

Abstract
The anaerobic and aerobic metabolism of acetone and butanone in the betaproteobacterium “Aromatoleum aromaticum” is initiated by their ATP-dependent carboxylation to acetoacetate and 3-oxopentanoic acid, respectively. Both reactions are catalyzed by the same enzyme, acetone carboxylase, which was purified and characterized. Acetone carboxylase is highly induced under growth on acetone or butanone and accounts for at least 5.5% of total cell protein. The enzyme consists of three subunits of 85, 75, and 20 kDa, respectively, in a (αβγ)2 composition and contains 1 Zn and 2 Fe per heterohexamer but no organic cofactors. Chromatographic analysis of the ATP hydrolysis products indicated that ATP was exclusively cleaved to AMP and 2 Pi. The stoichiometry was determined to be 2 ATP consumed per acetone carboxylated. Purified acetone carboxylase from A. aromaticum catalyzes the carboxylation of acetone and butanone as the only substrates. However, the enzyme shows induced (uncoupled) ATPase activity with many other substrates that were not carboxylated. Acetone carboxylase is a member of a protein family that also contains acetone carboxylases of various other organisms, acetophenone carboxylase of A. aromaticum, and ATP-dependent hydantoinases/oxoprolinases. While the members of this family share several characteristic features, they differ with respect to the products of ATP hydrolysis, subunit composition, and metal content.
INTRODUCTION
Short-chain ketones like acetone (dimethylketone) or butanone (methyl ethyl ketone) are ubiquitous organic compounds in natural environments. They are produced in several biological processes, for example, during solvent fermentation by many Clostridia, which produce acetone by decarboxylation of acetoacetate (6, 9). Moreover, both compounds are heavily used as solvents or synthetic precursors in industry and constitute significant groundwater contaminants due to their high solubility in water. Therefore, it is of interest to identify ketone-degrading microorganisms and the pathways used to metabolize these compounds to understand the mechanisms and prerequisites of their biological degradation.
Of three alternative pathways known for acetone degradation, two depend on molecular oxygen and only one is oxygen independent. The first pathway involves an initial hydroxylation of the methyl group of acetone (and probably also butanone) by a cytochrome P450-type monooxygenase to acetol (1-hydroxyacetone) in mammals (11, 12). The second known pathway is initiated by an FADH2- and NADPH-dependent monooxygenase in a Gordonia sp., which catalyzes a Bayer-Villiger rearrangement to yield methyl acetic ester (methylacetate) as the first intermediate (13). Finally, the third known pathway is initiated by carboxylating acetone to acetoacetate by a novel type of ATP-dependent carboxylase (see Fig. 2). Acetoacetate is then activated to acetoacetyl coenzyme A (acetoacetyl-CoA) by a CoA ligase or CoA transferase. Butanone may be metabolized by the same pathway, as suggested by the genome sequence (17).
Fig 2.

Proposed catabolic pathway of acetone. Acetone is carboxylated to acetoacetate (reaction 1) by acetone carboxylase. Activation of acetoacetate to acetoacetyl-CoA (reaction 2) may be catalyzed by either a CoA ligase or a CoA transferase. Reactions 1 and 2 are proposed to be ATP dependent. Acetoacetyl-CoA is then thiolytically cleaved (reaction 3) by a thiolase to form 2 acetyl-CoA.
Acetone carboxylation (Acx) was initially characterized in the aerobic acetone-degrading alphaproteobacterium Xanthobacter autotrophicus and in the anoxygenic phototrophic bacterium Rhodobacter capsulatus (1–3, 21–23). The enzymes catalyzing acetone carboxylation in these two species were purified and biochemically characterized. They consist of three subunits and exhibit very similar biochemical properties in coupling the carboxylation reaction to ATP hydrolysis without the need for an organic cofactor (see Table 4) (2, 22). The available biochemical and sequence data suggest that acetone carboxylase is the prototype of a specific class of carboxylases, which appear to be present in many different bacterial strains on the basis of their genome sequences (22).
Table 4.
Comparison of the characteristics of acetone carboxylases of A. aromaticum with acetone carboxylases of various organisms and acetophenone carboxylase of A. aromaticuma
| Characteristic | Acetone carboxylase of: |
Hydantoinase (ATP hydrolyzing) of a Pseudomonas sp. (16) | Acetophenone carboxylase of A. aromaticum (10) | ||
|---|---|---|---|---|---|
| A. aromaticum | X. autotrophicus (22) | R. capsulatus (22) | |||
| Subunit composition | (αβγ)2 (85/75/20)b | (αβγ)2 (85/78/20) | (αβγ)2 (85/78/20) | (αβ)2 (80/70) | (αββ′γ)2 + ε2 (87/75/70/15 + 34) |
| Sequence similarityc | 70/70/60 | 68/69/59 | 28/23 | 31/24/26/31 | |
| Nucleotide hydrolysis | 2ATP → 2AMP + 4Pi | ATP → ADP + Pi → AMP + 2Pi | ATP → AMP + 2Pi | ATP → ADP + Pi | 2ATP → 2ADP + 2Pi |
| Uncoupled ATPase activity with: | Ketones without HCO3 | Ketones without HCO3 | NDd | Ketones without HCO3 and with HCO3 without ketones | |
| Cofactor | 1 Zn/(αβγ)2, 2.2 Fe/(αβγ)2 | 1 Zn/(αβγ)2, 0.7 Fe/(αβγ)2, 1.3 Mn/(αβγ)2 | 0.31 Zn/(αβγ)2, 0.14 Fe/(αβγ)2, 1.5 Mn/(αβγ)2 | ND | 2 Zn/(αββ′γ)2 |
| Substrate specificity | Aliphatic ketones (acetone, butanone) | Aliphatic ketones (acetone, butanone) | ND | Hydantoin and N-methylhydantoin, e.g. | Acetophenone, some aromatic and alicyclic ketones |
| Carboxylation site | Methyl groups | Methyl groups | Methyl groups | No carboxylation | Methyl and methylene groups |
| Apparent Km value | Acetone, 7.5 ± 0.8 μM; butanone, 5.1 ± 0.6 μM; ATP, 1.0 ± 0.1 mM; HCO3, 6.9 ± 0.4 mM | Acetone, 7.8 ± 0.8 μM; ATP, 122 ± 14 μM; HCO3, 4.2 ± 0.7 mM | Acetone, 8.2 ± 0.5 μM; ATP, 98 ± 15 μM; HCO3, 2.3 ± 0.2 mMe | N-Methylhydantoin, 32 μM | Acetophenone, 9 ± 1 μM; ATP, 700 ± 200 μM; HCO3, 0.5 ± 0.15 mM |
Besides high sequence similarity, obvious differences in subunit composition, cofactor content, and ATP hydrolysis are observed. The numbers after the organism names are reference numbers.
Values in parentheses are molecular masses (in kilodaltons).
Sequence similarity is given as the percentage of identical amino acids of AcxA (β-subunit)/B (α subunit)/C (γ subunit) compared with the respective subunits of acetone carboxylase of A. aromaticum.
ND, not determined.
The original paper states a Km of 2.3 ± 0.2 nM, which is most probably a typing error.
Another recently identified member of this new enzyme family is acetophenone carboxylase of the denitrifying betaproteobacterial species “Aromatoleum aromaticum,” which is involved in anaerobic ethylbenzene degradation and catalyzes the ATP-dependent carboxylation of the intermediate acetophenone to benzoylacetate (10). In addition to the genes coding for acetophenone metabolism, the genome sequence of A. aromaticum indicated the presence of an apparent operon for acetone carboxylase (17), which was shown to be induced when the strain grows on either acetone or butanone as the sole substrate (24). Therefore, we investigated the biochemical properties of acetone carboxylase from this bacterium to compare them with those of other acetone carboxylases and acetophenone carboxylase.
MATERIALS AND METHODS
Growth of bacteria and preparation of cell extract (soluble fraction).
A. aromaticum strain EbN1 was grown aerobically or anaerobically on minimal medium (18) with acetone, butanone, acetate, or ethylbenzene as the sole carbon source and oxygen or nitrate as the electron acceptor, respectively, in 1-liter scale in flasks or 100-liter scale in fermentor cultures. Acetone and nitrate were found to be consumed in an approximately 1:2 molar stoichiometry. Therefore, carbon substrate and nitrate were added discontinuously in 5 mM and 10 mM aliquots, respectively. Growth was followed by determination of the increase of the optical density at 578 nm and the decrease of nitrate. When cultures were depleted of nitrate, additional aliquots of carbon substrate and nitrate were added. Cells were harvested during the exponential growth phase at an optical density of 4 to 5. The harvested cells were immediately frozen and stored at −80°C.
Frozen cells were suspended in 2 volumes of 10 mM Tris HCl (pH 7.5) containing 0.5 mg DNase I per ml. Cell suspensions were disrupted by sonication or passed through a French pressure cell at 137 MPa. Cell debris and membranes were removed by ultracentrifugation (100,000 × g) at 4°C for 1 h. The supernatant (soluble fraction) was used immediately or kept frozen at −20°C.
Acetone carboxylase assays.
Activity of acetone carboxylase was measured by either (i) incorporation of [14C]bicarbonate into nonvolatile acid-stable products (assay I), (ii) acetone-dependent ATP hydrolysis (assay II), (iii) determination of released inorganic phosphate (Pi) from ATP hydrolysis (assay III), or (iv) coupling the activity of acetone carboxylase to the activity of 3-hydroxybutyrate dehydrogenase (3HBDH) (assay IV). Unless otherwise indicated, the assays were performed aerobically using a standard assay mixture containing 50 mM HEPES KOH, pH 8.0, 10 mM MgCl2, 5 mM ATP, 20 mM NH4Cl, and 20 mM KHCO3.
(i) Assay I: substrate-dependent incorporation of [14C]bicarbonate.
Acetone carboxylase activity was measured via determination of substrate-dependent incorporation of radioactivity from 14C-labeled NaHCO3 into acid-stable products at 30°C. In addition to the standard assay mixture, 30 kBq of [14C]NaHCO3 per ml and enzyme (0.1 to 0.2 mg protein ml−1) were added. The reaction was initiated by addition of substrate (1 mM). Immediately after the initiation, a 300-μl control sample was withdrawn and the reaction was stopped by precipitation of the protein with 90 μl of 2.5 M NaHSO4. After 5 to 8 min, a second 300-μl sample was withdrawn and acidified to terminate the enzyme reaction. Volatile 14CO2 (nonfixed) was removed from the acidified sample by vigorously shaking the samples in open scintillation vials for 2 h. The remaining radioactivity in the samples was determined by liquid scintillation counting. Specific activity is given in nmol [14C]bicarbonate incorporated into acid-stable products per min and mg of protein.
(ii) Assay II: continuous, coupled spectrophotometric assay of ATPase activity.
ATP hydrolysis catalyzed by acetone carboxylase was determined spectrophotometrically by coupling ATP hydrolysis to the oxidation of NADH, using a modified coupled enzyme assay (25). The assay mixture (0.75 ml) contained 2 mM phosphoenolpyruvate, 0.4 mM NADH, 1.5 U ml−1 pyruvate kinase, 1 U ml−1 myokinase, and 4 U ml−1 lactate dehydrogenase, in addition to the standard assay mixture described above. Purified acetone carboxylase was added (0.1 to 0.2 mg) to the preincubated (30°C) assay mixture. After incubation for 3 min, the reaction was initiated by adding 1 mM substrate. Assays were monitored by determination of the decrease of absorbance at 365 nm. To determine decoupled ATP hydrolysis rates in the absence of HCO3−, HCO3− was omitted from the standard assay mixture and the buffer was flushed with nitrogen to remove dissolved CO2. Specific activities are given in nmol ATP hydrolyzed to AMP per min and mg of protein.
(iii) Assay III: determination of inorganic phosphate formation.
The time-dependent release of Pi from ATP hydrolysis was determined as described in reference 8. Specific activities are given in nmol Pi released (from ATP) per min and mg of protein.
(iv) Assay IV: determination of acetoacetate formation.
The formation of acetoacetate by acetone carboxylase was determined by coupling the activity of acetone carboxylase to the activity of 3HBDH. 3HBDH converts acetoacetate to 3-hydroxybutyrate in an NADH-dependent reaction. The consumption of NADH was followed by monitoring the decrease of absorbance at 365 nm. The assay mixture contained 0.4 mM NADH and 50 mU ml−1 3-hydroxybutyrate dehydrogenase (EC 1.1.1.30; Rhodobacter sphaeroides), in addition to the standard assay mixture.
The pH optimum of the reaction was determined using a HEPES KOH buffer system over the range of pH 6.0 to 9.0 (50 mM). Activity of acetone carboxylase was determined using assay II.
The apparent Km values for acetone and butanone were determined by assay II; the apparent Km value for ATP was determined by assay I. To study inhibition of acetone carboxylase activity by nonhydrolyzable ATP analogs, 2 mM each substance was added to the assay I mixture either instead of or in addition to ATP.
CoA ligase assays.
Acetoacetate-CoA ligase activity and benzoylacetate-CoA ligase activity were determined using an indirect photometric assay as described previously (19, 25).
Purification of acetone carboxylase.
Purification was performed at 4°C under oxic conditions. The soluble fraction from acetone-grown cells (2 to 2.5 g of protein) was applied to a DEAE-Sepharose column (diameter, 26 mm; volume, 60 ml; Amersham Biosciences), which had been equilibrated with buffer A (10 mM Tris HCl, pH 7.5, 2 mM MgCl2, 10% [vol/vol] glycerol). The column was washed with four column volumes of buffer A plus 150 mM KCl. Acetone carboxylase activity eluted with a step gradient with 225 mM KCl in buffer A at a constant flow rate of 3 ml min−1. Active fractions were pooled and concentrated by ultrafiltration (Centricon concentrator with a 30-kDa membrane). The concentrated pool was further purified by gel filtration on a Superdex 200 high-load column (diameter, 26 mm; volume, 320 ml; Amersham Biosciences), which had been equilibrated with buffer B (10 mM Tris HCl, pH 7.5, 100 mM KCl). Fractions containing acetone carboxylase were pooled and concentrated by ultrafiltration (Centricon concentrator with a 30-kDa membrane).
Substrate binding studies.
Binding of ATP (0 to 2.5 mM) and acetone (0 to 700 μM) to acetone carboxylase was studied by measuring the influence of increasing substrate concentration on the intrinsic tryptophan fluorescence of the enzyme. Buffer contained 50 mM HEPES KOH, pH 7.9, 20 mM potassium bicarbonate, 5 mM MgCl2, and 20 mM NH4Cl. Purified acetone carboxylase (500 nM) was added. After excitation at 280 nm, emission spectra were recorded (Varian Cary Eclipse fluorescence spectrophotometer) at 290- to 400-nm wavelengths after stepwise addition of substrate in portions of 50 to 100 μM.
Analytical methods.
Biotinylated proteins were detected with peroxidase-conjugated avidin as described in reference 15. ATP hydrolysis products formed in acetone carboxylase activity assays were analyzed by fast-performance liquid chromatography, using a Q Sepharose FF column (1 ml; Pharmacia) at a flow rate of 1 ml min−1. Acid-precipitated enzyme assays (500 μl) were applied on the column equilibrated with buffer C (50 mM potassium phosphate, pH 6.7). After washing the column with 3 ml of buffer C, ATP hydrolysis products were separated by using a linear gradient from 0 to 250 mM KCl in buffer C over 30 min. Retention times were as follows: AMP, 3 min; ADP, 13 min; ATP, 21 min. The formation of Pi and/or PPi was determined chromatographically by separation of the ATP hydrolysis products via paper chromatography on Whatman paper with 82.5% (vol/vol) 2-propanol, 17.5% H2O, 5% trichloroacetic acid, 0.1% NH3 as mobile phase (5) and staining of the separated phosphate species with solution P (8% [wt/vol] l-ascorbate and 0.7% [wt/vol] ammonium molybdate tetrahydrate in 0.7 M HCl). Rf values were 0.7 for Pi and 0.16 for PPi.
Other methods.
Protein concentrations were determined by Coomassie dye binding with bovine serum albumin as the standard (4) and by calculation from the absorption at 280 nm. Discontinuous SDS-PAGE was performed in 12.5% (wt/vol) polyacrylamide gels as described previously (14). Native molecular masses were estimated by gel filtration from the retention volumes of standard proteins (gel filtration HMW [high-molecular-weight] calibration kit; GE Healthcare) eluted under the same conditions. Purified enzyme (100 μM) was analyzed for the presence of metals by inductively coupled plasma emission spectroscopy (ICP-OES). Subunits were separated by discontinuous SDS-PAGE and identified by nano-high-pressure liquid chromatography electrospray ionization tandem mass spectrometry (nano-HPLC-Esi-MS/MS). UV-visible spectra of acetone carboxylase were recorded under aerobic conditions (0 to 2 mg ml−1 of protein). To picture phylogenetic alliances of acetone carboxylase subunits with database entries, sequences were clustered according to their distances from the query sequence by use of BLAST (NCBI, Bethesda, MD). The identification numbers of the sequences used and their original annotation are given in the supplemental material.
RESULTS
Growth characteristics with acetone and butanone.
Aromatoleum aromaticum was grown under oxic conditions with acetone or under denitrifying conditions with acetone, butanone, acetate, or ethylbenzene as sole carbon and energy sources, respectively. Similar growth rates with doubling times of 8 to 10 h were observed for all conditions (except for ethylbenzene-grown cells, with doubling times of 26 to 30 h). Typical cell yields with acetate and acetone under denitrifying conditions were 20 g and 35 g dry cell mass per mol substrate, respectively (yield factors, 0.42 and 0.37, respectively, based on the number of moles carbon assimilated in cell mass per number of moles carbon of consumed substrate). SDS-PAGE analysis of cell extracts showed similar patterns of highly induced proteins in cells grown on acetone and butanone, which were lacking in ethylbenzene- and acetate-grown controls (Fig. 1).
Fig 1.

SDS-PAGE (11% polyacrylamide) of soluble fraction proteins of A. aromaticum grown under denitrifying conditions on acetate (lane 1), acetone (lane 2), butanone (lane 4), and ethylbenzene (lane 5) or aerobically on acetone (lane 3). A molecular mass standard is shown on the left. Induced proteins in acetone- and butanone-grown cells are indicated by arrows.
Activities of induced enzymes.
Growth on acetone is assumed to proceed via the pathway shown in Fig. 2. Acetone is proposed to be initially carboxylated to acetoacetate by acetone carboxylase in an ATP-dependent reaction and subsequently activated to acetoacetyl-CoA by a CoA ligase or CoA transferase. Butanone may be metabolized analogously, possibly by the same pathway (17). Therefore, carboxylation activities were determined by 14CO2 incorporation assays (assay I) with acetone and butanone as substrates, using the soluble fraction of cell extracts of acetone- and butanone-grown cells (Table 1). Similar specific activities (30 to 40 nmol min−1 mg protein−1) were observed in extracts for carboxylation of acetone, regardless of aerobic or anaerobic cultivation or growth on acetone or butanone, respectively. In the same extracts, butanone was carboxylated at about 40 to 45% of the specific activity observed with acetone. Therefore, it was likely that the same enzyme carboxylates both ketones. The next enzyme of the proposed pathway, acetoacetate-CoA ligase, was also assayed in the same extracts (Table 1), yielding specific activities of 90 to 95 nmol min−1 mg−1. Control cells grown on ethylbenzene did not show detectable activity of acetone carboxylase and less than 50% of the specific activity of acetoacetate-CoA ligase (Table 1). In contrast, activity of a CoA ligase specific for benzoylacetate (7 nmol min−1 mg−1) was measured only in extracts of ethylbenzene-grown cells and was lacking in those of acetone- or butanone-grown cells.
Table 1.
Enzyme activities in extracts (soluble fraction) of acetone- and butanone-grown cells of A. aromaticuma
| Cell growth medium content | Carboxylase activity with: |
CoA ligase activity with: |
||
|---|---|---|---|---|
| Acetone | Butanone | Acetoacetate | Benzoylacetate | |
| Acetone, NO3− | 39 | 17 (43) | 93 | >1 |
| Acetone, O2 | 31 | 14 (45) | ND | ND |
| Butanone, NO3− | 33 | 14 (43) | 94 | >1 |
| Ethylbenzene, NO3− | <2 | ND | 40 | 7 |
A. aromaticum cells were grown under different conditions, and activities are in nmol min−1 mg protein−1. Carboxylase activities were determined by the [14C]bicarbonate incorporation assay (assay I). Acetoacetate- and benzoylacetate-CoA ligase (AMP-forming) activities were determined photometrically. Control cells were grown on ethylbenzene and nitrate. Values in parentheses are the percentage of activity (nmol min−1 mg protein−1) observed with acetone as substrate under the respective conditions. ND, not determined.
Purification of acetone carboxylase.
Acetone carboxylase (Acx) was purified from A. aromaticum cells grown anaerobically on acetone via two chromatographic steps (Fig. 3; Table 2). From DEAE-Sepharose, acetone carboxylase activity eluted between 160 and 225 mM KCl with a yield of 28% and a 2-fold enrichment. The second chromatographic step, Superdex 200 gel filtration, resulted in a symmetric protein peak containing the enzyme that corresponded to a molecular mass of 360 ± 20 kDa. A yield of 50% (14% in total) and a final enrichment of about 3-fold were obtained. Typically, specific acetone carboxylation activities (assay I) of 80 to 90 nmol [14C]bicarbonate fixed per min and mg of protein were observed with the purified protein.
Fig 3.

Purification of acetone carboxylase of A. aromaticum. Fractions were analyzed by SDS-PAGE (13.5% polyacrylamide). Lanes: 1, cell extract (soluble fraction); 2, DEAE fraction (160 to 225 mM KCl pool); 3, Superdex 200 gel filtration fraction. A molecular mass standard is shown on the left. The purified enzyme consisted of three subunits of 85, 75, and 20 kDa, respectively.
Table 2.
Purification of acetone carboxylase from cell extracts of A. aromaticuma
| Fraction | Sp act (nmol min−1 mg−1) | Purification (fold) | Yield (%) |
|---|---|---|---|
| Extract (soluble fraction) | 20.2 | 1 | 100 |
| DEAE pool | 38.6 | 2.0 | 28 |
| Gel filtration | 70.3 | 3.1 | 14 |
ATPase activity (AMP forming) was determined in a coupled photometric assay (assay II). Activities are in nmol min−1 mg protein−1.
Acetone carboxylase accounted for at least 5.5% of total cell protein (soluble fraction). This high expression level of Acx probably compensates for the rather low specific activity of the enzyme (see Discussion).
Molecular properties of acetone carboxylase.
Purified acetone carboxylase consisted of three subunits of 85, 75, and 20 kDa, respectively, in apparently equimolar ratios, as deduced from the SDS-PAGE. All subunits were analyzed by nano-HPLC-Esi-MS/MS and could be assigned to the expected genes encoding the subunits, acxABC, which are part of a larger gene cluster that also contains genes for a putative CoA ligase and a putative succinyl-CoA:3-ketoacid CoA transferase (Fig. 4), which could be involved in the activation of acetoacetate to acetoacetyl-CoA (Fig. 2). The native molecular mass of Acx was determined to be 360 ± 20 kDa by gel filtration, indicating an (αβγ)2 composition. Element analysis by ICP-OES showed a metal content of Acx of 1 zinc and 2.2 irons per native (αβγ)2 complex. No further cofactors were observed by UV-visible spectroscopy, and no biotin was detected in any of the subunits by blotting with peroxidase-conjugated avidin.
Fig 4.

Gene cluster encoding enzymes possibly involved in acetone degradation in A. aromaticum. The acxABC genes encode the three subunits of acetone carboxylase. The gene product of ebA4717 (labeled CoA Lig) shows similarity with known CoA ligases and may be the enzyme activating acetoacetate to acetoacetyl-CoA (Fig. 2). Alternatively, this reaction may be catalyzed by KctAB (labeled TF), which show sequence similarities to the subunits of succinyl-CoA:3-ketoacid-CoA transferases. The gene product of ebA4705 shows similarities to putative exporters of the RND superfamily (transporter class TC no. 2.6; tcdb.org). Open reading frames ebA4709, ebA4715, and ebA4720 code for putative proteins of unknown function. Gene ebA4712 encodes a putative LysR-like regulator. The gene acxR codes for a sigma 54-dependent transcriptional activator. Genes for similar sigma 54-dependent regulator proteins are also present in acetone metabolic gene clusters of other bacteria (22), while the other open reading frames are not conserved on a widespread basis. hyp., hypothetical protein; Reg., putative regulatory protein.
Catalytic properties of acetone carboxylase.
Routine assays of acetone carboxylase activity were based on either of three methods: substrate-dependent incorporation of [14C]bicarbonate into acid-stable products (assay I), determination of ATPase activity using a coupled spectrophotometric assay (assay II), or measurement of the release of Pi from ATP hydrolysis (assay III). Specific activities of the enzyme determined by assays II and III were not converted into rates of substrate carboxylation, because uncoupled ATPase activities were observed with many substrates.
Purified acetone carboxylase catalyzed the ATP- and HCO3−-dependent carboxylation of acetone to acetoacetate. Butanone was carboxylated with about 40 to 50% of the activity observed with acetone (assay I), as was observed in the extracts. No activity was recorded if ATP was replaced by GTP. The activity of acetone carboxylase was coupled to the activity of 3-hydroxybutyrate dehydrogenase (assay IV), confirming the formation of acetoacetate as the product of acetone carboxylation.
Activity was protein dependent and linear over the range of from 0.05 to 1 mg Acx per ml. Typical specific activities obtained with the purified enzyme were 80 to 90 nmol min−1 mg protein−1 for [14C]bicarbonate incorporation (net carboxylation activity of acetone, assay I), 180 to 200 nmol min−1 mg−1 for ATPase activity (AMP formation from ATP, assay II), and 320 to 340 nmol min−1 mg−1 for phosphate release (assay III). The obtained rates indicated a potential stoichiometry of 2 ATP and 4 released Pi per acetone carboxylated, which is further supported as follows: ATPase activity was measured only in the coupled photometric assay (assay II) with the addition of myokinase, indicating the formation of AMP rather than ADP. Chromatographic analysis of the ATP hydrolysis products indicated that ATP was exclusively cleaved to AMP, and no significant amounts of ADP were produced. Moreover, Pi rather than pyrophosphate was detected as the ATP hydrolysis product by paper chromatography. To directly assess for the stoichiometry of ATP hydrolyzed per acetone carboxylated, the amount of NADH oxidized after adding a limiting amount of acetone into the coupled photometric assay (assay II) was examined. From the observed oxidation of about 4 NADH per acetone added, a stoichiometry of 2 ATP hydrolyzed to 2 AMP and 4 Pi per acetone was determined (Fig. 5).
Fig 5.

Stoichiometry of ATP hydrolysis. A typical coupled photometric ATPase assay (assay II), which was started with a limiting amount of acetone (40 μM, indicated by an arrow), is shown. The corresponding amount of oxidized NADH (159 μM) was calculated from the absorbance difference (ΔA365) between the start of the reaction and its leveling off. Hydrolysis of 1 ATP to AMP leads to the oxidation of 2 NADH in the applied assay. Therefore, 80 μM ATP was hydrolyzed, resulting in an acetone:ATP stoichiometry of 1:2.
ATPase activity was detected not only in the presence of acetone and bicarbonate but also at a lower rate (about 15 to 20%) when bicarbonate was omitted. The hydrolysis product under these conditions was also exclusively AMP. This indicates a decoupled ATPase activity of the enzyme, which is similar but less prominent than that observed for acetophenone carboxylase and acetone carboxylases of alphaproteobacteria (10, 21, 22). This decoupled ATPase activity of Acx was increased up to 5-fold by addition of 1 mM MnCl2, whereas the same MnCl2 concentration inhibited net carboxylation activity by 25%. Interestingly, no decoupled ATPase activity was measured with bicarbonate as a sole substrate when acetone was omitted in the assays II.
The reaction followed Michaelis-Menten kinetics with the following apparent Km values of the various substrates: acetone, 7.5 ± 0.8 μM; butanone, 5.1 ± 0.6 μM; ATP; 1.0 ± 0.13 mM; HCO3−, 6.9 ± 0.4 mM. Addition of 20 mM NH4Cl increased the activity by about 80%, and NH4Cl was therefore routinely added to all enzyme assays. The pH optimum of the enzyme was 7.8 to 8.0, as determined by ATPase assay.
Inhibition of acetone carboxylase.
The inhibitory effects of nonhydrolyzable ATP analogs on net carboxylation activity (assay I) were studied. Neither AMP-CPP (α,β-methylene-ATP), AMP-PCP (β,γ-methylene-ATP), nor AMP-PNP (β,γ-imido-ATP) supported Acx activity. After preincubation with 2 mM AMP-CPP or AMP-PCP and initiation with 5 mM ATP, no inhibition of carboxylation activity was observed, indicating that the analogs are probably not able to bind to the enzyme. After preincubation with 2 mM AMP-PNP, 58% residual activity was determined, suggesting a competitive inhibitory effect. Remarkably, AMP-PNP completely abolished the activity of acetophenone carboxylase under the same conditions (10).
The observed stoichiometry of 2 ATP hydrolyzed per acetone carboxylated may indicate a transient activation of bicarbonate to carboxyphosphate, as proposed for acetophenone carboxylase. Yet, addition of 10 mM carbamoyl phosphate did not inhibit Acx activity, whereas acetophenone carboxylase was inhibited even at 5-fold lower carbamoyl phosphate concentrations (10).
Addition of EDTA (5 mM) to the enzyme assay II mixture completely inhibited ATPase activity, most probably due to binding and removal of Mg2+ ions necessary for ATP hydrolysis. Interestingly, preincubation of the enzyme with 10 mM EDTA for 1 h prior to assaying Acx did not inhibit activity, suggesting that the zinc and iron ions of the enzyme are bound very tightly.
Substrate spectrum of acetone carboxylase.
The substrate ranges for ATPase (assay II) and carboxylation (assay I) activities of acetone carboxylase were studied (Table 3). ATPase activity was observed with various tested substrates, but activity was decreasing with prolongation or branching of the substrate skeleton. Carboxylation activity was determined only with butanone, with about 50% of the activity observed with acetone. This indicates that a variety of substances can bind to acetone carboxylase, inducing a (decoupled) ATPase activity, but only acetone and butanone are actually carboxylated.
Table 3.
Substrate spectrum of acetone carboxylasea
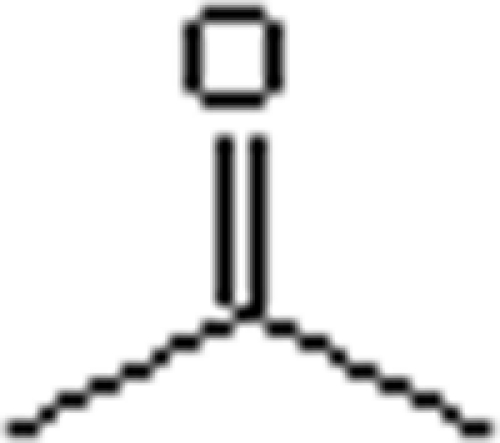
Mechanistically, carboxylation of acetone is supposed to be initiated via the abstraction of a hydrogen atom from the methyl group (Fig. 6). No significant kinetic isotope effect (KIE) was observed when [2H6]acetone was used instead of acetone in ATPase or carboxylation activity assays (Table 3). Moreover, no KIE was detected in decoupled ATPase activity assays where bicarbonate was omitted, in contrast to the observations with acetophenone carboxylase (10). Therefore, for acetone carboxylase hydrogen abstraction is not the rate-limiting step either of nonproductive substrate activation or of net carboxylation.
Fig 6.

Proposed catalytic mechanism of acetone carboxylation catalyzed by acetone carboxylase. Bicarbonate is activated to carboxyphosphate by hydrolysis of 1 ATP, while a second ATP is hydrolyzed to activate acetone to phosphoenol acetone. This activated intermediate can undergo hydrolysis in the absence of bicarbonate, which can be measured as decoupled ATPase activity. This activity appears to be stimulated by Mn2+.
Binding of ATP to acetone carboxylase was studied by quenching of the intrinsic tryptophan fluorescence of the enzyme. After excitation at 280 nm, fluorescence emission was monitored at 290 to 400 nm. Absorption intensity decreased with increasing concentrations of ATP added (Fig. 7A). ATP alone did not exhibit fluorescence emission (data not shown). Plotting the decrease in absorption at 343 nm (absorption maximum) against the ATP concentration in the assay allowed the calculation of an ATP binding constant (960 ± 20 μM), which was in a range comparable to the determined apparent Km for ATP (1.0 ± 0.15 mM) of the enzyme (Fig. 7B). No changes in the intrinsic fluorescence were observed with acetone (data not shown), indicating that binding of acetone does not induce conformational changes of the enzyme, that it is too small to efficiently shield fluorescence, or that ATP is needed for binding of the substrate.
Fig 7.

ATP kinetics of purified acetone carboxylase. (A) Changes in the intrinsic tryptophan fluorescence of Acx upon binding of ATP (excitation wavelength, 280 nm). Emission spectra of a buffer control [labeled (1)] and 500 nM Acx [labeled (2)] are shown along with emission spectra of Acx with increasing ATP concentrations (0.1 to 2.5 mM). (B) Kinetics of ATP binding (triangles) and enzyme activity (circles) of acetone carboxylase. ATP binding was determined as the concentration-dependent change in endogenous fluorescence at 343 nm in the absence of acetone, calculated from the data shown in panel A. ATP dependence of enzyme kinetics was determined by determining the [14C]bicarbonate incorporation (assay I) into reaction products. Standard deviations of triple assays were smaller than the graph symbols used.
DISCUSSION
The presence of acetone carboxylase was detected in cells grown on either acetone or butanone and regardless of aerobic or anaerobic conditions. Moreover, similar protein patterns and activity ratios with acetone and butanone were observed under all growth conditions, indicating that the same enzyme is responsible for acetone and butanone degradation. The enzyme is distinct from acetophenone carboxylase, because control cells grown on ethylbenzene, which contain active acetophenone carboxylase (10), did not exhibit carboxylation activity with acetone or butanone. The specific activities for net carboxylation (assay I) measured in the soluble fraction of cell extracts were about 50% of those needed to explain the observed growth rates of the respective cultures (doubling times of 8 to 10 h are equal to a minimum activity of 70 to 80 mU/mg). We regard this to be a good match, considering the difficult determination of reliable enzyme activity due to the intrinsically low specific activity of the isolated enzyme, the chemical instability of acetoacetate, and the absence of further metabolic steps that probably draw the reaction forward. Regarding the observed activation effect on acetone carboxylase by ammonium ions, it may also be speculated that further activation of the enzyme takes place in the cytoplasm, which is lost after preparation of extracts.
The further metabolism of acetone apparently follows the expected pathway, as evident from the presence of an induced acetoacetate-CoA ligase present in acetone- and butanone-grown cells with a rather high activity matching the theoretical minimum for the observed growth rate. Control cells grown on ethylbenzene contained only half of the acetoacetate-CoA ligase activity but showed an additional activity of a specific benzoylacetate-CoA ligase, which is needed for the metabolic pathway of acetophenone (10). The presence of a gene coding for a CoA ligase in a common gene cluster with the genes for acetone carboxylase is consistent with the simultaneous induction of both enzymes. A predicted CoA transferase encoded in the same gene cluster may also potentially contribute to acetoacetyl-CoA formation, especially since data on the induction of this enzyme in acetone-grown cells are available (24). A similar principle of a CoA transferase and a CoA ligase acting in parallel is known for several CoA-dependent degradation pathways, e.g., the degradation of carnitine to butyrobetaine (7). However, genes for CoA transferases or CoA ligases are not clustered with the acx genes in most other organisms (see Fig. S1 in the supplemental material).
The products of ATP hydrolysis by A. aromaticum acetone carboxylase are AMP and Pi, and a stoichiometry of 2 ATP hydrolyzed to 2 AMP and 4 Pi was determined for the overall reaction with acetone. This is supported by the chromatographic detection of AMP and Pi as the only hydrolysis products of ATP, the dependence of the coupled ATPase assay on myokinase, the stoichiometries determined in the ATPase assay (assay II) with limiting amounts of acetone, and the relative specific activities measured with the three different enzyme assays but differs from the values reported for other acetone carboxylases (Table 4). For example, the enzyme from X. autotrophicus was also reported to produce AMP as a final hydrolysis product from ATP, but in a stoichiometry of 1 ATP per acetone with transient formation of ADP (3, 21, 22). In contrast, acetophenone carboxylase was shown to produce ADP as the only hydrolysis product in a stoichiometry of 2 ATP hydrolyzed per acetophenone (10). The ATP stoichiometry determined for acetone carboxylation appears to be very high, considering the thermodynamic requirements for the reaction (ΔG°′ = 11.8 kJ/mol for acetone carboxylation with bicarbonate). However, the overall energy yield of acetone oxidation with nitrate reduction to nitrite is −1,116 kJ/mol (accounting for 14 ATP equivalents per acetone), and growth of the organism would therefore not be significantly affected by the energy costs of 4 ATP equivalents for the initial reaction.
Purified acetone carboxylase from A. aromaticum is extremely limited in its substrate range, catalyzing only the carboxylation of acetone and butanone. It may be inferred from the experiments shown that the enzyme actually accepts no other substrates, because neither diethyl ketone nor methyl alkyl ketones with anything larger than an ethyl side chain were carboxylated. Therefore, acetone carboxylase apparently carboxylates only methyl groups adjacent to carbonyl groups and accepts only a methyl or ethyl group as the second substituent of the ketone. The enzyme showed identical ATPase activity with acetone and butanone in the presence of bicarbonate but only about half of the carboxylating activity with butanone than with acetone. Therefore, the stoichiometry of ketone carboxylation to ATP hydrolysis apparently changes from 1:2 with acetone to 1:4 with butanone (Table 3). This may be explained by assuming that butanone may be bound either in a productive orientation (methyl group), leading to carboxylation, or in a nonproductive orientation (ethyl group), followed by abortion of the reaction with concomitant uncoupled ATP hydrolysis. If the chances of binding in either conformation are identical, this would lead to a doubling of the rate of ATP hydrolysis compared to the fully coupled situation with acetone as substrate. The enzyme also showed significant uncoupled ATPase activity with 2- and 3-pentanone, although these compounds were not carboxylated at all (Table 3). This is most probably due to the larger size of the compounds, which may prohibit productive binding in the active site.
Lower rates of uncoupled ATPase activities were seen with the productive substrates acetone and butanone in the absence of bicarbonate. Such uncoupled ATPase activities have also been reported previously for other acetone carboxylases (21, 22) and for acetophenone carboxylase (10). Therefore, the mechanism of these enzymes seems to involve separate modules for substrate activation by ATP and the actual carboxylation reaction.
Acetone carboxylase of A. aromaticum did not contain an organic cofactor, but it tightly bound metal ions in reproducible stoichiometries, namely, 1 Zn and 2 Fe per holoenzyme. The nature of the determined metals is in contrast to that for the previously described acetone carboxylases, which contained predominantly Mn, in addition to Zn and Fe ions (Table 4). This is in accordance with the observation that growth of A. aromaticum on acetone is not stimulated by added MnCl2 to the medium, as reported for R. capsulatus (2). Preliminary data on acetone carboxylase from Paracoccus denitrificans (data not shown), which is currently studied as a reference enzyme in our group, indicate that the metal content of this enzyme is comparable to that of the previously characterized enzymes from X. autotrophicus and R. capsulatus. Therefore, acetone carboxylases may occur in two different varieties that show similar apparent Km values for acetone and bicarbonate but differ in metal content and possibly ATP stoichiometry. The 10-fold higher apparent Km for ATP for the enzyme from A. aromaticum may also be related to the different ATPase stoichiometry, which represents a deviation in the reaction mechanism.
The differences between the acetone carboxylases of the studied strains (R. capsulatus, X. autotrophicus, and A. aromaticum) on the one hand and acetophenone carboxylase (from A. aromaticum), the only other characterized carboxylase of the enzyme family, on the other hand are more prominent than those between the different acetone carboxylases: (i) acetophenone carboxylase consists of more subunits than acetone carboxylases. One of these subunits is apparently derived from a duplication of the β subunit of acetone carboxylases; the fifth subunit is unique for acetophenone carboxylase (10). (ii) Acetophenone carboxylase also carboxylates the C-2 methylene group of the side chain of propiophenone, whereas acetone carboxylases carboxylate only the methyl groups of acetone or butanone. (iii) Acetophenone carboxylase contains Zn as the only metal cofactor, at a stoichiometry of 2 Zn per holoenzyme, but no Fe or Mn. (iv) ATP is hydrolyzed exclusively to ADP at a stoichiometry of 2 ATP per acetophenone carboxylated. (v) Uncoupled ATPase activity is recorded with either substrate (ketone or bicarbonate) in the absence of the other for acetophenone carboxylase, whereas acetone carboxylases show this activity only with ketones in the absence of bicarbonate, but not vice versa.
The observed uncoupled ATPase reactions and the ATP stoichiometry of acetophenone carboxylase were crucial in formulating a potential reaction mechanism (10). We propose an analogous mechanism for acetone carboxylase from A. aromaticum, which involves the hydrolysis of one ATP to activate acetone to the corresponding enolphosphate, while the other ATP is used to activate bicarbonate to carboxyphosphate (Fig. 6). In contrast to acetophenone carboxylase, only the activated acetone-enolphosphate would be released from the enzyme in the absence of bicarbonate, leading to the observed uncoupled ATPase activity, whereas carboxyphosphate either may be synthesized only in the presence of acetone or may remain tightly bound in the enzyme. Release and hydrolysis of acetone-enolphosphate are apparently stimulated by added Mn2+ ions in the assay, as indicated by the observed Mn2+-dependent increase in uncoupled ATPase but not in carboxylation activity. Because acetone carboxylase produces exclusively AMP and Pi as hydrolysis products of ATP, we postulate an activation mechanism that initially transfers a pyrophosphate group from ATP to the enzyme or the substrate(s) and hydrolyzes the pyrophosphate moiety at some point during the reaction. This postulated modified mechanism of acetone carboxylation compared to acetophenone carboxylation is also consistent with the different kinetic effects on uncoupled ATPase reactions recorded with deuterated substrates. Whereas the deprotonation of the methyl group was the rate-limiting step for acetophenone carboxylase (10), no isotope effect was recorded for acetone carboxylase with [2H6]acetone. This indicates that the rate of activation of acetone to the enolphosphate is limited by a different partial reaction lacking in the mechanism of acetophenone carboxylase, potentially pyrophosphate hydrolysis.
Comparison with other acetone and acetophenone carboxylases and hydantoinases.
Acetone and acetophenone carboxylases are members of a larger protein family that also contains ATP-dependent and ADP-forming hydantoinases/oxoprolinases (Pfam01968). The proposed mechanism of these hydantoinases parallels that of the carboxylases in phosphorylating the carbonyl groups of the substrates to form phosphoenamine intermediates that can be easily hydrolyzed (20). Bioinformatic analysis shows that the hydantoinase-associated members of this enzyme family consist of only two subunits, which are similar to the large α and β subunits of acetone carboxylases. For further features of these enzymes in comparison to acetone and acetophenone carboxylases, see Table 4. We have established that acetone carboxylase does not catalyze significant ATP hydrolysis with the typical hydantoinase substrate 2-pyrrolinidone or pyroglutamate.
A phylogenetic tree of ATP-dependent hydantoinases, acetone, and acetophenone carboxylases was constructed on the basis of the results of a BLAST search with the α subunit of A. aromaticum acetone carboxylase (acxB gene product), which is one of the conserved subunits present in all enzymes. The result indicates that the known acetone carboxylases indeed form a common branch within the enzyme family, which is divided into subbranches along the phylogenies of the respective organisms. Whereas the α subunits of the characterized enzymes of the alphaproteobacteria show 86 to 88% identity among each other, they are less than 70% identical to the α subunit of acetone carboxylase of the betaproteobacterium A. aromaticum or the orthologous gene products from other betaproteobacteria or other phylogenetic groups (Fig. 8). An apparent orthologous operon in the Geobacillus thermoglucosidasius genome (65% amino acid sequence identity of the α subunit with that of A. aromaticum) was confirmed to code for an acetone carboxylase on the basis of enzyme activity (K. Schühle, D. Kleinsorge, and J. Heider, unpublished data). The α subunit of acetophenone carboxylase from A. aromaticum clearly lies in a different branch, together with similar predicted gene products from various other bacteria, whereas the α subunits of hydantoinases form a third distinct branch. Closer inspection of the sequences of the acetone and acetophenone carboxylase branches indicates that predicted enzymes listed in some of the subbranches are so different from the characterized enzymes that some doubt about their annotation is warranted. For example, we noticed several enzymes in diverse bacterial species (labeled “acetophenone carboxylase-like” in Fig. 8) which contain subunits very similar to those of the core complex of acetophenone carboxylase (αββ′γ) but are associated with either a completely different fifth subunit (labeled φ instead of ε) or no apparent fifth subunit encoded in a common operon. Therefore, a function of those enzymes as acetophenone carboxylases is questionable.
Fig 8.

Phylogenetic tree of ATP-dependent hydantoinases, acetone, and acetophenone carboxylases based on the α subunit of A. aromaticum acetone carboxylase (acxB gene product) and the corresponding subunits in other enzymes. Acetone carboxylases of alphaproteobacteria cluster in two branches comprising the genera Xanthobacter, Rhodopseudomonas, Rhodobacter, and Paracoccus [labeled (1)] and genera Sinorhizobium and Mesorhizobium [labeled (2)]. Closely associated sequences are found in some beta- and epsilonproteobacteria (including A. aromaticum) and some Bacillus and Geobacillus species. Moreover, some Geobacillus species contain an additional operon for a more distantly related paralog of acetone carboxylase which is labeled as an acetone carboxylase-like enzyme [labeled (3)]. A different branch is occupied by known or putative acetophenone carboxylases from diverse species (including A. aromaticum); that branch also has an associated subbranch with more distantly related enzymes. In this subbranch (acetophenone carboxylase-like enzymes), a cluster of closely related sequences from phylogenetically diverse genera, including Xanthobacter, Azotobacter, Polaromonas, Geobacter, Rhodococcus, and Helicobacter, is indicated. Finally, the third major branch of the tree contains the known ATP-dependent hydantoinases. For accession numbers, see Table S1 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft, Germany, and by the LOEWE initiative of the state of Hessen (SynMicro LOEWE Centre, Marburg), Germany.
We thank U. Linne, Department of Chemistry, Marburg, Germany, for protein identification, and we thank A. Seubert, Department of Chemistry, Marburg, Germany, for performing ICP-OES analysis.
Footnotes
Published ahead of print 21 October 2011
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Birks SJ, Kelly DJ. 1997. Assay and properties of acetone carboxylase, a novel enzyme involved in acetone-dependent growth and CO2 fixation in Rhodobacter capsulatus and other photosynthetic and denitrifying bacteria. Microbiology 143:755–766 [DOI] [PubMed] [Google Scholar]
- 2. Boyd JM, Ellsworth H, Ensign SA. 2004. Bacterial acetone carboxylase is a manganese-dependent metalloenzyme. J. Biol. Chem. 279:46644–46651 [DOI] [PubMed] [Google Scholar]
- 3. Boyd JM, Ensign SA. 2005. ATP-dependent enolization of acetone by acetone carboxylase from Rhodobacter capsulatus. Biochemistry 44:8543–8553 [DOI] [PubMed] [Google Scholar]
- 4. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 5. Burianek J, Cifka J. 1965. Schnelle Bestimmung der radiochemischen Reinheit von Orthophosphat-32P. Z. Anal. Chem Fresenius 213:1–9 [Google Scholar]
- 6. Dürre P, et al. 1995. Solventogenic enzymes of Clostridium acetobutylicum: catalytic properties, genetic organisation, and transcriptional regulation. FEMS Microbiol. Rev. 17:251–262 [DOI] [PubMed] [Google Scholar]
- 7. Engemann C, et al. 2005. Identification and functional characterisation of genes and corresponding enzymes involved in carnitine metabolism of Proteus sp. Arch. Microbiol. 183:176–189 [DOI] [PubMed] [Google Scholar]
- 8. Gawronski JD, Benson DR. 2004. Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal. Biochem. 327:114–118 [DOI] [PubMed] [Google Scholar]
- 9. Hausinger RP. 2007. New insights into acetone metabolism. J. Bacteriol. 189:671–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jobst B, Schühle K, Linne U, Heider J. 2010. ATP-dependent carboxylation of acetophenone by a novel type of carboxylase. J. Bacteriol. 192:1387–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johansson I, Eliasson E, Norsten C, Ingelman-Sundberg M. 1986. Hydroxylation of acetone by ethanol- and acetone-inducible cytochrome P-450 in liver microsomes and reconstituted membranes. FEBS Lett. 196:59–64 [DOI] [PubMed] [Google Scholar]
- 12. Koop DR, Casazza JP. 1985. Identification of ethanol-inducible P-450 isozyme 3a as the acetone and acetol monooxygenase of rabbit microsomes. J. Biol. Chem. 260:13607–13612 [PubMed] [Google Scholar]
- 13. Kotani T, Yurimoto H, Kato N, Sakai Y. 2007. Novel acetone metabolism in a propane-utilizing bacterium, Gordonia sp. strain TY-5. J. Bacteriol. 189:886–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laemmli UK. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 15. Menendez C, et al. 1999. Presence of acetyl coenzyme A (CoA) carboxylase and propionyl-CoA carboxylase in autotrophic Crenarchaeota and indication for operation of a 3-hydroxypropionate cycle in autotrophic carbon fixation. J. Bacteriol. 181:1088–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ogawa J, et al. 1995. Purification and characterization of an ATP-dependent amidohydrolase, N-methylhydantoin amidohydrolase, from Pseudomonas putida 77. Eur. J. Biochem. 229:284–290 [DOI] [PubMed] [Google Scholar]
- 17. Rabus R, et al. 2005. The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch. Microbiol. 183:27–36 [DOI] [PubMed] [Google Scholar]
- 18. Rabus R, Widdel F. 1995. Anaerobic degradation of ethylbenzene and other aromatic-hydrocarbons by a new denitrifying bacteria. Arch. Microbiol. 163:96–103 [DOI] [PubMed] [Google Scholar]
- 19. Schühle K, et al. 2003. Benzoate-coenzyme A ligase from Thauera aromatica: an enzyme acting in anaerobic and aerobic pathways. J. Bacteriol. 185:4920–4929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seddon AP, Meister A. 1986. Trapping of an intermediate in the reaction catalyzed by 5-oxoprolinase. J. Biol. Chem. 261:1538–1543 [PubMed] [Google Scholar]
- 21. Sluis MK, Ensign SA. 1997. Purification and characterization of acetone carboxylase from Xanthobacter strain Py2. Proc. Natl. Acad. Sci. U. S. A. 94:8456–8461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sluis MK, et al. 2002. Biochemical, molecular, and genetic analyses of the acetone carboxylases from Xanthobacter autotrophicus strain Py2 and Rhodobacter capsulatus strain B10. J. Bacteriol. 184:2969–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sluis MK, Small FJ, Allen JR, Ensign SA. 1996. Involvement of an ATP-dependent carboxylase in a CO2-dependent pathway of acetone metabolism by Xanthobacter strain Py2. J. Bacteriol. 178:4020–4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wöhlbrand L, et al. 2007. Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1. Proteomics 7:2222–2239 [DOI] [PubMed] [Google Scholar]
- 25. Ziegler K, Braun K, Böckler A, Fuchs G. 1987. Studies on the anaerobic degradation of benzoic acid and 2-aminobenzoic acid by a denitrifying Pseudomonas strain. Arch. Microbiol. 149:62–69 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.