

Abstract
Mutants of the fungus Ustilago maydis defective in the RecQ helicase Blm are highly sensitive to killing by the DNA replication stressor hydroxyurea. This sensitivity or toxicity is dependent on the homologous recombination (HR) system and apparently results from formation of dead-end HR DNA intermediates. HU toxicity can be suppressed by deletion of the gene encoding Brh2, the BRCA2 ortholog that serves to regulate HR by mediating Rad51 filament formation on single-stranded DNA. Brh2 harbors two different DNA binding domains that contribute to HR function. DNA-binding activity from a single domain is sufficient to provide Brh2 functional activity in HR, but to enable HU-induced killing two functional DNA-binding domains must be present. Despite this stringent requirement for dual functioning domains, the source of DNA-binding domains is less critical in that heterologous domains can substitute for the native endogenous ones. The results suggest a model in which the nature of the DNA lesion is an important determinant in the functional response of Brh2 action.
INTRODUCTION
Repairing damaged DNA by homologous recombination (HR) is a mechanism for maintaining stability and restoring integrity of the genetic material common throughout the domains of life. The HR repair pathway can accommodate damaged DNA channeled from a number of different sources. DNA broken by the action of exogenous genotoxins, ionizing radiation, replication-associated events, or programmed cellular processes can be restored through the HR pathway (Budzowska & Kanaar, 2009, Mimitou & Symington, 2009, San Filippo et al., 2008). During DNA replication the HR system can be utilized for repair of a broken sister chromatid arising from fork passage over a nicked template and for processing stalled or collapsed replication forks. Repair of single-stranded DNA (ssDNA) gaps formed as a consequence of replication fork skipping past a lesion on the template strand can also be processed through the HR system (Lehmann & Fuchs, 2006, Lopes et al., 2006). Aberrant DNA structures formed under replication stress or resulting from encounter of obstacles during DNA synthesis can be generated and also resolved by the HR system (Liberi et al., 2005, Mankouri et al., 2011, Saintigny et al., 2001).
In eukaryotes DNA substrates directed into the HR system are engaged by Rad51, which assembles on exposed single-stranded regions to form orderly filamentous structures active for homologous pairing and DNA strand invasion (Heyer et al., 2010). Access of Rad51 to DNA is regulated by mediator proteins (San Filippo et al., 2008). In many taxa the primary mediator appears to be of the BRCA2 protein family, the eponymous member being the product of a hereditary breast cancer predisposition gene in human (Holloman, 2011, Thorslund & West, 2007). BRCA2 is an extremely large protein (>3000 amino acids) with characteristic features that include crucial sites for interaction with Rad51 and DNA. BRCA2 interacts with Rad51 through the BRC repeat, a conserved motif of about 35 amino acid residues, reiterated eight times in the mammalian proteins. An additional site of Rad51 interaction unrelated in sequence to the BRC motif is present at the extreme C-terminus of BRCA2. The BRC repeats promote the nucleation and growth of the Rad51 filament on ssDNA to stimulate DNA strand exchange (Carreira et al., 2009, Shivji et al., 2009), while the C-terminal Rad51-interaction site helps coordinate the filament assembly with the cell cycle (Ayoub et al., 2009). The C-terminal region of BRCA2 contains a conserved DNA-binding domain consisting of a helix-rich module followed by three tandem OB (oligonucleotide/oligosaccharide-binding) folds that are structurally related to the binding folds in the ubiquitous single-strand DNA-binding protein RPA (Yang et al., 2002), which is essential for numerous processes in DNA replication, recombination and repair. BRCA2 promotes the activity of Rad51 in HR through the coordinated action of these different domains. It enforces delivery of Rad51 to ssDNA, attenuates association of Rad51 with double-stranded DNA (dsDNA), and accelerates the RPA displacement from ssDNA by Rad51 (Jensen et al., 2010). Through its ability to promote and maintain formation of the Rad51 filament, BRCA2 performs a critical role in homology-directed repair of broken DNA molecules. Moreover, BRCA2 provides a protective role for replication forks–outside of its repair function–in helping prevent degradation of newly replicated strands at stalled replication forks (Hashimoto et al., 2010, Schlacher et al., 2011). This protective function involves recruitment of Rad51 to stalled, uncoupled replication forks so as to inhibit nucleolytic processing of nascent strands.
Brh2 is the BRCA2 ortholog in the fungus Ustilago maydis but appears as a more streamlined version. It is one-third the size of BRCA2 and contains only a single Rad51-binding BRC motif located in the N-terminal region of the protein (Kojic et al., 2002, Kojic et al., 2003). Mapping studies revealed that a region at the extreme C-terminus structurally unrelated to the C-terminal Rad51-binding motif in BRCA2 also binds Rad51 (Zhou et al., 2007). In addition, genetic analysis in U. maydis and co-precipitation methodology established that an element related to a DMC1-interaction motif in BRCA2 also contributes to Rad51 filament assembly (Kojic et al., 2011), DMC1 being a meiosis-specific RAD51-related strand exchange protein. In Brh2 two separate regions in the protein interact with DNA. Within the C-terminus is the domain (CTD) that is structurally related to the DNA-binding domain of mammalian BRCA2 although it appears to lack the third OB fold based on sequence alignment (Kojic et al., 2003). Within the N-terminal part of the protein is an empirically determined DNA-binding domain (NBD) that preferentially binds to D-loops or branched structures with protruding single-stranded regions (Zhou et al., 2009a).
BLM, the Bloom’s syndrome RecQ family DNA helicase, could be considered able to provide a counterbalance to BRCA2 function due to its ability to dissociate the Rad51-ssDNA filament as well as D-loop strand invasion intermediates (Bachrati et al., 2006, Bugreev et al., 2007, van Brabant et al., 2000). But besides this anti-recombinational activity, BLM or orthologs such as Sgs1 from budding yeast have been shown to perform multiple additional functions in overcoming obstacles to DNA replication (Liberi et al., 2005, Mankouri et al., 2011), preparing DNA ends for recombinational repair (Mimitou & Symington, 2009), and dissolving double Holliday junction intermediates (Wu & Hickson, 2003). In U. maydis, blm loss-of-function mutants are crippled in growth when cultured in the presence of the replication stressor hydroxyurea (HU), but viability is substantially restored when Brh2 is deleted (Mao et al., 2009). This observation suggests that processing of HU-induced DNA structures by HR in the course of repair can lead to formation of intermediates, which become toxic in the absence of Blm action, as previously proposed in studies with budding and fission yeasts (Gangloff et al., 2000, Hope et al., 2005).
U. maydis mutants lacking Brh2 are defective in double-strand break repair, heteroallelic recombination, and meiosis, and are extremely sensitive to DNA damaging agents including ionizing radiation, alkylating chemicals, and UV light (Kojic et al., 2002). Since UV damage is thought to lead to single-stranded gaps in daughter strands during DNA replication as the replication fork occasionally skips past lesions, the UV sensitivity of brh2 mutants is likely a consequence of genome instability due to failed recombinational repair of the single-stranded gaps.
We showed previously that certain mutant variant forms of Brh2 with deletions of the entire C-terminal domain or with a heterologous DNA-binding domain in place of the C-terminal domain provide a level of activity that permits cells expressing these variants to survive UV irradiation as well as wild type (Kojic et al., 2005). This observation implies that these Brh2 variants are active in promoting repair activity and enable processing UV damage-derived DNA structures via the HR pathway. Nevertheless, the action of these Brh2 variants should not be categorized as functionally normal in that strains expressing these variants exhibit elevated levels of both double-strand gap repair and allelic recombination, pointing to some dysfunction in how recombination is executed. Taken together, however, these findings suggest a model for Brh2 in which the N-terminal and C-terminal regions cooperate in some as yet unknown way to promote recombination in a precise and appropriately regulated manner (Zhou et al., 2009b).
Here we were interested in exploring how the N-terminal and C-terminal regions of Brh2 contribute to functional activity by studying mutant variants deleted of either the NBD or CTD, or else containing heterologous DNA-binding modules in place of the endogenous domains (Kojic et al., 2011). The aim was to assess the contribution of the Brh2 domains in response to UV-induced damage or HU-induced replication fork stalling.
RESULTS
Recombination proficiency and HU toxicity in the absence of Blm function
We reported previously that lethality of a U. maydis blm mutant strain caused by the DNA replication stressor HU could be attenuated by deleting the gene encoding Brh2 required for HR (Mao et al., 2009). To determine whether the observed attenuation was a general characteristic of the HR system of U. maydis or a peculiar response to deletion of the Brh2 gene, as it is formally possible that a specific HU-induced activity of Brh2 is abrogated by Blm, we tested survival of the blm mutant after deleting other recombination genes. We measured HU sensitivity, and also resistance to UV as an indirect measure of recombinational repair proficiency. UV is a potent inducer of recombination and inactivation of HR genes results in extreme sensitivity to UV in U. maydis (Holloman et al., 2008). When the gene for Brh2, Rad51, Rec2 (a Rad51 paralog), or Rad54 (a chromatin remodeling translocase that stimulates Rad51) was deleted, there was loss of resistance to UV and, in every case, concomitant resistance to killing by HU (Fig. 1A). Therefore, in accord with other systems, it would appear that HR proficiency in U. maydis is a general feature that sets the stage for HU-induced lethality in a Blm-deficient mutant.
Figure 1.

HU poisoning and homologous recombination dependence. A. Survival of mutant strains after UV irradiation (120 J/m2) or exposure to HU (4 mM). Strains indicated were grown in liquid medium, adjusted to a density of 2 × 107 per ml spotted on solid medium as serial 10-fold dilutions from left to right. Images were recorded after 2–3 days of incubation. B. CHEF gels of genomic DNA complements of indicated strains. HU (20 mM) was added to exponentially growing cells at a density of 2 × 107 per ml for a period of 3hrs. For recovery the cells were collected, washed free of HU and resuspended in fresh medium. Aliquots removed at the indicated times were prepared for CHEF gel electrophoresis. M--chromosomal DNAs from Saccharomyces cerevisiae were run as size standards. C. Mutant strains including those indicated expressing RusA were tested for survival after UV irradiation (120 J/m2), exposure to MMS (0.01%), or HU (4 mM).
For simplicity in work described below, the phrase “HU-induced lethality” or variations such as “HU toxicity,” etc., will refer specifically to HU-induced toxicity in the context of the Blm-deficient mutant.
DNA structure resulting from HU stress
Single-stranded breaks and gaps generated following UV irradiation, likely by replication fork-restart downstream of sites of UV-induced DNA photodamage (Lehmann & Fuchs, 2006, Lopes et al., 2006), are substrates for HR action as evinced by the strong induction of HR by UV (Roman & Jacob, 1958) and the sensitivity of HR mutants to UV (Holloman et al., 2008). However, the nature of the structure of the putative HU-induced DNA intermediate that becomes toxic in the absence of functional Blm activity is unclear. HU specifically inhibits ribonucleotide reductase resulting in depletion of nucleotide pools and as a consequence DNA replication is disturbed (Timson, 1975). HU treatment is presumed to provoke generation of DNA structures with single-stranded gaps as replication forks stall or as the coordination of leading and lagging strand synthesis breaks down (Fabre et al., 2002), although certain off-target secondary events might also contribute to DNA damage (Davies et al., 2009). But in contrast to the processing of UV-induced DNA damage, resolution of DNA intermediates caused by HU treatment depends on the action of Blm related RecQ helicases rather than through the homology-directed repair activity of the HR system (Liberi et al., 2005).
As an approach for gaining some insight into the nature of the DNA lesion(s) induced by HU stress, we examined the appearance of chromosomes in strains under investigation here by pulsed field gel electrophoresis (Fig. 1B). U. maydis has 23 chromosomes ranging in size from 0.34 to 2.5 Mb (http://mips.helmholtz-muenchen.de/genre/proj/ustilago/). We were surprised to note that in all the strains HU tested treatment for even a relatively brief duration caused the disappearance of the entire complement of chromosomal DNAs from the gel. However, the observations that no extensive DNA fragmentation was detectable in the gels and that the chromosomes reappeared shortly after HU was removed from the medium suggest that the DNA had not been degraded. Rather, it seems that the chromosome-sized DNA molecules failed to enter the gel, presumably due to being rendered into conformational state that precludes electrophoresis. In the example of wild type, after removal of HU and addition of fresh culture medium, cells were able to resume growth after a short recovery period with little loss of viability (data not shown) indicating that no lasting damage had been done. After a recovery period of 1–2 hours the entire chromosome complement once again became evident after electrophoresis suggesting that HU-induced lesions had been repaired (Fig. 1B). Similarly, the chromosome complement from brh2 mutant cells disappeared from the gel after HU treatment and then reappeared, but even faster than in wild type. In contrast, the chromosome complement from blm mutant cells became evident more slowly after recovery from HU treatment, while chromosomes from the blm brh2 double mutant appeared to be restored slightly faster than the blm single mutant. Two conclusions can be drawn from this analysis. First, at the gross level of DNA structure discernible by pulsed field gel electrophoresis, it appears that HU treatment results in a global change in the genome structure. Second, chromosomal integrity can be restored upon HU removal but restoration appears less efficient in blm mutant compared to brh2 mutant.
We considered the possibility that the HU-induced change causing disappearance of the chromosomal DNAs might be due to generation of Holliday-junction-containing X-forms. Since Blm orthologs from yeast and human have been shown capable of removing double Holliday junctions (Mankouri et al., 2011, Wu & Hickson, 2003), the absence of such an activity might account for the sensitivity of the blm mutant to HU. However, when the Holliday junction resolvase RusA was expressed in blm cells there was little or only incremental improvement in survival after HU treatment (Fig. 1C). On the other hand complementation of the extreme UV and methylmethane sulfonate (MMS) phenotype of the blm mus81 double mutant by RusA was remarkably complete indicating that it could substitute for Mus81 in resolving MMS-induced, but not HU-induced HR intermediates. Because it has been established in other systems that MMS treatment causes formation of Holliday junctions or X-forms (Liberi et al., 2005, Mankouri et al., 2011), our findings suggest that some kind of HR DNA intermediate is generated during HU treatment, but evidently not containing just X-forms, if at all. Apparently something more than generation of X-forms takes place. Stated another way, while an X-form structure could well be produced as an intermediate(s) during the HR process, it appears that the HU toxicity in the blm mutant does not result from an inability to resolve X-forms, but rather to some other as yet undefined dead-end structure.
Contribution of the DNA-binding domains to HU-induced toxicity
Two different DNA-binding domains contribute to Brh2 functional activity–the NBD located in the N-terminal region, and the OB fold-containing CTD (Zhou et al., 2009a). This latter is the site for interaction with the regulator protein Dss1 (Zhou et al., 2009b). A surprising observation made previously was that a strain expressing a truncated form of Brh2 deleted of the entire CTD (e.g., Brh2ΔC551) was proficient in recombination (Kojic et al., 2005). It exhibited resistance to UV, proficiency in allelic recombination and double-strand break repair, and independence of the requirement for Dss1 as a cofactor. Similarly, a strain expressing a chimeric Brh2, consisting of the N-terminal region fused to a heterologous DNA-binding domain comprised of the RPA70 single-strand binding protein subunit (Brh2ΔC551::RPA70) was resistant to UV and exhibited elevated recombination activity regardless of Dss1 status (Kojic et al., 2005).
Given the recombination proficiency of these two different Brh2 variants, it seemed reasonable to conclude that both would cause HU-induced lethality in the absence of Blm activity. Nevertheless, expression of these Brh2 variants in the blm mutant resulted in different outcomes in response to DNA replication stress (Fig. 2A). Expression of mutant variant Brh2ΔC551 lacking the CTD did not reverse the HU resistance, while expression of the chimeric hybrid Brh2ΔC551::RPA70 restored HU sensitivity causing cell killing. Therefore, recombination proficiency per se does not necessarily lead to HU-induced toxicity in the absence of Blm. In other words, it appears that Brh2 deleted of the CTD is still capable of providing sufficient functional activity for recombinational repair of UV-induced lesions, but cannot enable generation of the presumed HR intermediate induced by replication stress that is toxic in the absence of Blm activity. In contrast, coupling an additional DNA-binding domain, as in the Brh2ΔC551::RPA70 hybrid, potentiates the activity of the N-terminal region and enables processing of putative HU-induced DNA intermediates to a toxic form.
Figure 2.
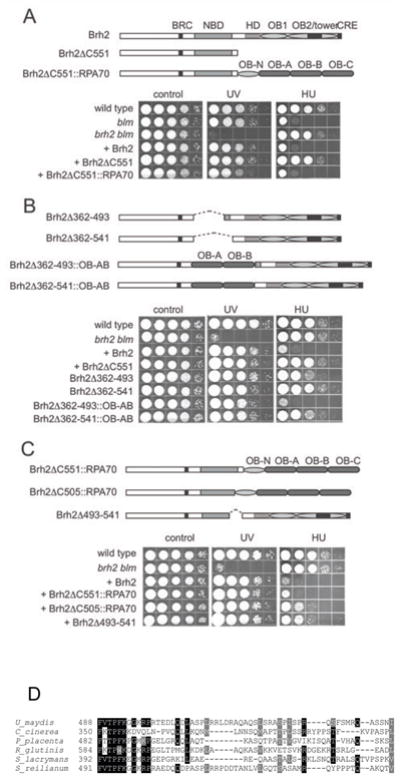
DNA-binding domain contribution to Brh2-promoted UV survival or HU poisoning. A–C. Survival of brh2 blm mutant strains expressing Brh2 variants or hybrids with RPA70 OB-folds, as illustrated schematically, after UV irradiation (120 J/m2) or exposure to HU (4 mM). D. Sequence alignment of the 49 residue spacer region of Brh2 in fungal orthologs. As an anchor for orientation the conserved FVTP motif important for Dmc1 interaction is shown, but the spacer region begins at U. maydis Brh2 K493. Accession numbers of Brh2 orthologs: Coprinopsis cinerea EAU92556, Postia placenta EED79697, Rhodotorula glutinis EGU12633, Serpula lacrymans ENG93090, Sporisorium reilianum CBQ73595.
In the reciprocal experiment we examined whether the same response was true for Brh2 forms lacking the NBD or containing a substitute DNA-binding domain. DNA-binding activity of the N-terminal region of Brh2 was previously mapped by direct biochemical assay associated with overlapping polypeptides in conjunction with genetic complementation of the brh2 mutant phenotype by truncations of the N-terminal fragment (Zhou et al., 2009a). The region was localized approximately to residues 359–503 although the precise boundaries remain uncertain in the absence of more rigorous structural information. While the DNA-binding activity associated with the NBD had been previously determined to be more avid than that associated with the isolated CTD, deletion of NBD from full length Brh2 did not result in loss of repair capacity as measured by UV resistance (Fig. 2B). Two Brh2 forms (Brh2Δ362–493 and Brh2Δ362–541) differing by 49 residues at the distal endpoint of the internal deletion were tested and appeared comparable in conferring DNA repair activity. However, brh2 mutant strains expressing these two forms were not poisoned by HU in the absence of Blm activity, similar to the finding with Brh2ΔC551, which retains the NBD but is deleted of the CTD. These observations suggest that a single DNA-binding domain suffices for Brh2 to promote proficiency in recombinational repair after UV damage, but that both the N-terminal and C-terminal DNA-binding domains of Brh2 must be in place to establish the appropriate recombination intermediate for HU-induced toxicity in the absence of Blm activity.
In a test for the degree of stringency in DNA-binding-domain structure required for functional activity, the NBD region from the two Brh2 mutant isoforms was replaced by a heterologous module consisting of OB-A and OB-B folds (OB-AB) from RPA70 (Fig. 2B). Complementation of UV sensitivity was conferred by expression of these chimeric Brh2 variants. However, restored sensitivity to HU was evident with only one of the Brh2 variants (Brh2Δ362–493::OB-AB), not the other (Brh2Δ362–541::OB-AB). This observation shows that the OB-AB domain can substitute for the NBD to a certain extent for functional proficiency, but that the region between residues 493 and 541 contributes some additional property that is important for promoting formation of the HU-induced toxic intermediate. In the absence of more concrete information about its structure or function, we shall designate this 49 amino acid region as the “spacer” in discussion below because it links the N-terminal and C-terminal domains.
Two-DNA-binding-domain model for HU toxicity
With the spacer region present it seemed apparent that substituting the NBD with the OB-AB module was sufficient to form a chimeric Brh2 active in generating a HU-inducible toxic intermediate in the blm mutant. To examine this spacer feature in an alternative context, we tested the requirement for the spacer in the reciprocal configuration by swapping the CTD for the RPA70 OB-fold array retaining or deleting the spacer region (Fig. 2C). In agreement with the NBD replacement studies, the Brh2ΔC551::RPA70 and Brh2ΔC505::RPA70 chimeric proteins both promoted recombinational repair activity after UV damage, but the Brh2ΔC551::RPA70 chimeric fused with the spacer region was more potent in HU-induced toxicity than the fusion without the spacer. The results suggest that severe toxicity caused by HU exposure to the blm mutant is caused when a Brh2 isoform harboring two DNA-binding domains separated by the spacer region is present.
No protein motifs are recognizable within the spacer sequence and there is considerable divergence even among fungal orthologs, although a sprinkling of conserved residues is evident (Fig. 2D). A noteworthy feature, however, is the excess number of basic residues to the extent that the calculated pI of the spacer sequence is 11.8.
Domain interplay
Previously, using pull-down methodology and protein crosslinking procedures, we obtained evidence that Brh2 could interact with itself to form dimeric and higher order structures (Zhou et al., 2007). Intermolecular interaction appeared to be mediated through the region spanning the helical domain and OB1 fold. Genetic evidence supporting protein-protein interaction came from the observation that two different inactive mutant forms could cooperate to complement the UV phenotype when co-expressed in the brh2 mutant. One inactive variant was deleted at the N-terminus to remove the Rad51-interacting BRC motif while the other was truncated at the C-terminus to remove a short tract of residues encompassing the Rad51-interacting region there. Separately the mutant variants were inactive, but when expressed together they were able to restore UV resistance. This same cooperation was evident also in the absence of Blm activity (Fig. 3A). These findings suggest that Brh2 mutant forms cooperate through intermolecular interaction in recombinational repair of DNA lesions caused by UV. However, when we tested whether the same inactive Brh2 variants could cooperate in response to HU, we found that the cells did not become sensitive to HU challenge (Fig. 3A). These results suggest that Brh2 cooperation, which presumably reflects domain interaction, responds to the nature of the DNA substrate. Response of Brh2 to stalled replication forks appears more demanding in terms of precise domain architecture than response to UV-induced DNA lesions.
Figure 3.

Domain interplay. A. Intermolecular interaction. N- and C-terminal truncated forms of Brh2 shown schematically were introduced into the brh2 blm mutant and tested for intermolecular complementation with UV (120 J/m2) and HU (4 mM). B. Survival of brh2 blm mutant strains expressing Brh2-Rad52 DBD hybrid after UV irradiation (120 J/m2) or exposure to HU (4 mM).
In all the different variants of Brh2 above in which the interplay between two DNA binding domains was tested for activity in either the intramolecular or intermolecular configuration, the C-terminal binding domain was composed of an OB-fold array. To test whether an OB-fold was a requirement, we constructed a Brh2 variant with a different C-terminal DNA binding, namely the DNA-binding/self-association domain from Rad52 (Kojic et al., 2008). This fold bears no resemblance to the OB-fold (Kagawa et al., 2002, Singleton et al., 2002). Nevertheless, the Brh2ΔC551::Rad52DBD did not impair UV complementation and was active in supporting the restoration of HU sensitivity (Fig. 3B). We conclude that there is plasticity in the structural requirements for recognizing and presumably processing the HU-induced DNA intermediates, and that tandem modular arrangement of two different binding domains separated by a spacer region can meet a minimum standard.
Two DNA-binding domains in interhomolog versus intersister recombination
Resistance to UV and sensitivity of the blm mutant to HU mediated by Brh2 and variant isoforms as used above are metrics of functional proficiency in HR, but are indirect measurements. We wished to determine by direct measurement the functional contribution of the Brh2 isoforms to HR (Fig. 4). For this purpose two different reporter systems were utilized for direct readout of recombination proficiency.
Figure 4.

Activity of Brh2 variants in supporting damage-induced recombination. A. Schematic illustration of Brh2 variants tested for recombination proficiency. B. Allelic recombination at nar1 as depicted was tested in brh2/brh2 diploid strains expressing the indicated Brh2 variants after UV irradiation (40 J/m2) or HU treatment (20 mM for 6 hrs). Nar+ recombinants per survivor was determined on 3–5 independent cultures. Error bars represent standard deviations. C. brh mutant strains with the integrated recombination reporter containing a duplicated internal MscI-NaeI (M–N) sequence and expressing the Brh2 variants were tested for recombination as resistance to geneticin (GenR). The black bar represents a short oligonucleotide linker with a stop codon at the proximal SphI (S) site. Cells were irradiated with UV (40 J/m2) or treated with HU (20 mM) then plated on medium containing geneticin. Recombinants appeared as geneticin resistant colonies. Error bars represent standard deviations from 3–5 independent determinations. Spontaneous recombination frequency was subtracted from the total frequency after irradiation to derive the induced value.
First, allelic recombination was assayed. In this assay recombination between mutant alleles of nar1 (nitrate reductase) residing on homologous chromosomes can reconstruct an active gene enabling growth on nitrate as the sole nitrogen source (Holliday, 1966). Therefore, we expressed the Brh2 variants in brh2 diploids and measured heteroallelic recombination at nar1 in response to UV irradiation or HU block. Conditions of treatment, i.e., dose of UV and time in HU medium, were standardized in pilot studies using a wild type control so that the level of induction of Nar+ recombinants was about the same and killing was kept to a minimum. The Brh2 variants deleted of either the N-terminal NBD (Brh2Δ362–541) or C-terminal DBD (Brh2ΔC551) were both about a third as active as Brh2 in restoring UV-induced recombination (Fig. 4B). The chimeric Brh2Δ362541::OB-AB with heterologous binding domain in place of NBD was also reduced to about the same extent. However, the chimeric Brh2ΔC551::RPA70 with CTD swapped out was hyperactive suggesting that the heterologous DNA-binding domain enabled recombination with UV damaged DNA to a significantly higher level than the endogenous DNA-binding domain. The profile with HU-stressed cells was somewhat different. In this case all the Brh2 mutant forms were defective in recombination compared to wild type Brh2, although Brh2ΔC551::RPA70 retained considerable activity, consistent with its ability to promote killing of HU-treated blm mutant cells.
The second assay was designed to model recombination as envisioned between sister chromatids in the wake of a stalled replication fork and used a modified drug resistance marker as a reporter for recombination events. A neomycin phosphotransferase gene adapted for expressing geneticin (G418) resistance in U. maydis was engineered to contain a 421 bp repeated sequence within the 795 bp open reading frame. Strains containing this construct integrated into the genome at a defined locus are sensitive to G418. Nevertheless, by unequal sister-chromatid recombination, resistant clones can be obtained in which the repeated sequence has recombined out and the open reading frame has been restored (Fig. 4C). It should be noted that the assay is not specific for sister-chromatid recombination events. Due to the direct repeat configuration it is possible that other events including single-strand annealing and primer-template slippage would contribute to the signal.
When the Brh2 variants were assayed for recombination proficiency after UV irradiation, the pattern was similar to that above, but the magnitude of the differences was more exaggerated. All version were active in supporting recombination, with Brh2ΔC551::RPA70 exhibiting more activity than the natural Brh2 isoform. However, when assayed for recombination after HU treatment, all the variant forms were found to be reduced in activity to a much greater degree than in the case with UV irradiation. These results suggest that DNA structure disturbed as a consequence of replication stalling by HU stress presents a more formidable problem for the recombination system as mediated by Brh2 than damage resulting from UV. For an appropriate response, both the NBD and CTD domains of Brh2 must be intact.
DISCUSSION
UV irradiation is a potent inducer of HR. It is thought that photoproducts in DNA are occasionally skipped over during DNA replication leaving single-strand gaps in the wake of the fork, and that these gaps are repaired by the recombination machinery using an undamaged homologous sequence, preferably from the sister chromatid, as a template. HU, on the other hand, corrupts DNA replication by inhibiting ribonucleotide reductase causing depletion of nucleotide pools, which leads to uncoupling of leading and lagging strand synthesis, accumulation of single-strand gaps, replication fork stalling, and finally collapse. DNA structures generated as a consequence of HU treatment can be acted upon by the HR system and processed into intermediates. Normally these structures can be resolved or removed by the action of additional downstream components of the HR system. However, in the absence of their resolution or removal the structures have the potential to trigger a pathological response. Such unprocessed dead-end intermediates can become toxic presumably due to a failure in replication or in chromosome segregation during mitosis.
Here we assessed how the different domains of Brh2 contribute to proficiency in recombinational repair in response to UV irradiation or to generating the pathological recombination intermediates manifested by HU poisoning in the absence of Blm function. There are several noteworthy findings. First, Brh2 variants deleted of either the NBD or CTD domains are competent for HR and survival after UV irradiation, but unlike the natural Brh2 form do not cause a pathological state in blm mutant cells treated with HU. This finding suggests that at least one of those Brh2 domains is required to support a threshold activity in recombinational repair proficiency, but that both domains synergistically contribute to processing intermediates formed when DNA replication is stressed. Second, either of the domains can be replaced by a heterologous DNA-binding domain from RPA70 with little consequence on survival after UV damage. However, any combination of two DNA-binding domains arranged in an appropriate configuration is necessary for Brh2 to become pathological in generating toxic recombination intermediates during replication stress. That a small stretch of residues, or spacer region, connecting the DNA-binding regions is important for the poisonous response to HU replication stress suggests the possibility that a precise architectural arrangement of these domains is required. Third, two defective Brh2 variants with inactivating truncations in the N-terminal and C-terminal regions can cooperate to enable survival after UV irradiation, but not to confer HU-induced killing. In the previously proposed model of Brh2 as a dimer, it was suggested that proficiency in recombinational repair requires two functional regions of Brh2, which could be contributed from two different molecules of Brh2 in a heterodimeric complex (Zhou et al., 2007). In the studies performed here, attainment of the pathological state for HU-induced killing could only be achieved when functional regions of Brh2 are arranged in cis, implying that Brh2-mediated processing of certain DNA intermediates requires a more stringent constellation of functional domains.
The particular functional requirement for two DNA-binding domains in Brh2 to ensure successful processing of the HU-induced DNA structures raises an intriguing question of the specific molecular events involved in the process. A primary function of Brh2 in HR is to control Rad51 (Yang et al., 2005). This protein conducts the search for DNA sequence homology and promotes strand invasion after polymerization on exposed single-stranded DNA to form a nucleoprotein filament. As demonstrated in studies on human BRCA2, Rad51-filament status is regulated through the BRC motif, which promotes assembly of Rad51 on single-stranded DNA while slowing association on double-stranded DNA, and which attenuates ATP hydrolysis of Rad51 when bound to single-stranded DNA, thereby stabilizing it in an extended state suitable for strand invasion (Carreira et al., 2009, Jensen et al., 2010, Shivji et al., 2009). A second Rad51-binding region located at the C-terminus of BRCA2 also contributes to filament stability through specific interaction with the polymerized rather than monomeric form (Davies & Pellegrini, 2007, Esashi et al., 2007).
Accumulating evidence from studies on Rad51 and BRCA2 in replication stressed cells using vertebrate systems (Lomonosov et al., 2003, Petermann et al., 2010, Saintigny et al., 2001) suggests two mechanistically different situations in which these proteins function together in replication fork protection and DNA repair. During replication it appears that BRCA2 is recruited to sites of fork stalling where it serves to stabilize the fork by promoting formation of Rad51 filaments on exposed ssDNA to protect nascent strands from degradation (Hashimoto et al., 2010, Schlacher et al., 2011). After prolonged fork stalling resulting from continuous exposure to HU, forks collapse coincident with DSB formation (Petermann et al., 2010). The HR system can be brought into action again to enable DSB repair and restore the integrity of the genome. Mutational analysis of BRCA2 has revealed that the BRCA2 C-terminal Rad51-interaction site is essential for fork protection activity but is dispensable for HR (Schlacher et al., 2011).
Although it seems entirely possible that Brh2 would have capabilities similar to BRCA2 in nucleating Rad51 filament formation on ssDNA and stabilizing the filament, these activities have not yet been formally distinguished experimentally with Brh2 nor have been ascribed to different protein domains. Nonetheless, as investigated here, the responses to UV irradiation or HU treatment of U. maydis cells expressing the variant forms of Brh2 suggest different functional activities can be attributed to the NBD and CTD protein domains. In contrast, however, to the findings with BRCA2 and the Rad51 filament, the observations that Brh2 variants active in DNA repair and in promoting HU toxicity in the absence of Blm can be created by substituting OB-fold DNA binding modules from RPA70 for either the NBD or CTD domains indicate the primacy of DNA-binding activity, rather than Rad51 filament stabilization, in mediating these responses. Perhaps the conformation or disposition of the Rad51 filament along DNA requires precise ordering by the two DNA-binding domains of Brh2 for optimal activity in processes that channel stalled replication forks through Blm. Alternatively, a maturation process for DNA intermediates arising during fork stalling might explain the requirement for two precisely arranged Brh2 DNA-binding regions.
We imagine that the nature of the DNA lesion is a determinant in deployment of appropriate Brh2 elements. In the case of post-replication repair after UV irradiation, an actionable response requires only the stripped-down, fundamental mediator activity provided by a BRC motif coupled to a ssDNA binding domain. Stalled replication forks, however, present a more formidable structural problem that requires the advent of two spatially coordinated DNA-binding domains for adequate response. This dual domain requirement suggests a two-step process. The first might entail the mediator activity of helping Rad51 gain access to DNA while the second might require a transition that involves DNA handling by the other binding domain for appropriate positioning suitable for subsequent processing. In this scenario it is the DNA-binding-domain architecture of Brh2 rather than the disposition of the Rad51 filament that establishes competence in elaborate processing of the stalled replication forks. The specific possibility of a transitional step is interesting to contemplate. The additional major hurdle in understanding molecular events performed by Brh2 during HU-induced stress is, of course, to identify the specific DNA structure(s) formed during the process. Our hope is that understanding the genetic and molecular events will provide intellectual insights into the broader question of why such an elaborate process is required for dealing with challenge to the most pivotal cellular function of DNA-replication.
EXPERIMENTAL PROCEDURES
U. maydis strains and genetic methods
Manipulations with U. maydis, culture methods, gene disruption and gene transfer procedures, survival after DNA damage by UV or HU treatment, synthesis of diploids, and measurement of allelic recombination, etc., have been described previously [see (Kojic et al., 2011, Zhou et al., 2007) and references therein]. For survival in brief, cultures of exponentially growing cells were diluted to 2 × 107 per ml, then aliquots (10 μl) of serial 10-fold dilutions were spotted on solid medium, and irradiated with 254 nm UV (120 J/m2) or else were plated on medium containing HU. Plates were incubated at 30°C for 2–3 days until colonies developed. The blm mutant strain used in these studies was inactivated for helicase activity by mutation in the Walker A box (K443R) as described previously (Mao et al., 2009). A rad54 null mutant was constructed by standard gene disruption methodology. The gene was identified as entry um02083 in the annotated U. maydis database (see http://mips.helmholtz-muenchen.de/genre/proj/ustilago/) and the entire open reading frame was replaced in haploid strain UCM350 (pan1-1 nar 1–6 a1b1) by a cassette consisting of the strong constitutive otef promoter from plasmid p123 (Aichinger et al., 2003) [kindly provided by Regine Kahmann (Max Planck Institute, Marburg)] fused to a neomycin phosphotransferease gene expressing resistance to geneticin (Gibco/BRL Life Technologies, Rockville, MD). Null mutants in the structural genes for Brh2, Rad51, and Rec2 were previously described (Holloman et al., 2008). pan, met, nar, and ab indicate auxotrophic requirements for pantothenate, inability to utilize nitrate, and mating type loci, respectively. Multiple sequence alignment was performed using the T-Coffee alignment tool [http://www.ebi.ac.uk/Tools/msa/tcoffee/]. Molecular karyotype analysis was performed by contour-clamped homogeneous electric field (CHEF) gel electrophoresis using a CHEF DRII drive module (BioRad Laboratories) essentially as described (Kojic et al., 2002) for 15 hrs with a switch time of 60 sec followed by 11 hrs with a switch time of 90 sec. Protoplasts from 109 cells were prepared using Glucanex (Sigma) in place of Novozyme234, embedded in agarose plugs, then digested with Proteinase K at 30° for 24 hr in a solution containing 10 mM Tris-HCl, pH 8.0, 100 mM EDTA, 1% sodium N-lauroylsarcosine, 0.2% sodium deoxycholate.
Brh2 variants and plasmid construction
Self-replicating plasmids expressing genes encoding Brh2 or derivatives driven by gap promoter contained the hygromycin phosphotransferase gene or carboxin resistance gene for selection and maintenance. Brh2 mutant variants deleted of fragments of the N-terminal DNA-binding domain were constructed as described previously (Kojic et al., 2011). Internal deletions and insertions were constructed so that the distal coding region would be translated in frame. Brh2 variants with RPA70 OB-AB modules in place of the entire NBD (residues 362–541) were constructed using codons 141–434 of RPA70 encoding the OB-AB fragment inserted in frame with Brh2 coding sequence. A Brh2-Rad52 DBD hybrid containing the highly conserved DNA-binding/self-association domain of Rad52 (DBD, residues 79–317) was constructed by fusing the 239 amino acid Rad52 domain (Kojic et al., 2008) in frame to Brh2ΔC551. A vector for expression of RusA under control of the gap promoter was constructed by transferring an NdeI-BamHI fragment containing the rusA gene from plasmid pKR6980 (Bastin-Shanower et al., 2003) kindly provided by Steve Brill (Rutgers University) into a self-replicating plasmid expressing CbxR for selection in U. maydis.
Recombination assays
Allelic recombination at the nar1 locus was measured as described previously (Holliday, 1966, Kojic et al., 2002) by determining Nar+ prototroph formation in brh2 homozygous diploids expressing Brh2 mutant variants after induction with a UV dose of 40 J/m2 or else after holding in medium containing 20 mM HU for 7 hr. A reporter for measuring sister chromatid recombination was constructed by duplicating an internal sequence in the transposon Tn5 neomycin phosphotransferase gene expressing geneticin resistance (GenR) in U. maydis. A 421 bp MscI-NaeI fragment from the middle of the open reading frame was introduced into the 795 bp gene opened at the NaeI site generating a reporter with an internal tandem duplication of this sequence. A short linker with a stop codon in frame was inserted at the SphI site (at position 534 in the orf) within the proximal MscI-NaeI repeat to ablate residual drug resistance. This construct was placed under control of the otef promoter in an integrative plasmid bearing an allele of the iron-sulfur subunit sdh2 of the succinic dehydrogenease gene (um00844) expressing resistance to carboxin (CbxR) in U. maydis. CbxR transformed brh2 haploid strains with the integrated recombination reporter that were sensitive to geneticin (100 μg per ml) were chosen for analysis. For UV induced recombination, strains were cultured overnight in rich medium, washed, then resuspended in water at a density of 2 × 107 per ml, spread directly on plates with solid medium containing geneticin (Gibco/BRL Life Technologies) at 100 μg per ml, then irradiated with 40 J/m2 of UV light. For HU induction, freshly growing cells at 2 × 107 per ml were cultured in medium spiked with 20 mM HU for 7 hrs, then washed, collected and plated on geneticin-containing solid medium. GenR colonies were counted after incubating plates three days at 28°.
Acknowledgments
We thank Lorraine Symington, Columbia University, for stimulating discussion and laboratory members Qingwen Zhou and Jeanette Sutherland for contributing help and ideas. This work received financial support from NIH grants GM042482 and GM079859.
References
- Aichinger C, Hansson K, Eichhorn H, Lessing F, Mannhaupt G, Mewes W, Kahmann R. Identification of plant-regulated genes in Ustilago maydis by enhancer-trapping mutagenesis. Mol Genet Genomics. 2003;270:303–314. doi: 10.1007/s00438-003-0926-z. [DOI] [PubMed] [Google Scholar]
- Ayoub N, Rajendra E, Su X, Jeyasekharan AD, Mahen R, Venkitaraman AR. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol. 2009;19:1075–1085. doi: 10.1016/j.cub.2009.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006;34:2269–2279. doi: 10.1093/nar/gkl258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol Cell Biol. 2003;23:3487–3496. doi: 10.1128/MCB.23.10.3487-3496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzowska M, Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem Biophys. 2009;53:17–31. doi: 10.1007/s12013-008-9039-y. [DOI] [PubMed] [Google Scholar]
- Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–1043. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol Cell. 2009;36:845–860. doi: 10.1016/j.molcel.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies OR, Pellegrini L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol. 2007;14:475–483. doi: 10.1038/nsmb1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi F, V, Galkin E, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A. 2002;99:16887–16892. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. Studies on mitotic gene conversion in Ustilago. Genet Res. 1966;8:323–337. doi: 10.1017/s0016672300010181. [DOI] [PubMed] [Google Scholar]
- Holloman WK. Unraveling the mechanism of BRCA2 in homologous recombination. Nat Struct Mol Biol. 2011 doi: 10.1038/nsmb.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloman WK, Schirawski J, Holliday R. The homologous recombination system of Ustilago maydis. Fungal Genet Biol. 2008;45(Suppl 1):S31–39. doi: 10.1016/j.fgb.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope JC, Maftahi M, Freyer GA. A postsynaptic role for Rhp55/57 that is responsible for cell death in Δrqh1 mutants following replication arrest in Schizosaccharomyces pombe. Genetics. 2005;170:519–531. doi: 10.1534/genetics.104.037598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, Shibata T, Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol Cell. 2002;10:359–371. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- Kojic M, Kostrub CF, Buchman AR, Holloman WK. BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2002;10:683–691. doi: 10.1016/s1097-2765(02)00632-9. [DOI] [PubMed] [Google Scholar]
- Kojic M, Mao N, Zhou Q, Lisby M, Holloman WK. Compensatory role for Rad52 during recombinational repair in Ustilago maydis. Mol Microbiol. 2008;67:1156–1168. doi: 10.1111/j.1365-2958.2008.06116.x. [DOI] [PubMed] [Google Scholar]
- Kojic M, Yang H, Kostrub CF, Pavletich NP, Holloman WK. The BRCA2-interacting protein DSS1 is vital for DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2003;12:1043–1049. doi: 10.1016/s1097-2765(03)00367-8. [DOI] [PubMed] [Google Scholar]
- Kojic M, Zhou Q, Fan J, Holloman WK. Mutational analysis of Brh2 reveals requirements for compensating mediator functions. Mol Microbiol. 2011;79:180–191. doi: 10.1111/j.1365-2958.2010.07440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic M, Zhou Q, Lisby M, Holloman WK. Brh2-Dss1 interplay enables properly controlled recombination in Ustilago maydis. Mol Cell Biol. 2005;25:2547–2557. doi: 10.1128/MCB.25.7.2547-2557.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, Fuchs RP. Gaps and forks in DNA replication: Rediscovering old models. DNA Repair (Amst) 2006;5:1495–1498. doi: 10.1016/j.dnarep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005;19:339–350. doi: 10.1101/gad.322605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Mankouri HW, Ashton TM, Hickson ID. Holliday junction-containing DNA structures persist in cells lacking Sgs1 or Top3 following exposure to DNA damage. Proc Natl Acad Sci U S A. 2011;108:4944–4949. doi: 10.1073/pnas.1014240108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao N, Kojic M, Holloman WK. Role of Blm and collaborating factors in recombination and survival following replication stress in Ustilago maydis. DNA Repair (Amst) 2009;8:752–759. doi: 10.1016/j.dnarep.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman H, Jacob F. A comparison of spontaneous and ultraviolet-induced allelic recombinaiton with reference to the recombination of outside markers. Cold Spring Harbor Symp Quant Biol. 1958;23:155–160. doi: 10.1101/sqb.1958.023.01.019. [DOI] [PubMed] [Google Scholar]
- Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-Strand Break Repair-Independent Role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MK, Mukund SR, Rajendra E, Chen S, Short JM, Savill J, Klenerman D, Venkitaraman AR. The BRC repeats of human BRCA2 differentially regulate RAD51 binding on single- versus double-stranded DNA to stimulate strand exchange. Proc Natl Acad Sci U S A. 2009;106:13254–13259. doi: 10.1073/pnas.0906208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. Structure of the single-strand annealing domain of human RAD52 protein. Proc Natl Acad Sci U S A. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, West SC. BRCA2: a universal recombinase regulator. Oncogene. 2007;26:7720–7730. doi: 10.1038/sj.onc.1210870. [DOI] [PubMed] [Google Scholar]
- Timson J. Hydroxyurea. Mutat Res. 1975;32:115–132. doi: 10.1016/0165-1110(75)90002-0. [DOI] [PubMed] [Google Scholar]
- van Brabant AJ, Ye T, Sanz M, German IJ, Ellis NA, Holloman WK. Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry. 2000;39:14617–14625. doi: 10.1021/bi0018640. [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433:653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Kojic M, Cao Z, Lisby M, Mazloum NA, Holloman WK. Dss1 interaction with Brh2 as a regulatory mechanism for recombinational repair. Mol Cell Biol. 2007;27:2512–2526. doi: 10.1128/MCB.01907-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Kojic M, Holloman WK. DNA-binding domain within the Brh2 N terminus Is the primary interaction site for association with DNA. J Biol Chem. 2009a;284:8265–8273. doi: 10.1074/jbc.M809226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Mazloum N, Mao N, Kojic M, Holloman WK. Dss1 regulates interaction of Brh2 with DNA. Biochemistry. 2009b;48:11929–11938. doi: 10.1021/bi901775j. [DOI] [PMC free article] [PubMed] [Google Scholar]