (0·5) | 0·2 |
| Psychiatric disorders | 27 (0·7) | 16 (0·4) | 0·3 | 4 (0·2) | 8 (0·4) | −0·2 |
| General disorders and administration site conditions | 112 (2·7) | 109 (2·6) | 0·1 | 61 (2·7) | 67 (3·0) | −0·2 |
| Skin and subcutaneous tissue disorders | 55 (1·3) | 55 (1·3) | 0·01 | 23 (1·0) | 15 (0·7) | 0·4 |
| Surgical and medical procedures | 5 (0·1) | 7 (0·2) | −0·05 | 1 (0·0) | 9 (0·4) | −0·4† |
| Immune system disorders | 1 (0·0) | 5 (0·1) | −0·1 | 1 (0·0) | 1 (0·0) | 0·0 |
| Ear and labyrinth disorders | 10 (0·2) | 16 (0·4) | −0·1 | 9 (0·4) | 8 (0·4) | 0·1 |
| Events of interest | ||||||
| Lower respiratory illness | 93 (2·2) | 110 (2·6) | −0·4 | 51 (2·3) | 60 (2·6) | −0·4 |
| Wheezing illness | 59 (1·4) | 68 (1·6) | −0·2 | 19 (0·9) | 30 (1·3) | −0·5 |
LAIV, Ann Arbor strain live attenuated influenza vaccine; TIV, trivalent inactivated influenza vaccine.
*LAIV rate minus TIV rate.
† P < 0·05, unadjusted for multiplicity.
In placebo‐controlled studies, the percentage of subjects reporting ≥1 AE was similar in the LAIV and placebo groups in days 0–10 after dosing in years 1 and 2 (Table 4). A statistically significant rate difference for general disorders (2·2%; P < 0·01) was observed after dose 1. This difference was attributable to an increased rate of pyrexia among LAIV recipients (rate difference, 2·1%; P < 0·01). After dose 2 in year 1, fewer LAIV recipients reported ear disorders (rate difference, −0·3%; P = 0·02), which was because of more reports of ear pain among placebo recipients (rate difference, −0·2%; P = 0·04). Fewer lower respiratory illnesses were reported among LAIV recipients (rate difference, −1·0%; P = 0·03). In year 2, rates of reported AEs were similar between LAIV and placebo recipients.
Table 4.
Adverse events (AEs) after LAIV and placebo administration in placebo‐controlled studies
| AEs | Year 1, dose 1 | Year 1, dose 2 | Year 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| LAIV, n (%) | Placebo, n (%) | Rate difference* | LAIV, n (%) | Placebo, n (%) | Rate difference* | LAIV, n (%) | Placebo, n (%) | Rate difference* | |
| Total number of events | 1380 | 841 | 893 | 657 | 955 | 482 | |||
| Subjects reporting ≥1 event | 975 (29·7) | 559 (27·6) | 2·2 | 616 (24·3) | 460 (26·5) | −2·2 | 624 (27·2) | 336 (26·8) | 0·4 |
| Events by organ class with absolute rate difference ≥0·10% | |||||||||
| General disorders and administration site conditions | 318 (9·7) | 152 (7·5) | 2·2† | 147 (5·8) | 119 (6·9) | −1·1 | 206 (9·0) | 97 (7·7) | 1·3 |
| Injury, poisoning and procedural complications | 25 (0·8) | 8 (0·4) | 0·4 | 13 (0·5) | 6 (0·3) | 0·2 | 4 (0·2) | 5 (0·4) | −0·2 |
| Psychiatric disorders | 30 (0·9) | 12 (0·6) | 0·3 | 15 (0·6) | 9 (0·5) | 0·1 | 21 (0·9) | 6 (0·5) | 0·4 |
| Nervous system disorders | 13 (0·4) | 4 (0·2) | 0·2 | 5 (0·2) | 2 (0·1) | 0·1 | 4 (0·2) | 0 (0·0) | 0·2 |
| Reproductive system and breast disorders | 5 (0·2) | 0 (0·0) | 0·2 | 2 (0·1) | 0 (0·0) | 0·1 | 1 (0·0) | 0 (0·0) | 0·04 |
| Metabolism and nutrition disorders | 11 (0·3) | 6 (0·3) | 0·04 | 13 (0·5) | 8 (0·5) | 0·1 | 15 (0·7) | 3 (0·2) | 0·4 |
| Immune system disorders | 3 (0·1) | 2 (0·1) | −0·01 | 0 (0·0) | 2 (0·1) | −0·1 | 0 (0·0) | 0 (0·0) | |
| Ear and labyrinth disorders | 7 (0·2) | 5 (0·2) | −0·03 | 1 (<0·1) | 6 (0·3) | −0·3† | 6 (0·3) | 6 (0·5) | −0·2 |
| Infections and infestations | 388 (11·8) | 243 (12·0) | −0·2 | 288 (11·4) | 228 (13·1) | −1·8 | 230 (10·0) | 124 (9·9) | 0·1 |
| Eye disorders | 14 (0·4) | 13 (0·6) | −0·2 | 18 (0·7) | 5 (0·3) | 0·4 | 8 (0·3) | 3 (0·2) | 0·1 |
| Skin and subcutaneous tissue disorders | 17 (0·5) | 16 (0·8) | −0·3 | 15 (0·6) | 11 (0·6) | −0·04 | 13 (0·6) | 2 (0·2) | 0·4 |
| Gastrointestinal disorders | 98 (3·0) | 67 (3·3) | −0·3 | 70 (2·8) | 34 (2·0) | 0·8 | 46 (2·0) | 30 (2·4) | −0·4 |
| Respiratory, thoracic and mediastinal disorders | 285 (8·7) | 188 (9·3) | −0·6 | 198 (7·8) | 137 (7·9) | −0·1 | 255 (11·1) | 135 (10·7) | 0·4 |
| Events of interest | |||||||||
| Lower respiratory illness | 58 (1·8) | 37 (1·8) | −0·1 | 48 (1·9) | 51 (2·9) | −1·0† | 40 (1·7) | 19 (1·5) | 0·2 |
| Wheezing illness | 22 (0·7) | 14 (0·7) | 0·0 | 16 (0·6) | 18 (1·0) | −0·4 | 17 (0·7) | 7 (0·6) | 0·2 |
LAIV, Ann Arbor strain live attenuated influenza vaccine.
*LAIV rate minus placebo rate.
† P < 0·05, unadjusted for multiplicity.
When all AEs were analyzed by severity, the majority of AEs reported in TIV‐ and placebo‐controlled studies were mild. Adverse event severity was similar between LAIV and the corresponding TIV or placebo comparison groups.
SAEs days 0–42 and 0–180 post‐vaccination
Rates of SAEs occurring through day 42 after the last dose of vaccine were low and similar among LAIV, TIV, and placebo recipients. In TIV‐controlled studies, 0·75% of LAIV recipients and 1·01% of TIV recipients reported any SAE. Similarly, in placebo‐controlled studies, 0·5% of LAIV and 0·6% of placebo recipients reported any SAE in year 1 and 0·5% of LAIV and 0·6% of placebo recipients reported any SAE in year 2. When SAEs were summarized by system organ class, no statistically significant differences were seen between LAIV and TIV or placebo groups in year 1 or year 2; all rate differences were <0·025%. No significant differences were seen for lower respiratory or wheezing illness; rates were either similar or lower in LAIV recipients compared with TIV or placebo recipients.
Similarly, when the available data were analyzed through day 180 after the last dose, rates of any SAE were similar among LAIV and TIV or placebo recipients. In TIV‐controlled studies, 2·3% of LAIV recipients and 2·5% of TIV recipients reported any SAE. In placebo‐controlled studies, 2·9% of LAIV and 2·7% of placebo recipients reported any SAE in year 1, and 2·1% of LAIV and 1·7% of placebo recipients reported any SAE in year 2. When analyzed by system organ class, the only statistically significant difference was an increase in the rate of injuries/poisonings among LAIV versus placebo recipients in year 1 (LAIV, 0·3%; placebo, 0·0%; P = 0·05). No statistically significant differences were seen for lower respiratory or wheezing illness.
Subgroup analyses
For children aged 24–35 months, similar rates of lower respiratory and wheezing illness were observed among LAIV recipients and their corresponding TIV or placebo recipients (Table 5). The only exception was that in placebo‐controlled studies after dose 2 in year 1, the incidence of lower respiratory illness was significantly lower among LAIV recipients (rate difference, −1·0%; P = 0·03). Thus, there was no evidence for increased lower respiratory or wheezing illness among LAIV recipients aged 24–35 months. When analyzed by subject region and gender, the patterns of REs, AEs, and SAEs were consistent with those observed in the overall population.
Table 5.
Adverse Events (AEs) and SAEs because of lower respiratory illness and wheezing in children 24–35 months of age
| AEs, days 0–10 | Year 1 | Year 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Dose 1 | Dose 2 | ||||||||
| LAIV, n (%) | Comparator, n (%) | Rate difference* | LAIV, n (%) | Comparator, n (%) | Rate difference* | LAIV, n (%) | Comparator, n (%) | Rate difference* | |
| TIV‐controlled studies | |||||||||
| Lower respiratory illness | 33 (2·0) | 45 (2·7) | −0·7 | 27 (2·5) | 28 (2·6) | −0·1 | NA | NA | NA |
| Wheezing illness | 23 (1·4) | 27 (1·6) | −0·2 | 9 (0·8) | 13 (1·2) | −0·4 | NA | NA | NA |
| Placebo‐controlled studies | |||||||||
| Lower respiratory illness | 58 (1·8) | 37 (1·9) | −0·1 | 48 (1·9) | 51 (2·9) | −1·0† | 23 (2·2) | 9 (1·6) | 0·6 |
| Wheezing illness | 22 (0·7) | 14 (0·7) | 0·0 | 16 (0·6) | 18 (1·0) | −0·4 | 9 (0·8) | 5 (0·9) | 0·0 |
| SAEs | Year 1 | Year 2 | ||||
|---|---|---|---|---|---|---|
| LAIV, n (%) | Comparator, n (%) | Rate difference* | LAIV, n (%) | Comparator, n (%) | Rate difference* | |
| TIV‐controlled studies, days 0–42 | ||||||
| Lower respiratory illness | 9 (0·55) | 9 (0·55) | 0·00 | NA | NA | NA |
| Wheezing illness | 3 (0·18) | 2 (0·12) | 0·06 | NA | NA | NA |
| TIV‐controlled studies, days 0–180 | ||||||
| Lower respiratory illness | 29 (1·29) | 19 (1·17) | 0·12 | NA | NA | NA |
| Wheezing illness | 6 (0·37) | 3 (0·18) | 0·18 | NA | NA | NA |
| Placebo‐controlled studies, day 0–42 | ||||||
| Lower respiratory illness | 13 (0·41) | 8 (0·42) | −0·01 | 4 (0·38) | 3 (0·52) | −0·15 |
| Wheezing illness | 7 (0·22) | 4 (0·21) | 0·01 | 1 (0·09) | 0 (0·00) | 0·09 |
| Placebo‐controlled studies, day 0–180 | ||||||
| Lower respiratory illness | 28 (1·17) | 15 (0·97) | 0·20 | 7 (0·66) | 4 (0·70) | −0·04 |
| Wheezing illness | 9 (0·38) | 5 (0·32) | 0·05 | 2 (0·19) | 0 (0·00) | 0·19 |
LAIV, Ann Arbor live attenuated influenza vaccine; AE, adverse event; SAE, serious AE; TIV, trivalent inactivated influenza vaccine.
*LAIV rate minus comparator rate.
† P < 0·05, unadjusted for multiplicity.
Discussion
This integrated safety analysis of Ann Arbor strain LAIV supports the overall safety of the vaccine in children aged 2–17 years. The large, integrated study population provided increased statistical power to detect small differences associated with initial vaccination and revaccination across multiple studies and multiple seasonal formulations. The analysis also provides a consensus assessment of the expected events post‐vaccination. This analysis, in which data were integrated from multiple studies across multiple years using multiple vaccine strains, is based on the assumption that LAIV and TIV reactogenicity and safety are consistent across different annual formulations of the vaccine. Results from individual studies have demonstrated that the safety profiles of the vaccines are consistent across formulations; however, minor differences in reactogenicity could exist between different annual formulations, and as a result, the safety profile of any particular formulation could differ slightly from the aggregated experience reported here.
Runny/stuffy nose was the primary factor contributing to the overall occurrence and distribution of reactogenicity for LAIV subjects in TIV‐ and placebo‐controlled studies. LAIV recipients had an approximate 10% increased incidence of runny/stuffy nose post‐vaccination. Other reactogenicity events more common in LAIV recipients than in placebo recipients were headache, tiredness/decreased activity, and decreased appetite; however, these events were not increased compared with TIV recipients, perhaps because TIV also induced systemic reactogenicity. Solicited AEs that occurred more commonly in the LAIV group were those similar to the reactogenicity events associated with LAIV, namely headache, nasal congestion/rhinorrhea, and rhinitis; pyrexia was also associated with LAIV. Ear pain and lower respiratory illness were decreased among LAIV recipients. A consistent trend across all reactogenicity events and AEs was that rate differences were lower after revaccination in years 1 and 2 compared with the initial vaccination, a phenomenon that has been noted previously in children and adults receiving LAIV. 14 , 15 , 16
Fever, collected by daily temperature readings as a solicited reactogenicity event, was not statistically increased in LAIV recipients in this integrated analysis; however, more LAIV recipients than placebo recipients reported pyrexia as an unsolicited AE term after the first dose. Although a few individual studies noted a statistically significant increase in low‐grade fevers in LAIV subjects compared with placebo or TIV recipients after dose 1, 3 , 17 , 18 fever rates were similar in other studies. 4 , 5 , 19 , 20 , 21 In addition, an early study of LAIV reported increased rates of vomiting or abdominal pain in LAIV recipients; however, the incidence of abdominal pain was not actively solicited from subjects in that study. 18 In other studies, some of which actively solicited the incidence of abdominal pain, the rates of vomiting and abdominal pain were similar between LAIV and TIV or placebo recipients. 3 , 4 , 5 , 17 , 19 , 20 , 21 In this integrated analysis, no statistically significant increase in vomiting or abdominal pain was observed among LAIV recipients.
There was no evidence of an increase in any potential vaccine‐related SAE in LAIV recipients compared with TIV or placebo recipients. Additionally, there was no evidence that lower respiratory illness or wheezing illness occurred at a higher rate in LAIV subjects; this was true for children aged 2–17 years and those aged 24–35 months. This finding is consistent with a study that prospectively tracked the incidence of medically attended wheezing. 3 Although an increased rate of wheezing was seen among LAIV recipients aged 6–23 months through 42 days after the last dose (LAIV, 5·9%; TIV, 3·8%; P < 0·01), no increase was seen in children aged 24–59 months (LAIV, 2·1%; TIV, 2·5%; P = 0·38), even when the incidence was analyzed by individual‐month cohorts. 14
In conclusion, the current integrated analysis provides a broad assessment of the overall safety and tolerability of Ann Arbor strain LAIV in children aged 2–17 years across multiple populations, seasons, and formulations. The results support the safety of Ann Arbor strain LAIV in eligible children aged 2–17 years and provide healthcare providers with a valuable consensus summary of events that can be expected after vaccine administration.
Re‐use of this article is permitted in accordance with the Terms and Conditions set out at http://wileyonlinelibrary.com/onlineopen#OnlineOpen_Terms.
References
- 1. Maassab HF. Plaque formation of influenza virus at 25°C. Nature 1968; 219:645–646. [DOI] [PubMed] [Google Scholar]
- 2. Block SL, Reisinger KS, Hultquist M, Walker RE. Comparative immunogenicities of frozen and refrigerated formulations of live attenuated influenza vaccine in healthy subjects. Antimicrob Agents Chemother 2007; 51:4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Belshe RB, Edwards KM, Vesikari T et al. Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med 2007; 356:685–696. [DOI] [PubMed] [Google Scholar]
- 4. Ashkenazi S, Vertruyen A, Aristegui J et al. Superior relative efficacy of live attenuated influenza vaccine compared with inactivated influenza vaccine in young children with recurrent respiratory tract infections. Pediatr Infect Dis J 2006; 25:870–879. [DOI] [PubMed] [Google Scholar]
- 5. Fleming DM, Crovari P, Wahn U et al. Comparison of the efficacy and safety of live attenuated cold‐adapted influenza vaccine, trivalent, with trivalent inactivated influenza virus vaccine in children and adolescents with asthma. Pediatr Infect Dis J 2006; 25:860–869. [DOI] [PubMed] [Google Scholar]
- 6. Belshe RB, Gruber WC, Mendelman PM et al. Efficacy of vaccination with live attenuated, cold‐adapted, trivalent, intranasal influenza virus vaccine against a variant (A/Sydney) not contained in the vaccine. J Pediatr 2000; 136:168–175. [DOI] [PubMed] [Google Scholar]
- 7. Nichol KL, Mendelman PM, Mallon KP et al. Effectiveness of live, attenuated intranasal influenza virus vaccine in healthy, working adults: a randomized controlled trial. JAMA 1999; 282:137–144. [DOI] [PubMed] [Google Scholar]
- 8. Wiggs‐Stayner KS, Purdy TR, Go GN et al. The impact of mass school immunization on school attendance. J Sch Nurs 2006; 22:219–222. [DOI] [PubMed] [Google Scholar]
- 9. King JC Jr, Cummings GE, Stoddard J et al. A pilot study of the effectiveness of a school‐based influenza vaccination program. Pediatrics 2005; 116:e868–e873. [DOI] [PubMed] [Google Scholar]
- 10. King JC Jr, Stoddard JJ, Gaglani MJ et al. Effectiveness of school‐based influenza vaccination. N Engl J Med 2006; 355:2523–2532. [DOI] [PubMed] [Google Scholar]
- 11. Mears CJ, Lawler EN, Sanders LD 3rd, Katz BZ. Efficacy of LAIV‐T on absentee rates in a school‐based health center sample. J Adolesc Health 2009; 45:91–94. [DOI] [PubMed] [Google Scholar]
- 12. Davis MM, King JC Jr, Moag L, Cummings G, Magder LS. Countywide school‐based influenza immunization: direct and indirect impact on student absenteeism. Pediatrics 2008; 122:e260–e265. [DOI] [PubMed] [Google Scholar]
- 13. Carpenter LR, Lott J, Lawson BM et al. Mass distribution of free, intranasally administered influenza vaccine in a public school system. Pediatrics 2007; 120:e172–e178. [DOI] [PubMed] [Google Scholar]
- 14. Belshe RB, Ambrose C, Yi T. Safety and efficacy of live attenuated influenza vaccine in children 2–7 years of age. Vaccine 2008; 26S:D10–D16. [DOI] [PubMed] [Google Scholar]
- 15. Ohmit SE, Victor JC, Teich ER et al. Prevention of symptomatic seasonal influenza in 2005–2006 by inactivated and live attenuated vaccines. J Infect Dis 2008; 198:312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piedra PA, Gaglani MJ, Riggs M et al. Live attenuated influenza vaccine, trivalent, is safe in healthy children 18 months to 4 years, 5 to 9 years, and 10 to 18 years of age in a community‐based, nonrandomized, open‐label trial. Pediatrics 2005; 116:e397–e407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tam JS, Capeding MR, Lum LC et al. Efficacy and safety of a live attenuated, cold‐adapted influenza vaccine, trivalent against culture‐confirmed influenza in young children in Asia. Pediatr Infect Dis J 2007; 26:619–628. [DOI] [PubMed] [Google Scholar]
- 18. Belshe RB, Mendelman PM, Treanor J et al. The efficacy of live attenuated, cold‐adapted, trivalent, intranasal influenzavirus vaccine in children. N Engl J Med 1998; 338:1405–1412. [DOI] [PubMed] [Google Scholar]
- 19. Breiman RF, Brooks WA, Goswami D et al. A multinational, randomized, placebo‐controlled trial to assess the immunogenicity, safety, and tolerability of live attenuated influenza vaccine coadministered with oral poliovirus vaccine in healthy young children. Vaccine 2009; 27:5472–5479. [DOI] [PubMed] [Google Scholar]
- 20. Bracco Neto H, Farhat CK, Tregnaghi MW et al. Efficacy and safety of 1 and 2 doses of live attenuated influenza vaccine in vaccine‐naive children. Pediatr Infect Dis J 2009; 28:365–371. [DOI] [PubMed] [Google Scholar]
- 21. Vesikari T, Fleming DM, Aristegui JF et al. Safety, efficacy, and effectiveness of cold‐adapted influenza vaccine‐trivalent against community‐acquired, culture‐confirmed influenza in young children attending day care. Pediatrics 2006; 118:2298–2312. [DOI] [PubMed] [Google Scholar]