

Abstract
The multi-functional apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1) DNA repair and redox signaling protein has been shown to have a role in cancer growth and survival, however, little has been investigated concerning its role in inflammation. In this study, an APE1 redox-specific inhibitor (E3330) was used in lypopolysaccharide (LPS)-stimulated macrophages (RAW264.7). E3330 clearly suppressed secretion of inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin (IL-6) and IL-12 and inflammatory mediators nitric oxide (NO) as well as prostaglandin E2 (PGE2) from the LPS-stimulated RAW264.7 cells. These data were supported by the down-regulation of the LPS-dependent expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) genes in the RAW264.7 cells. The effects of E3330 were mediated by the inhibition of transcription factors nuclear factor-κB (NF-κB) and activator protein 1 (AP-1) in the LPS-stimulated macrophages, both known targets of APE1. In conclusion, pharmacological inhibition of APE1 by E3330 suppresses inflammatory response in activated macrophages and can be considered as a novel therapeutic strategy for the inhibition of tumor-associated macrophages.
Keywords: APE1, Ref-1, macrophages, inflammatory response
Apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1 or APE1) is a multifunctional protein which functions as the DNA base excision repair protein and as a redox signaling factor keeping transcription factors in an active reduced state (1, 2). APE1 stimulates the DNA binding activity of a number of transcription factors involved in cancer and inflammation (e.g. activator protein 1 [AP-1], nuclear factor-κB [NF-κB], hypoxia-inducible transcription factor 1α [HIF-1α], cAMP response-element binding [CREB], p53 and others); thus, APE1 has been suggested as a therapeutic and chemopreventive target (1–8). Indeed, E3330, a small-molecule inhibitor of the APE1 redox domain, suppressed proliferation and migration of pancreatic cancer cells and inhibited growth of pancreatic cancer-associated endothelial cells, respectively (9–11). APE1 was significantly increased in the colon epithelium in patients with ulcerative colitis, a precursor of colon cancer (12). Moreover, APE1 modulated inflammatory responses through Toll-like receptor 2 (TLR2) by the activation of NF-κB and HIF-1α resulting in the expression of inflammatory cytokines and chemokines tumor necrosis factor-α (TNF-α) and chemokine (C-X-C motif) ligand 8 (CXCL8) in primary keratinocytes (13).
The tumor microenvironment contains stromal cells such as fibroblasts, endothelial cells, and macrophages. In spite of the importance of macrophages in the host-defense mechanism and inflammation (14, 15), the overproduction of inflammatory mediators by macrophages has also been implicated in cancer (16), whereas anti-inflammatory drugs reduce the risk of cancer (17). Further, a high density of tumor-associated macrophages correlates with poor prognosis in a majority of published studies, including those on breast, prostate, bladder, kidney, endometrial and esophageal carcinomas (18, 19). Thus, the inflammatory stimuli from macrophages promote cancer growth (20–22), whereas the reduction of macrophages results in the decrease of tumor growth (22). Macrophage activations result in the secretion of a number of different inflammatory mediators, including TNF-α, interleukin-6 (IL-6), reactive oxygen species (ROS), prostaglandin E2 (PGE2) and nitric oxide (NO) (23–29). Lipopolysaccharide (LPS), a constituent of the gram-negative bacterial cell wall, interacts with TLR4, which is expressed on macrophages (30, 31). The interaction between LPS and the TLR4 receptor complex activates intracellular signaling through myeloid differentiation primary response gene 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) pathways leading to the activation of transcription factors NF-κB and AP-1 and the expression of TNF-α and IL-6 (32, 33). In addition, LPS induces expression of IL-12 in macrophages through NF-κB (27, 34), and AP-1 (35). Moreover, LPS-dependent expression of cyclooxygenase-2 (COX-2), which controls production of PGE2, and inducible nitric oxide synthase (iNOS), which controls production of NO, is also regulated through NF-κB and AP-1 (36).
In the present study, the anti-inflammatory activity of E3330, a small-molecule inhibitor of APE1 redox signaling function, was investigated in LPS-challenged RAW264.7 macrophages.
Materials and Methods
Cell culture
RAW264.7 cells, a murine macrophage cell line (American Type Culture Collection, Manassas, VA, USA), were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin. The RAW264.7 cells were plated at a density of 2–3×106 and cultivated at 37°C in a humidified atmosphere containing 5% CO2. For all the experiments, the cells were grown to 80–90% confluence. The RAW264.7 cells were stimulated with 1 μg/ml LPS (Sigma, St. Louis, MO, USA). The cells were incubated with or without E3330 in the serum free medium for 1 hour before exposure to LPS. E3330 was synthesized as previously published (4, 37, 38), and used as described in the individual experiments.
Expression of pro-inflammatory mediators in RAW264.7 cells
The RAW264.7 cells were preincubated with 0–25 μg/ml E3330 for 24 hours followed by an additional 24 hours’ incubation with LPS (1 μg/ml). The levels of TNF-α, IL-6, IL-12p40 (Biolegend, San Diego, CA, USA) and PGE2 (R&D Systems, Minneapolis, MN, USA) were determined by ELISA; the release of NO was determined by Griess reagent (Sigma) as previously described (39).
Preparation of whole-cell and nuclear extracts
After E3330 and LPS treatments (the whole-cell and nuclear extracts were prepared as described previously (39). The protein concentration in the whole-cell and nuclear extracts was determined according to the manufacturer’s protocol (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot analysis
Western blot analysis was performed as previously described (39), on RAW264.7 cells pretreated with E3330 (0–25 μg/ml) for 24 hours followed by LPS (1 μg/ml) for 24 hours. Briefly, the total cell lysates were separated by SDS–PAGE and were transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% bovine serum albumin in TBS-Tween 20 solution and was further incubated with the corresponding antibody: COX-2 (BD Biosciences, San Jose, CA, USA), iNOS and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Reactive bands were visualized with horseradish peroxidase (HRP)-coupled secondary antibody via an enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia Biotech, Little Chalfont, UK) according to the manufacturer’s procedures.
Electrophoretic mobility shift assay (EMSA)
The nuclear extracts from RAW264.7 cells pretreated with E3330 (0–25 μg/ml) for 24 hours followed by LPS (1 μg/ml) for 30 minutes were used. Oligonucleotide probes containing consensus sequences for AP-1-and NF-κB-binding sites were purchased from Promega (Madison, WI, USA). EMSA for AP-1 and NF-κB was performed with a 32P-AP-1 or 32P-NF-κB probe in gel shift binding buffer with 5 μg of nuclear protein as previously described (39). The specificity was confirmed by competitive EMSA with cold AP-1 or NF-κB oligonucleotide or unrelated DNA (URL).
Statistical analysis
Data are presented as mean±S.D and were analyzed by Student t-test. Results were considered significant if p≤0.05.
Results
Effect of E3330 on LPS-dependent production of pro-inflammatory cytokines
As expected, LPS stimulation markedly induced the production of TNF-α from the RAW264.7 cells, whereas pretreatment with E3330 at doses 12.5 and 25 μg/ml significantly suppressed TNF-α production from these macrophage cells (Figure 1A). In addition, E3330 (6.25–25 μg/ml) also significantly suppressed IL-6 production from LPS-stimulated RAW264.7 cells (Figure 1B). Moreover, LPS-dependent production of IL-12 in macrophages was also markedly suppressed by E3330 in a dose-dependent manner (Figure 1C).
Figure 1.

Effect of E3330 on LPS-induced production of pro-inflammatory cytokines in RAW264.7 cells. (A) TNF-α, (B) IL-6 (C) IL-12 production in cell culture media from RAW264.7 cells pretreated with E3330 for 24 hours followed by LPS for an additional 24 hours. Each value is the mean±S.D. of two independent experiments, repeated minimally twice. *p<0.05 control vs. LPS alone, *p<0.05 LPS vs. LPS plus E3330.
Effect of E3330 on LPS-dependent secretion of PGE2 and NO and on the regulation of expression of COX-2 and iNOS
E3330 treatment markedly decreased the secretion of the LPS-induced inflammatory mediators PGE2 (Figure 2A) and NO (Figure 2B) in a dose-dependent manner. Because the production of PGE2 and NO is controlled by COX-2 and iNOS, respectively, the expression of COX-2 and iNOS was evaluated by Western blot. As shown in Figure 2C, LPS induced the expression of COX-2 in the RAW264.7 cells, whereas E3330 pretreatment suppressed this LPS-dependent expression. In addition, the LPS-induced expression of iNOS was also markedly reduced by the E3330 in the macrophages (Figure 2D).
Figure 2.

Effect of E3330 on LPS-induced PGE2, NO, COX-2 and iNOS expression in RAW264.7 cells. RAW264.7 cells were pretreated with E3330 for 24 hours followed by LPS for 24 hours. (A) PGE2 and (B) NO in cell culture media. Each value is the mean±S.D. of two independent experiments, repeated minimally twice. p<0.05 control vs. LPS alone, *p<0.05 LPS vs. LPS plus E3330. (C) COX-2 and (D) iNOS determined in lysates by Western blot analysis. The equal protein loading was verified with anti-β-actin antibody. The results are representative of three separate experiments.
Effect of E3330 on LPS-inducible NF-κB and AP-1 activation
NF-κB and AP-1 DNA-binding activity was evaluated in nuclear extracts by gel shift analysis. As shown in Figure 3A, LPS markedly induced the binding activity of nuclear extracts to the NF-κB DNA consensus sequence, whereas pretreatment of the macrophages for 24 hours with E3330 (12.5 and 25 μg/ml) suppressed the LPS-dependent increase of NF-κB binding. As expected, the binding activity of AP-1 in nuclear extracts was also induced by the LPS treatment, and E3330 pretreatment suppressed this LPS-dependent binding activity of AP-1 in the RAW264.7 cells (Figure 3B).
Figure 3.
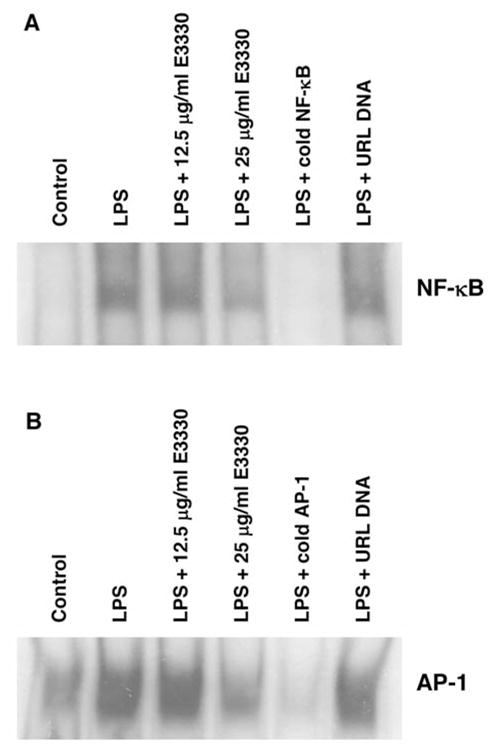
Effect of E3330 on NF-κB and AP-1 DNA-binding. RAW264.7 cells were pretreated with E3330 for 24 hours followed by the LPS (1 μg/ml) treatment for 30 min. Nuclear extracts were subjected to gel shift analysis with (A) [32P]-labeled NF-κB probe or [32P]-labeled AP-1 probe. The specificity was confirmed by the competitive gel shift in LPS-stimulated cells with cold NF-κB or AP-1 or unrelated DNA (URL). The data are representative of three separate experiments.
Discussion
The inhibition of APE1 redox signaling function by the APE1-specific small molecule E3330 suppressed the LPS-dependent production of pro-inflammatory cytokines, TNF-α, IL-6, and IL-12, and inflammatory mediators, NO and PGE2, in the murine macrophages RAW264.7 cells. The inhibition of secretion of NO and PGE2 by E3330 was mediated through the down-regulation of expression of iNOS and COX-2, respectively. In addition, E3330 inhibited the LPS-dependent induction of NF-κB and AP-1. Mechanistically, E3330 inhibits or blocks the redox function of APE1 resulting in the cysteine residues located in the DNA binding domain of p50 (NF-κB) and Fos and Jun (AP-1) not being converted from an oxidized to a reduced status. This results in the inability of NF-κB and AP-1 to bind to their respective DNA target sequences (Figure 4) (40, 41).
Figure 4.

Schematic illustration of the relationship of affected molecules and pathways following inhibition of Ref-1 (APE1) redox function with E3330. Blocking Ref-1 with the small molecule Ref-1 redox inhibitor E3330 inhibits NF-κB function and most likely other transcription factors that are downstream of Ref-1 such as AP-1 and CREB leading to a decrease in synthesis of COX2, IL-12, IL-6, TNF-α and iNOS. Decreased levels of iNOS result in decreased NO produced. Decreased production of COX2 leads to fewer AA being converted by COX2 and eventually to PGE2 which could result in a signaling cascade decrease that further suppresses iNOS production via CREB suppression. NFAT, Nuclear factor of activated T-cells; PPAR, peroxisome proliferator-activated receptor; CREB, cAMP response element-binding; PR, prostagladin receptors; C/EBP, CCAAT-enhancer binding proteins. The red lettering signifies molecules measured in the present study.
The present study was in congruence with original reports demonstrating the suppression of LPS-dependent production of TNF-α by E3330 through the inhibition of NF-κB in macrophages and monocytes (42, 43). The inhibition of TNF-α by E3330 has also been associated with the protection against endotoxin-mediated hepatitis and alcoholic liver injury (44, 45). However, the data presented here demonstrates, for the first time, that the inhibition of NF-κB and AP-1 by E3330 can directly suppress production of IL-6, IL-12, PGE2 and NO and down-regulate expression of COX-2 and iNOS in activated macrophages.
APE1 has been the focus of a number of studies, from use as a diagnostic aid in cancer screening (46) to a suitable target for the prevention or treatment of cancer (1). However, more intensive studies have focused on both repair and redox functions of APE1 as a novel therapeutic target in cancer (3–5). Although recent studies have demonstrated a direct effect of E3330 on a variety of cancer cells (1, 9, 37, 38–34, 47–51), the inhibition of the inflammatory response by E3330 in activated macrophages is of particular interest.
The inhibition of NF-κB and AP-1 signaling in activated macrophages by E3330 could have a potential therapeutic effect as NF-κB and AP-1 are necessary for the inflammatory response leading to the production of TNF-α, IL-6, IL-12, NO and PGE2 and other inflammatory mediators in these cells (23–29). As mentioned above, a link between activated macrophages and cancer has been demonstrated (18, 20–22), and a recent study further confirmed that an increased number of tumor-associated macrophages was strongly associated with shortened survival of cancer patients (19). Although the depletion of macrophages with clodronate inhibited tumor recovery after irradiation (52), this strategy would also deplete other non-tumor-associated macrophages and would therefore interrupt the homeostatic function of macrophages in the gut, liver, lung and spleen in humans (53). Therefore, direct targeting of tumor-associated macrophages is a more plausible strategy. Blocking TNF-α with TNF-α receptor or inhibiting macrophage recruitment by macrophage antibodies inhibited tumor regrowth after tumor irradiation, respectively (52, 54). Another possible strategy would be the inhibition of secretion of pro-inflammatory cytokines and mediators from macrophages by E3330 as presented in the current study.
In summary, E3330 exerts an anti-inflammatory effect by blocking the ability of APE1 to convert NF-κB and AP-1 from an oxidized to a reduced state. These two transcription factors have been implicated in signaling in LPS-activated macrophages, Thus E3330 treatment results in the suppression of the production of pro-inflammatory cytokines and mediators. The inflammatory response in LPS-stimulated macrophages controlled by APE1 and has a potential clinical applicability to affect the tumor microenvironment as having direct effects on tumor cells. The demonstration of an anti-inflammatory activity of E3330 provides further proof-of-concept data promoting the development of E3330 as an anti-inflammatory agent for the inhibition of the detrimental function of tumor-associated macrophages in cancer treatment. Furthermore, it continues to illustrate the important function of APE1 redox signaling in the control of the tumor microenvironment.
Acknowledgments
This study was supported by the Methodist Research Institute, Indian University Health. MRK is supported by the National Institutes of Health, National Cancer Institute [CA94025, CA106298, CA114571 and CA121168 to M.R.K.] and the Riley Children’s Foundation. We also thank Dr. Michael Vasko, Department of Pharmacology for editing Figure 4.
Footnotes
This article is freely accessible online.
References
- 1.Fishel ML, Kelley MR. The DNA base excision repair protein APE1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Georgiadis M, Luo M, Gaur R, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo M, He H, Kelley MR, Georgiadis M. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, II, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small molecule inhibition of Ape1’s redox function. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bapat A, Fishel ML, Georgiadis M, Kelley MR. Going ape as an approach to cancer therapeutics. Antioxid Redox Signal. 2009;11:651–668. doi: 10.1089/ars.2008.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bapat A, Glass LS, Luo M, Fishel ML, Long EC, Georgiadis MM, Kelley MR. Novel small molecule inhibitor of Ape1 endonuclease blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther. 2010;334:988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tell G, Zecca A, Pellizzari L, Spessotto P, Colombatti A, Kelley MR, Damante G, Pucillo C. An ‘environment to nucleus’ signaling system operates in B lymphocytes: redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation. Nucleic Acids Res. 2000;28:1099–1105. doi: 10.1093/nar/28.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008;7:2012–2021. doi: 10.1158/1535-7163.MCT-08-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou GM, Karikari C, Kabe Y, Handa H, Anders RA, Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009;219:209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- 11.Jimeno A, Feldmann G, Suárez-Gauthier A, Rasheed Z, Solomon A, Zou GM, Rubio-Viqueira B, García-García E, López-Ríos F, Matsui W, Maitra A, Hidalgo M. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther. 2009;8:310–314. doi: 10.1158/1535-7163.MCT-08-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, Hussain SP, Roth RB, Zhou X, Mechanic LE, Zurer I, Rotter V, Samson LD, Harris CC. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112:1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HM, Yuk JM, Shin DM, Yang CS, Kim KK, Choi DK, Liang ZL, Kim JM, Jeon BH, Kim CD, Lee JH, Jo EK. Apurinic/apyrimidinic endonuclease 1 is a key modulator of keratinocyte inflammatory responses. J Immunol. 2009;183:6839–6848. doi: 10.4049/jimmunol.0901856. [DOI] [PubMed] [Google Scholar]
- 14.Laskin DL, Pendino KJ. Macrophages and inflammatory mediators in tissue injury. Annu Rev Pharmacol Toxicol. 1995;35:655–677. doi: 10.1146/annurev.pa.35.040195.003255. [DOI] [PubMed] [Google Scholar]
- 15.Hawiger J. Innate immunity and inflammation: a transcriptional paradigm. Immunol Res. 2001;23:99–109. doi: 10.1385/IR:23:2-3:099. [DOI] [PubMed] [Google Scholar]
- 16.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Bingle L, Brown NJ, Lewis CE. The role of tumor-associated macrophages in tumor progression: implication for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 19.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Jaffe ES, Lenz G, Connors JM, Staudt LM, Chan WC, Gascoyne RD. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagemann T, Robinson SC, Schulz M, Trumper L, Balkwill FR, Binder C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-alpha dependent up-regulation of matrix metalloproteases. Carcinogenesis. 2004;25:1543–1549. doi: 10.1093/carcin/bgh146. [DOI] [PubMed] [Google Scholar]
- 21.Lin CY, Lin CJ, Chen KH, Wu JC, Huang SH, Wang SM. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. 2006;580:3042–3050. doi: 10.1016/j.febslet.2006.04.049. [DOI] [PubMed] [Google Scholar]
- 22.Kimura YN, Watari K, Fotovati A, Hosoi F, Yasumoto K, Izumi H, Kohno K, Umezawa K, Iguchi H, Shirouzu K, Takamori S, Kuwano M, Ono M. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007;98:2009–2018. doi: 10.1111/j.1349-7006.2007.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nature Rev Immunol. 2002;2:787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- 24.Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24:25–29. doi: 10.1016/s1471-4906(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 25.Bosca L, Zeini M, Traves PG, Hortelano S. Nitric oxide and cell viability in inflammatory cells: a role for NO in macrophage function and fate. Toxicology. 2005;208:249–258. doi: 10.1016/j.tox.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 26.Lapa e Silva JR, Possebon da Silva MD, Lefort J, Vargaftig BB. Endotoxins, asthma, and allergic immune responses. Toxicology. 2000;152:31–35. doi: 10.1016/s0300-483x(00)00289-4. [DOI] [PubMed] [Google Scholar]
- 27.Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- 28.Bogdan C. Nitric oxide and the immune response. Nature Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 29.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 31.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 32.Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol. 2008;181:1849–1858. doi: 10.4049/jimmunol.181.3.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, Wahl LM, Fenton MJ, Vogel SN. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem. 2007;282:16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol. 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu C, Gagnidze K, Gemberling JH, Plevy SE. Characterization of an activation protein-1-binding site in the murine interleukin-12 p40 promoter. Demonstration of novel functional elements by a reductionist approach. J Biol Chem. 2001;276:18519–18528. doi: 10.1074/jbc.M100440200. [DOI] [PubMed] [Google Scholar]
- 36.Cho HJ, Seon MR, Lee YM, Kim J, Kim JK, Kim SG, Park JHY. 3,3′-Diindolylmethane suppresses the inflammatory response to lipopolysaccharide in murine macrophages. J Nutr. 2008;138:17–23. doi: 10.1093/jn/138.1.17. [DOI] [PubMed] [Google Scholar]
- 37.Kelley MR, Luo M, Reed A, Su D, Delaplane S, Borch R, Nyland RL, Gross ML, Georgiadis M. Functional analysis of new and novel analogs of E3330 that block the redox signaling activity of the multifunctional AP endonuclease/redox signaling enzyme APE1/Ref-1. Antioxid Redox Signal. 2011 Jan 4; doi: 10.1089/ars.2010.3410. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/Ref-1) J Med Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dudhgaonkar S, Thyagarajan A, Sliva D. Suppression of the inflammatory response by triterpenes isolated from the mushroom Ganoderma lucidum. Int Immunopharmacol. 2009;9:1272–1280. doi: 10.1016/j.intimp.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 40.Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, Aizawa S, Tanaka H, Kataoka K, Watanabe H, Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 41.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyamoto K, Nagakawa J, Hishinuma I, Hirota K, Yasuda M, Yamanaka T, Katayama K, Yamatsu I. Suppressive effects of E3330, a novel quinone derivative, on tumor necrosis factor-alpha generation from monocytes and macrophages. Agents Actions. 1992;37:297–304. doi: 10.1007/BF02028123. [DOI] [PubMed] [Google Scholar]
- 43.Goto M, Yamada K, Katayama K, Tanaka I. Inhibitory effect of E3330, a novel quinone derivative able to suppress tumor necrosis factor-alpha generation, on activation of nuclear factor-kappa B. Mol Pharmacol. 1996;49:860–873. [PubMed] [Google Scholar]
- 44.Nagakawa J, Hishinuma I, Miyamoto K, Hirota K, Abe S, Yamanaka T, Katayama K, Yamatsu I. Protective effects of (2E)-3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propenoic acid on endotoxin-mediated hepatitis in mice. J Pharmacol Exp Ther. 1992;262:145–150. [PubMed] [Google Scholar]
- 45.Nanji AA, Sadrzadeh SM, Khettry U, Thomas P, Yamanaka T. Protective effects of a novel quinone derivative, (2E)-3-[5-(2,3 dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propanoic acid on experimental alcoholic liver injury. J Pharmacol Exp Ther. 1993;266:1085–1090. [PubMed] [Google Scholar]
- 46.Evans AR, Limp-Foster M, Kelley MR. Going APE overref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 47.Jiang Y, Zhou S, Sandusky GE, Kelley MR, Fishel ML. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest. 2010;28:885–895. doi: 10.3109/07357907.2010.512816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fishel ML, Colvin ES, Luo M, Kelley MR, Robertson KA. Inhibition of the redox function of APE1/Ref-1 in myeloid leukemia cell lines result in enhanced sensitivity to retinoic acid-induced differentiation and apoptosis. Exp Hematol. 2010;38:1178–1188. doi: 10.1016/j.exphem.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang Y, Guo C, Fishel ML, Wang ZY, Vasko MR, Kelley MR. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: use of APE1 small-molecule inhibitors to delineate APE1 functions. DNA Repair. 2009;8:1273–1282. doi: 10.1016/j.dnarep.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saitou Y, Shiraki K, Yamanaka T, Miyashita K, Inoue T, Yamanaka Y, Yamaguchi Y, Enokimura N, Yamamoto N, Itou K, Sugimoto K, Nakano T. Augmentation of tumor necrosis factor family-induced apoptosis by E3330 in human hepatocellular carcinoma cell lines via inhibition of NF kappa B. World J Gastroenterol. 2005;11:6258–6261. doi: 10.3748/wjg.v11.i40.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Curtis CD, Thorngren DL, Ziegler YS, Sarkeshik A, Yates JR, Nardulli AM. Apurinic/apyrimidinic endonuclease 1 alters estrogen receptor activity and estrogen-responsive gene expression. Mol Endocrinol. 2009;23:1346–1359. doi: 10.1210/me.2009-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, Weichselbaum RR. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qualls JE, Murray PJ. A double agent in cancer: stopping macrophages wounds tumors. Nature Med. 2010;16:863–864. doi: 10.1038/nm0810-863. [DOI] [PubMed] [Google Scholar]
- 54.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. 2010;107:8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]