

Abstract
Histone deacetylase inhibitors (HDACi) represent a promising avenue for cancer therapy. We applied mass spectrometry (MS) to determine the impact of clinically relevant HDACi on global levels of histone acetylation. Intact histone profiling revealed that the HDACi SAHA and MS-275 globally increased histone H3 and H4 acetylation in both normal diploid fibroblasts and transformed human cells. Histone H3 lysine 56 acetylation (H3K56ac) recently elicited much interest and controversy due to its potential as a diagnostic and prognostic marker for a broad diversity of cancers. Using quantitative MS, we demonstrate that H3K56ac is much less abundant than previously reported in human cells. Unexpectedly, in contrast to H3/H4 N-terminal tail acetylation, H3K56ac did not increase in response to inhibitors of each class of HDACs. In addition, we demonstrate that antibodies raised against H3K56ac peptides cross-react against H3 N-terminal tail acetylation sites that carry sequence similarity to residues flanking H3K56.
Eukaryotic DNA is packaged into a nucleoprotein structure known as chromatin. The primary function of this structure is to compact DNA within the cell nucleus. The fundamental repeating unit of chromatin is the nucleosome core particle (NCP), which is composed of 147 base pairs of DNA wrapped nearly twice around the surface of an octamer of small basic proteins known as histones1. This octamer is formed from two molecules each of four types of core histones: H2A, H2B, H3, and H4. Although the structure of chromatin is inherently dynamic, it also considerably restricts access to genetic information2. Cells have evolved mechanisms to control access to DNA packaged into chromatin. These include ATP-driven nucleosome remodeling machines, histone variants and histone post-translational modifications (PTMs)3. Each core histone consists of a conformationally flexible N-terminal extension, commonly referred to as “histone tail”, and a globular domain that mediates protein-protein interactions and DNA binding within NCPs1. The N-terminal tails of core histones protrude beyond the DNA gyres of NCPs, and are therefore readily accessible to histone-modifying enzymes1. These tail domains contain multiple residues that are covalently modified by a wide diversity of PTMs3. Advances in mass spectrometry (MS) have also uncovered a number of PTMs in the globular domains of core histones4,5,6. Histone PTMs occur either alone or in combination, leading to the notion of a “histone code”, a generic term used to describe how specific patterns of histone PTMs influence the binding of effector proteins to NCPs and, as a result, control a variety of processes that require access to DNA7.
One of the most intensively studied histone PTMs is the acetylation of lysine residues3. The addition and removal of an acetyl group on lysine residues, respectively mediated by histone acetyltransferases (HATs) and deacetylases (HDACs), is a highly dynamic and regulated process that contributes to transcriptional activation and silencing8. In humans, there are 18 known HDAC enzymes that are subdivided into four classes9. The seminal discovery that the histone deacetylase inhibitor (HDACi) trichostatin A (TSA) triggered cell cycle arrest and differentiation of Friend erythroleukaemic cells promoted numerous subsequent studies on various types of cancer cells10. In diseases such as cancer, HDACs contribute to oncogenesis via at least two distinct mechanisms: overexpression of individual HDACs11, and aberrant recruitment of HDACs to specific chromosomal loci by oncogenic fusion proteins (e.g. PML-RARα)12. These perturbations of HDAC function often lead to silencing of tumor suppressor genes. Changes in the abundance of specific histone PTMs also occur in cancer cells. For example, a study of numerous cancer cell lines, normal tissues and primary tumors revealed that a global decrease in H4K16 acetylation and H4K20 methylation is a recurring feature of many types of cancer cells13.
The involvement of HDACs in cancer has raised hope that these enzymes may represent valuable targets in drug discovery programs. Recent clinical trials demonstrated that, at least for hematological cancers14,15,16, small molecules that inhibit HDACs can be effective pharmacological agents, either when administered alone or in combination with other drugs17. HDACi exhibit a number of anti-proliferative effects, such as cell cycle arrest, differentiation, angiogenesis inhibition and apoptosis.9,18 A significant number of HDACi, such as suberoylanilide hydroxamic acid (SAHA, also known as vorinostat or Zolinza), entinostat (MS-275), romidepsin (Istodax), and belinostat (PXD-101) are at various stages of drug development. At present, only SAHA19 and romidepsin20 have been approved by the US food and drug administration for treatment of cutaneous T-cell lymphoma. HDACi such as SAHA are non-selective21. There is a growing debate over the use of HDACi that cause non-specific inhibition of several distinct HDACs versus development of HDACi that would target specific classes of enzymes or even perhaps a single enzyme. In addition to histones, HDACs deacetylate many important protein substrates, such as transcription regulators (e.g. p53, Rb, E2F1 and nuclear hormone receptors)18. Thus, inhibiting multiple HDACs could be cytotoxic to normal cells and lead to undesirable side effects. On the other hand, it can be argued that non-selective HDACi are effective at killing cancer cells precisely because they interfere with the deacetylation of multiple substrates. Currently, it is not known whether inhibition of histone deacetylation, as opposed to other protein substrates, plays a major role in the anti-neoplastic effects of non-selective HDACi. As a first step to address these difficult questions, it is important to determine to what extent HDACi impair histone deacetylation in normal and cancer cells.
The ability to determine stoichiometric changes in histone acetylation elicited by HDACi is of paramount importance to identify their enzyme targets and therapeutically relevant protein substrates. Moreover, a better understanding of HDACi-induced changes in the abundance of histone acetylation at specific lysine residues could facilitate biomarker discovery and monitoring of the therapeutic effects of HDACi22. Due to its sensitivity, selectivity and wide dynamic range, mass spectrometry (MS) has become the method of choice to detect and establish the stoichiometry of histone PTMs. Recent applications of MS also enabled the identification of histone modifications from precise mass measurements on intact protein ions and from on-line tandem MS spectra of the precursor ions23,24. In this report, we conducted a comprehensive label-free quantitative proteomics study to profile histone modifications in normal and cancer cell lines treated with different HDACi. Our experimental strategy included a two pronged-approach that we previously developed and validated to identify histone PTMs whose abundance changes between wild-type and HAT mutant yeast strains25. Global changes in histone modifications are first determined from the mass profiles of intact histones. In a second step, the modification sites and their respective stoichiometric changes are established by a combination of propionylation, trypsin digestion and LC-MS/MS analyses. Bioinformatics tools are exploited to rapidly sift through vast amounts of raw MS data. In this study, we also designed two highly quantitative assays to accurately determine the degree of modification of specific histone lysine residues, even when the stoichiometry of acetylation is very low.
Results
Cell viability and cell cycle progression in response to HDACi
HDACi can trigger apoptosis or cell cycle arrest in transformed cell lines9,18. Recent studies performed on leukemic cell lines indicated that TSA globally increased H4 acetylation, an observation that has been correlated with the induction of apoptosis in vitro and in vivo26,27. However, we wanted to analyze changes in histone acetylation under conditions where HDACi-treated cells did not lose viability or cease to proliferate. We felt that the latter was important because newly synthesized histones H3 and H4 are acetylated at several lysine residues and incorporated into chromatin throughout the genome during S-phase28,29,30,31. Because new H3/H4 molecules are very abundant in proliferating cells (50% of canonical histones present in G2 cells have been newly synthesized during S phase), they likely make a significant contribution to global levels of acetylation. Accordingly, when their deacetylation is blocked by HDACi, the acetylation of new histones would ultimately represent a significant fraction of global acetylation levels.
We first examined whether HDACi treatment caused cell cycle arrest using propidium iodide staining of DNA and flow cytometry (FACS). Asynchronously growing K562 (human erythroleukemic) cells exhibit three distinct levels of propidium iodide fluorescence intensity, corresponding to DNA contents typical of cells in G1, S, and G2/M phase (Fig. 1a). From the relative proportion of the peaks, we found that no more than 20 % of control cells (i.e. cells that were not treated with HDACi) were in S-phase. No significant change in the FACS profile was observed for K562 cells exposed to 1 μM MS-275 or SAHA (data not shown) for a period of up to 24 h (Fig. 1a). Thus, at this concentration, these two HDACi did not trigger any obvious cell cycle arrest. Consistent with this, K562 cell numbers doubled during a 24 h period of exposure to either 1 μM MS-275 or 1 μM SAHA (data not shown). Cellular viability and energy status were monitored using alamar blue, a non-fluorescent chemical that is converted into fluorescent resorufin upon metabolic reduction32. Control cells and cells treated with either 1 μM MS-275 or 1 μM SAHA for 48 to 72 h showed no major decrease in viability or energy status (Fig. 1b and Supplementary Figs. 3–4). Thus, at the time when histones were extracted for MS analysis (24 h), neither MS-275 nor SAHA had caused appreciable proliferation arrest or lethality in K562 cells. Our results are consistent with the fact that the same concentrations of MS-275 or SAHA were not cytotoxic to several other human cancer cell types21. Normal human diploid fibroblasts proliferated more slowly than K562 cells. IMR90 and WI38 cells had doubling times of 48 and 72 h, respectively (Supplementary Fig. 1). However, no decrease in cell viability was observed when SAHA or MS-275 was added every 24 h for up to 72 hours (Supplementary Figs. 3–4).
Figure 1. Cell cycle distribution and viability of K562 cells are not affected by treatment with MS-275 or SAHA.

(a) Propidium iodide (PI) staining of DNA detected by flow cytometry of K562 cells treated for 6 or 24 hours with 1 μM of the class I HDAC inhibitor MS-275. (b) Viability of K562 cells after continuous exposure to either 1µM SAHA or 1µM MS-275 (dissolved in DMSO) for 6 or 24 hours. The y-axis reflects the degree of reduction of alamar blue by live cells monitored using fluorescence emission (see Experimental Section). Dead cells cannot reduce alamar blue. Error bars indicate three independent alamar blue assays performed with each cell sample. CTL: Control with DMSO only.
Global histone acetylation probed by intact mass profiling
We first examined the changes in the molecular mass distribution of intact histones following HDACi treatment using LC-MS on an Agilent 6520 Q-TOF mass spectrometer. In each case, a total of 500 ng of acid extracts enriched in histones were injected onto the LC-MS system. The total ion chromatogram (TIC) of histones from K562 cells that were not treated with HDACi shows that core histones H4, H2B, H2A and H3 eluted successively between 50 and 65 min under our chromatographic conditions (Supplementary Fig. 5). For convenience, mass spectra of core histones were transformed into reconstructed molecular mass profiles and the deconvoluted spectra obtained from extracts of cells treated with HDACi for 0, 6 or 24 h were overlaid for comparison (Figs. 2a and 2b). The most significant changes in the intact mass profiles were observed for histones H3 and H4, rather than H2A or H2B (Figs. 2a and 2b). In eukaryotes, the initiator methionine is removed from the four major types of core histones: H2A, H2B, H3 and H433. Unlike H2A, H2B and H3, which exist as multiple sequence variants, there is a single H4 protein encoded by several genes34. Essentially all H4 molecules are acetylated on their N-terminal amino groups33. The N-terminally acetylated form of H4 is observed at 11278 Da (Mcalc.: 11278.2 Da), along with peaks at 11306 Da (+28 Da, dimethylation) and up to three additional acetylated forms at 11348, 11390, and 11432 Da. All these H4 peaks were detected in both normal and transformed cells (Fig. 2 and Supplementary Fig. 4). A rapid increase in mono-, di- and tri-acetylation was observed as early as 6 h after addition of SAHA to K562 cells. Similar changes in acetylation patterns were observed in K562 cells treated with MS-275, although the more highly acetylated forms of H4 were only apparent after a 24 h exposure. Unlike histone H2A and H4, virtually all the H3 molecules lack acetylation of the N-terminal amino group39. In rapidly proliferating cells, the replication-coupled histone H3 variants (H3.1 and H3.2) are more abundant than the replication-independent H3.3 protein35. In K562 and normal diploid fibroblasts, the unmodified form of H3.2 (Mcalc.: 15256.8 Da) was not detected, but that of H3.1 (Mcalc.: 15272.9 Da) was observed in low abundance at 15273 Da (Fig. 2 and Supplementary Fig. 4). The intact mass profile of H3.1 revealed a heterogeneous distribution of molecular species with incremental shifts of +14 and +42 Da, corresponding to multiple forms of mono-methylation, acetylation and/or tri-methylation. Treatment of K562 cells with SAHA or MS-275 caused an overall shift to more highly acetylated forms. However, as observed for H4, the most highly acetylated forms of H3 accumulated more rapidly with SAHA (6 h) than with MS-275 (24 h). Based on intact mass profiling, both SAHA and MS-275 predominantly increased the acetylation of H3 and H4, whereas acetylation of H2A and H2B was affected to a much lesser extent.
Figure 2. LC-MS analyses of intact histones derived from transformed cells.
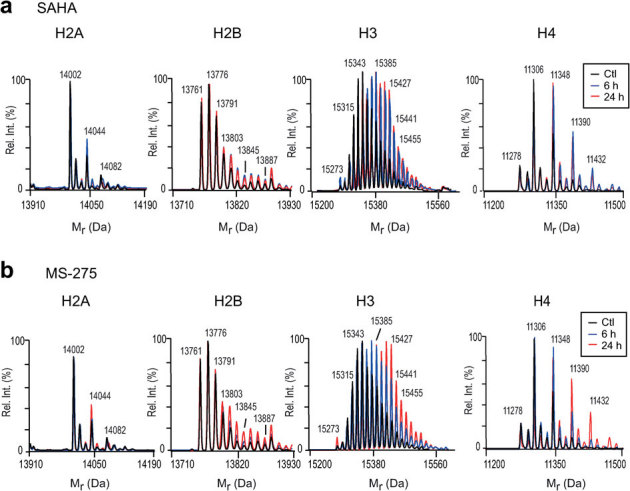
Intact histone profiles from K562 cells treated with SAHA (a) or MS-275 (b). Color traces indicate histones purified from control cells (black) or cells treated for 6 h (blue) or 24 h (red) with either 1 μM SAHA or 1 μM MS-275 (both dissolved in DMSO). Ctl: Control cells were treated with DMSO only.
To determine whether HDACi had similar effects on normal cells, we treated human diploid fibroblasts (IMR90 and WI-38 cells) with the same concentrations of SAHA or MS-275 that were used for K562 cells. A major fraction of histone acetylation likely occurs in new histone molecules that are synthesized during S-phase. Therefore, to ensure that normal and transformed cells went through a single round of S-phase in the presence of HDACi, it was necessary to treat IMR90 (48 h) and WI-38 (72 h) cells for longer times than K562 cells (24 h). This is because normal cells proliferate more slowly than K562 cells (Supplementary Fig. 1). As observed with the K562 cell line, the viability of IMR90 or WI-38 cells was not affected by HDACi concentrations of up to at least 1 μM (Supplementary Fig. 2 and 3). The intact mass profiles of histones derived from HDACi-treated IMR90 and WI-38 cells showed global increases in the acetylation patterns of H3 and H4 (Supplementary Fig. 4 and data not shown). In addition, over a range of concentrations where cells remained fully viable (from 0.125 μM up to 1 μM SAHA or 1μM MS-275), both HDACi caused global increases in H3 and H4 acetylation that were comparable in normal cells and transformed cells (data not shown).
Mapping modification sites by propionylation, tryptic digestion and LC-MS/MS
To determine the degree of acetylation of specific lysine residues following HDACi treatment, we purified individual histones by reversed-phase HPLC and propionylated free lysine residues prior to tryptic digestion and LC-MS/MS analyses25. The amount of protein in acid extracts containing histones was quantitated using the Micro-BCA assay, and a total of 15 μg was separated by HPLC to isolate individual histones. A typical chromatographic separation of histone extracts from K562 control cells resulted in near baseline separation of the four major core histones (Supplementary Figs. 6–7). We focused primarily on the identification of modification sites on H3 and H4, since these core histones showed the most significant global changes upon treatment with either SAHA or MS-275. H3 and H4 histones from all cells and HDACi conditions were derivatized with propionic anhydride, digested with trypsin, and analyzed by LC-MS/MS on an LTQ-Orbitrap XL. Internal calibration using the polysiloxane ion at m/z 445.120029 (protonated (Si(CH3)2O))6) provided m/z measurements within 10 ppm of the theoretical mass and facilitated unambiguous identification of modifications with closely related masses (e.g. acetylation versus tri-methylation).
The injection of tryptic digests of individual histones, rather than a complex acid extract, maximized the number of histone peptides detected and sequenced. This approach resulted in the identification of a multitude of tryptic peptides including histones H3 and H4 peptides that originally contained free or acetylated lysine residues in vivo. Maps representing the properties of all peptide ions (e.g. m/z, charge, LC retention time, peak intensity) were obtained from each raw LC-MS data file and aligned across replicates and HDACi treatment conditions to assess changes in abundance of each identified peptide ion. A list of all identified peptides and their abundance is presented in Supplementary Tables 1 to 10. The relative standard deviation observed for peptide ion intensities detected in all three replicate LC-MS/MS analyses varied between 17 and 24 %, and was consistent with the reproducibility attained by other quantitative proteomics approaches25,36. For all identified H3 and H4 tryptic peptides, the statistical significance and fold change in abundance caused by either SAHA or MS-275 treatment is depicted in volcano plots (Figs. 3a and 3b). The fold change was determined by calculating the abundance ratio of the same peptide in histones from control and HDACi-treated cells. A large proportion (∼90% of all identified peptides) were unaffected by treatment with either SAHA or MS-275. These are clustered near the origin of the volcano plots (p-values > 0.05, fold change < 2). Two quadrants are delineated by dashed boxes in the volcano plots. These contain peptides showing a statistically significant fold change in either control or HDACi-treated cells for triplicate technical measurements (p-values < 0.05, fold change > 2). MS/MS sequencing with either CID or ETD fragmentation revealed that all the histone H3 residues that increased in acetylation following HDACi treatment were located within the first 36 residues of H3. Most notably, H3 K9, K14, K18 and K23 were primary sites of hyperacetylation upon treatment with either SAHA or MS-275 (Supplementary tables 1–4 and 9). For example, Fig. 3c shows the CID MS/MS spectrum of precursor peptide m/z 493.272+, corresponding to the tryptic peptide K9(ac)STGGK14(ac)APR, which is highlighted by arrows in Fig. 3a. This peptide showed a 5- to 17-fold increase in abundance upon SAHA or MS-275 treatment. Unexpectedly, the dimethylated tryptic peptide K27(me2)SAPATGGVK36(me2)K37(pr)PHR identified by ETD fragmentation increased approximately 2- to 4-fold in abundance under the same conditions (Fig. 3a). The physiological importance of this finding is unclear, but our result nonetheless indicates that HDACi can also increase the abundance of histone lysine methylation. The combined use of CID and ETD fragmentation facilitated the localization of modification sites and enabled the identification of 41 differentially modified histone H3 peptides following addition of SAHA or MS-275 to asynchronously growing K562 cells. The use of decision tree driven MS/MS sequencing was used to maximize protein sequence coverage and identify any peptides that might have been missed if only one fragmentation mode had been used37. The majority of peptide ions (70%) were identified using CID as doubly charged species, whereas ETD detected 30 % of ions as triply or more highly charged species.
Figure 3. Histone H3 and H4 peptide ion intensity profiling using volcano plots.

K562 cells were either left untreated (CTL) or treated with either 1 μM SAHA (a) or 1 μM MS-275 (b). Arrows show examples of modified histone peptides whose abundance is significantly affected by HDACi. Tandem MS sequencing of an N-terminal histone H3 peptide (c), and an N-terminal histone H4 peptide (d), revealed acetylation sites that increased in abundance in response to HDACi treatment.
Volcano plots were also generated for histone H4 tryptic peptides to identify residues showing the most significant changes in acetylation after SAHA (Fig. 3a) or MS-275 treatment (Fig. 3b). Following propionylation, tryptic digestion of H4 yielded an N-terminal 14-amino acid peptide (from residues 4 to 17) with four lysine residues (K5, K8, K12, and K16). This 14-residue peptide was detected in multiple forms corresponding to different modification patterns. As observed for H3, amino acids located within the N-terminal tail of H4 were the most prominent targets of acetylation. Upon HDACi treatment, we observed a gradual increase in the di- (K8, K12 or K12, K16 or K8, K16), tri- (K8, K12, K16) and tetra-acetylated (K5, K8, K12, K16) forms with the fully acetylated peptide clearly dominating after exposure to MS-275 (7- and 20-fold increase at 6 h and 24 h, respectively). The mono-acetylated (K16 or K12) forms did not show a significant change in abundance after exposure to any HDACi (Supplementary tables 5–8 and 10). Fig. 3d shows the MS/MS spectrum for the tetra-acetylated tryptic peptide. Careful examination of the fold changes in acetylation triggered by SAHA and MS-275 revealed interesting differences between these two inhibitors. Indeed, treatment with 1 μM SAHA yielded a 17-fold increase in the tetra-acetylated H4 tail peptide at 6 h with a subsequent reduction to 7-fold at 24 h, suggesting a limited long-term inhibitory activity of SAHA compared with MS-275. The fact that MS-275 caused a more robust long-term hyper-acetylation than SAHA was also obvious from the intact mass profiles of H4 (Figs. 2a and 2b).
Abundance of H3K56ac measured by targeted MRM and absolute quantification
Our profiling of tryptic peptides by LC-MS/MS confirmed that lysine residues located within the N-terminal tails of H3 and H4 were the most prominent acetylation sites following HDACi treatment. Indeed, we observed very few acetylated tryptic peptides within the globular domains of H3 and H4. This prompted us to determine the stoichiometry of acetylation of some of these sites using more sensitive and quantitative approaches. We were particularly interested to look for H3K56ac because several recent publications have implicated this modification in replication-coupled nucleosome assembly, regulation of gene expression and the DNA damage response38,39,40,41,42,43,44,45,46,47,48,49,50. In addition, based on immunohistochemistry, it has been argued that H3K56ac is a diagnostic and prognostic marker of several different types of tumours39. However, studies of H3K56ac in human cells have been fraught with controversy, particularly with regards to the role of this histone modification in the DNA damage response38,39,44,47,48,50, but also the identity of the enzymes that acetylate or deacetylate H3K5639,40,41,42,43,44,45,46,48,50.
To detect H3K56ac and determine its stoichiometry, we used two approaches: MRM on an AB Sciex 4000 Q-Trap hybrid triple quadrupole-linear ion trap mass spectrometer, and a high-resolution quantitative approach on a Thermo Orbitrap-LTQ XL instrument. An absolute quantitation method was developed using both an H3K56ac synthetic peptide and an isotopically labeled standard of the target tryptic peptide (YQK56(ac)STELLI*R), where I* is a [13C615N1]-labeled isoleucine residue. The unmodified (K56 propionylated) ion was detected in high abundance in the non-targeted LC-MS/MS analyses described earlier. An MRM method was built based on the MS/MS spectra of both synthetic peptides (Figs. 4a and 4b). Four precursor/fragment ion pairs were selected to design an MRM method for detection of the corresponding tryptic peptides (see Supplementary information for details). We also monitored the unmodified form (YQK56(pr)STELLIR), together with another H3 peptide (YRPGTVALR) to normalize for the amount of H3 present in different samples. Following HPLC fractionation of acid extracts (50 μg of starting material), our MRM assay was used to screen for the presence of H3K56ac in K562 and HeLa cells treated with various HDACi, including sodium butyrate, SAHA, MS-275, and nicotinamide. The modified K56ac residue was detected at low abundance in all cell types and conditions tested, consistent with the LC-MS/MS analyses described in the previous section. Figure 4c shows LC-MS/MS results for H3K56ac in K562 cells treated with SAHA for 6 h. Four MRM transition channels corresponding to the formation of diagnostic y- and b-type fragment ions were monitored. This strategy unambiguously identified a K562 cell-derived peptide that contained H3K56ac and eluted at 26 min (Figs. 4c and 4d). First, the K56-acetylated peptide co-eluted with the [13C615N1-isoleucine]-labeled internal standard (Figs. 4c and 4d). Second, as expected, fragment ions from the K562 cell-derived H3K56ac peptide were 7 Th lighter than the corresponding fragment ions from the internal standard containing [13C615N1]-isoleucine (Figs. 4a and 4b). The stoichiometry of acetylation was calculated using the peak areas of the ion signal corresponding to the K56ac peptide divided by the sum of the areas of the K56ac and K56pr peptides. The stoichiometry of H3K56ac in transformed human cells (K562 or HeLa) is shown in Table 1. The stoichiometry of K56ac determined by MRM was 0.03% in all conditions tested, and did not change upon prolonged incubation with any of the HDACi that we tested.
Figure 4. Identification and quantitation of H3K56ac from K562 cells using targeted MRM.
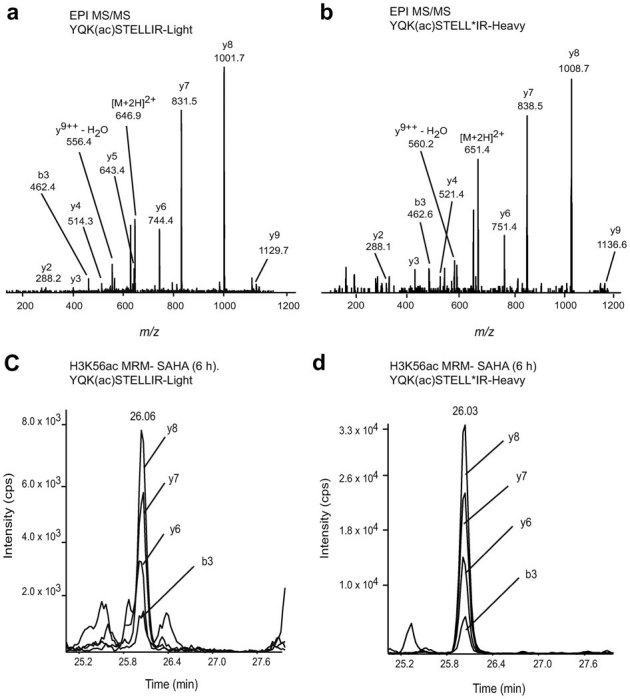
Tandem MS spectra of the light (a) and heavy (b) YQK56(ac)STELLIR peptide standards at 10 μg/mL using enhanced product ion (EPI) scan. LC-MS/MS analysis (MRM) of the native H3K56ac peptide (c) and the [13C615N1-isoleucine]-labeled YQK56(ac)STELLI*R internal peptide standard (d) from a digest of histones isolated from K562 cells treated with SAHA for 6 h.
Table 1. Stoichiometry and amounts of H3K56ac detected in transformed cells exposed to different HDACi.
| Conditions | Amount of H3 K56(ac) peptide detected LTQ-Orbitrap (attomol) | Stoichiometry of H3 K56(ac) (%) LTQ-Orbitrap | Stoichiometry of H3 K56(ac) (%) MRM |
|---|---|---|---|
| K562, control (0 h) | 160 | 0.05 | 0.03 |
| K562, control (6 h) | 198 | 0.05 | 0.03 |
| K562, control (24 h) | 224 | 0.05 | 0.03 |
| K562, MS-275 (6 h) | 169 | 0.05 | 0.03 |
| K562, MS-275 (24 h) | 297 | 0.05 | 0.03 |
| K562, SAHA (6 h) | 206 | 0.05 | 0.03 |
| K562, SAHA (24 h) | 164 | 0.05 | 0.03 |
| K562, nicotinic acid (12 h) | 201 | 0.05 | 0.03 |
| K562, nicotinamide (12 h) | 205 | 0.05 | 0.03 |
| K562, nicotinamide (24 h) | 291 | 0.07 | 0.03 |
| HeLa, butyrate (24 h) | 367 | 0.08 | 0.03 |
We also performed high-resolution LC-MS analyses on the LTQ-Orbitrap XL mass spectrometer to determine the stoichiometry of H3K56ac in the same samples. The absolute amount of H3K56ac in transformed cells was determined using a calibration curve spiked at levels ranging from 5 to 1000 pg/mL, containing a constant amount of the [13C615N1]-labeled internal standard. In order to mimic, as closely as possible, the reaction conditions used to process human histones prior to MS, recombinant yeast histone H3 was propionylated and digested first, then spiked with increasing amounts of the synthetic peptide standard. The rationale was to generate reaction mixtures containing similar sets of peptides and chemical species as those produced during analysis of human cell-derived histones. Although recombinant yeast histone H3 is less complex and devoid of PTM33, the conditions of propionylation and trypsin digestion are similar, which helps tackle challenges to quantitation, such as chemicals that co-elute with the H3K56ac peptide of interest and might suppress its ionization. This is particularly important in this case because of the very low abundance of the H3K56ac peptide. The first residue of the yeast H3 tryptic peptide containing K56 is a Phe, instead of a Tyr residue in human H3. Hence, H3K56 tryptic peptides derived from yeast histones do not interfere with our quantitative analysis. Our calibration curve showed excellent linearity (r2 = 0.9993) over the range of concentrations examined (Fig. 5a). We detected the authentic H3K56ac peptide at a level approaching the instrumental detection limit in all the human histone samples. An example of the precursor ion mass spectrum for the H3K56ac peptide derived from K562 cells treated with SAHA for 24 h is shown in Fig. 5b. The masses measured for the light and heavy labeled doubly protonated peptides were respectively within 1 ppm and 5 ppm of those calculated from their corresponding theoretical masses. An MS/MS spectrum that confirms the identity of the precursor ion corresponding to the human K56ac peptide is shown in Fig. 5c. The H3K56ac stoichiometry was determined from the ratio of the peak height of the doubly protonated K56ac tryptic peptide divided by the sum of the peak heights for the K56ac and K56pr peptides. The results from high-resolution LC-MS analyses are shown in Table 1, together with the absolute amount of tryptic peptide detected. The amount of H3K56ac present in all conditions ranged from 150 to 370 attomol, while the acetylation stoichiometry varied from 0.05–0.08%. Consistent with our MRM experiments, no significant change in the stoichiometry of H3K56ac was detected under any of the conditions examined, and even after prolonged incubation (up to 24 h) of K562 or HeLa cells with HDACi that target different classes of deacetylases (Table 1). Thus, based on two different quantitative MS experiments, we find that the stoichiometry of H3K56ac (roughly 0.04%) is considerably lower than previously reported (1%)39,49. In addition, unlike other sites of acetylation, H3K56ac did not increase in response to several HDACi (Table 1). This was very intriguing because two of the HDACi that we used in our MS experiments, namely sodium butyrate and nicotinamide, were previously shown to increase H3K56ac based on immunoblotting39,48. This prompted us to investigate the specificity of three commercially available antibodies that were previously used to detect H3K56ac in human cells38,39,42,43,44,45,46,47,48. For these experiments, the same histone samples were analyzed either by MS or immunoblotting. The amounts of acid-extracted histones often used in immunoblots are sufficiently large to be readily detected by Ponceau S staining after histone transfer to nitrocellulose or PVDF membranes39,46. As previously reported39,46,48, we found that, under those conditions, sodium butyrate treatment of HeLa cells strongly increased the signal obtained with antibodies supposedly specific for H3K56ac (Figs. 6a and 6b). However, with amounts of histones detectable by Ponceau S or Coomassie blue staining, the same antibodies also detected a considerable signal in histones purified from yeast rtt109Δ mutant cells (Figs. 6a and 6c). In yeast, Rtt109 is the main enzyme that acetylates H3K56. Consistent with this, as judged by MS, H3K56ac is virtually absent from rtt109 null mutant cells25. Therefore, the immunoblotting signals detected by commercial antibodies in histones purified from rtt109Δ mutant yeast cells (Figs. 6a and 6c) must be derived from modifications other than H3K56ac. Intriguingly, three of the six known acetylation sites located in the N-terminal domain of histone H3 share some sequence similarity with residues flanking H3K56 (Fig. 6d). Based on this, we hypothesized that commercial antibodies raised against H3K56-acetylated peptides might show some degree of cross-reactivity against abundant acetylation sites that bear similarity to residues flanking H3K56. To address this possibility, we obtained a set of six synthetic peptides, each containing a single acetylated lysine residue. These peptides represent all the known acetylation sites within the first 30 amino acid residues of histone H3 (Supplementary Fig. 7). Each peptide was incubated with commercial antibodies raised against H3K56ac to determine whether they would block the ability of the antibodies to detect signals in immunoblots of histones purified from human cells. As control for a peptide that should block each antibody, we used a K56-acetylated synthetic peptide. As shown in Fig. 6e, in addition to the K56-acetylated peptide, three of the competitor peptides derived from the N-terminal domain of H3 either abolished or significantly reduced the immunoblotting signals obtained with histones purified from butyrate-treated HeLa cells. The K9ac peptide was the best competitor, but the K4ac and K27ac peptides also blocked some of the commercial antibodies that were reportedly H3K56ac-specific (Fig. 6e). This was true for two monoclonal antibodies (Fig. 6e) and a polyclonal antiserum from Millipore (data not shown). The latter antibody is no longer commercially available but was previously used in several publications. Interestingly, the acetylated peptides that blocked the commercial antibodies were those that shared sequence similarity with residues flanking H3K56. In contrast, peptides acetylated at H3K14, H3K18 or H3K23, which lack similarity with residues that flank H3K56, all failed to block the antibodies in our competition experiments (Figs. 6d and 6e). In cells treated with SAHA, we estimated by MS that the stoichiometry of H3K9ac (about 33%) is roughly 800-fold higher than that of H3K56ac (0.04%). These findings, combined with the fact that two quantitative MS approaches did not reveal an increase in H3K56ac, even after extended periods of incubation with several HDACi (Table 1), lead us to conclude that several commercial antibodies previously used to study H3K56ac in human cells cross-react with sites of acetylation other than histone H3K56 and, particularly, H3K9. It is noteworthy that several studies reported that the abundance of H3K9ac and H3K56ac changed in the same direction under a variety of physiological conditions41,43,44,45,46. Given the large stoichiometric excess of H3K9ac over H3K56ac, and the fact that antibodies raised against H3K56ac cross-react with H3K9ac, we suggest that these manuscripts described changes in the abundance of H3K9ac, rather than H3K56ac.
Figure 5. LC-MS/MS analysis of H3K56ac from K562 cells using an LTQ-Orbitrap XL mass spectrometer.

(a) Samples (see Experimental Section) were prepared by mixing solutions of increasing concentrations (from 5 to 1000 pg/mL) of [12C-isoleucine]-containing K56-acetylated peptide with a fixed amount (100 pg/mL) of internal standard (IS) peptide (YQK56(ac)STELLI*R), where I* is a [13C615N1]-labeled isoleucine residue). The y-axis is the peak height abundance ratio (as determined by LC-MS) of the YQK56(ac)STELLIR peptide (containing only the [12C] isotope) to that of the [13C615N1]-labeled internal peptide standard (IS). (b) Precursor ion mass spectrum (from LC-MS/MS analysis of histones derived from K562 cells treated with 1 μM SAHA for 24 h) showing the detection of the native YQK56(ac)STELLIR peptide along with the [13C615N1-isoleucine]-labeled internal standard. (c) MS/MS spectrum of precursor m/z 646.862+ from the analysis shown in (b). This spectrum confirms the identity of the natural peptide containing H3K56ac.
Figure 6. Antibodies supposedly specific for H3K56ac cross-react against other acetylated lysine residues of histone H3.

(a) Immunoblot of core histones purified from control and butyrate-treated HeLa cells (NaBut), as well as WT and rtt109Δ yeast cells, probed with an antibody from Upstate/Millipore (07-677). (b) Immunoblot of core histones purified from control and butyrate-treated HeLa cells probed with Epitomics antibody 2134-1. Comparable amounts of histones from control and butyrate-treated cells were loaded as evidenced from the Coomassie Blue-stained gel. (c) Immunoblot of core histones purified from yeast cells lacking the H3K56 acetyltransferase Rtt109 (rtt109Δ) or WT cells. In panels (a) to (c), the “H3K???ac” label indicates that the immunoblotting signal could not be ascribed to a specific acetylated lysine(s). (d) Amino acid sequences flanking histone H3K56 and known acetylated lysines in the histone H3 N-terminal tail. Identical residues neighbouring acetylated lysines are highlighted. (e) Immunoblot of core histones purified from butyrate-treated HeLa cells were probed with monoclonal antibodies from Epitomics (2134-1) or Active Motif (61061) that were pre-incubated with different competitor peptides acetylated at specific lysine residues (histone H3 acetylated peptides). Ø: No competitor peptide. Equal loading of histones from HeLa cells was verified either by stripping the blot and reprobing it with an antibody that detects unmodified histone H3 (top panel) or by Ponceau S staining (bottom panel). (*) In addition to histone H3, the Active Motif antibody recognized a band that migrated faster than H3. Detection of this faster migrating species was blocked by H3K9ac and H3K56ac competitor peptides, suggesting that it is related to histone H3. In the same histone sample preparation, the Epitomics antibody did not recognize the band that migrated faster than H3.
Discussion
Our two-pronged approach to identify global changes in histone acetylation that arise from HDAC inhibition has led to three unexpected findings about the dynamics of histone acetylation in human cells. First, HDACi with either hydroxamic acid (SAHA) or benzamide (MS-275) inhibitory moieties both trigger global hyperacetylation of H3 and H4 in normal diploid fibroblasts and several transformed cell lines (K562, HeLa, HEK293T, and U937). Second, even when SAHA and MS-275 elicit global increases in H3 and H4 N-terminal tail acetylation in normal diploid fibroblasts, the cells did not lose viability (Supplementary Figs. 2, 3 and 4). In the future, it will be interesting to determine if this is also the case in cancer cells and whether the cytotoxicity of SAHA and MS-275 has anything to do with their ability to globally enhance histone H3 and H4 acetylation9. Third, the development of two robust MS-based quantitative assays to determine the stoichiometry of histone H3K56ac in control and HDACi-treated cells has revealed some highly unexpected properties of this modification in human cells that are distinct from those of H3K56ac in yeast.
In S. cerevisiae and other yeasts, H3K56ac is very abundant during S-phase and G2. This is because essentially all the new H3 molecules are K56-acetylated prior to their deposition into nascent chromatin and subsequently deacetylated in late G2 or M-phase4,51. Our data demonstrate that this is clearly not the case for H3K56ac in human cells. In contrast to the acetylation of many other residues in the H3 and H4 N-terminal domains52, it is puzzling that prolonged exposure to either class I/II or class III HDACi did not increase the stoichiometry of H3K56ac in transformed human cells. We cannot exclude the possibility that H3K56ac might slightly increase above 0.03% upon HDACi treatment. However, the very low stoichiometry of H3K56ac in human cells makes it unlikely that this modification can significantly contribute to genome-wide deposition of newly synthesized histone molecules. Our findings certainly do not rule out the possibility that H3K56ac may have an important function in human cells. A stoichiometry of 0.03% corresponds to roughly one K56-acetylated H3 molecule every 1665 nucleosomes (or roughly every 333 Kb, assuming an average nucleosome repeat length of 200 bp). Given the size of the genome of a diploid human cell (roughly 6×109 bp), there would be approximately 18000 K56-acetylated H3 molecules per cell. Despite the intriguing absence of H3K56 deacetylation, some of these molecules may occur at specific chromosomal loci where H3K56ac might play important functions in either the DNA damage response or the regulation of gene expression36,39,40,42,43,44,45,46,47,48,49,50,53. However, we would like to emphasize that reports of the presence of H3K56ac at specific loci (sites of DNA damage or gene regulatory elements), as determined by immunofluorescence or chromatin immunoprecipitation (ChIP) assays, are questionable because the H3K9 and H3K56 epitopes are related in sequence and H3K9ac is far more abundant than H3K56ac in human cells. Our study underscores the fact that even partial cross-reactivity of antibodies can lead to erroneous data interpretation when epitopes contain modified amino acids whose flanking residues share sequence similarity with other segments of the same protein. This may be an important issue for lysine residues in the N-terminal domain of histone H4, where three of the four known acetylated lysines reside in the sequence context GKG. As we demonstrated for H3K9ac and H3K56ac, this is particularly problematic when the stoichiometry of modification of the residue that should not be recognized by the antibody is much higher than the extent of modification of the intended target residue. To test whether this might be the case, and avoid false conclusions due to antibody cross-reactivity, we recommend quantitative mass spectrometry as a means to address the stoichiometry of modification of residues that are flanked by similar amino acid sequences. This is particularly important for studies of vertebrate cells, where it is not currently possible to mutate specific histone residues in order to rigorously prove antibody specificity in immunofluorescence or ChIP experiments. This is also a major issue in immunohistochemistry. Overall, our findings demonstrate the importance of quantitative MS to determine the stoichiometry of specific protein modifications under a variety of physiological conditions and use this information to formulate hypotheses regarding the function(s) of these modifications.
Methods
Immunoblotting
Histone proteins were run through SDS-polyacrylamide gels and transferred onto PVDF membranes. After transfer, the presence of equal amounts of core histones was assessed using either Ponceau S staining or by immunoblotting using an antibody against non-modified histone H4. The membranes were then probed with primary antibodies raised against H3K56ac peptides (Active Motif mouse monoclonal antibody 61061, Upstate/Millipore rabbit polyclonal antibody 07-677 or Epitomics rabbit monoclonal antibody 2134-1) overnight at 4°C. After a 12–14 h incubation with primary antibodies, blots were washed extensively in Tris-buffered saline (25 mM Tris-HCl pH 8.0, 137 mM NaCl, 3 mM KCl). After incubation with secondary antibodies (anti-rabbit IgG coupled to horseradish peroxidase) for one hour at room temperature, the blots were washed extensively in Tris-buffered saline (25 mM Tris-HCl pH 8.0, 137 mM NaCl, 3 mM KCl) containing 0.1% (w/v) Tween-20, and antibody binding was detected by enhanced chemiluminescence (GE Healthcare). Synthetic peptides were mixed at a 1000:1 molar ratio with purified antibodies to determine whether they blocked the ability of antibodies to detect acetylated histones.
Sample preparation and protein digestion
The total protein content of histone-enriched acid extracts was measured with the Micro-BCA assay. Approximately 15 μg of starting material was separated using an Agilent 1100 HPLC system equipped with a micro-fraction collector. Separations were performed on a Jupiter microbore C18 column (3 μm, 300 Å; 150×1 mm i.d.) with a solvent system consisting of 0.1% TFA in water (v/v) (solvent A), and 0.1% TFA in acetonitrile (v/v) (solvent B). Gradient elution was performed from 5 to 70% B in 60 min at 15 µL/min. Fractions were collected in a 96-well plate at 20 second intervals and fractions representing individual histone peaks were pooled together. For the targeted analysis of H3K56ac, approximately 50 μg of starting material was separated using an Agilent 1200 HPLC system equipped with a fraction collector. Separations were performed on an ACE standard bore C8 column (5 μm, 300 Å; 150×4.6 mm i.d.) at 0.7 mL/min. Histone H3 fractions were collected in 2 mL Eppendorf tubes at 1 minute intervals. Multiple fractions were pooled together as described above. Histones were prepared and digested as previously described25. LC fractions were dried in a Speed-Vac evaporator and resuspended in 0.1 M ammonium bicarbonate buffer (without pH adjustment). Intact histones were derivatized in a 1:1 volume ratio with a propionic anhydride reagent. After a 30 minute derivatization period at room temperature, samples were evaporated a second time, resuspended in 0.1 M ammonium bicarbonate, and digested overnight at 37° C using 1 µg of trypsin. Samples were acidified with 5% TFA in water (v/v) prior to LC-MS analysis. Under our conditions, propionylation proceeded nearly to completion, as judged by negligible trypsin cleavage at non-propionylated lysine residues, which we verified by MS. Near complete propionylation of non-modified lysine residues is important to determine the accurate stoichiometry of histone acetylation because trypsin would otherwise cleave after non-modified lysines. This would obviously lead to an over-estimation of the fraction of histone molecules that are acetylated at a given site.
Intact histone profiling
Intact histone analysis was performed on an Agilent 6520 Q-TOF mass spectrometer equipped with a nanoelectrospray interface. The instrument was operated in positive ion mode (2800 V at interface) and scanned from m/z 400 to 1600. Equivalent amounts of histone acid extracts were diluted down to a concentration of 100 ng/μL with the initial LC-MS mobile phase (see below). An aliquot was injected onto an in-house packed capillary C18 trapping column (4 mm × 360 µm I.D.) for 3 min at 10 µL/min using a loading solvent (95:5 water:acetonitrile containing 0.2% formic acid (v/v)). Histones were eluted onto the C18 analytical column (100 mm × 150 µm i.d.) using 0.2% formic acid in water (v/v) (solvent A), and 0.2% formic acid in acetonitrile (v/v) (solvent B). Gradient elution was performed from 5 to 90% B in 60 min at 600 nL/min. Intact histone mass profiles were generated from acquired mass spectra using the MaxEnt1 deconvolution algorithm of the Mass Hunter software.
Ion profiling and peptide sequencing by LC-MS/MS
LC-MS/MS analyses were performed using an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a nanoelectrospray interface. Tryptic digests were injected in triplicate using an Eksigent nano 2D-LC system. The same columns and solvent systems were used as described above in the intact mass profiling section. The instrument was operated in positive ion mode and scanned from m/z 300 to 1600, resolution set to 60 000, with a target value of 1.0 × 106. For tandem MS/MS sequencing, the LTQ ion trap was scanned from m/z 50 to 2000, with a target value of 1.0 × 104. One full survey scan in the Orbitrap was followed by data dependant MS/MS acquisition on the three most intense ions. A decision tree based on peptide m/z ratio and charge state was used to trigger either CID or ETD fragmentation37. A normalized collision energy of 35 was used to generate fragment ions in the LTQ by CID, while a 100 ms activation time was used for ETD ion-ion reactions. All MS/MS spectra of modified histone peptides are provided as supplementary Figures.
All raw data files (.raw) from triplicate injections on the LTQ-Orbitrap XL mass spectrometer were converted into peptide maps using Mass Sense (version 2.5), an in-house peptide detection software used to identify peptides based on m/z values, retention time, abundance, and charge state. An intensity threshold of 10 000 counts was set as cutoff for precursor ion peak detection. Segmentation analysis was performed by clustering the peptide ions identified in each treatment condition based on their respective m/z ratio, charge, and LC retention time using an m/z tolerance of ± 0.02 Th and a time difference of 1 min. Normalization of LC retention time is then performed on the initial peptide cluster list using a dynamic and nonlinear correction. Once clustered, all peptide intensities were averaged and a mean ratio was calculated to generate a correction factor used to normalize for changes in abundance and differences in amount of peptide loaded. Normalized intensities are then used to generate a volcano plot for differential histone PTM expression profiling. Raw data files were converted to Mascot generic files (.mgf) using Mascot Distiller (version 2.2.1) and Mascot Daemon (version 2.2.2). A mass tolerance of 0.02 Th was used for the precursor ions, and 0.5 Th for the fragment ions. A maximum of 9 missed cleavages was permitted during the Mascot searches. The following variable modifications were included in the search parameters: acetylation, methylation+propionylation, dimethylation, and trimethylation, and propionylation of lysine residues, methylation of arginine residues, phosphorylation of serine, threonine and tyrosine residues, and oxidation of methionine residues. Manual MS/MS spectra verification was performed on each modified peptide with Mascot scores greater than 20 to confirm the sequence assignment. Mascot searches were downloaded from the Mascot server into an Excel spreadsheet, from which redundant and low scoring peptides were removed. All MS/MS spectra corresponding to modified histone tryptic peptides are provided as supplementary information.
Targeted analysis of H3K56ac by multiple reaction monitoring and absolute quantitation
Stock solutions at 1 mg/mL in deionized water were prepared for two K56-acetylated synthetic peptides: a peptide containing only [12C] and a peptide where a leucine was [13C, 15N]-labeled (see below). Unmodified yeast histone H3 was expressed in E. coli, purified as described54, and resuspended in 0.1 M ammonium bicarbonate buffer. Approximately 3 µg of recombinant yeast histone H3 was propionylated, digested, and spiked with increasing amounts of synthetic human H3K56ac peptide (YQK(ac)STELLIR) ranging from 5 to 1000 pg/mL. To each solution, a constant amount of the isotopically labeled internal standard (IS) peptide (YQK(ac)STELLI*R, wherein I* is a [13C615N1]-labeled isoleucine), was added to obtain a final concentration of 100 pg/mL IS in each vial. A 20-µL aliquot of each mixture was injected onto the LTQ-Orbitrap XL mass spectrometer, and analyzed using the same parameters as described in the LC-MS/MS analysis section.
A multiple reaction monitoring (MRM) method was designed to detect and quantify the human H3K56ac tryptic peptide using an AB Sciex 4000 Q-trap hybrid triple quadrupole-linear ion trap mass spectrometer equipped with a Nanospray II interface. Working solutions of human K56ac peptide and its labeled analogue were prepared at a concentration of 10 µg/mL in 50∶50 water:methanol with 0.2% formic acid (v/v/v) for nanoelectrospray infusion. Source parameters were optimized by constant infusion of the working solutions at 600 nL/min. The electrospray voltage was set to 3800 V, with a declustering potential of 120 V, and an optimized collision energy of 32 V. The four most intense ions were chosen to build MRM transitions within the Analyst software. All MRM transitions used in this study are provided in the Supplementary Methods. Both H3K56ac and its non-modified counterpart (H3K56pr) were monitored in parallel; another H3 tryptic peptide (YRPGTVALR) was also monitored to normalize for the amount of peptide loaded and fluctuations in the electrospray response.
Author Contributions
P.D. and V.V. carried out experiments, analysed the data, prepared figures and wrote the first draft of the manuscript. C.P., E.-H. L., and V. B. carried out cell culture and cell viability experiments, analysed the data, and prepared figures. E.B. participated in the MS analyses. G.F. provided cell cultures and discussed results. A.V. and P.T. designed the study, analysed data, discussed results and wrote the manuscript. All authors reviewed the manuscript.
Supplementary Material
Supplementary file
Acknowledgments
This work was supported by funding from the Collaborative Health Research Program of the Natural Sciences and Engineering Research Council (NSERC) and the Canadian Institutes for Health Research (CIHR) to A.V. and P.T and the Canadian Cancer Research Society to G.F. The Institute for Research in Immunology and Cancer receives infrastructure support from the Canadian Center of Excellence in Commercialization and Research, the Canadian Foundation for Innovation, and the Fonds de la Recherche en Santé du Québec.
References
- Davey C. A., Sargent D. F., Luger K., Maeder A. W. & Richmond T. J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol 319, 1097–1113 (2002). [DOI] [PubMed] [Google Scholar]
- Luger K. Dynamic nucleosomes. Chromosome Res 14, 5–16 (2006). [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- Masumoto H., Hawke D., Kobayashi R. & Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436, 294–298 (2005). [DOI] [PubMed] [Google Scholar]
- van Leeuwen F., Gafken P. R. & Gottschling D. E. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756 (2002). [DOI] [PubMed] [Google Scholar]
- Ye J. et al. Histone H4 lysine 91 acetylation: a core domain modification associated with chromatin assembly. Mol Cell 18, 123–130 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B. D. & Allis C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- Morales V. et al. Chromatin structure and dynamics: functional implications. Biochimie 83, 1029–1039 (2001). [DOI] [PubMed] [Google Scholar]
- Bolden J. E., Peart M. J. & Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5, 769–784 (2006). [DOI] [PubMed] [Google Scholar]
- Yoshida M., Kijima M., Akita M. & Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265, 17174–17179 (1990). [PubMed] [Google Scholar]
- Halkidou K. et al. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59, 177–189 (2004). [DOI] [PubMed] [Google Scholar]
- Minucci S., Nervi C., Lo Coco F. & Pelicci P. G. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene 20, 3110–3115 (2001). [DOI] [PubMed] [Google Scholar]
- Fraga M. F. et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37, 391–400 (2005). [DOI] [PubMed] [Google Scholar]
- Lee M. J., Kim Y. S., Kummar S., Giaccone G. & Trepel J. B. Histone deacetylase inhibitors in cancer therapy. Curr Opin Oncol 20, 639–649 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince H. M., Bishton M. J. & Harrison S. J. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15, 3958–3969 (2009). [DOI] [PubMed] [Google Scholar]
- Stimson L., Wood V., Khan O., Fotheringham S. & La Thangue N. B. HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol 20, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Bots M. & Johnstone R. W. Rational combinations using HDAC inhibitors. Clin Cancer Res 15, 3970–3977 (2009). [DOI] [PubMed] [Google Scholar]
- Dokmanovic M., Clarke C. & Marks P. A. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5, 981–989 (2007). [DOI] [PubMed] [Google Scholar]
- Mann B. S., Johnson J. R., Cohen M. H., Justice R. & Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12, 1247–1252 (2007). [DOI] [PubMed] [Google Scholar]
- Campas-Moya C. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today (Barc) 45, 787–795 (2009). [DOI] [PubMed] [Google Scholar]
- Beckers T. et al. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer 121, 1138–1148 (2007). [DOI] [PubMed] [Google Scholar]
- Stimson L. & La Thangue N. B. Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett 280, 177–183 (2009). [DOI] [PubMed] [Google Scholar]
- Durbin K. R. et al. Intact mass detection, interpretation, and visualization to automate Top-Down proteomics on a large scale. Proteomics 10, 3589–3597 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuti N. & Kelleher N. L. Efficient readout of posttranslational codes on the 50-residue tail of histone H3 by high-resolution MS/MS. Anal Biochem 396, 180–187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogaris P., Wurtele H., Masumoto H., Verreault A. & Thibault P. Comprehensive profiling of histone modifications using a label-free approach and its applications in determining structure-function relationships. Anal Chem 80, 6698–6707 (2008). [DOI] [PubMed] [Google Scholar]
- Ninios Y. P., Sekeri-Pataryas K. E. & Sourlingas T. G. Differential sensitivity of human leukemic cell lines to the histone deacetylase inhibitor trichostatin A. Leukemia Res 34, 786–792 (2010). [DOI] [PubMed] [Google Scholar]
- Johnstone R. W. Histone deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discovery 1, 287–299 (2002). [DOI] [PubMed] [Google Scholar]
- Benson L. J. et al. Modifications of H3 and H4 during chromatin replication, nucleosome assembly, and histone exchange. J Biol Chem 281, 9287–9296 (2006). [DOI] [PubMed] [Google Scholar]
- Loyola A., Bonaldi T., Roche D., Imhof A. & Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell 24, 309–316 (2006). [DOI] [PubMed] [Google Scholar]
- Sobel R. E., Cook R. G., Perry C. A., Annunziato A. T. & Allis C. D. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci U S A 92, 1237–1241 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verreault A., Kaufman P. D., Kobayashi R. & Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 87, 95–104 (1996). [DOI] [PubMed] [Google Scholar]
- Anoopkumar-Dukie S. et al. Resazurin assay of radiation response in cultured cells. Br J Radiol 78, 945–947 (2005). [DOI] [PubMed] [Google Scholar]
- Song O. K., Wang X., Waterborg J. H. & Sternglanz R. An N-α-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J Biol Chem 278, 38109–38112 (2003). [DOI] [PubMed] [Google Scholar]
- Marzluff W. F., Gongidi P., Woods K. R., Jin J. & Maltais L. J. The human and mouse replication-dependent histone genes. Genomics 80, 487–498 (2002). [PubMed] [Google Scholar]
- Orsi G. A., Couble P. & Loppin B. Epigenetic and replacement roles of histone variant H3.3 in reproduction and development. Int J Dev Biol 53, 231–243 (2009). [DOI] [PubMed] [Google Scholar]
- Falick A. M. et al. ABRF-PRG07: advanced quantitative proteomics study. J Biomol Tech 22, 21–26 (2011). [PMC free article] [PubMed] [Google Scholar]
- Swaney D. L., McAlister G. C. & Coon J. J. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods 5, 959–964 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battu A., Ray A. & Wani A. A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 39, 7931–7945 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C., Lucia M. S., Hansen K. C. & Tyler J. K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S. et al. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J Biol Chem 286, 16967–16975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. And-1 is required for the stability of histone acetyltransferase Gcn5. Oncogene [Epub ahead of print] PMID: 21987584 (2011). [DOI] [PMC free article] [PubMed]
- Lo K. A. et al. Genome-wide profiling of H3K56 acetylation and transcription factor binding sites in human adipocytes. PLoS One 6, e19778 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E. et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K. M. et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 17, 1144–1151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B. et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci U S A 107, 21790–21794 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjeertes J. V., Miller K. M. & Jackson S. P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J 28, 1878–1889 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vempati R. K. & Haldar D. DNA damage in the presence of chemical genotoxic agents induce acetylation of H3K56 and H4K16 but not H3K9 in mammalian cells. Mol Biol Rep 39, 303–308 (2012). [DOI] [PubMed] [Google Scholar]
- Vempati R. K. et al. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem 285, 28553–28564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. et al. Histone H3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell 33, 417–427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Pu M., Zhang Z. & Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. .Cell Cycle 8, 1747–1753 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celic I. et al. The sirtuins Hst3 and Hst4p preserve genome integrity by controlling histone H3 lysine 56 deacetylation. Curr Biol 16, 1280–1289 (2006). [DOI] [PubMed] [Google Scholar]
- Waterborg J. H. Dynamics of histone acetylation in vivo. A function for acetylation turnover? Biochem Cell Biol 80, 363–378 (2002). [DOI] [PubMed] [Google Scholar]
- Battu A., Ray A. & Wani A. A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 39, 7931–7945 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer P. N. et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375, 23–44 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file