

Abstract
Retinoblastoma is the most common intraocular tumor in children. The diagnosis is usually established by the ophthalmologist on the basis of fundoscopy and US. Together with US, high-resolution MRI has emerged as an important imaging modality for pretreatment assessment, i.e. for diagnostic confirmation, detection of local tumor extent, detection of associated developmental malformation of the brain and detection of associated intracranial primitive neuroectodermal tumor (trilateral retinoblastoma). Minimum requirements for pretreatment diagnostic evaluation of retinoblastoma or mimicking lesions are presented, based on consensus among members of the European Retinoblastoma Imaging Collaboration (ERIC). The most appropriate techniques for imaging in a child with leukocoria are reviewed. CT is no longer recommended. Implementation of a standardized MRI protocol for retinoblastoma in clinical practice may benefit children worldwide, especially those with hereditary retinoblastoma, since a decreased use of CT reduces the exposure to ionizing radiation.
Keywords: Retinoblastoma, MRI, CT, US
Introduction
Retinoblastoma is the most common intraocular tumor in children. The incidence is one in 17,000 births. Mean age at clinical presentation is 2 years in unilateral forms (60% of cases) and 1 year in bilateral forms [1, 2]. All bilateral forms, as well as 15% of unilateral forms are related to a constitutional (hereditary or de novo) mutation of the RB-1 gene, localized on chromosome 13q14 [2]. Usually the patients present with leukocoria (white pupil reflection) or a squint.
Retinoblastoma is curable. If detected while still confined to the globe and if there are no metastatic risk factors, the child will nearly always survive following appropriate treatment [3, 4]. The preservation of visual function depends on ocular preservation, initial tumor volume, the anatomical relationships of the tumors to the macula and optic disk and the adverse effects of the treatments (cataracts, vitreous hemorrhage) [5]. In the presence of metastatic risk factors, adjuvant treatment regimens are usually applied to prevent life-threatening relapse [6, 7].
Diagnosis of retinoblastoma is usually made by fundoscopy (Fig. 1) (under general anesthesia) and US. The ophthalmologist usually performs both investigations. In almost all cases classic intratumoral calcifications can be detected by US providing high confidence rate regarding diagnosis. Various tumor parameters (laterality; number, location and size of tumors; tumor seeding to vitreous, subretinal space or anterior segment) can be evaluated with these techniques. These are important for grouping the retinoblastoma and to guide therapeutic decisions. Further diagnostic imaging plays a crucial role in determining the local extent and for detecting associated brain abnormalities, i.e. intracranial tumor extension, possible midline intracranial primitive neuroectodermal tumor (PNET) and brain malformations in patients with 13q deletion syndrome [8–10].
Fig. 1.
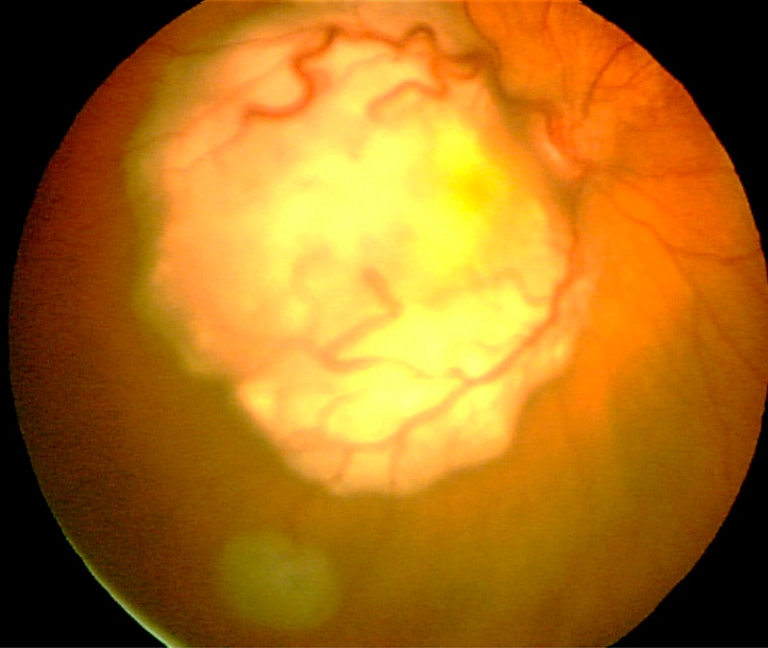
Fundus photograph shows two tumors. The large white mass with prominent feeder vessels is located at the macula causing reduced visual acuity. A smaller tumor is located in the inferolateral part of the retina. Image courtesy Annette C. Moll, Amsterdam
PNETs are associated with hereditary retinoblastoma, a combination known as trilateral retinoblastoma, which occurs in 5–15% of children in the hereditary subgroup [10, 11]. Besides the pineal region (pineoblastoma), tumors may also occur in the suprasellar or parasellar regions. Trilateral retinoblastoma has been lethal in virtually all cases reported in literature; however, early detection and intensive (chemo-)therapy may be lifesaving for some patients [10, 12, 13].
Conservative treatment strategies (avoidance of enucleation and external beam radiation therapy) can be successful in the early stages of retinoblastoma and in some patients with advanced intraocular disease [14]. The options for eye-preserving therapy have significantly improved during recent years and are mainly based on tumor reduction with chemotherapy, and are usually combined with laser coagulation, cryotherapy or radioactive plaque. Recently, selective ophthalmic artery infusion of a chemotherapeutic agent became available as an additional treatment option for locally advanced disease [15]. As a consequence, more children are treated without histopathological confirmation and, what is more important, without assessment of risk factors for disease dissemination and prognosis. Therefore, imaging is very important in local staging.
Whereas imaging is increasingly used for diagnosis and as a basis for treatment decisions in retinoblastoma, there is a lack of standardization for choosing among modalities and for the minimum quality of MRI. The purpose of this report is to present a guideline for diagnostic imaging of retinoblastoma including a standardized MRI protocol using conventional pulse sequences. The potential role of advanced imaging techniques for lesion characterization and detection of tumor extent (such as 3D T1-weighted sequences, diffusion-weighted imaging, diffusion tensor imaging and MR-spectroscopy) is beyond the scope of this guideline.
Risk factors for metastasis and local recurrence
Retinoblastoma may spread either by hematogenous dissemination or by direct extension either through the bulbar wall into the orbit or via the optic nerve and its meningeal sheath [16]. Therefore, current risk factors for metastasis and local recurrence include invasion of the optic nerve posterior to the lamina cribrosa (in particular if there is tumor at the surgical resection margin), anterior eye segment (AES), or extensive invasion of the ocular coats (massive choroidal and scleral invasion) [17–20]. Pathology remains the gold standard to assess high-risk features of retinoblastoma. The rate of postlaminar optic nerve invasion in patients treated by primary enucleation has been estimated at 7–8% [21, 22]. Choroidal invasion is present in 23–42% of enucleated eyes, out of which the invasion is massive in about 9–11% [19, 23–25]. The exact incidence of massive choroidal invasion is unknown, especially since the definition of “massive” differs among pathologists. Recently, new consensus criteria were proposed by a worldwide collaboration of pathologists and pediatric oncologists [26]. Tumor invasion into the AES is very rare, being present in approximately 2% of primary enucleated eyes [24, 27–30]. Children with histopathological risk factors for metastatic disease require adjuvant chemotherapy to reduce the risk of relapse [18–20, 31].
Radiation exposure and second primary malignancies in hereditary retinoblastoma
Unlike survivors of non-hereditary retinoblastoma, survivors of hereditary retinoblastoma have an elevated risk of developing second (or even more) malignancies with a cumulative mortality rate of 17% [32–34]. In patients with hereditary retinoblastoma, the cumulative incidence of a second primary malignancy within 40 years of the initial retinoblastoma is 28% [34]. Chemotherapy has been reported to increase the risk of leukemia in survivors of retinoblastoma [35]. Radiotherapy is associated with an increased risk of soft-tissue sarcomas in survivors of hereditary retinoblastoma, with a reported significant association of radiation dose with the risk of second primary (or more) cancers [34, 36]. Assuming a linear relation between radiation dose and stochastic risk, several studies have demonstrated a theoretical increased risk of CT-associated radiation-induced fatal cancers in children [37]. Although low, this risk is likely to be further increased in patients with hereditary retinoblastoma, who are known to be genetically unstable, due to the inherited germ cell mutation in the RB-1 tumor-suppressor gene. To minimize the development of subsequent cancers, survivors of retinoblastoma are advised to avoid unnecessary radiation. Furthermore, children undergoing radiotherapy for retinoblastoma may experience abnormalities in the growth and maturation of their craniofacial skeleton, resulting in mid-face deformities [38]. For these reasons, external beam radiotherapy was dramatically reduced for the conservative treatment options, and the principle of minimizing the exposure to ionizing radiation should also be applied to imaging. US and MRI should be used instead of CT. The radiation from interventional procedures, e.g., selective ophthalmic artery chemotherapy infusion, is also important [39]. The radiation dose should be optimized, precisely measured and clearly reported in future publications to facilitate balancing risks and benefits both in imaging and therapy.
Choice of imaging modality
US, CT and MRI are the mainstay for imaging of head and neck tumors in children. US is particularly useful for examining superficial masses, such as retinoblastoma, whereas CT and MRI are used to delineate deeper lesions, particularly those involving the skull base and the central nervous system. Nowadays, diagnostic evaluation of retinoblastoma consists primarily of US and MRI. Positron emission tomography has become an important modality for cancer imaging in general; however, its value in retinoblastoma imaging is currently limited [40].
US
The human eye, with its superficial position and its fluid-filled structures, is ideally suited for US. Ocular US is usually performed by the ophthalmologist while the child is under general anesthesia, but can also quite easily be performed without sedation. In retinoblastoma, US demonstrates an irregular mass, more echogenic than the vitreous body, with fine calcifications (highly reflective foci mostly with characteristic acoustic shadowing) [41] (Fig. 2). Histologically, there is calcification present in approximately 95% of tumors [42]. Calcification is key to differentiating retinoblastoma from other mass lesions in a young child. US detects calcifications in 92–95% of cases where it is present histopathologically [42, 43]. Retinal detachment may also be observed, which is an important feature to define tumor growth pattern, either endophytic or exophytic, or a combination of both. Endophytic tumors arise from the inner layers of the retina and grow into the vitreous body. Frequently, small clusters of tumor cells detach from an endophytic mass, producing multiple floating tumor islands; this process is known as vitreous seeding. Exophytic tumors originate in the outer layers and grow in the subretinal space, which causes retinal detachment with subretinal exudate and possible subretinal tumor seeding. Tumors with exophytic growth more frequently have choroidal infiltration compared to endophytic tumors [44]. Diffusely infiltrating retinoblastoma is a rare histological form characterized by diffuse infiltration of the retina without a tumor mass [45]. Tumor height and diameter are usually measured at US, as these measurements are used for choice of treatment. Color Doppler can be useful for differentiating a vascularized tumor mass from echogenic effusions and for differentiation against developmental abnormalities such as persistent hyperplastic primary vitreous (PHPV; also known as persistent fetal vasculature, PFV), with the characteristic persisting hyaloid artery.
Fig. 2.

US reveals a hyperechoic tumor occupying the posterior segment of the globe. Calcium deposits, seen as highly reflective foci (arrows), are pathognomonic for retinoblastoma in a young child
US is not the imaging modality of choice for direct evaluation of metastatic risk factors. Tumoral calcifications commonly obscure visualization of the optic nerve [41]. Indirect detection of optic nerve invasion by measurement of optic nerve diameter with a 3D-US technique has been reported in a single case report [46], hence its value remains unknown.
CT
On CT, retinoblastoma is typically a mass of high density compared with the vitreous body, usually calcified and moderately enhancing after iodinated contrast medium administration. CT detection of calcifications in retinoblastoma has a sensitivity of 81–96%, and an even higher specificity [47]. However, delineation of intraocular soft-tissue detail is limited. The evidence from surveys suggests that CT is still regarded an obligatory imaging tool for evaluation of leukocoria, primarily because CT is supposed to be the best imaging modality for detection of intraocular calcifications [47–49]. However, justification of the irradiation of a large group of retinoblastoma patients requires a base of evidence of the procedure’s clinical effectiveness and possibly also radiation-effectiveness [50] for supplying (1) valuable additional information leading toward the diagnosis of retinoblastoma and (2) valuable additional information, compared to non-ionizing radiation modalities in detection of tumor extent.
CT was the first imaging modality used to detect optic nerve invasion [51–53] and is historically assumed to be precise in detection of tumor extent [54–56]. However, this assumption is based on conflicting outdated literature, without thorough evidence by radiologic-pathologic correlation studies. The sensitivity of CT in detection of optic nerve invasion is actually very low, even in patients with extensive optic nerve invasion (length of invaded nerve segment > 2 mm) [21, 51, 53]. The specificity, accuracy and negative predictive value of CT remain artificially high because of the relatively low incidence of optic nerve invasion in normal-size nerves. An enlarged nerve due to massive tumor infiltration is rare in developed countries. Assuming retinoblastoma invasion into the optic nerve produces distortion of the anastomotic vascular network in the anterior optic nerve region, Jacquemin and Karcioglu [52] considered that non-visualization of the central retinal vessels is a reliable indicator of optic nerve invasion. However, these results were not confirmed by other studies [21].
MRI
Diagnostic MRI evaluation of a suspected retinoblastoma requires much more than performing a routine MR imaging examination of the orbit. High-resolution contrast-enhanced MRI is the technique of choice and should be used whenever possible to answer the key clinical questions (to evaluate an intraocular mass and to determine disease extent; Fig. 3). MRI has proved to be the most sensitive technique for evaluating retinoblastoma, especially regarding tumor infiltration of the optic nerve, extraocular extension and intracranial disease [21, 22, 44, 57, 58]. A major factor influencing the success of MRI is the use of appropriate hardware and optimized pulse sequences with appropriate spatial resolution for ocular MRI [21, 22, 44, 57–61].
Fig. 3.

Transaxial T2-weighted (TR/TE, 3,460/116 ms) (a) and T1-weighted (TR/TE, 374/14 ms) precontrast (b) and postcontrast (c) MRI of exophytically growing retinoblastoma with secondary retinal detachment. Retinoblastoma typically has low signal intensity compared to the vitreous body on T2-weighted images and intermediate signal intensity on precontrast T1-weighted images, and it demonstrates marked contrast enhancement
Imaging strategy
Diagnosis of retinoblastoma
Examination under general anesthesia with fundoscopy and US almost inevitably leads to the diagnosis. As US detects foci of calcification in almost all retinoblastomas, there is now little benefit of routine CT for detection of calcifications in suspected retinoblastoma. Due to technical development, US and MRI are currently almost as accurate as CT for detection of calcifications. Recently, Galluzzi et al. [42] showed that when data from ophthalmoscopy, US and MRI are put together, no calcifications detected on CT were missed. A high-resolution gradient-echo T2-weighted sequence showed promising results regarding detection of calcifications and has been shown to be more effective than spin-echo techniques [42, 44].
The differential diagnosis of retinoblastoma includes several non-neoplastic lesions that also cause leukocoria. After retinoblastoma, which accounts for 47–58% of cases of leukocoria in children, other causes in decreasing order of frequency include PHPV, Coats disease, larval granulomatosis (Toxocara canis), retinopathy of prematurity, and retinal astrocytic hamartoma [48]. Calcification is the most important differentiating feature of retinoblastoma. However, when clinical diagnosis remains uncertain, US and MRI help characterize and differentiate intraocular abnormalities, especially when ophthalmological evaluation is limited due to opaque ocular refractive media, as may occur in all of these conditions. The role of CT in the detection of (sometimes subtle) characteristic findings is limited due to its low soft-tissue contrast [48, 55].
Detection of tumor extent
In the past, CT was used to determine tumor size, retro-orbital spread and intracranial growth; however, spread within the optic nerve past the cribriform lamina, and infiltration of choroid and sclera, which are important prognostic factors, are not reliably assessed with CT [51, 53]. Because of its superior soft-tissue contrast, MRI is more sensitive and specific than CT in detection of tumor extent and metastatic risk factors. MR imaging using high-resolution protocols is currently considered to be the most accurate and valuable tool in pretreatment staging of retinoblastoma, without known biological side effects.
Standardized retinoblastoma MRI protocol
Although individual examinations should always be tailored to the specific queries in individual patients (laterality, disease extent, therapy options), there are general recommendations for MRI in retinoblastoma. In the following paragraphs we discuss the minimum requirements for diagnostic evaluation of retinoblastoma or mimicking lesions according to the consensus reached among members of the European Retinoblastoma Imaging Collaboration (ERIC). If these recommendations cannot be followed because of technical limitation, ERIC members recommend to refer the patient to the nearest (or national) reference center for retinoblastoma, where a multidisciplinary team of specialized physicians (ophthalmology, pediatric oncology, radiology, pathology, radiotherapy, clinical genetics, psychology) and specialized nurses will ensure that practice conforms to the best standards of care.
Patient handling
Although the technical success of MRI usually depends on the cooperation of the patient, in retinoblastoma appropriate sedation techniques or general anesthesia are nowadays widely used, with a high yield of diagnostic scans. Nevertheless, in our experience the success rate of intravenous sedation is highly dependent on the presence of trained anesthetists and of the choice of radiofrequency coils. Especially, the use of small surface coils decreases the success rate, because these coils need to be accurately positioned close to the eye. The depth of sedation may be insufficient for accurate patient positioning. Therefore, general anesthesia is recommended for MRI in children with retinoblastoma. Another advantage of general anesthesia is the possibility to ensure that the eyelids are fully closed and to avoid uncontrolled eye movements by putting pads (fixed with tape) on the closed eyelids. Thereby susceptibility artifacts caused by air-tissue interface or by air bubbles under the eyelids is avoided, which is favorable for the image quality in the anterior eye segment. If possible, the MR examination can be combined with fundoscopy and US under the same general anesthetic.
Hardware
Clinical MRI of orbit and eyes is mostly acquired using the current standard field strengths up to 1.5 T. Since adequate imaging of retinoblastoma requires high spatial resolution, the field strength of the MR system should be at least 1.5 T. The performance of scans at 1.5 T using a head coil is the most practical approach for evaluation of retinoblastoma. However, this combination usually gives insufficient signal-to-noise ratio (SNR) at required in-plane image resolution and section thickness. Therefore, scanning at 1.5 T should always be performed with one or two small surface coils (diameter ≤ 5 cm) to reliably detect small lesions and metastatic risk factors (Fig. 4). Indeed, the use of surface coils in ocular tumors has been reported to increase the diagnostic accuracy [44, 62].
Fig. 4.

High-resolution MRI in retinoblastoma. The child is under general anesthesia. A small circular surface coil (arrow) is accurately positioned close to the affected eye
The main advantage of higher field strengths (3 T) is the increased SNR. Publications on ocular 3-T MRI are still limited [63, 64]. At 3 T with multi-channel head coil or surface coils one can achieve high-resolution images similar to those obtained at 1.5 T with surface coils (Fig. 5). We think the diagnostic accuracy for detection of tumor extent might improve, but there is currently no published evidence for this.
Fig. 5.

Transaxial contrast-enhanced T1-weighted (TR/TE, 722/14 ms) image obtained at 3.0 T using a 32-channel head coil shows an exophytic retinoblastoma with secondary total retinal detachment and proteinacious subretinal effusion
Imaging protocol
MRI protocols vary because of differences in available equipment and individual preferences. However, despite differences, certain basic elements are common to most imaging protocols for retinoblastoma. The minimal requirements for diagnostic evaluation of retinoblastoma or mimicking lesions according to the consensus reached among members of the European Retinoblastoma Imaging Collaboration (ERIC) are presented below and summarized in Table 1. A typical MR imaging protocol for retinoblastoma should always include high-resolution imaging of the affected eye(s) and imaging of the entire brain.
Table 1.
MRI protocol in retinoblastomaa
| Requirements |
|---|
| Scanner and coils |
| Field strength above 1 T |
| 1.5-T system combined with one or two small surface coils (diameter < 5 cm) |
| 3.0-T system combined with multichannel head coil |
| Sequences (minimum requirements) |
| Orbits |
| Transaxial T2-W (slice thickness ≤ 2 mm) |
| Optional: Transaxial CISS (Siemens) / FIESTA (GE) / DRIVE (Philips) |
| Eye(s) and optic nerve(s) |
| In-plane pixel size < 0.5 × 0.5 mm; slice thickness ≤ 2 mm |
| Unilateral disease (or bilateral disease with only one eye strongly affected) |
| Precontrast T1-W; at least one plane: transaxial or sagittal oblique |
| T2-W; at least one plane: transaxial or sagittal oblique |
| Postcontrast T1-W, no FS; transaxial and sagittal oblique |
| Bilateral disease (both eyes strongly affected) |
| Precontrast T1-W (transaxial) |
| T2-W (transaxial) |
| Postcontrast T1-W, no FS; sagittal oblique of both eyes and transaxial |
| Brain |
| Transaxial T2-W (slice thickness ≤ 4 mm) |
| Postcontrast T1-W (2D SE with slice thickness ≤ 3 mm or 3D GRE ≤ 1 mm) |
FS fat-saturation, SE spin-echo, GRE gradient-echo
a Consensus among members of the European Retinoblastoma Imaging Collaboration (ERIC)
Orbits
Regardless of laterality, at least one transaxial thin-slice (≤ 2 mm) T2-weighted sequence should cover both orbits. For T2-weighted imaging based on a (fast) spin-echo technique, it is recommended to use a long TE (heavily T2-weighted; TE ≥ 120 ms) for generating the image contrast necessary to provide an optimal differentiation of retinoblastoma and surrounding vitreous or subretinal fluid (Fig. 6). Fat saturation combined with T2-weighted imaging is not recommended. When fat suppression is used, the resulting loss in SNR should be compensated (e.g., by increasing the number of acquisitions).
Fig. 6.

a Thin-slice transaxial T2-weighted (TR/TE, 4,430/102 ms; section thickness, 2 mm) image demonstrates retinoblastoma of the right eye with secondary retinal detachment. b Transaxial constructive interference steady-state (TR/TE, 14/7 ms) image of bilateral retinoblastoma with secondary retinal detachment. Notice the shallow anterior chamber of the right eye, a sign of increased intraocular pressure
T2-W spin-echo may be replaced by gradient-echo T2-weighted sequences such as 3D steady-state free precession sequences with slice thickness ≤ 1 mm (Fig. 6; vendor-specific acronyms: CISS [Constructive Interference in Steady State, Siemens], FIESTA [Fast Imaging Employing Steady State Acquisition, GE Healthcare]; DRIVE [Driven Equilibrium, Philips]) (slice thickness, ≤ 1 mm). These pulse sequences provide detailed images of both orbits and eyes, and allow accurate comparison of eye size, anterior chamber depth and laterality. Very small tumors can be depicted with these techniques.
Eye and distal optic nerve
Increased spatial resolution will improve the accuracy of MRI in assessing the anatomical details of the papilla, lamina cribrosa and pre- and postlaminar segments of the optic nerve [65]. The continuous improvement of MR units and the use of small fields-of-view with either multi-channel head coils or surface coils now allows much higher image resolution. High spatial resolution means section thickness ≤ 2 mm and in-plane pixel size ≤ 0.5 × 0.5 mm. For optimal detection of optic nerve invasion, the image plane through the orbit (transaxial and sagittal oblique) should align with the orientation of the distal (1 cm) end of the nerve, just posterior to the lamina cribrosa (Fig. 7). One section in each of these sequences should be precisely aligned within the distal part of the optic nerve at the level of the middle of the optic disk. Although the use of the fat-saturation technique is highly recommended for contrast-enhanced MR imaging in orbital pathology, its use in high-resolution contrast-enhanced T1-weighted MRI in retinoblastoma is declining [44, 58]. In the minimal requirements for diagnostic evaluation of retinoblastoma or mimicking lesions according to the consensus reached among members of the ERIC, the use of fat saturation in contrast-enhanced T1-weighted sequences is no longer recommended.
Transaxial or sagittal oblique T1-weighted spin-echo helps detection of intraocular blood and subretinal fluid with high protein content. Retinoblastoma is slightly hyperintense with respect to the vitreous body.
Transaxial or sagittal oblique heavily T2-weighted spin-echo provides detailed information about the classic low signal intensity of retinoblastoma and presence of retinal detachment.
Transaxial and sagittal oblique contrast-enhanced T1-weighted spin-echo provides information about the enhancement of lesions, optic nerve- and ocular wall invasion, and anterior eye segment enhancement.
Fig. 7.

Recommended slice positions. One T2-weighted section in the sagittal oblique plane and one in the transaxial plane should be precisely aligned at the middle of the optic disk and the distal (at least 1 cm) end of the optic nerve. a Correct alignment of transaxial sections. b Correct alignment of sagittal oblique sections. c Detailed view of the distal optic nerve (line segment: imaging axis; distal 1 cm of the optic nerve between red dots)
The recommended protocol for high-resolution MR imaging of the eye(s) and distal optic nerve(s) differs slightly between unilateral or bilateral disease (Table 1, Figs. 8–9). Incidence of metastatic risk factors is highly dependent on tumor location and tumor size. Therefore, a distinction is made between bilateral disease with only one eye strongly affected (high-resolution MRI can be performed in the worst affected eye only) and extensive disease in both eyes (high-resolution MRI of both eyes).
Fig. 8.

In unilateral retinoblastoma (or bilateral disease with only one eye strongly affected), high-resolution MRI is done in the (most) affected eye only. Imaging example of a left unilateral lesion. a Sagittal oblique T2-weighted (TR/TE 3,460/110 ms) image. b Transaxial precontrast T1-weighted (TR/TE 360/13 ms) image. c Transaxial postcontrast T1-weighted (TR/TE 360/13 ms) image. Notice the inhomogeneous enhancement pattern, which is common in retinoblastoma
Fig. 9.

MRI of bilateral retinoblastoma with extensive disease in both eyes should be performed with two surface coils. The field of view should be slightly increased to cover both eyes in the transaxial plane. Imaging example of bilateral lesions: a Precontrast transaxial T1-weighted (TR/TE, 360/13 ms) image. b Postcontrast transaxial T1-weighted (TR/TE, 360/13 ms) image
Brain
The brain should always be imaged in retinoblastoma patients for analysis of midline structures in order to depict trilateral retinoblastoma (i.e. PNET located mainly in the pineal gland, or more rarely in suprasellar area) or leptomeningeal spread. For a patient presenting with leukocoria suspicious of having retinoblastoma or already diagnosed as retinoblastoma based on clinical findings and US, the baseline evaluation should include an MR imaging of the brain that meets the standardized protocol. Imaging of the brain is performed with (multi-channel) headcoil only and should at least include the following or similar types of sequences:
Transaxial fast spin-echo T2-weighted sequence (slice thickness, ≤ 4 mm). This sequence provides an overview of the brain anatomy and structural abnormalities (patients with 13q deletion syndrome).
Transaxial or sagittal contrast-enhanced T1-weighted sequence (2D spin-echo T1-weighted with slice thickness ≤ 3 mm; or 3D gradient-echo with slice thickness ≤1 mm). This sequence provides information about enhancement of the pineal gland, presence of a midline PNET, leptomeningeal metastases and extensive optic nerve invasion.
Coronal and sagittal high-resolution T2-weighted sequence (slice thickness, 1.5 mm). These sequences are optional but should be added to the protocol in case of an atypical pineal gland (partially cystic, irregular, enlarged).
Image analysis checklist for MR reporting (Table 2)
Table 2.
Retinoblastoma: Checklist for MRI radiology reports
| Parameters |
|---|
| Tumor characteristics |
| SI relative to the vitreous body; moderately high on T1-W and low on T2-W |
| Laterality |
| Growth pattern |
| Tumor size and location; in contact with optic nerve |
| Buphthalmia |
| Tumor extension |
| Optic nerve and meningeal sheath invasion |
| Ocular wall invasion (choroid and sclera) |
| Extraocular extension |
| Anterior eye segment |
| Anterior chamber depth |
| Enhancement |
| Tumor invasion; ciliary body |
| Brain |
| Trilateral retinoblastoma; pineal gland and supra- or parasellar region |
| Leptomeningeal metastases |
| Malformations |
SI signal intensity
Tumor size and location
Compared to the vitreous body, retinoblastoma has moderately higher signal intensity on T1- and lower on T2-weighted images. Increased size of the globe, globe deformation and reduced anterior chamber depth are signs of increased intraocular pressure and are usually associated with buphtalmia (Fig. 10). These signs should be mentioned since they are associated with a higher risk of globe rupture and secondary orbital seeding during enucleation. Laterality and growth pattern should be mentioned as well as the location of the tumor, with respect to the equator of the eye (anterior, posterior or combined) and with respect to the papilla (the optic nerve disk) and the macula. One should in particular identify tumor close to the optic disk, because this may invade the nerve [22] (Fig. 11).
Fig. 10.

Retinoblastoma with signs of increased intraocular pressure following subretinal hemorrhage (fluid–fluid levels). a Bilateral retinoblastoma with increased size of the left eye (buphthalmus) and a shallow anterior chamber (arrow) seen on T2-weighted (TR/TE, 4,430/102 ms) image. b Bilateral retinoblastoma with focal bulging of the posterior eye segment (arrow) of the right eye and a shallow anterior chamber seen on T2-weighted (TR/TE, 4,430/102 ms) image
Fig. 11.

Pre- (a) and postcontrast (b) transaxial T1-weighted (374/14) MR images show small nodular enhancement at the optic nerve disk (arrow), which represents superficial optic nerve invasion by intraocular tumor seeding (predilection site). c Abnormal contrast-enhancement of the anterior eye segment combined with macroscopic tumor seedings (arrow)
Optic nerve and meningeal sheath invasion
In normal-size optic nerves, the direct radiological criterion used to diagnose postlaminar nerve invasion is the presence of abnormal contrast enhancement (enhancement ≥ 2 mm in diameter) in the distal nerve [21] (Fig. 12). Interruption of the normal linear enhancement at the optic nerve disk (choroidoretinal complex) supports a suggestion of optic nerve invasion [22]. Postlaminar optic nerve or optic nerve menigeal sheath invasion should raise suspicion of leptomeningeal metastases. In such situations, additional contrast-enhanced sagittal T1-weighted imaging of the whole spine is recommended.
Fig. 12.

Postcontrast transaxial T1-weighted (TR/TE, 374/14 ms) MRI. Abnormal enhancement of the distal optic nerve in continuity with tumor is a sign of postlaminar optic nerve invasion
Ocular wall invasion and extraocular extension
Discontinuity of the normal choroidal enhancement is the leading criterion for infiltration [22, 24] (Fig. 13). Massive choroidal invasion presents as focal choroidal thickening (Fig. 13). Increased enhancement and thickening of the entire uveal tract (choroid, ciliary body, iris) is a sign of uveitis, usually secondary to massive (sub) total tumor necrosis [22]. Protrusion of enhancing tissue through the thickened choroid into the (low signal-intensity) sclera or beyond is a sign of scleral invasion or extraocular extension, respectively.
Fig. 13.

Postcontrast transaxial T1-weighted (TR/TE, 305/15 ms) MRI. Intraocular enhancing retinoblastoma combined with focal choroidal thickening and a discontinuity of the linear enhancement pattern of the choroid (arrowheads) adjacent to the tumor mass is suspicious for tumor invasion. Histopathological examination of this eye showed massive choroidal invasion
Anterior eye segment
Anterior eye segment enhancement occurs frequently in retinoblastoma and is usually a sign of iris angiogenesis [27, 30]. Tumor invasion into the anterior eye segment (Fig. 11) is an infrequent finding, usually associated with anteriorly located retinoblastoma. Enhancement of the tumor extending into the ciliary body or beyond should raise suspicion of anterior eye segment invasion. Transaxial T2-weighted images of both orbits can be used to depict a decreased anterior chamber depth (Fig. 10).
Brain
Careful analysis of midline structures should be performed to depict trilateral retinoblastoma (i.e. PNET located mainly in the pineal gland, or rarely in the suprasellar area) (Fig. 14) or leptomeningeal spread (if patient shows extensive postlaminar optic nerve invasion enhancement). Congenital brain malformations occur mainly in patients with 13q- deletion syndrome [10]. Benign pineal cysts should not be misinterpreted as pineal PNET, even in children with retinoblastoma [10, 66]. Thin-section T2-weighted and contrast-enhanced T1-weighted slices are helpful for differential diagnoses. In doubtful cases, close follow-up with MRI is recommended.
Fig. 14.

Pineoblastoma in a patient with hereditary unilateral retinoblastoma. Postcontrast axial T1-weighted (TR/TE, 650/10 ms) MRI shows a cystic enhancing mass of the pineal gland (arrow) suspicious for pineoblastoma (trilateral retinoblastoma)
Conclusion
Together with US, high-resolution MR imaging has emerged as the most important imaging modality in the assessment of retinoblastoma—for diagnostic confirmation and for determination of local tumor extent and associated intracranial abnormalities. CT is no longer indicated in children with leukocoria because of (1) ionizing radiation and (2) no added diagnostic value. US combined with MRI using our suggested standardized retinoblastoma MRI protocol provides state-of-the-art pretreatment diagnostic evaluation in children with retinoblastoma.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Grant support: P.d.G. is financially supported in part by grants from the ODAS Foundation, Delft, The Netherlands; ZonMw (Netherlands Organization for Health Research and Development) ‘s Gravenhage, The Netherlands; the National Foundation for the Blind and Visually Impaired, Utrecht, The Netherlands; the Blindenhulp Foundation, ‘s Gravenhage, The Netherlands; the Dutch Eye Fund (Grant 2004-23), Utrecht, The Netherlands. European Retinoblastoma Imaging Collaboration (ERIC) is financially supported by a grant from the ODAS Foundation, Delft, The Netherlands.
Presented at the Oncology Task Force Session; Annual Meeting of the European Society of Pediatric Radiology 2010, Bordeaux, France.
References
- 1.Moll AC, Kuik DJ, Bouter LM, et al. Incidence and survival of retinoblastoma in The Netherlands: a register based study 1862–1995. Br J Ophthalmol. 1997;81:559–562. doi: 10.1136/bjo.81.7.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doz F, Brisse HJ, Stoppa-Lyonnet D, et al. Retinoblastoma. In: Pinkerton R, Plowman P, Pieters R, et al., editors. Paediatric oncology. London: Arnold; 2004. pp. 323–338. [Google Scholar]
- 3.Shields CL, Shields JA. Retinoblastoma management: advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Curr Opin Ophthalmol. 2010;21:203–212. doi: 10.1097/ICU.0b013e328338676a. [DOI] [PubMed] [Google Scholar]
- 4.Abramson DH. Retinoblastoma in the 20th century: past success and future challenges the Weisenfeld lecture. Invest Ophthalmol Vis Sci. 2005;46:2683–2691. doi: 10.1167/iovs.04-1462. [DOI] [PubMed] [Google Scholar]
- 5.Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, et al. Retinoblastoma. Orphanet J Rare Dis. 2006;1:31. doi: 10.1186/1750-1172-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunkel IJ, Khakoo Y, Kernan NA, et al. Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatr Blood Cancer. 2010;55:55–59. doi: 10.1002/pbc.22504. [DOI] [PubMed] [Google Scholar]
- 7.Dunkel IJ, Chan HS, Jubran R, et al. High-dose chemotherapy with autologous hematopoietic stem cell rescue for stage 4B retinoblastoma. Pediatr Blood Cancer. 2010;55:149–152. doi: 10.1002/pbc.22491. [DOI] [PubMed] [Google Scholar]
- 8.Baud O, Cormier-Daire V, Lyonnet S, et al. Dysmorphic phenotype and neurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. Clin Genet. 1999;55:478–482. doi: 10.1034/j.1399-0004.1999.550614.x. [DOI] [PubMed] [Google Scholar]
- 9.Ballarati L, Rossi E, Bonati MT, et al. 13q Deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J Med Genet. 2007;44:e60. doi: 10.1136/jmg.2006.043059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodjan F, de Graaf P, Moll AC, et al. Brain abnormalities on MR imaging in patients with retinoblastoma. AJNR Am J Neuroradiol. 2010;31:1385–1389. doi: 10.3174/ajnr.A2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17:1829–1837. doi: 10.1200/JCO.1999.17.6.1829. [DOI] [PubMed] [Google Scholar]
- 12.Dunkel IJ, Jubran RF, Gururangan S, et al. Trilateral retinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010;54:384–387. doi: 10.1002/pbc.22336. [DOI] [PubMed] [Google Scholar]
- 13.Wright KD, Qaddoumi I, Patay Z, et al. Successful treatment of early detected trilateral retinoblastoma using standard infant brain tumor therapy. Pediatr Blood Cancer. 2010;55:570–572. doi: 10.1002/pbc.22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lumbroso-Le Rouic L, Aerts I, Levy-Gabriel C, et al. Conservative treatments of intraocular retinoblastoma. Ophthalmology. 2008;115(8):1410. doi: 10.1016/j.ophtha.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Abramson DH, Dunkel IJ, Brodie SE, et al. Superselective ophthalmic artery chemotherapy as primary treatment for retinoblastoma (chemosurgery) Ophthalmology. 2010;117:1623–1629. doi: 10.1016/j.ophtha.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Shields CL, Shields JA, Baez K, et al. Optic nerve invasion of retinoblastoma. Metastatic potential and clinical risk factors. Cancer. 1994;73:692–698. doi: 10.1002/1097-0142(19940201)73:3<692::aid-cncr2820730331>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Chantada GL, Dunkel IJ, Antoneli CB, et al. Risk factors for extraocular relapse following enucleation after failure of chemoreduction in retinoblastoma. Pediatr Blood Cancer. 2007;49:256–260. doi: 10.1002/pbc.21067. [DOI] [PubMed] [Google Scholar]
- 18.Chantada GL, Casco F, Fandino AC, et al. Outcome of patients with retinoblastoma and postlaminar optic nerve invasion. Ophthalmology. 2007;114:2083–2089. doi: 10.1016/j.ophtha.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 19.Uusitalo MS, Van Quill KR, Scott IU, et al. Evaluation of chemoprophylaxis in patients with unilateral retinoblastoma with high-risk features on histopathologic examination. Arch Ophthalmol. 2001;119:41–48. [PubMed] [Google Scholar]
- 20.Honavar SG, Singh AD, Shields CL, et al. Postenucleation adjuvant therapy in high-risk retinoblastoma. Arch Ophthalmol. 2002;120:923–931. doi: 10.1001/archopht.120.7.923. [DOI] [PubMed] [Google Scholar]
- 21.Brisse HJ, Guesmi M, Aerts I, et al. Relevance of CT and MRI in retinoblastoma for the diagnosis of postlaminar invasion with normal-size optic nerve: a retrospective study of 150 patients with histological comparison. Pediatr Radiol. 2007;37:649–656. doi: 10.1007/s00247-007-0491-4. [DOI] [PubMed] [Google Scholar]
- 22.de Graaf P, Barkhof F, Moll AC, et al. Retinoblastoma: MR imaging parameters in detection of tumor extent. Radiology. 2005;235:197–207. doi: 10.1148/radiol.2351031301. [DOI] [PubMed] [Google Scholar]
- 23.Shields CL, Shields JA, Baez KA, et al. Choroidal invasion of retinoblastoma: metastatic potential and clinical risk factors. Br J Ophthalmol. 1993;77:544–548. doi: 10.1136/bjo.77.9.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khelfaoui F, Validire P, Auperin A, et al. Histopathologic risk factors in retinoblastoma: a retrospective study of 172 patients treated in a single institution. Cancer. 1996;77:1206–1213. [PubMed] [Google Scholar]
- 25.Chantada GL, Dunkel IJ, de Davila MT, et al. Retinoblastoma patients with high risk ocular pathological features: who needs adjuvant therapy? Br J Ophthalmol. 2004;88:1069–1073. doi: 10.1136/bjo.2003.037044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sastre X, Chantada GL, Doz F, et al. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med. 2009;133:1199–1202. doi: 10.5858/133.8.1199. [DOI] [PubMed] [Google Scholar]
- 27.Galluzzi P, Cerase A, Hadjistilianou T, et al. Retinoblastoma: abnormal gadolinium enhancement of anterior segment of eyes at MR imaging with clinical and histopathologic correlation. Radiology. 2003;228:683–690. doi: 10.1148/radiol.2283020466. [DOI] [PubMed] [Google Scholar]
- 28.Pe'er J, Neufeld M, Baras M, et al. Rubeosis iridis in retinoblastoma. Histologic findings and the possible role of vascular endothelial growth factor in its induction. Ophthalmology. 1997;104:1251–1258. doi: 10.1016/s0161-6420(97)30150-x. [DOI] [PubMed] [Google Scholar]
- 29.Saket RR, Mafee MF. Anterior-segment retinoblastoma mimicking pseudoinflammatory angle-closure glaucoma: review of the literature and the important role of imaging. AJNR Am J Neuroradiol. 2009;30:1607–1609. doi: 10.3174/ajnr.A1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Graaf P, van der Valk P, Moll AC, et al. Contrast-enhancement of the anterior eye segment in patients with retinoblastoma: correlation between clinical, MR imaging, and histopathologic findings. AJNR Am J Neuroradiol. 2010;31:237–245. doi: 10.3174/ajnr.A1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chantada GL, Guitter MR, Fandino AC et al (2008) Treatment results in patients with retinoblastoma and invasion to the cut end of the optic nerve. Pediatr Blood Cancer 52(2):218–222 [DOI] [PubMed]
- 32.Marees T, van Leeuwen FE, Schaapveld M, et al. Risk of third malignancies and death after a second malignancy in retinoblastoma survivors. Eur J Cancer. 2010;46:2052–2058. doi: 10.1016/j.ejca.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 33.Marees T, van Leeuwen FE, de Boer MR, et al. Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer. 2009;45:3245–3253. doi: 10.1016/j.ejca.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 34.Marees T, Moll AC, Imhof SM, et al. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100:1771–1779. doi: 10.1093/jnci/djn394. [DOI] [PubMed] [Google Scholar]
- 35.Gombos DS, Hungerford J, Abramson DH, et al. Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology. 2007;114:1378–1383. doi: 10.1016/j.ophtha.2007.03.074. [DOI] [PubMed] [Google Scholar]
- 36.Wong FL, Boice JD, Jr, Abramson DH, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278:1262–1267. doi: 10.1001/jama.278.15.1262. [DOI] [PubMed] [Google Scholar]
- 37.Brenner D, Elliston C, Hall E, et al. Estimated risks of radiation-induced fatal cancer from pediatric CT. AJR Am J Roentgenol. 2001;176:289–296. doi: 10.2214/ajr.176.2.1760289. [DOI] [PubMed] [Google Scholar]
- 38.Imhof SM, Mourits MP, Hofman P, et al. Quantification of orbital and mid-facial growth retardation after megavoltage external beam irradiation in children with retinoblastoma. Ophthalmology. 1996;103:263–268. doi: 10.1016/s0161-6420(96)30706-9. [DOI] [PubMed] [Google Scholar]
- 39.Vijayakrishnan R, Shields CL, Ramasubramanian A, et al. Irradiation toxic effects during intra-arterial chemotherapy for retinoblastoma: should we be concerned? Arch Ophthalmol. 2010;128:1427–1431. doi: 10.1001/archophthalmol.2010.258. [DOI] [PubMed] [Google Scholar]
- 40.Moll AC, Hoekstra OS, Imhof SM, et al. Fluorine-18 fluorodeoxyglucose positron emission tomography (PET) to detect vital retinoblastoma in the eye: preliminary experience. Ophthalmic Genet. 2004;25:31–35. doi: 10.1076/opge.25.1.31.29001. [DOI] [PubMed] [Google Scholar]
- 41.Kaste SC, Jenkins JJ, III, Pratt CB, et al. Retinoblastoma: sonographic findings with pathologic correlation in pediatric patients. AJR Am J Roentgenol. 2000;175:495–501. doi: 10.2214/ajr.175.2.1750495. [DOI] [PubMed] [Google Scholar]
- 42.Galluzzi P, Hadjistilianou T, Cerase A, et al. Is CT still useful in the study protocol of retinoblastoma? AJNR Am J Neuroradiol. 2009;30:1760–1765. doi: 10.3174/ajnr.A1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roth DB, Scott IU, Murray TG, et al. Echography of retinoblastoma: histopathologic correlation and serial evaluation after globe-conserving radiotherapy or chemotherapy. J Pediatr Ophthalmol Strabismus. 2001;38:136–143. doi: 10.3928/0191-3913-20010501-06. [DOI] [PubMed] [Google Scholar]
- 44.Lemke AJ, Kazi I, Mergner U, et al. Retinoblastoma - MR appearance using a surface coil in comparison with histopathological results. Eur Radiol. 2007;17:49–60. doi: 10.1007/s00330-006-0197-2. [DOI] [PubMed] [Google Scholar]
- 45.Brisse HJ, Lumbroso L, Freneaux PC, et al. Sonographic, CT, and MR imaging findings in diffuse infiltrative retinoblastoma: report of two cases with histologic comparison. AJNR Am J Neuroradiol. 2001;22:499–504. [PMC free article] [PubMed] [Google Scholar]
- 46.Finger PT, Khoobehi A, Ponce-Contreras MR, et al. Three dimensional ultrasound of retinoblastoma: initial experience. Br J Ophthalmol. 2002;86:1136–1138. doi: 10.1136/bjo.86.10.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beets-Tan RG, Hendriks MJ, Ramos LM, et al. Retinoblastoma: CT and MRI. Neuroradiology. 1994;36:59–62. doi: 10.1007/BF00599199. [DOI] [PubMed] [Google Scholar]
- 48.Chung EM, Specht CS, Schroeder JW. From the archives of the AFIP: Pediatric orbit tumors and tumorlike lesions: neuroepithelial lesions of the ocular globe and optic nerve. Radiographics. 2007;27:1159–1186. doi: 10.1148/rg.274075014. [DOI] [PubMed] [Google Scholar]
- 49.James SH, Halliday WC, Branson HM. Best cases from the AFIP: Trilateral retinoblastoma. Radiographics. 2010;30:833–837. doi: 10.1148/rg.303095142. [DOI] [PubMed] [Google Scholar]
- 50.Golding SJ. Radiation exposure in CT: what is the professionally responsible approach? Radiology. 2010;255:683–686. doi: 10.1148/radiol.10100449. [DOI] [PubMed] [Google Scholar]
- 51.John-Mikolajewski V, Messmer E, Sauerwein W, et al. Orbital computed tomography. Does it help in diagnosing the infiltration of choroid, sclera and/or optic nerve in retinoblastoma? Ophthalmic Paediatr Genet. 1987;8:101–104. doi: 10.3109/13816818709028525. [DOI] [PubMed] [Google Scholar]
- 52.Jacquemin C, Karcioglu ZA. Detection of optic nerve involvement in retinoblastoma with enhanced computed tomography. Eye (Lond) 1998;12:179–183. doi: 10.1038/eye.1998.44. [DOI] [PubMed] [Google Scholar]
- 53.Olivecrona H, Agerberg PA, Huaman A. CT diagnosis of retinoblastoma with histopathologic correlations. Eur Radiol. 1994;4:307–313. [Google Scholar]
- 54.Mafee MF, Mafee RF, Malik M, et al. Medical imaging in pediatric ophthalmology. Pediatr Clin North Am. 2003;50:259–286. doi: 10.1016/s0031-3955(03)00002-6. [DOI] [PubMed] [Google Scholar]
- 55.Apushkin MA, Apushkin MA, Shapiro MJ, et al. Retinoblastoma and simulating lesions: role of imaging. Neuroimaging Clin N Am. 2005;15:49–67. doi: 10.1016/j.nic.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Barkovich AJ. Pediatric neuroimaging. Philadelphia: Lippincott Williams & Wilkins; 2005. [Google Scholar]
- 57.Gizewski ER, Wanke I, Jurklies C, et al. T1 Gd-enhanced compared with CISS sequences in retinoblastoma: superiority of T1 sequences in evaluation of tumour extension. Neuroradiology. 2005;47:56–61. doi: 10.1007/s00234-004-1316-x. [DOI] [PubMed] [Google Scholar]
- 58.Schueler AO, Hosten N, Bechrakis NE, et al. High resolution magnetic resonance imaging of retinoblastoma. Br J Ophthalmol. 2003;87:330–335. doi: 10.1136/bjo.87.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ainbinder DJ, Haik BG, Frei DF, et al. Gadolinium enhancement: improved MRI detection of retinoblastoma extension into the optic nerve. Neuroradiology. 1996;38:778–781. doi: 10.1007/s002340050346. [DOI] [PubMed] [Google Scholar]
- 60.Barkhof F, Smeets M, van der Valk P, et al. MR imaging in retinoblastoma. Eur Radiol. 1997;7:726–731. doi: 10.1007/BF02742934. [DOI] [PubMed] [Google Scholar]
- 61.Wilson MW, Rodriguez-Galindo C, Billups C, et al. Lack of correlation between the histologic and magnetic resonance imaging results of optic nerve involvement in eyes primarily enucleated for retinoblastoma. Ophthalmology. 2009;116:1558–1563. doi: 10.1016/j.ophtha.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 62.Lemke AJ, Hosten N, Bornfeld N, et al. Uveal melanoma: correlation of histopathologic and radiologic findings by using thin-section MR imaging with a surface coil. Radiology. 1999;210:775–783. doi: 10.1148/radiology.210.3.r99fe39775. [DOI] [PubMed] [Google Scholar]
- 63.Mafee MF, Rapoport M, Karimi A, et al. Orbital and ocular imaging using 3- and 1.5-T MR imaging systems. Neuroimaging Clin N Am. 2005;15:1–21. doi: 10.1016/j.nic.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 64.Lemke AJ, Alai-Omid M, Hengst SA, et al. Eye imaging with a 3.0-T MRI using a surface coil–a study on volunteers and initial patients with uveal melanoma. Eur Radiol. 2006;16:1084–1089. doi: 10.1007/s00330-005-0087-z. [DOI] [PubMed] [Google Scholar]
- 65.Brisse HJ. Retinoblastoma imaging. Ophthalmology. 2010;117:1051. doi: 10.1016/j.ophtha.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 66.Beck PM, Balmer A, Maeder P, et al. Benign pineal cysts in children with bilateral retinoblastoma: a new variant of trilateral retinoblastoma? Pediatr Blood Cancer. 2006;46:755–761. doi: 10.1002/pbc.20464. [DOI] [PubMed] [Google Scholar]