

Abstract
Mitochondrial DNA (mtDNA) mutations were reported in different cancers. However, the nature and role of mtDNA mutation in never-smoker lung cancer patients including patients with EGFR and KRAS gene mutation are unknown. In the present study, we sequenced entire mitochondrial genome (16.5 kb) in matched normal and tumors obtained from 30 never-smoker and 30 current-smoker lung cancer patients, and determined the mtDNA content. All the patients’ samples were sequenced for KRAS (exon 2) and EGFR (exon 19 and 21) gene mutation. The impact of forced overexpression of a respiratory complex-I gene mutation was evaluated in a lung cancer cell line. We observed significantly higher (P=0.006) mtDNA mutation in the never-smokers compared to the current-smoker lung cancer patients. MtDNA mutation was significantly higher (P=0.026) in the never-smoker Asian compared to the current-smoker Caucasian patients’ population. MtDNA mutation was significantly (P=0.007) associated with EGFR gene mutation in the never-smoker patients. We also observed a significant increase (P=0.037) in mtDNA content among the never-smoker lung cancer patients. The majority of the coding mtDNA mutations targeted respiratory complex-I and forced overexpression of one of these mutations resulted in increased in vitro proliferation, invasion and superoxide production in lung cancer cells. We observed a higher prevalence and new relationship between mtDNA alterations among never-smoker lung cancer patients and EGFR gene mutation. Moreover, a representative mutation produced strong growth effects after forced overexpression in lung cancer cells. Signature mtDNA mutations provide a basis to develop novel biomarkers and therapeutic strategies for never-smoker lung cancer patients.
Keywords: Lung cancer, never-smokers, MtDNA mutation, Respiratory Complex-I, EGFR mutation
Introduction
Lung cancer is a deadly disease killing more than 1 million people worldwide and smoking is by far the most important risk factor (Sato et al., 2007). In the United States, there will be over 222,550 cases of lung cancer and an estimated 157,300 deaths in 2010 (www.cancer.gov). Despite significant improvement in therapeutic modalities including surgery, platinum based chemotherapy and radiotherapy alone or in combination, the overall 5-year survival rate is only 15% (Jonsson et al., 2008). Moreover, affected patients remain at significant risk for the development of a second primary tumor throughout their lifetime. Although lung cancer is more frequent among tobacco smokers, around 15% of all lung cancers do not appear to be associated with tobacco smoking (Sun et al., 2007). Epidemiological studies identified numerous non-smoking related risk factors, but the major genetic causes of lung cancer among never smokers are yet to be identified (Sun et al., 2007; Subramanian and Govindan 2007). Specific activating mutations in the epidermal growth factor receptor (EGFR) were found to be associated with lung adenocarcinoma subtypes, East Asian ethnicity, and female gender among the never-smokers (Sun et al., 2007). These activating EGFR mutations are heterozygous and include exons 18 to 21 encoding the kinase domain (Pao et al., 2010; Rudin et al.. 2010). The most commonly found mutations in the kinase domain are exon 19 deletion that eliminate a leucine-arginine-glutamate-alanine (LREA) motif and a point mutation at nucleotide position 858 in exon 21 that substitutes arqinine for leucine (L858R) (Pao et ai., 2010; Rudin et al., 2010). These mutations affect the ATP binding site of the tyrosine kinase domain of EGFR rendering a constitutively active and ligand independent receptor state fRudin et al., 2010). Other major drug sensitive EGFR mutations such as G719X (exon 18), T790M (exon 2) and L861Q (exon 21) was also reported in other studies (Pao et al., 2010; Rudin et al., 2010). Upon ligand binding, EGFR undergoes autophosphorylation and transduces signals through several cascades mediated by adaptors that bind to C-terminal phosphotyrosines and recruit proteins involved in downstream signaling events. Those cascades involve signal transducer and activator of transcription (STAT), phosphatidylinositol 3′-kinase (PI3K)/Akt and Ras/Raf/extracellular signal-regulated kinase 1/2 (Erk1/2) pathways (Dobashi et al 2011). Interestingly, in a recent study deregulation of E-cadherin was shown to activate EGFR by triggering downstream signaling pathway (Mateus et al 2007). Missense mutations clustering in the extracellular domain region of E-cadherin exhibited increased phosphorylation of EGFR upon EGF stimulation leading to enhanced cell motality via RhoA GTPase activation which was abrogated by EGFR inhibition (Mateus et al 2007). Thus, EGFR appears to play a significant role in non-smoker lung cancer.
In addition to nuclear genetic alterations, mitochondrial DNA (mtDNA) mutations were reported in different cancers and implicated in their development (Chatterjee et al., 2011; He et al., 2010). However, no information is available to date on the pattern of mtDNA alteration and its role in never-smoker lung cancer patients and particularly in patients with EGFR mutation.
The human mitochondrial DNA (mtDNA) is a 16.5 kb double stranded closed circular molecule which codes for the 12S and 16S rRNAs, 22 tRNAs and 13 proteins essential for the mitochondrial respiratory complex (Chatterjee et al., 2011). Most human cells contain hundreds of copies of mitochondrial DNA (mtDNA) and nearly all of these mtDNA copies are identical i.e. nearly homoplasmic at birth (Chatterjee et al., 2011). Due to the lack of protective histones and introns, mtDNA is susceptible to damage by environmental carcinogens as well as endogenous reactive oxygen species (ROS), a by product of the oxidative phosphorylation system (OXPHOS) (Chatterjee et al., 2011). Maybe as a result, the mutation rate in mtDNA is approximately 10 times higher than nuclear genomic DNA (nDNA) (Chatterjee et al., 2011; Dasgupta et al., 2010). Thus, presence of high copy number of mutated mtDNA in cancer cells either in a heteroplasmic or homoplasmic state (Chatterjee et al., 2011), affecting specific respiratory complex of the OXPHOS system could be an attractive target for developing mtDNA based strategies for cancer detection and monitoring.
In the present study, we attempted to analyze the pattern of mtDNA alteration (mutation and absolute content), its contribution in lung cancer cell growth, and correlation with key mutations in never smoker lung cancer patients. We observed a significant correlation between mtDNA alteration and EGFR gene mutation in never-smoker compared to current-smoker lung cancer patients. The majority of the coding mtDNA mutations targeted respiratory complex-I and forced overexpression of one of these mutations resulted in increased in vitro proliferation, invasion and superoxide production in lung cancer cells.
Materials and Methods
Patient’s History and Tumor Samples
We obtained adjacent matched normal, tumor tissues from 60 lung cancer patients after signed informed consent from a University of British Columbia IRB approved protocol. Of the 60 patients, 30 were categorized as “never-smoker” who never smoked or had any previous smoking history. The rest of the 30 were categorized as “current smoker” who was currently smoking (25–120 pack/year) at the time of disease diagnosis. Detailed demographic information of all the patients along with the frequency of mtDNA and nuclear DNA (nDNA) alterations is shown in Table 1–2.
Table 1.
Current-smoker lung cancer patients with mtDNA and KRAS/EGFR alterations
| Patient ID1 | Age/Sex | Ethnicity2 | Primary site3 | Stage | Number of mtDNA mutation | MtDNA content4 | KRAS/EGFR mutation5 |
|---|---|---|---|---|---|---|---|
| 07L66 | 74/F | C | RUL | IA | 0 | SI | KRAS |
| 07L51 | 53/F | C | LLL | IA | 0 | NC | KRAS |
| 07L5 | 64/F | c | RUL | IB | 0 | NC | KRAS |
| 07L36 | 69/F | A1 | LLL | IB | 0 | NC | NM |
| 07L31 | 47/F | A | RUL | IA | 0 | NC | NM |
| 07L25 | 72/F | C | LUL | IB | 0 | NC | KRAS |
| 06L57 | 53/F | C | N | IIIA | 0 | SI | KRAS |
| 06L54 | 73/M | C | RML | IA | 0 | SI | NM |
| 06L46 | 73/F | C | RUL | IB | 0 | NC | KRAS |
| 06L3 | 67/F | C | LUL | IIB | 0 | NC | NM |
| 06L20 | 70/F | C | RML | IB | 0 | SI | KRAS |
| 06L14 | 57/F | C | N | IIB | 0 | SI | KRAS |
| 06L11 | 67/F | C | N | IA | 0 | NC | KRAS |
| 06L10 | 72/F | C | RLL | IB | 0 | NC | NM |
| 05L19 | 68/F | C | RUL | IV | 0 | SI | NM |
| 0003030 | 68/F | C | RLL | N | 0 | SI | KRAS |
| 0003024 | 63/M | C | RUL | IIIA | 0 | SI | KRAS |
| 0003023 | 74/M | C | LUL | N | 0 | NC | NM |
| 0003022 | 73/M | C | RUL | N | 0 | NC | NM |
| 0003014 | 69/F | C | RUL | N | 0 | NC | KRAS |
| 0003012 | 58/F | C | LUL | N | 0 | SI | NM |
| 0003003 | 61/F | C | LLL | N | 0 | SI | KRAS |
| 07L21 | 67/M | C | RLL | IB | 1 | NC | KRAS |
| 07L32 | 71/F | C | LUL | IIB | 1 | SI | KRAS |
| 06L50 | 73/F | C | LUL | IA | 1 | NC | KRAS |
| 05L9 | 71/F | C | LLL | IA | 1 | NC | KRAS |
| 0003021 | 66/M | C | RUL | - | 1 | SI | NM |
| 0003013 | 64/F | C | RUL | - | 1 | NC | KRAS |
| 05L43 | 64/F | C | LUL | IB | 3 | NC | KRAS |
| 05L32 | 65/M | C | LUL | IIB | 2 | NC | NM |
All except one patient (07L36, large cell carcinoma) were adenocarcinoma.
Caucasian, A: Asian, AI: American Indian or Alaska;
RUL: Right upper lobe, RML: Right middle lobe, RLL: Right lower lobe, LUL: Left upper lobe, LLL: Left lower lobe;
MtDNA content was quantitatively determined by Q-PCR (in triplicate) using mitochondria encoded COX-I and nuclear encoded Beta-actin (control) gene. SI: Significant Increase; NC: No significant change in mtDNA content.
KRAS/EGFR mutation was examined by sequencing the exon 2 of KRAS and exon 19 and 21 of EGFR; KRAS: KRAS mutation present; NM: No mutation detected in KRAS or EGFR gene.
Table 2.
Never-smoker lung cancer patients with mtDNA and KRAS/EGFR alterations
| Patient ID1 | Age/Sex | Ethnicity2 | Primary site3 | Stage | Number of MtDNA mutation | MtDNA content4 | KRAS/EGFR mutation5 |
|---|---|---|---|---|---|---|---|
| 07L6 | 71/M | A | RUL | IIIB | 0 | SI | EGFR |
| 06L71 | 68/F | C | RUL | IA | 0 | SI | NM |
| 06L64 | 78/F | C | LUL | IA | 0 | SI | EGFR |
| 06L29 | 71/F | A | RUL | IB | 0 | NC | NM |
| 05L6 | 75/F | A | LLL | IB | 0 | SI | NM |
| 05L49 | 77/F | A | LLL | IB | 0 | NC | NM |
| 05L4 | 82/F | A | RLL | MB | 0 | NC | EGFR |
| 05L34 | 73/F | A | LUL | IB | 0 | NC | EGFR |
| 0003036 | 64/F | A | LUL | IV | 0 | SI | EGFR |
| 0003033 | 58/F | C | N | N | 0 | SI | KRAS |
| 0003031 | 74/F | C | LLL | N | 0 | NC | EGFR |
| 07L38 | 60/F | A | RUL | IIIB | 1 | SI | EGFR |
| 07L28 | 65/F | C | LLL | MIA | 1 | SI | EGFR |
| 07L22 | 85/M | A | RUL | IA | 1 | SI | EGFR |
| 07L19 | 73/F | A | RUL | IB | 1 | SI | EGFR |
| 07L16 | 81/M | C | RUL | IA | 1 | SI | NM |
| 05L7 | 72/M | A | RUL | IB | 1 | NC | EGFR |
| 05L54 | 77/F | A | LLL | IA | 1 | SI | KRAS |
| 05L47 | 63/F | A | LLL | MB | 1 | SI | NM |
| 05L45 | 39/F | A | RUL | IA | 1 | SI | NM |
| 0003032 | 55/F | C | LUL | - | 1 | SI | NM |
| 05L40 | 52/F | A | LLL | IA | 2 | NC | EGFR |
| 05L39 | 86/F | C | LLL | IB | 2 | NC | KRAS |
| 05L13 | 74/M | A | RUL | IB | 2 | SI | EGFR |
| 05L12 | 66/M | A | LLL | IMA | 2 | SI | EGFR |
| 0003035 | 63/M | C | RUL | - | 2 | SI | NM |
| L2 | 70/F | A | - | MIA | 3 | SI | EGFR |
| L15 | 72/F | A | RLL | IB | 3 | SI | EGFR |
| 05L27 | 77/F | A | LLL | IIB | 3 | SI | EGFR |
| 0003039 | 80/F | C | - | - | 1 | NC | NM |
AII except one patient (05L45, bronchoalveolar carcinoma) were adenocarcinoma.
Caucasian, A: Asian;
RUL: Right upper lobe, RLL: Right lower lobe, LUL: Left upper lobe, LLL: Left lower lobe;
MtDNA content was quantitatively determined by Q-PCR (in triplicate) using mitochondria encoded COX-I and nuclear encoded Beta-actin (control) gene. SI: Significant increase; NC: No significant change in mtDNA content.
KRAS/EGFR mutation was examined by sequencing the exon 2 of KRAS and exon 19 and 21 of EGFR; KRAS: KRAS mutation present; EGFR: EGFR mutation present; NM: No mutation detected in KRAS or EGFR gene.
Mitochondrial Whole Genome Amplification
Genomic DNA was extracted according to our standard protocol from the microdissected tumor and adjacent normal tissues (Zhou et al., 2006). Whole mitochondrial genomic DNA was amplified in three overlapping long PCR fragments, with each reaction containing 50 ng of genomic DNA as per the Affymetrix Mitochip resequencing array v2,0 protocol (Affymetrix.com) (Zhou et al., 2006). The primers used to amply mitochondrial DNA are Mito-1F ACATAGCACATTACAGTCAAATCCCTTCTCGTCCC,1R-TGAGATTGTTTGGGCTACTGCTCGCAGTGC (3968 bp); Mito 2F-TACTCAATCCTCTGATCAGGGTGAGCATCAAACTC, 2R-GCTTGGATTAAGGCGACAGCGATTTCTAGGATAGT (5513bp); Mito 3F-TCATTTTTATTGCCACAACTAACCTCCTCGGACTC, 3R-CGTGATGTCTTATTTAAGGGGAACGTGTGGGCTAT (7814 bp). The above 3 primer sets amplify a 3968, 5513 and 7814 base pair mtDNA fragments covering the entire mitochondrial genome. The PCR reaction was comprised of: 94 degree for 2 mins × 1 cycle: 94 degree for 15 sec, 68 degree for 1 min × 30 cycles and 68 degree for 10 mins (GeneAmp® PCR System 9700). The amplified DNA obtained from three PCR reactions (3968bp + 5513bp + 7814 bp) was pooled together.
Digestion of the mtDNA
The pooled DNA fragments was digested with DNase I for 15 minutes in a 50uI reaction containing Affymetrix fragmentation reagent (0.2 U of DNase I/uo DNA), 5 ul of OnePhorAll buffer (Amersham Life Sciences, Arlington Heights, IL), and EB buffer. Fragmented DNA was labeled by adding 2.0 ul of GeneChip DNA labeling reagent and 3.4 ul of 30 U/ul terminal deoxynucleotidyl transferase (Affymetrix).
Mitochip v2.0 Sequencing Array Analysis
Prehybridization, hybridization, washing, and scanning of the MitoChip were performed as described in the Affymetrix CustomSeq Resequencing protocol. The prehybridization was performed for 15 minutes in 80-ul solution containing 3 mol/L tetramethylammonium chloride, 0.1% Tween 20, and 10 mmol/L Tris, pH 7.8. The chips were hybridized for 16 hours at 48°C with 60 rpm rotation in a hybridization solution containing 3 mol/L tetramethylammonium chloride, 100 ug/ml herring sperm DNA, 500 ug/ml bovine serum albumin, 10 mmol/L Tris, pH 7.8, 0.01% Tween 20, and 200 pmol/L control oligo. The chips were washed on the Affvmetrix fluidics station using the preprogrammed CustomSeq Resequencing wash protocols. The chips were scanned using the Gene Chip Scanner 3000 on a Gene Chip Operating Software 1.4 (GCOS 1.4) supported platform.
Data Analysis
Data analysis was performed using Affymetrix GSEQ software. The revised Cambridge Reference Sequence (rCRS) was used as the reference sequence. All the resulting mtDNA sequences were interrogated at different Human Mitochondrial Genome Databases as described earlier to identify specific mtDNA sequence variants (Dasgupta et al 2010; Zhou et al., 2006). The web resources used for data analysis are listed below.
Determination of MtDNA Mutations in Tumors
Somatic mtDNA sequence variants were identified as base pair changes in mtDNA of tumor when compared with mtDNA sequence of matched normal control (Dasgupta et al 2010; Zhou et al., 2006). In each case, mtDNA sequences were interrogated at the available Human Mitochondrial Genome Databases for defining each sequence variant as described earlier (Dasgupta et al 2010; Zhou et al., 2006).
Quantitative Real-Time PCR
To examine mtDNA content, we used the 7900HT sequence detection system (Applied Biosystems, Foster City, CA) to amplify nuclear DNA (nDNA) encoded β-Actin and mtDNA encoded Cytochrome C oxidase I (COI) as described earlier (Dasgupta et al 2009). This assay quantitatively determines the relative rate of amplification of mtDNA sequence (COL) compared to a reference sequence of nDNA (Beta-actin). The primer and probe sequence for COI used is as follows. Forward primer ttcgccqaccgttgactattctct (nt 6007–6030); reverse primer aagattattacaaatgcatgggc ( nt 6103–6081). The Taqman probe used is aacgaccacatctacaacgttatcgtcac ( nt 6051–6079). The primer and probe sequence for beta-actin is as follows. Forward primer tcacccacactgtgcccatctacga (2141–2165 bp), reverse primer, cagcggaaccgctcattgccaatgg (2435–2411 nt). The probe sequence is atgccctcccccatgccatcctgcgt (2171–2196.)
The mitochondrial Col TaqMan probe was labeled with 5′-FAM (6-carboxyfluorescein, fluorescent reporter) and 3′-TAMRA (6-carboxy-tetramethylrhodamine, fluorescence quencher) whereas the β-actin probe was labeled with 5′-Vic (fluorescent reporter), and 3′-TAMRA (quencher). Duplex PCR amplifications for COI and β-actin were carried out in buffer containing 16.6 mM ammonium sulfate, 67 mM Tris base, 2.5 mM MgCl2 10 mM 2-mercaptoethanol, 0.1% DMSO, 0.2 mM each of dATP, dCTP, dGTP, and dTTP, 600 nM each of forward and reverse primers, 200 nM TaqMan probe. 0.6 U Platinum Tag polymerase, and 2% Rox reference dye. The real-time PCR reactions were carried out in triplicate for each gene and standard curves were obtained by using cell line DNA. The mtDNA content was determined as a ratio of the mitochondrial Co I signal by the corresponding β-actin signal in each sample.
EGFR/KRAS Mutation Analysis
Mutational analysis was performed for EGFR includging exons 19 and 21 and exon 2 of KRAS. Mutation analysis was done by PCR amplification of genomic DNA followed by direct sequencing of the PCR products. All PCR reactions were performed in 25 μL volumes containing 50 –100 ng of DNA using the Applied Biosystems GeneAmp PCR System 9700. PCR products were sequenced using Applied Biosystems Big Dye Terminator 3.1 chemistry and Applied Biosystems capillary instrumentation. The following primers were used for sequencing. EGFR; exon 19: F 5′CCAGATCACTGGGCAGCATGTGGCACC 3′; R 5′AGCAGGGTCTAGAGCAGAGCAGCTGCC 3′; EGFR exon 21: F 5′ TCAGAGCCTGGCATGAACATGACCCTG 3′ R 5′ GGTCCCTGGTGTCAGGAAAATGCTGG 3′; KRAS exon 2 F 5′ GTATTAACCTTATGTGTGACA 3′, R 5′ GTCCTGCACCAGTAATATGC 3′ (Shigematsu et al., 2005).
MtDNA-ND5 Constructs and Transfection
One mtDNA mutation in the ND5 gene from respiratory complex-I (G13289A) was selected for further analysis (Table S3). The ND5 gene was converted into nuclear format, and both the wild type (wt) and mutant (mt) genes (G13289A) were synthesized using long-range gene synthesis (Genescript Corp.) as described earlier (Dasgupta et al., 2008). The mt and wt genes were then subcloned into Sall and Notl sites of the phosphoryfated cytomegalovirus (pCMV)/myc/mito plasmid as described earlier (Dasgupta et al., 2008). The resultant plasmids were resequenced using the ABI BigDye cycle sequencing kit (Applied Biosystems) for verification of the insert sequences. In transfections, SW900 lung cancer cells were transfected with mt and wt ND5 plasmids in the presence of the FuGene 6 transfection regent. Stable clones were selected in presence of 400 μg/ml of G418. Three independent clones were analyzed for all the subsequent experiments.
BrdU Incorporation Assay
The proliferation capability of the transfected cells was determined by the BrdU incorporation assay kit (Roche Diagnostics) as per the manufacturer’s protocol. Briefly, the cells were incubated at 37°C, 5% C02 for about 15–60 min in BrdU labeling medium. After thorough washing, cells were fixed in ethanol fixative for 20 min at −20°C.The cells were then incubated with Anti-BrdU working solution for 30 min at 37°C. After rewashing, cells were incubated for 30 min at 37°C with anti-mouse-lg-AP solution supplied by the manufacturer. Color-substrate solution (NBT+BCIP) was then be added to the cells. After 15–30 min incubation at room temperature, the cells were analyzed directly under a light microscope. At least ten fields were chosen randomly for evaluation and data were represented as Mean±SE of 2 independent experiments.
Invasion Assay
We performed the invasion assay in 24-well Matrigel invasion chambers as per manufacturer’s specification (BD Biosciences). This assay was performed in an Invasion Chamber, a 24-well tissue culture plate with 12 cell culture inserts. The inserts contain an 8 um pore size polycarbonate membrane, over which a thin layer of ECM matrix is dried. Specifically, the ECM layer in the testing chamber was rehydrated with 300 ul pre-warmed serum-free medium and 300 ul of the cell suspension in serum-free medium (1 × 106) was placed onto the up-chamber while 500 ul of cell-free medium containing 10% FBS was added to the bottom chamber. After 48 hours, the ECM membrane was fixed in 100% methanol and stained with 0.05% crystal violet and cell number was counted. The assay was performed in triplicate. At least 10 fields were randomly selected for counting cells that invaded through the membrane from each group (Dasgupta et al., 2008). Data were represented as Mean±SE of 2 independent experiments.
Superoxide Production Assay
Transfected cells were incubated with 5 μM MitoSOX™ reagent (Invitrogen) in 1 ML of appropriate cell culture medium for 10 minutes at 37°C as per manufacturer’s specifications. Cells were then washed with PBS 3 times and examined immediately under the fluorescent microscope. At least 10 fields were randomly selected for counting positively stained cells from each group. Data were represented as Mean±SE of 2 independent experiments.
Statistical Analysis
The prevalence of DNA mutations in different age groups, ethnicity, never-smokers and current-smokers was summarized to report the group specific proportions. Association of these phenotypes was modeled using a linear model with normal error assumption. The p-value for the association coefficient was obtained from the analysis of variance of the linear model. Proportion of the binary variables was shown in a mosaic plot with the rectangles of dimension proportional to the observed proportion. Distribution of the number of mitochondrial mutations is displayed in a bar chart for categorical variables and a box plot for continuous variables. Pearson’s Chi-squared test with Yates’ continuity Correction or Student’s t test was performed when appropriate. All P values are two sided, and all confidence intervals are at the 95% level.
Results
Pattern of MtDNA Mutation in the Never-Smoker and Current-Smoker Lung Cancer Patients
To understand the frequency and nature of mtDNA mutations in never-smoker lung cancer patients, we sequenced the entire mitochondrial genome in every specimen obtained from 30 never-smoker and 30 current smoker lung cancer patients. All the mtDNA sequencing was performed in parallel on a reliable mtDNA resequencing platform (Zhou et al., 2006). The average call rate on the Mitochip version 2.0 was 92.5%. Forty five percent (27/60) of the lung tumors harbored at least 1 somatic mtDNA mutation (Table 1–2; Table S1). The mtDNA mutations were mostly heteroplasmic in nature and often nucleotide transitions (A↔G; T↔C) (Supplementary Table S1). A total of 41 somatic mtDNA mutations were detected spanning both the coding and non-coding region of the mitochondrial genome (Table 1–2 and Table S1). The number of mutations was markedly higher (28 vs13) in the coding compared to the non-coding regions of the mtDNA. (Table 1–2 and Table S1). Of the 41 mutations, 31.7% (13/41) were from the non-coding regions of the mtDNA; 3 from the 12SrRNA; and 6 from 16SrRNA and 4 from tRNAs. The remainder of the 28 mutations was found in the coding regions; 2 each from from ND1, ND2 and ND4 and 10 were from ND5 (Complex I); 5 were from CYTB (Complex III); 5 were from COXI and 2 from COXII (Complex IV). No mutation was detected in complex V. Of the coding mtDNA mutations, 71.4% (20/28) were non-synonymous in nature (Table S1).
Association between Clinical Characteristics and MtDNA Mutation
We also compared the pattern of mtDNA mutation with clinical characteristics such as smoking, ethnicity, age and gender to understand their impact on lung tumorigenesis. MtDNA mutation was significantly higher (P=0.006) among the non-smokers compared to the smoker lung cancer patients (Fig. 1A). The frequency of mtDNA mutation was also significantly higher (P=0.026) in the never-smoker Asian compared to the current-smoker Caucasian patient population (Fig. 1B). No association was observed between mtDNA mutation and age (P=0.631) or gender (P=0.253) (Fig. S1 and S2).
Figure 1.
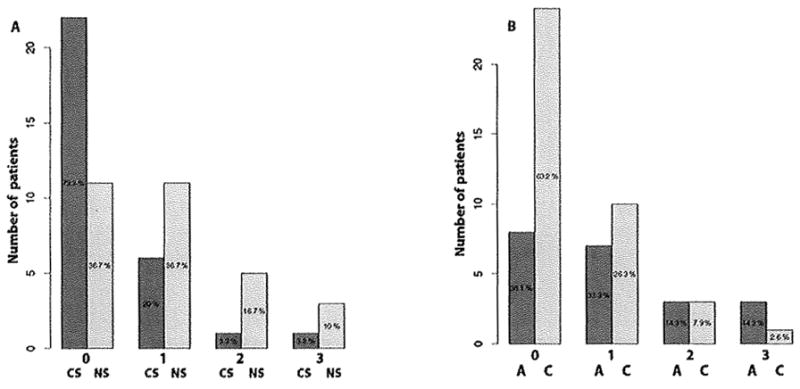
Distribution of somatic mtDNA mutation among different smoking and ethnic groups. A. Number of mtDNA mutation (0–3) was significantly higher (P=0.006) in the never-smoker (NS) compared to the current-smoker (CS) group. Percent of patients without mutation (0 mutation) or with mutation (1–3 mutation) is indicated for both the current-smoker and never-smoker group. B. Number of mtDNA mutations (0–3) was significantly higher (P=0.026) in the Asian (A) compared to the Caucasian (C) group. Percent of patients without mutation (0 mutation) or with mutation (1–3 mutation) is indicated for both the Asian and Caucasian populations.
Coding MtDNA Mutations affect Mitochondrial Respiratory Complex-I
To understand the impact of mtDNA mutation on different respiratory complexes, we examined the load of mtDNA mutation for each complex (Dasgupta et at., 2009). Only non-synonymous mtDNA mutations were considered for calculating the mutation load. Of the 20 non-synonymous coding mtDNA mutations, 14 were from complex-I, 2 from complex-III and 4 were from complex-IV (Table S1). MtDNA mutation load was significantly higher in complex-I compared to complex-III (P=0.013) and complex-IV (P=0.033) (Fig. 2A).
Figure 2.
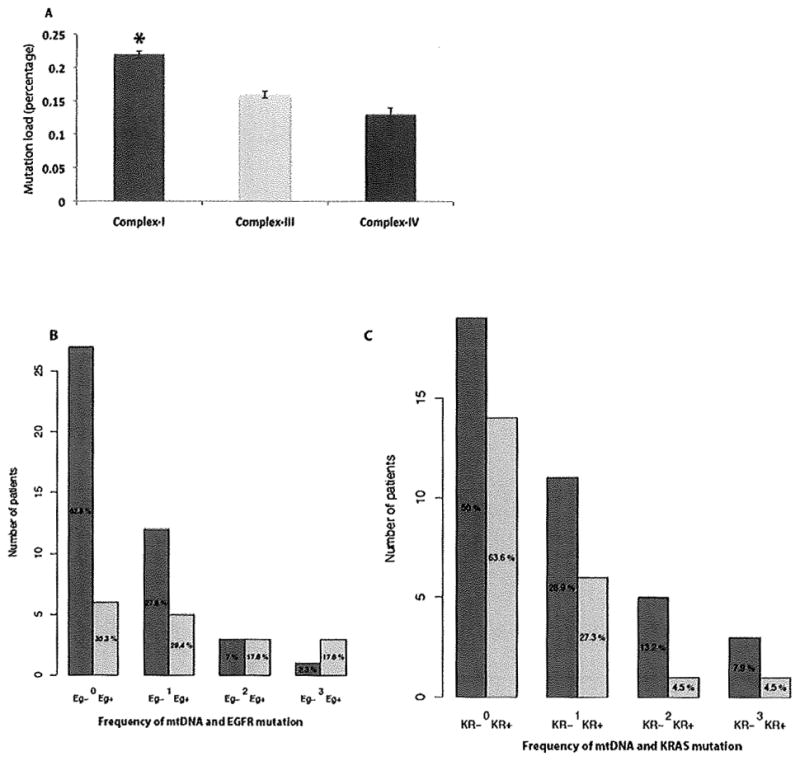
The effect of mtDNA mutation on different mitochondria encoded respiratory complexes and association between EGFR/KRAS and MtDNA mutation in the smoker and never-smoker lung cancer patients. A. Mutational load was significantly higher in Complex I (Indicated by asterisk) compared to complex-III (P=0.013) and complex-IV (P=0.033). B. Somatic mtDNA mutation was significantly higher (P=0.007) in never-smoker patients with EGFR mutation (Eg+) compared to the never-smoker patients without EGFR (Eg−) mutation. C. No significant association (P=0.238) was found between mtDNA and KRAS mutation in the current-smoker lung cancer patients.
EGFR/KRAS Gene Mutation in the Lung Cancer Patients and its Association with MtDNA Mutation
In earlier studies, alteration of EGFR and KRAS signaling pathways was implicated in never-smoker and smoker lung adenocarcinoma respectively (Sun et al., 2007; Subramanian and Govindan 2007). In recent studies, EGFR was also found to be localized and interact with respiratory complex-IV in the mitochondria (Boerner et al., 2004; Demory et al., 2009). To examine the extent of EGFR and KRAS mutation in both the patient cohorts, sequencing of exon 19 and 21 of EGFR and exon 2 of KRAS gene was performed. We also examined whether there was any correlation between mtDNA and EGFFi/KRAS gene mutation in both the patient cohorts. EGFR gene mutation was detected in 56.6% (17/30) cases of non-smoker patients only (Table 1–2 and Table S2) and found to be significantly associated (P=0.007) with mtDNA mutation (Fig. 2B). On the other hand, KRAS mutation was present in 63.3% (19/30) of the current-smoker and 10% (3/30) of the non-smoker patients (Table 1–2; Table S3), We did not detect any significant association (P=0.238) between KRAS and mtDNA mutation in the current-smoker group (Fig. 2C) where KRAS mutation was predominant.
Pattern of MtDNA Content in the Lung Cancer Patients
Alteration in the mtDNA content reflects a commonly observed increase in the number and copy of mtDNA in tumors (Jiang et al., 2005). We performed a quantitative real-time PCR to determine mtDNA content in patients’ tumors, and normal tissues from all the 60 lung cancer patients. This assay determines the relative rate of amplification of mtDNA sequence (Cox-I) compared to a sequence of nDNA (Beta-actin). We observed a significant increase (P=0.037) in the mtDNA content among the never-smoker compared to the smoker lung cancer patients (Fig. 3). The average fold-increase of the mtDNA ranges between 2.23 to 4.79 in the tumors compared to the corresponding normal. No association of mtDNA content was observed with age (P=0.358) (Figure S3), gender (P=0.623) (Figure S4), or ethnicity (P=0,336) (Figure S5).
Figure 3.

Pattern of mtDNA content in the never-smoker (NS) and current-smoker (CS) lung cancer patients. MtDNA content was determined by real-time PCR using nuclear encoded β-Actin and mtDNA encoded Cytochrome C oxidase I (COI) that quantitatively determines the relative rate of amplification of mtDNA sequence. MtDNA content was significantly higher (P=0.037) in the never-smokers compared to the current-smoker lung cancer patients.
Effect of ND5 Mutation on In Vitro Cell Proliferation and Invasion
In this study, fifty seven percent (8/14) of the non-synonymous complex-I mtDNA mutations were detected in the ND5 gene (Table S1). We have chosen one ND5 mutation from one of the two early stage (1A) patients with ND5 mutations (Table S1) to investigate its effect on cell growth and ROS production. We subsequently cloned this mutation from the ND5 gene (G13289A) and examined the impact of its forced overexpression in SW900 lung cancer cell line, which does not harbor mtDNA mutation in ND5 Gene (data not shown). The presence of the exogenous fusion protein in the mitochondria of the ND5 transfected cells was detected using anti-myc antibody by immunofluorescence (Fig. 4A, arrow). Compared to the ND5-wt transfected cells, we observed a significant increase (P=0.0031) of In vitro proliferation (Fig. 4B). The average number of BrdU positive cells in the ND5-mt group was 47±13 compared to the 12±3 cells in the ND5-wt transfected group. We also observed a significant increase (P=0.0002) in the number of invaded cells in the ND5-mt group (44±8) compared to the ND5-wt transfected group (8±3) (Fig. 4C). Representative photomicrograph is shown in the right panel.
Figure 4.
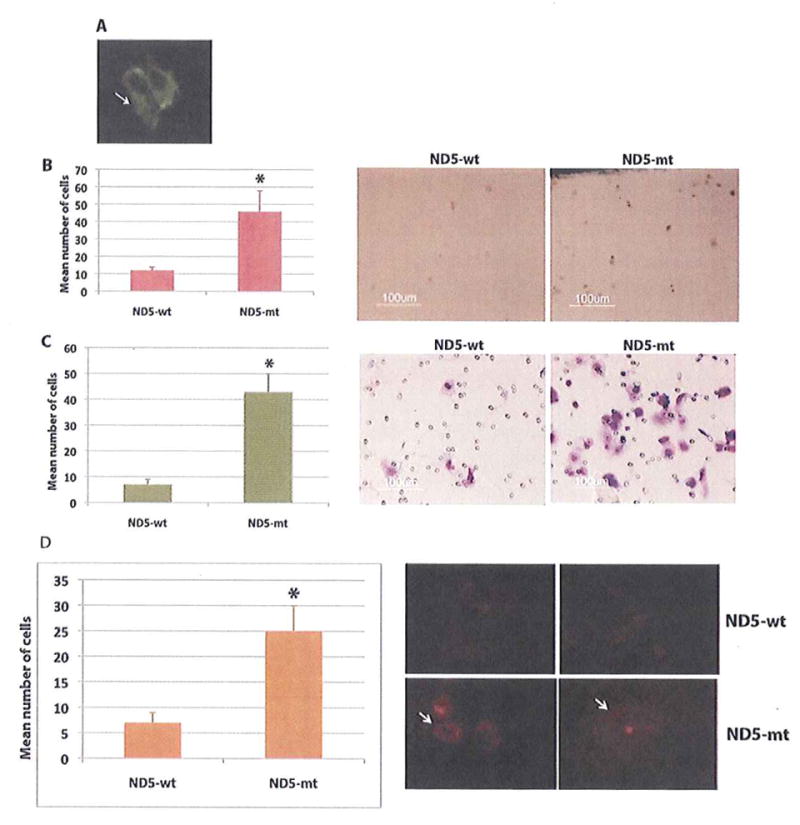
Impact of forced overexpression of ND5 gene on in vitro proliferation, invasion and superoxide production of lung cancer cells SW-900. A. The presence of the exogenous fusion protein in the mitochondria of the ND5 transfected cells was detected using anti-myc antibody by immunofluorescence (arrow). B. Number of proliferating cells was significantly higher (P=0.0031, indicated by asterisk) in the ND5 mutant (ND5-mt) transfected cells compared to the wild type (ND5-wt) transfected cells (Left panel). Representative photomicrograph from each experimental group is shown in the right panel. Magnification X 200. C. Number of invasive cells was also significantly higher (P=0.0002, indicated by asterisk) in the ND5 mutant (ND5-mt) transfected cells compared to the wild type (ND5-wt) transfected cells (Left panel). Representative photomicrograph from each experimentai group is shown in the right panel. Magnification X 400 C. D. Number of superoxide producing cells was determined by the MitoSox superoxide assay. Number of positive cells was significantly higher (P=0.0049, indicated by arrow) in the ND5 mutant (ND5-mt) transfected group compared to the wild type (ND5-wt) transfected group (Left panel). Representative photomicrograph from each experimental group is shown in the right panel. Magnification X 200.
Superoxide Production by the ND5 Transfected Cells
Earlier studies demonstrated that cancer cells with mtDNA mutations have increased ROS production including superoxide radicals with respiratory complex-I and III representing the major sites of ROS production (Dasgupta et al., 2010 and 2008; Zhou et al., 2007). To examine the extent of superoxide production in the ND5 transfected lung cancer cells, we performed the MitoSOX-Red mitochondrial superoxide indicator assay, which selectively and specifically detects superoxides in the mitochondria. We observed a significant increase (P=0.0049) in the number of positively stained cells in the ND5-mt transfected group (25±6) compared to the control ND5-wt transfected cells (7±3) (Fig. 4D). Representative photomicrograph is shown in the right panel.
Discussion
The “prime intellect” of a cancer cell is governed not only by nuclear genetic changes but also mitochondrial genetic alterations as envisioned long ago by Otto Warburg (Warburg et al., 1927; Warburg 1956). Only a few decades after Warburg’s emphatic contribution, a strong link appears to exist between mitochondrial dysfunction and tumorigenesis, reflected in the numerous somatic and rare germ line mtDNA mutations observed in different tumors (Chatterjee et al., 2011; Koppenol et al., 2011). Further emphasizing their role in tumor progression, recent studies also demonstrated a bona fide functional role for mitochondrial genetic alterations in different human cancers (Dasgupta et al 2008; Zhou et al., 2007), while also pointing out a potential application in biomarker development (He et al., 2010).
Lung cancer among never-smoker remains as an enigma with distinct molecular and biological features compared to the smoker lung cancer patients (Sun et al., 2007; Subramanian and Govindan 2007). Specific nuclear genetic alterations such as activation of the EGFR signaling pathway through EGFR mutation plays a key role in the development of lung cancer among a minority of never-smokers Sun et al., 2007). Prior to activation, EGFR remains as inactive monomer in the plasma (Fiorelli et al., 2011). Upon liqand binding to a monomeric unit, EGFR induces the formation of receptor homodimers. This dimerization of EGFR triggers intrinsic intracellular protein-tyrosine (Y) kinase activity that results in autophosphorylation of several tyrosine residues in the C-terminal domain. These include Y992, Y1045, Y1068, Y1148 and Y1173 (Downward et al., 1984). This autophosphorylation triggers downstream activation and signaling by several other proteins that associate with the phosphorylated tyrosines through their own phosphotyrosine-binding SH2 domains. These downstream signaling proteins initiate multiple signal transduction pathways, mainly the MAPK, Akt and JNK pathways, that lead to DNA synthesis and cell proliferation (Oda et al., 2005). Such proteins can modulate cell migration, adhesion, and proliferation as well. In addition, EGFR also induces Src activation, DNA synthesis and cytoskeleton changes in steroid dependent tumor cells (Mioliaccio et al., 2000: Castoria et al., 2001 and 2004).
However, to date, the nature and role of mitochondrial genetic alterations was not examined in never-smoker lung cancer patients and its association with EGFR gene mutation. Given the fact that EGFR was found to translocate to the mitochondria and interact with cytochrome c oxidase II (Cox-II; Complex-IV) (Boerner et al., 2004; Demory et al., 2009), we felt it important to understand the nature and functional consequences of mtDNA alterations in never-smokers, particularly in patients with EGFR gene mutation.
The mtDNA sequencing in all specimens was done in a blinded fashion without any prior information on the patients. Considering the polymorphic nature of mtDNA, appropriate methodologies and analytical tools were employed for data analysis and interpretation (Chatterjee et al., 2011; Dasgupta et al., 2010). Strikingly, mtDNA mutations were significantly higher among the never-smokers compared to the current-smoker lung cancer patients and targeted respiratory complex-I. Thus, there appear to be a smoking independent pathway for acquiring specific mtDNA mutation in these patients. Notably, two studies have reported somatic mtDNA mutations in complex-I (ND genes) in a small number of predominantly smoker lung cancer patients (Dasgupta et al., 2009a; Jakupciak et al 2005). Therefore, complex-I appears to be a frequent target for alteration in lung cancer patients, but now we know that this is true particularly among never-smokers with lung cancer. The mtDNA mutations were also prevalent among the never-smoker Asian compared to the smoker Caucasian patient population, which suggests an increased susceptibility of this population for acquiring mtDNA mutation, perhaps similar to acquisition of EGFR gene mutations (Sun et al., 2007; Subramanian and Govindan 2007). Moreover, forced overexpression of a complex-I specific mutant targeted to the mitochondria triggered increased superoxide production, in vitro proliferation and invasion of lung cancer cells. We analyzed three different stable clones and observed similar results on the ceil growth and proliferation. These results continue to support the notion that many mtDNA mutations have a functional role and are unlikely to be due to a passenger “bystander” effect. Although, we examined the impact of forced overexpression of only one mtDNA mutation in this study, it would be interesting to examine the impact of other mtDNA mutations obtained from patients with different clinical stages in the future. That could further enhance our understanding on the contribution of specific mtDNA mutation driving tumor progression in concert with nuclear genetic changes. To our knowledge, this is the first study to demonstrate the spectrum of somatic mtDNA mutation in never-smoker lung cancer patients and the impact of forced overexpression of a mtDNA mutation in lung cancer cells, detected in a never-smoker lung cancer patient.
Along with targeted mtDNA mutations, key n uclear DNA (nDNA) alterations are necessary for driving and maintaining progressive tumor growth. Notably, EGFR gene mutation was found to be a key alteration pathway and specific among never-smoker lung cancer patients of Asian ethnicity with adenocarcinoma histology; where as KRAS mutation was found predominantly among the smokers (Sun et al., 2007). Our result on the pattern of EGFR and KRAS gene mutation among the never-smoker and current-smokers is completely consistent with that reported earlier (Sun et al., 2007), thereby supporting the accuracy of the smoking histories of these patients. Of note, in this study most patients of smokers were Caucasian while most patients of non-smokers were Asian in origin. The presence of a significant number of mtDNA mutations in the non-smoker Asian patients with adenocarcinoma subtype carrying frequent EGFR gene mutation suggests a possible functional coordination between different respiratory complexes and EGFR activation favoring tumor growth. A recent study demonstrated translocation of EGFR into the mitochondria and associated reduction of Cox-II (complex-IV) activity and cellular ATP production, which suggested a new mechanism for EGFR in driving oncogenesis in concert with mtDNA alteration (Demory et al., 2009). Thus, an interplay between perturbed mitochondrial complex-I and EGFR activation is possible, where EGFR translocates to the mitochondria and regulates complex-I activity for sustained tumor growth. In this light, it is it is possible that the never smoker Asians with EGFR gene mutation are more susceptible to acquire mtDNA mutation compared to the smoker Caucasian patients with KRAS gene mutation for establishing a growth favorable EGFR -mitochondrial signaling loop (Demory et al., 2009). At the same time it remains to be determined that other than EGFR gene mutation, whether the difference of mtDNA mutation among the smoker and never smoker groups was derived from smoking status or due to the difference in ethnicity. One possibility could be a different life style and nutritional habit of the Asian with the habit of taking different chewing products such as betel nuts, paan, chaalia, gutka, naswar and areca that are widely used in the Asian subcontinent, South East Asia and South Pacific Islands (Dasgupta et al., 2011).
An interesting possibility is that increased endogenous ROS production through perturbed complex-I assembly and altered function due to targeted mutation(s) in complex-I predispose Asian populations to subsequent specific EGFR mutations (Dasgupta et al., 2008; Nitta et al., 2010). This might help explain why Asian lung cancer patients in general harbor such a high frequency of these EGFR mutations. In support of this notion, we observed significant superoxide production in the ND5-mutant (complex-I) transfected lung cancer cells. The increased ROS in turn could diffuse into the cytoplasm or nucleus and select for altered growth regulatory genes for tumor growth maintenance by inhibiting apoptosis as observed in bladder cancer (Dasgupta et al., 2008, Dasgupta et al., 2009b). Moreover, common EGFR mutations are often T to G transversions (L858R) and could be due to high ROS levels in the nucleus and mispairing of GO lesions with adenine during replication. However, it remains to be determined whether EGFR mutation occurs first and directly interacts with and regulates complex-I activity in the mitochondria, thus selecting for mtDNA mutations among these patients. In support of this hypothesis, one recent study revealed that EGFRvIII over-expression in glioblastoma cells caused increased levels of ROS, DNA strand break accumulation, and genome instability (Nitta et al 2010).
Increased mtDNA content is also an indicator of increased mitochondrial function necessary for favoring tumor growth (Jiang et al., 2005). Increased mtDNA content was reported in different tumors, but to our knowledge not in the never-smoker lung cancer patients (Dasgupta et al., 2009a; Jiang et al., 2005; Mambo et al., 2005). In the present study, concurrence of increased mtDNA content and frequent coding mtDNA mutations with a complex specific bias among the never-smoker indicates that these events occurred simultaneously. Measuring the mtDNA content index could also be a useful and easy detector for monitoring altered mitochondrial function.
Our study demonstrates the potential role of mitochondrial genetic alterations among never-smokers, and raises new questions about the relationship with common EGFR mutations often seen in the same lung cancer patients. Identification of specific or signature mtDNA mutations and understanding their biological role in concert with nDNA alterations will help us understand and develop new strategies for detecting and managing lung cancer that occur among non-smokers. Importantly, non-smokers or patients that smoked only for a few years in the distant past are rapidly becoming a common presentation of new lung cancer cases in the clinic.
Supplementary Material
Acknowledgments
This work was supported by U01 CA 084986 (DS); US-Egypt Joint Science and Technology fund-58-3148-169 and A D Williams fund-646299 (SD).
Footnotes
Conflict of interest
None
Additional Supporting Information may be found in the online version of this article.
Literature cited
- Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y485 on the epidermal growth factor receptor mediated binding to the mitochondrial protein cytochrome c oxidase subunit II. Moll Cell Biol. 2004;24:7059–7071. doi: 10.1128/MCB.24.16.7059-7071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Dasgupta S, Sidransky D. Mitochondrial subversion in cancer. Cancer Prev Res. 2011;4:638–654. doi: 10.1158/1940-6207.CAPR-10-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria G, Migliaccio A, Bllancio A, Pi Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimuiated MCF-7 cells. EMBO J. 2001;20:6050–6059. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoria G, Migliaccio A, Di Domenico M, Lombardi M, de Falco A, Varricchio L, Bilancio A, Barone MV, Auricchio F. Role of atypical protein kinase C in estradiol-triggered G1/S progression of MCF-7 cells. Mol Cell Biol. 2004;24:7643–7653. doi: 10.1128/MCB.24.17.7643-7653.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Dash R, Das SK, Sarkar D, Fisher PB. Emerging strategies for the early detection and prevention of head and neck squamous cell cancer. J Cell Physiol. 2011 Apr 4; doi: 10.1002/jcp.22767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008;68:700–706. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Forced cytochrome B gene mutation expression induces mitochondrial proliferation and prevents apoptosis in human uroepithelial SV-HUC-1 cells. Int J Cancer. 2009b;125:2829–2835. doi: 10.1002/ijc.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Koch R, Westra WH, Califano JA, Ha PK, Sidransky D, Koch WM. Mitochondrial DNA mutation in margins and tumors of recurrent HNSCC patients. Can Prev Res. 2010;3:1205–1211. doi: 10.1158/1940-6207.CAPR-10-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Yung RC, Westra WH, Rini DA, Brandes J, Sidransky D. Following mitochondrial footprints through a long mucosal path to lung cancer. PLoS One. 2009a;4:e6533. doi: 10.1371/journal.pone.0006533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009;284:36592–36604. doi: 10.1074/jbc.M109.000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobashi Y, Suzuki S, Kimura M, Matsubara H, Tsubochi M, Imoto I, Qoi A. Paradigm of kinase-driven pathway downstream of epidermal growth factor receptor/Akt in human lung carcinomas. Human Pathol. 2011;42:214–226. doi: 10.1016/j.humpath.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Downward J, Parker P, Waterfield MP. Autophosphorylation sites on the epidermal growth factor receptor. Nature. 1984;311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- Fiorelli A, Ricciardi C, Pannone G, Santaro A, Bufo P, Santini M, Serpico R, Rullo R, Pierantoni GM, Pi Domrnico M. Interplay between steroid receptors and neoplastic progression in sarcoma tumors. J Cell Physiol. 2011 Feb 1; doi: 10.1002/jcp.22645. [DOI] [PubMed] [Google Scholar]
- He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA, Jr, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakupciak JP, Maragh S, Markowitz ME, Greenberg AK, Hoque MO, Maitra A, Barker PE, Wagner PD, Rom WN, Srivastava S, Sidransky D, O’Connell CD. Performance of mitochondrial DNA mutations detecting early stage cancer. BMC Cancer. 2005;8:285. doi: 10.1186/1471-2407-8-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang WW, Masayesva B, Zahurak M, Carvalho AL, Rosenbaum E, Mambo E, Zhou S, Minhas K, Benoit N, Westra WH, Alberg A, Sidransky D, Koch W, Califano J. Increased mitochondrial DNA content in saliva associated with head and neck cancer. Clin Cancer Res. 2005;11:2486–2491. doi: 10.1158/1078-0432.CCR-04-2147. [DOI] [PubMed] [Google Scholar]
- Jonsson S, Varella-Garcia M, Miller YE, Wolf HJ, Byers T, Braudrick S, Kiatsimkul P, Lewis M, Kennedy TC, Keith RL, Bjornsson J, McWilliams A, Lam S, Hirsch FR, Franklin WA. Chromosomal aneusomy in bronchial high-grade lesions is associated with invasive lung cancer. Am J Respir Crit Care Med. 2008;177:342–347. doi: 10.1164/rccm.200708-1142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Mambo E, Chatterjee A, Xing M, Tallini G, Haugen BR, Yeung SC, Sukumar S, Sidransky D. Tumor-specific changes in mtDNA content in human cancer. Int J Cancer. 2005;116:920–924. doi: 10.1002/ijc.21110. [DOI] [PubMed] [Google Scholar]
- Mateus AR, Seruca R, Machado JC, Keller G, Oiiveira MJ, Suriano G, Luber B. EGFR regulates RhoA-GTP dependent cell motality in E-cadherin mutant cells. Human Mol Genet. 2007;16:1639–1647. doi: 10.1093/hmg/ddm113. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, Castoria G, Pi Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406–5417. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta M, Kozono D, Kennedy R, Stommel J, Ng K, Zinn PO, Kushwaha D, Kesari S, Furnari F, Hoadley KA, Chin L, DePinho RA, Cavenee WK, D’Andrea A, Chen CC. Targeting EGFR induced oxidative stress by PARP1 inhibition in glioblastoma therapy. PLoS One. 2010;5:e10767. doi: 10.1371/journal.pone.0010767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Moi Syst Biol. 2005;1:1–17. doi: 10.1038/msb4100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nature Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Avila-Tang E, Harris CC, Herman JG, Hirsch FR, Pao W, Schwartz AG, Vahakangas KH, Samet JM. Lung cancer in never-smokers: molecular profiles and therapeutic implications. Clin Can Res. 2009;15:5646–5661. doi: 10.1158/1078-0432.CCR-09-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Shames DS, Gazdar AF, Minna JD. A Translational View of the Molecular Pathogenesis of Lung Cancer. J Thoracic Oncol. 2007;2:327–343. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- Subramanian J, Govindan R. Lung cancer in never smokers-a review. J Clin Oncol. 2007;25:561–569. doi: 10.1200/JCO.2006.06.8015. [DOI] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers- a different disease. Nature Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–134. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- www.cancer.gov
- Zhou S, Kachhap S, Sun W, Wu G, Chuang A, Poeta L, Grumbine L, Mithani SK, Chatterjee A, Koch W, Westra WH, Maitra A, Glazer C, Carducci M, Sidransky D, McFate T, Verma A, Califano JA. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc Natl Acad Sci U S A. 2007;104:7540–7545. doi: 10.1073/pnas.0610818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Kassauei K, Cutler DJ, Kennedy GC, Sidransky D, Maitra A, Califano J. An oligonucleotide microarray for high-throughput sequencing of the mitochondrial genome. J Mol Diagnostics. 2006;24:476–482. doi: 10.2353/jmoldx.2006.060008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.