

Abstract
The type IV secretion system encoded by the virB operon is required for full virulence of Brucella sp., and the present study links the RNA chaperone Hfq to wild-type expression of virB in Brucella abortus 2308. Studies employing virB-lacZ fusions, quantitative reverse transcription-PCR, and immunoblot analysis showed that both transcription and translation of virB are decreased in an isogenic hfq mutant compared to those in the parental strain. These results led to the hypothesis that Hfq regulation of virB is mediated through an intermediate transcriptional regulator. Subsequent experiments determined that expression of the gene encoding the putative Brucella quorum-sensing regulator BabR (also known as BlxR), a known virB regulator, is also controlled by Hfq at the posttranscriptional level, and a cis-acting element in the 5′ untranslated region of the babR transcript responsible for this regulation was identified. Consistent with its role as a virB regulator, recombinant Brucella BabR binds to the virB promoter region in electrophoretic mobility shift assays. However, experiments employing a babR mutant strain determined that BabR is a repressor, not an activator, of virB transcription. These findings suggest that Hfq regulates virB expression through both BabR-dependent and BabR-independent pathways.
INTRODUCTION
The Brucella spp. are part of the α2 subclass of proteobacteria (28), and these bacteria infect wild and domestic animals, such as cattle, goats, and sheep, causing abortions in pregnant females and sterility in males. The Brucella spp. can also infect humans, and brucellosis is described as the most common zoonosis worldwide, with an estimated 500,000 human brucellosis cases being reported each year (29). The bulk of these infections occur in areas where Brucella is endemic in the food animal populations, and humans become infected most often by consuming unpasteurized dairy products or handling animals and products from Brucella-infected animals (28). Brucellosis in humans is characterized by nonspecific symptoms, including a recurring high fever, extreme fatigue, and a variety of other “flu-like” symptoms that in aggregate are extremely debilitating (12).
During the course of chronic infection, the brucellae reside within macrophages, where they replicate in a specialized compartment associated with the endoplasmic reticulum, and the ability of the brucellae to survive and replicate within macrophages is essential to their virulence (4). While the brucellae do not produce classical virulence factors, such as toxins or adhesins (39), several genetic determinants of infection have been identified that aid the bacterium in infecting and surviving within the host (36). One of these determinants of Brucella virulence is the RNA chaperone Hfq (33).
Hfq is a bacterial protein that mediates RNA-RNA interactions that, in turn, regulate gene expression at the posttranscriptional level (25, 37). Hfq serves as a modulator of gene expression by acting as a scaffold for interactions between small regulatory RNAs (sRNAs) and their target mRNA molecules. These sRNA-mRNA interactions are often formed through short stretches of imperfect base pairing, and therefore, Hfq is required to bridge this limited complementarity between sRNA and mRNA. In many instances, sRNA-mRNA interactions facilitated by Hfq inhibit gene expression, usually by occluding the ribosome-binding site (RBS) and/or decreasing the stability of the mRNA, leading to degradation of the transcript (1). However, Hfq and sRNAs can also activate gene expression by alleviating secondary structure in the RBS region of the mRNA and allowing translation to proceed or by binding to the 3′ end of the mRNA, which stabilizes the transcript (13). Additionally, even though Hfq levels change slightly during the transition from exponential to stationary phase, there is generally an abundance of Hfq in the cell, and thus, the growth phase-dependent and stimulus-driven gene expression mediated by Hfq can usually be attributed to differential expression of sRNAs involved in these regulatory events, rather than changes in Hfq levels (52). Hfq is also required for the normal expression of some target genes in the absence of an sRNA by binding to mRNAs and affecting mRNA stability (10, 53); however, this mechanism of Hfq-mediated gene regulation has not been widely described.
An isogenic hfq mutant derived from Brucella abortus 2308 displays a pleiotropic phenotype compared to the parental strain. The B. abortus hfq mutant exhibits increased sensitivity to oxidative stress, decreased survival at low pH, and increased sensitivity to nutrient deprivation (33). While the full repertoire of Hfq-dependent genes responsible for these deficiencies has not been elucidated, the proper expression of some Brucella genes has been linked to Hfq, including those encoding the periplasmic superoxide dismutase SodC (14) and the acid resistance protein HdeA (51). Aberrant production of these proteins may contribute to the inability of a B. abortus hfq mutant to adapt to oxidative and pH stresses, respectively, but it is likely that other genes involved in the adaptive response to stress conditions also require Hfq for their proper expression.
Hfq has been linked to the virulence of several pathogenic bacteria, including Pseudomonas aeruginosa (44), Vibrio cholerae (7), Salmonella enterica serovar Typhimurium (43), Francisella tularensis (23), Neisseria meningitidis (9), and Escherichia coli (42). Likewise, an isogenic hfq mutant derived from B. abortus 2308 exhibits severe attenuation in experimentally infected mice (33), and a B. melitensis hfq mutant derived from strain 16M is attenuated in goats (35) and nonhuman primates (R. Borschel and M. J. Nikolich, personal communication). The present study was undertaken to identify the molecular mechanisms underlying the link(s) between Hfq and wild-type gene expression in B. abortus 2308 and to determine how Hfq is linked to virulence in Brucella.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
B. abortus 2308 and derivative strains were routinely grown on Schaedler agar (BD, Franklin Lakes, NJ) containing 5% defibrinated bovine blood (Quad Five, Ryegate, MT) (SBA) or in brucella broth (BD). In some cases, Brucella strains were grown on tryptic soy agar (BD) containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal; 100 μg/ml). In order to induce maximal expression of the virB operon, Brucella strains were grown in a low-pH (pH 4.5) defined medium (21). E. coli strains were grown routinely on tryptic soy agar (BD) or in Luria-Bertani broth. When appropriate, growth media were supplemented with ampicillin (100 μg/ml), kanamycin (45 μl/ml), or chloramphenicol (30 μl/ml; E. coli strains only).
Construction of virB1- and babR-lacZ fusions.
All of the transcriptional and translational lacZ fusions used in these studies were plasmid borne, and the plasmids used (pMR10 [13] and pMR15 [3]) contain the same vector backbone (e.g., pGL10, an RK2 derivative). These plasmids are maintained at 2 to 4 copies per genome in Brucella (34). Additionally, the nucleotide sequences of the gene fusions were verified by DNA sequence analyses.
The promoter regions of the Brucella virB and babR genes were fused to a lacZ reporter as transcriptional fusions. For the virB promoter fusion construct (pC3003), approximately 450 bp of the virB1 upstream region was amplified by PCR using primers virB-Tsc-For and virB-Tsc-Rev, B. abortus 2308 genomic DNA as a template, and Taq polymerase. For the babR promoter fusion (pC3006), approximately 180 bp of the babR upstream region was amplified by PCR using primers babR-Tsc-For and babR-Tsc-Rev (Table 1) and B. abortus 2308 genomic DNA as a template. The amplified DNA fragments were sequentially digested with BamHI and HindIII and subsequently ligated into BamHI/HindIII-digested pMR15, which contains a promoterless lacZ gene. pC3003 and pC3006 were introduced into B. abortus 2308 and derivatives of this strain by electroporation.
Table 1.
Oligonucleotide primers used in this study
| Primer | Sequence (5′→3′)a |
|---|---|
| virB-Tsc-For | GCAAGCTTATGACAGGCATATTTCAACGCGAC |
| virB-Tsc-Rev | GCGGATCCCGATTATTATGATAGCCTTAATTATAG |
| virB-Tsl-For | GCGGATCCATGACAGGCATATTTCAACGCGAC |
| virB-Tsl-Rev | GCAAGCTTCACCATAGGATCGTCTCCTTCTC |
| babR-Tsc-For | GCAAGCTTATTATCTAATTTAGAATGAAG |
| babR-Tsc-Rev | GCGGATCCATAAATATGAATTCTTGG |
| babR-Tsl-For | GCGGATCCCTAATTTAGAATGAAGTTATATTC |
| babR-Tsl-Rev | GCAAGCTTTTTCATTGCAGACATCCCAG |
| babR-Trunc-Up-For | GCGGATCCAAAAGACCACCTTGTTCCG |
| babR-Trunc-Up-Rev | CTTTCATCATAATTAGCGATTT |
| babR-Trunc-Down-For | TGAAAGATATGCTAGGAATGTTT |
| babR-Trunc-Down-Rev | GCCTGCAGACAAAGCTCAAGAGGCCGA |
| hfq-For | CATCAATGGTGATGACTGGCCAAA |
| hfq-Rev | CGAGCGTGGCGAAAAGCATA |
| Inverse-hfq-For | GCCTGACGCTTTCCGATATTG |
| Inverse-hfq-Rev | AGCCATTGTTTTTATTCCTT |
| hfq-comp-For | AAAGCATAAGGAAGTCGTGTCG |
| hfq-comp-Rev | CGCTTTTTTATCCTTGGGTT |
| 16S-RT-For | TCTCACGACACGAGCTGACG |
| 16S-RT-Rev | CGCAGAACCTTACCAGCCCT |
| virB1-RT-For | TGGCAGCACCTACGCACAAG |
| virB1-RT-Rev | GCCTTGTTCCTGACCGGGCA |
| babR-RT-For | AAGAATTATGCGCATGACCT |
| babR-RT-Rev | GTTCCCACCCATCTGGAAAT |
| babR-seq-For | TTATGTCCCTGGGGCATCGAAAGAC |
| babR-seq-Rev | AATAGACATGACATAGGATACATAC |
| babR-PE | CGTTTCAAAATAAATATGAATTCTTGGAAT |
| ΔbabR-Up-For | GCGGATCCGATATCCGCCTCTTTACGGA |
| ΔbabR-Up-Rev | TTTCATTGCAGACATCCCAGTAGCGTTTC |
| ΔbabR-Down-For | CTCTGACCTTTTCCAGTGTCCCTCGGCGG |
| ΔbabR-Down-Rev | GCCTGCAGATCGGCAGATAAGAAGAGGA |
| babR-comp-For | AACTGCAGGAGGAAACGGCAAAACAGCCCT |
| babR-comp-Rev | AATGTGTAGCCTTTTTCAGGGAAAG |
| babR-UTR-T7-For | TAATACGACTCACTATAGGGTAATCTTCGTAGAAGAAATATGAA |
| babR-UTR-T7-Rev | TGTTTCCCATTTCATTGCAGAC |
| babR-20-UTR-For | TAATACGACTCACTATAGGGTGAAAGATATGCTAGGAATGTTTAG |
| rHfq-For | GCGGTCTCCGCGCATGGCTGAACGATCGCAAATCT |
| rHfq-Rev | GCGGTCTCCTATCAGGCTTCCTCGCCTTCAAACAT |
| rBabR-For | CATGAAATGGGAAACATTTTATG |
| rBabR-Rev | AGGAGACCAAATGTACGGGCC |
| PvirB-For | CTGACTGGGCGTCATTCACAC |
| PvirB-Rev | CGTCTCCTTCTCAGAGAATGGACG |
| babR-RACE-Rev | CAGGTCATGCGCATAATTCTTTACT |
Underlined sections indicate restriction endonuclease recognition sequences.
For construction of virB-lacZ and babR-lacZ translational fusions, the upstream regions of the corresponding Brucella genes to the second codon of the coding region were cloned in frame with lacZ in pLKC-481 (48) using the HindIII and BamHI restriction sites. The virB fragment was amplified by PCR using B. abortus 2308 genomic DNA as a template and primers virB-Tsl-For and virB-Tsl-Rev (Table 1), and the babR fragment was amplified with primers babR-Tsl-For and babR-Tsl-Rev (Table 1). Following successful cloning of the fragments into pLCK-481, the intact gene fusions were liberated from the plasmid by digestion with PstI and SmaI. These fragments were then cloned into the Brucella-compatible vector pMR10 (14) for introduction into B. abortus 2308 and its derivatives.
In order to truncate the 5′ untranslated region (UTR) of the mRNA of the babR-lacZ translational fusion, an approximately 500-bp DNA fragment of the babR upstream region to the transcriptional start site of babR was amplified by PCR using primers babR-Trunc-Up-For and babR-Trunc-Up-Rev (Table 1), genomic DNA from B. abortus 2308 as a template, and Pfx polymerase (Invitrogen). Using similar PCR conditions and primers babR-Trunc-Down-For and babR-Trunc-Down-Rev (Table 1), an ∼500-bp DNA fragment beginning with the +21 position of the babR 5′ UTR was amplified. The upstream fragment was digested with BamHI, while the downstream fragment was digested with PstI, and both fragments were treated with polynucleotide kinase. Both of the DNA fragments were included in a single ligation mixture with BamHI/PstI-digested pUC19 (50). The resulting plasmid (pC3012) was used as a template for PCR with primers babR-Tsl-For and babR-Tsl-Rev, and a babR-lacZ translational fusion harboring a truncated 5′ UTR was constructed exactly as the babR-lacZ translational fusion described in the previous paragraph to generate pC3014.
B. abortus strains harboring the lacZ fusion constructs were grown in brucella broth or low-pH (pH 4.5) defined medium, and β-galactosidase assays were performed as described previously (24).
Construction and genetic complementation of a B. abortus hfq mutant.
The hfq gene (BAB1_1134) in B. abortus 2308 was mutated using a nonpolar, unmarked gene excision approach. The hfq gene, along with approximately 1 kb of each flanking sequence, was amplified by PCR using primers hfq-For and hfq-Rev (Table 1), B. abortus 2308 genomic DNA as a template, and Taq polymerase, and the amplified DNA fragment was ligated into pGEM-T Easy (Promega). The resulting plasmid (pJMG001) was used as a template for inverse PCR with primers Inverse-hfq-For and Inverse-hfq-Rev. Inverse PCR was performed using Herculase Hotstart DNA polymerase (Stratagene). A DNA fragment of approximately 5 kb was generated from inverse PCR, and following gel purification, the ends of the DNA fragment were phosphorylated using polynucleotide kinase. The free ends of the DNA fragment were then ligated together to yield plasmid pJMG002, which contains an in-frame deletion hfq allele and approximately 1 kb of flanking DNA sequence on each side. The in-frame nature of the hfq mutation was confirmed by DNA sequencing. The mutated hfq gene and the flanking DNA were liberated by treatment with NotI, and following gel purification, the liberated DNA fragment was treated with the Klenow fragment of DNA polymerase. This fragment of DNA was then ligated into SmaI-digested pEX100T (38), which contains an ampicillin resistance marker and a sacB gene, and the resulting plasmid was named pJMG003. pJMG003 was introduced into B. abortus 2308 by electroporation, and merodiploid clones were obtained by selection on SBA plus ampicillin. A single ampicillin-resistant clone was grown overnight in brucella broth and then plated onto SBA containing 10% sucrose. Genomic DNA from sucrose-resistant, ampicillin-sensitive colonies was isolated and screened by PCR for loss of the hfq gene. Following confirmation of its genotype by Southern blotting, the isogenic hfq mutant derived from B. abortus 2308 was named B8.
Genetic complementation of the hfq mutation was achieved by in-trans complementation using pBBR1MCS-4 (20). An approximately 800-bp DNA fragment containing the hfq gene was amplified by PCR using primers hfq-comp-For and hfq-comp-Rev (Table 1), genomic DNA from B. abortus 2308 as a template, and Pfx polymerase (Invitrogen). The resulting DNA fragment was digested with StyI and then treated with polynucleotide kinase, and the digested and treated fragment was ligated into XbaI/HincII-digested pBBR1MCS-4 to create pJMG004. pJMG004 was introduced into B. abortus hfq mutant strain B8 by electroporation.
Construction and genetic complementation of a B. abortus babR mutant.
The babR gene (BAB1_0190) of B. abortus 2308 was mutated using a nonpolar, unmarked gene excision strategy. An approximately 1-kb fragment of the babR upstream region to the second codon of the babR coding region was amplified by PCR using primers ΔbabR-Up-For and ΔbabR-Up-Rev (Table 1), genomic DNA from B. abortus 2308 as a template, and Pfx polymerase. Similarly, a fragment containing the last two codons of the babR coding region to approximately 1 kb downstream of the babR open reading frame was amplified with primers ΔbabR-Down-For and ΔbabR-Down-Rev (Table 1). The upstream fragment was digested with BamHI, while the downstream fragment was digested with PstI, and both fragments were treated with polynucleotide kinase in the presence of ligase buffer. Both of the DNA fragments were included in a single ligation mixture with BamHI/PstI-digested pUC19 (54). The nucleotide sequence of the ligated Brucella DNA fragments in the resulting plasmid (pC3009) was determined to confirm the desired mutant genotype. The mutated babR gene with 1-kb flanking regions was then liberated from the vector by digestion with BamHI and PstI, and the liberated fragment was ligated into pNPTS138 (46), which contains a kanamycin resistance marker and the sacB gene for counterselection with sucrose. The resulting plasmid (pC3010) was introduced into B. abortus 2308 and the hfq mutant B8 by electroporation, and merodiploid clones were obtained by selection on SBA plus kanamycin. A single kanamycin-resistant clone was grown overnight in brucella broth and then plated onto SBA containing 10% sucrose. Genomic DNA was isolated from sucrose-resistant, kanamycin-sensitive colonies and screened by PCR for loss of the babR gene. The isogenic babR mutant derived from B. abortus 2308 was named CC058.
Genetic complementation of the babR mutation was achieved by in-trans complementation using pBBR1MCS-4 (20). A DNA fragment containing the babR gene and approximately 300 bp of upstream and 100 bp of downstream DNA was amplified by PCR using primers babR-comp-For and babR-comp-Rev (Table 1) and genomic DNA from B. abortus 2308 as a template. The resulting DNA fragment was digested with PstI and then treated with polynucleotide kinase, and the digested and treated fragment was ligated into PstI/HincII-digested pBBR1MCS-4 to create pC3011. pC3011 was introduced into the B. abortus babR mutant strain by electroporation.
Expression and purification of recombinant proteins.
The Strep-tagII system (IBA, Göttingen, Germany) was used to produce recombinant B. abortus proteins in E. coli strain BL21. For recombinant BabR (rBabR), the coding region of the babR gene (BAB1_0190) was amplified using primers rBabR-For and rBabR-Rev (Table 1), B. abortus 2308 chromosomal DNA as a template, and Taq polymerase. The amplified DNA fragment was treated with polynucleotide kinase and blunt end ligated into PshAI-digested, phosphatase-treated pASK-IBA6, which encodes an amino-terminal Strep-tagII on the protein of interest. The plasmid encoding recombinant Hfq (rHfq) was constructed in a similar fashion; however, the hfq coding region (BAB1_1134) was amplified with primers rHfq-For and rHfq-Rev (Table 1). The amplified DNA fragment was digested with BsaI and ligated into BsaI-digested pASK-IBA6. The resulting plasmids, prBabR and prHfq, were transformed into E. coli strain BL21, and the strain harboring the recombinant protein expression plasmid was grown to an optical density at 600 nm of approximately 0.6 before protein expression was induced by the addition of anhydrotetracycline (200 μg/ml [final concentration]). Following 2 h of incubation at 37°C, the cells were collected by centrifugation (4,200 × g for 10 min at 4°C) and lysed by treatment with CelLyticB (Sigma, St. Louis, MO) in the presence of the protease inhibitor phenylmethylsulfonyl fluoride. The supernatant from the suspension of lysed cells was collected by centrifugation (14,000 × g for 10 min at 4°C), and the collected supernatant was passed through an affinity column packed with Strep-Tactin Sepharose. The column was washed extensively with buffer W (100 mM Tris-HCl, 300 mM NaCl, 1 mM EDTA, pH 8.0), and recombinant protein was eluted with 2.5 mM desthiobiotin in buffer W. The degrees of purity of recombinant BabR and Hfq were high, as judged by SDS-PAGE.
Real-time reverse transcription (RT)-PCR.
Total RNA was isolated from cultures of B. abortus 2308, the hfq mutant B8, and the complemented mutant grown to the appropriate phase of growth in brucella broth. An equal volume of 1:1 ethanol-acetone was added to the cultures, and the mixture was stored at −80°C. Cultures were thawed, and the cells were collected by centrifugation at 18,000 × g for 5 min at room temperature. RNA was extracted from the cells using the TRIzol reagent (Invitrogen, Carlsbad, CA), and following ethanol precipitation of the RNA, genomic DNA was removed by treatment with RQ1 DNase I (Promega, Madison, WI) following the manufacturer's instructions. cDNA was generated from the final RNA preparation using the SuperScript III cDNA Synthesis System (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol, and this cDNA was used for real-time PCR employing SYBR green PCR SuperMix (Roche, Mannheim, Germany). For these experiments, primers for 16S RNA were used as a control, while gene-specific primers were used for evaluating relative mRNA levels (Table 1). Parameters for PCR included a single denaturing step of 5 min at 95°C, followed by 40 cycles of amplification consisting of denaturation for 15 s at 95°C, annealing for 15 s at 50°C, and extension for 15 s at 72°C. Fluorescence from SYBR green incorporation into double-stranded DNA was measured with an iCycler machine (Bio-Rad), and the relative abundance of mRNA was determined using the Pfaffl equation (30).
Immunoblot analysis.
Whole-protein lysates were collected from Brucella cultures harvested during exponential growth or after entry into stationary phase. Cultures were collected by centrifugation at 15,000 × g for 10 min at 4°C, and the cell pellet was suspended in 1 ml of protein sample buffer (0.3% SDS, 200 mM dithiothreitol, 22 mM Tris base, 28 mM Tris-HCl). The cells were then boiled for 1 h and subjected to 10 cycles (setting 4, 20 s per cycle) of disintegration in Lysing Matrix B (MP Biomedicals, Solon, OH) using a Bio 101 FastPrep FP120 cell disintegrator (Thermo Scientific Savant; Thermo Fisher Scientific Inc., Waltham, MA). Protein concentrations were determined using a Bradford assay with bovine serum albumin (BSA) standards (Bio-Rad, Hercules, CA). Ten micrograms of total protein per strain was separated by 15% SDS-PAGE, and MagicMark and SeeBluePlus2 standards (Invitrogen, Carlsbad, CA) were included on the gel. Proteins were transferred to a nitrocellulose membrane by electroblotting at approximately 400 mA for 45 min. Membranes were blocked at room temperature in 5% skim milk in TBST (Tris-buffered saline [50 mM Tris, 150 mM NaCl, pH 7.4]–0.05% Tween 20), and primary antibodies (anti-VirB1 [1:1,000 dilution]) were incubated with the membranes at room temperature in 5% skim milk. Secondary antibodies (anti-rabbit–horseradish peroxidase [HRP]) were used at a 1:10,000 dilution. All washing steps were performed with TBST. Development of the HRP signal was performed using WestPico (Pierce [Thermo Scientific], Rockford, IL). The anti-VirB1 antiserum was kindly provided by Christian Baron, Department of Biochemistry, Université de Montréal.
Determination of the babR transcriptional start site.
Total RNA was extracted from B. abortus as described above. The oligonucleotide primer babR-PE was end labeled with [γ-32P]ATP (Perkin-Elmer, San Jose, CA) and polynucleotide kinase (Promega, Madison, WI), and the labeled primer was annealed to 50 μg of Brucella RNA. Primer extension analysis was carried out using the avian myeloblastosis virus reverse transcriptase primer extension system (Promega, Madison, WI) according to the manufacturer's instructions. DNA sequence analysis was performed using the same radiolabeled primer and the SequiTherm EXCEL II DNA sequencing kit (Epicentre, Madison, WI) following the manufacturer's protocol. Primer extension products and DNA sequencing reaction products were separated on 6% denaturing polyacrylamide gels and visualized by autoradiography.
5′ rapid amplification of cDNA ends (RACE) was carried out using the FirstChoice RLM-RACE kit (Ambion, Austin, TX) according to the manufacturer's instructions. B. abortus 2308 was grown in brucella broth to late exponential phase, and RNA was isolated and treated with DNase I as described above. The babR gene-specific primer babR-RACE-Rev was used in the PCR steps of the 5′ RACE protocol where Taq polymerase was also employed. The babR 5′ RACE product (see Fig. 3B) was gel purified and cloned into pGEM-T Easy. Plasmid DNA was purified from potential clones, and DNA sequencing analysis was performed to identify the babR transcriptional start site.
Fig 3.

Mapping and characterization of the babR promoter region. (A) Schematic of the chromosomal region containing babR. The plus strand sequence of DNA corresponding to the region between the hyp gene and babR is shown below the schematic, and the identified transcriptional start site (+1) is labeled. This transcriptional start site is 119 nucleotides upstream of the babR start codon. The first 20 nucleotides of the 5′ UTR are boxed to indicate that this region was removed for the experiments shown in Fig. 5. The −10 and −35 elements of the babR promoter are also labeled. The genes in this region are metH (B12-dependent methionine synthase; BAB1_0188), hyp (hypothetical protein; BAB1_0189), babR (LuxR-type transcriptional regulator; BAB1_0190), and atf (aminotransferase; BAB1_0191). (B) Identification of the transcriptional start site of the babR mRNA. Primer extension (left panel) was performed using total RNA isolated from Brucella strains (wild-type 2308 and the hfq mutant) grown in brucella broth. The primer extension reaction products, along with DNA sequencing reaction products produced with the same radiolabeled primer, were separated on 6% denaturing polyacrylamide gels and visualized by autoradiography. 5′ RACE was also employed (right panel) using RNA isolated from B. abortus 2308 grown in brucella broth. Molecular weight standards and the 5′ RACE product were separated by 2.0% agarose gel electrophoresis. The indicated DNA band was extracted from the gel, cloned, and sequenced to determine the transcriptional start site. (C) Secondary structure predictions of the RNA containing the babR 5′ UTR and the first five codons of the babR coding region. The RBS of each predicted structure is underlined, while a box is placed around the start codon (AUG) of each predicted structure.
Electrophoretic mobility shift assays (EMSAs). (i) rBabR EMSAs.
All rBabR EMSA experiments were carried out in a 20-μl total reaction volume containing a binding buffer composed of 10 mM Tris-HCl (pH 7.4), 50 mM KCl, 1 mM dithiothreitol, 6% glycerol, 0.5 mM EDTA, 50 μg/ml BSA, and 50 μg/ml salmon sperm DNA. A 150-bp DNA fragment corresponding to the promoter region of the B. abortus 2308 virB operon was amplified by PCR using primers PvirB-For and PvirB-Rev (Table 1) and chromosomal DNA from B. abortus 2308 as a template. The amplified DNA fragment was purified by agarose gel electrophoresis, and the fragment was end labeled with [γ-32P]ATP (Perkin-Elmer, San Jose, CA) and polynucleotide kinase (Promega, Madison, WI). Increasing amounts of recombinant BabR were mixed with the radiolabeled PvirB DNA in binding buffer, and the reaction mixtures were incubated at room temperature for 20 min. As a control, a 50× molar concentration of nonradiolabeled PvirB DNA (specific competitor) or nonradiolabeled BAB2_0612 DNA (nonspecific competitor) was added to some reaction mixtures. The binding reaction products were subjected to electrophoresis on 6% native polyacrylamide gels in 0.5× TBE running buffer for approximately 1 h at 4°C. Following electrophoresis, gels were dried onto Whatman 3MM filter paper using a vacuum gel dryer system and visualized by autoradiography.
(ii) rHfq EMSAs.
All rHfq EMSA experiments were performed in a 20-μl total reaction volume containing binding buffer composed of 10 mM Tris-HCl (pH 7.4), 50 mM KCl, 1 mM dithiothreitol, 6% glycerol, 0.5 mM EDTA, 50 μg/ml BSA, and 50 μg/ml baker's yeast tRNA. In vitro transcription using the MAXIscript system (Ambion, Austin, TX) was employed to generate the single-stranded RNA probe for these experiments. This system relies on the T7 RNA polymerase promoter to drive the transcription of the desired ribonucleotide molecule. PCR utilizing chromosomal DNA from B. abortus 2308 as a template and primers babR-UTR-T7-For and babR-UTR-T7-Rev (Table 1) was used to amplify an approximately 150-bp DNA fragment corresponding to the 5′ UTR and first 5 codons of the babR mRNA. A DNA fragment coding for a mutated form of the babR 5′ UTR (missing the first 20 nucleotides) was also generated, and for this, a PCR was performed as described above with primers babR-20-UTR-For and babR-UTR-T7-Rev (Table 1). The single-stranded RNA probes were then generated according to the manufacturer's recommendations. Radiolabeled probe was generated using uniform incorporation with [α-32P]UTP (Perkin-Elmer, San Jose, CA). Nonradiolabeled probe was also generated in order to serve as a specific competitor in the EMSA experiments. For the binding assays, increasing amounts of rHfq were mixed with labeled babR-UTR RNA probe in binding buffer, and the reaction mixtures were incubated at 37°C for 30 min. The binding reaction products were then subjected to electrophoresis on 5% native polyacrylamide gels in 0.5× TBE running buffer for ∼2 h at room temperature on ice. Following electrophoresis, gels were dried onto Whatman 3MM filter paper using a vacuum gel dryer system and visualized by autoradiography.
RESULTS
Hfq is required for wild-type production of the VirB1 protein in B. abortus 2308.
To better define the Hfq regulon in B. abortus 2308, two-dimensional gel electrophoresis was performed on whole-protein lysates from the parental strain and an isogenic hfq mutant. One of the components of the VirB type IV secretion system, VirB1, was found to be produced at approximately 6-fold higher levels in parental strain 2308 than in the hfq mutant (data not shown). VirB1 is encoded by the first gene in the virB operon, virB1, and to date, the sole promoter described for this operon is located upstream of virB1 (5, 27). In order to gain insight into Hfq-mediated regulation of VirB1 protein levels, a pMR10-based virB-lacZ translational fusion was constructed, and β-galactosidase activity produced by this fusion was assessed in B. abortus 2308, the isogenic hfq mutant B8, and a derivative of B8 carrying a plasmid-borne copy of hfq.
β-Galactosidase activity from the virB-lacZ translational fusion was significantly reduced in the hfq mutant compared to that in strain 2308, and genetic complementation of B. abortus B8 with plasmid-borne hfq restored the β-galactosidase activity of this strain to wild-type levels (Fig. 1A). These data indicate that Hfq has a positive influence on VirB1 production in the parental 2308 strain, and these data also confirm the results of the original two-dimensional gel electrophoresis analyses that first identified the relationship between Hfq and VirB1. Immunoblot analysis of Brucella protein lysates with anti-VirB1 antisera also revealed a significant difference between the VirB1 protein levels in B. abortus 2308 and the hfq mutant (Fig. 1B). VirB1 protein levels were greatly reduced in the hfq mutant compared to those in parental strain 2308, and this reduction was observed during exponential growth of the bacteria; however, no significant difference in VirB1 levels was seen during stationary phase. Importantly, the levels of VirB1 protein in the hfq mutant were restored to wild-type levels by the expression of hfq in trans in the mutant. Altogether, these data demonstrate that Hfq is required for wild-type production of the VirB1 protein in B. abortus 2308.
Fig 1.

Hfq is required for maximal expression of the VirB system. Plasmid constructs pC3005 (A) and pC3003 (C) (Table 2) were transformed into B. abortus 2308, the hfq isogenic mutant (hfq), and the hfq mutant strain expressing hfq from a plasmid (hfq/Com), and following cultivation in low-pH (pH 4.5) defined medium, β-galactosidase activity was assessed. The asterisk denotes a statistically significant difference between the β-galactosidase activities of parental strain 2308 and the hfq mutant strain (P < 0.01). (B) Immunoblot analysis of VirB1 protein levels. Brucella strains were cultivated in low-pH minimal E medium. GroEL levels are shown as a protein loading control, and molecular weight markers are shown to the left. (D) Real-time RT-PCR analysis of virB1 transcript levels. Brucella strains were grown in low-pH (pH 4.5) defined medium, and total RNA was isolated. Oligonucleotide primers specific for virB1 or 16S rRNA were used to amplify the target genes by PCR, and quantification of the amplified DNA fragments was performed using SYBR green incorporation. The values represent the relative abundances of specific mRNAs, with the level of mRNA from parental strain 2308 designated 1.000.
Table 2.
Plasmids used in this study
| Plasmid | Description | Reference |
|---|---|---|
| pUC19 | Cloning vector; Ampr | 54 |
| pGEM-T Easy | Cloning vector; Ampr | Promega |
| pEX100T | Cloning vector; contains sacB gene; Ampr | 38 |
| pBBR1MCS-4 | Broad-host-range cloning vector; Ampr | 20 |
| pNPTS138 | Cloning vector; contains sacB gene; Kanr | 46 |
| pLKC-481 | Cloning vector containing promoterless lacZ gene; Ampr Kanr | 48 |
| pMR10 | Broad-host-range cloning vector; Kanr | 14 |
| pMR15 | Broad-host-range vector containing promoterless lacZ gene; Kanr | 3 |
| pC3003 | virB-lacZ transcriptional fusion in pMR15 | This study |
| pC3004 | virB-lacZ translational fusion in pLKC-481 | This study |
| pC3005 | virB-lacZ translational fusion fragment from pC3004 in pMR10 | This study |
| pC3006 | babR-lacZ transcriptional fusion in pMR15 | This study |
| pC3007 | babR-lacZ translational fusion in pLKC-481 | This study |
| pC3008 | babR-lacZ translational fusion fragment from pC3007 in pMR10 | This study |
| pC3009 | In-frame deletion of babR plus 1 kb of each flanking region in pUC19 | This study |
| pC3010 | Mutated babR locus plus flanking regions from pC3009 in pNPTS138 | This study |
| pC3011 | babR locus including entire promoter region in pBBR1MCS-4 | This study |
| pC3012 | babR locus with truncated 5′ UTR in pUC19 | This study |
| pC3013 | babR-lacZ translational fusion with truncated 5′ UTR in pLKC-481 | This study |
| pC3014 | Truncated babR-lacZ translational fusion fragment from pC3013 in pMR10 | This study |
| pJMG001 | Full-length hfq gene plus 1 kb of each flanking region in pGEM-T Easy | This study |
| pJMG002 | Religation of pJMG001 following inverse PCR to delete hfq coding region | This study |
| pJMG003 | Mutated hfq locus plus flanking regions from pJMG002 in pEX100T | This study |
| pJMG004 | hfq locus including entire promoter region in pBBR1MCS-4 | This study |
It is possible that Hfq can affect the levels of a specific protein by directly interacting with the mRNA encoding that protein and altering translation and/or transcript stability; however, the possibility also exists that Hfq indirectly alters target protein levels by altering the abundance of a transcriptional regulator that will result in decreased transcription of the target gene. To determine if Hfq affects the transcription of the virB promoter, a virB-lacZ transcriptional fusion was constructed on the same plasmid backbone as the virB-lacZ translational fusion (e.g., pMR10), and for this construct, the region from −450 to +1 relative to the virB transcriptional start site was cloned in front of lacZ. The β-galactosidase activity of the strains carrying this fusion was reduced in the B. abortus hfq mutant compared to that in parental strain 2308, and promoter activity was restored to wild-type levels by in-trans complementation of the mutant with a plasmid-borne copy of hfq (Fig. 1C). Real-time RT-PCR was employed as an independent means of assessing the relationship between Hfq and virB transcription, and virB mRNA was found to be significantly less abundant in the hfq mutant than in strain 2308 (Fig. 1D). Genetic complementation of the hfq mutant restored virB mRNA abundance in this strain to a level above that observed in strain 2308. These data indicate that in addition to affecting the levels of VirB1 protein, Hfq is also involved in the regulation of virB1 transcript levels. Thus, it was concluded that Hfq likely regulates virB expression through an indirect mechanism, and this led to the hypothesis that Hfq is influencing the levels of a regulator of virB transcription.
Production of the virB regulator BabR is altered by Hfq.
Several transcriptional regulators that control the expression of the virB operon have been identified (16, 31), and the hypothesis that one of these regulators is the link between Hfq and virB expression in B. abortus 2308 was tested. In order to test this hypothesis, the promoter regions of the arsR6 (BAB1_1605), gntR4 (BAB1_1894), araC2 (BAB2_0136), vjbR (BAB2_0118), and babR/blxR (BAB1_0190) genes were cloned in frame with lacZ using the same plasmid-based strategy that was employed to construct the virB-lacZ translational fusion. β-Galactosidase assays with parental B. abortus 2308, the isogenic hfq mutant strain, and the in-trans-complemented hfq mutant strain harboring these plasmids revealed no significant differences in β-galactosidase activity between the strains for the lacZ fusions to arsR6, gntR4, araC2, and vjbR (data not shown). However, the β-galactosidase activity of the babR-lacZ translational fusion was significantly reduced in the hfq mutant strain compared to that in parental strain 2308, and genetic complementation of the hfq mutant elevated β-galactosidase activity production by this strain to levels above those observed in strain 2308 (Fig. 2A). Moreover, when the Brucella strains harboring the babR-lacZ translational fusion were grown on agar containing the β-galactosidase indicator X-Gal, strain 2308 and the complemented hfq mutant formed blue colonies but the hfq mutant formed white colonies, indicating that babR expression is severely inhibited in the latter strain (Fig. 2B).
Fig 2.
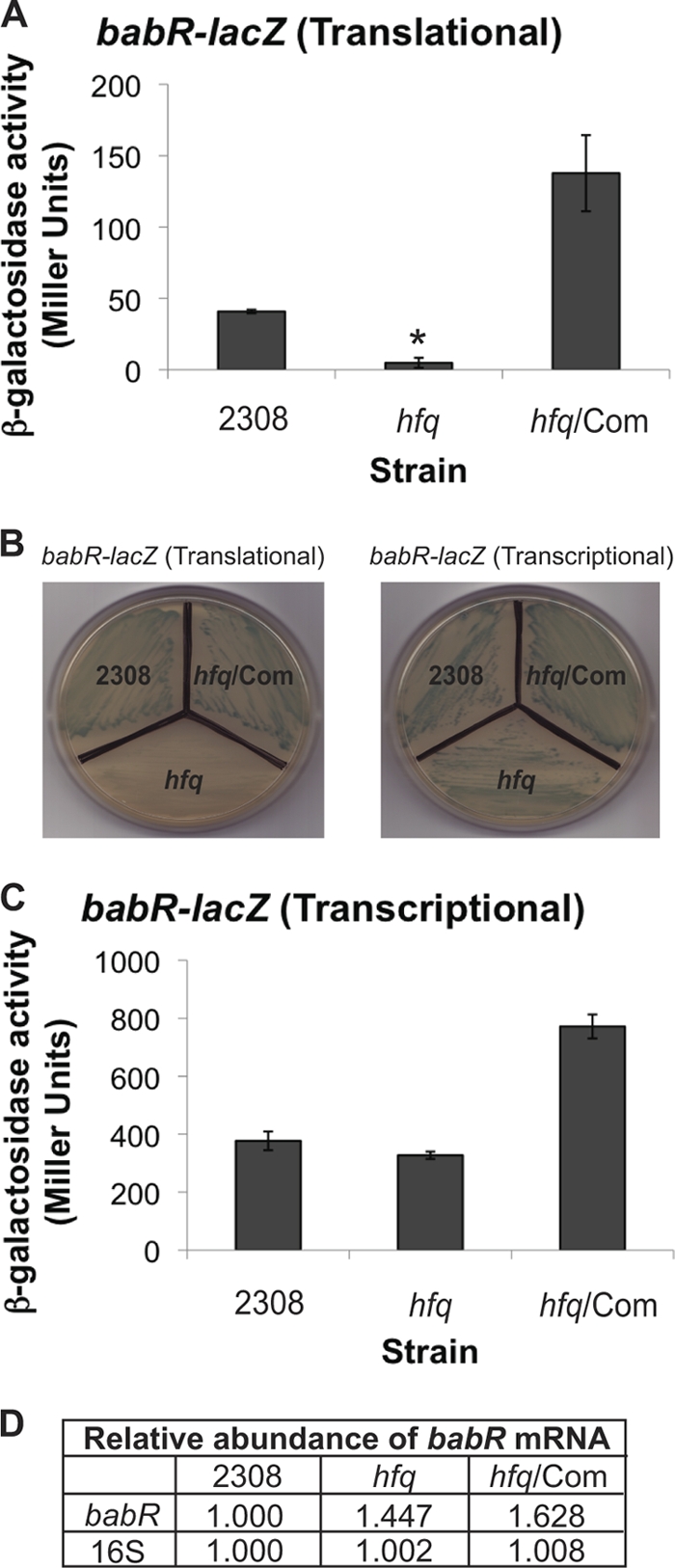
Identification of an Hfq-regulated transcription regulator, BabR. Plasmid constructs pC3008 (A) and pC3006 (C) (Table 2) were transformed into B. abortus 2308, the hfq isogenic mutant (hfq), and the hfq mutant strain expressing hfq from a plasmid (hfq/Com), and following cultivation in brucella broth, β-galactosidase activity was assessed. The asterisk denotes statistically significant differences between the β-galactosidase activities of parental strain 2308 and the hfq mutant strain (P < 0.01). (B) Pictures of Brucella stains carrying either pC3008 (left) or pC3006 (right) grown on tryptic soy agar containing X-Gal. (D) Real-time RT-PCR analysis of babR transcript levels. Brucella strains were grown in brucella broth, and total RNA was isolated. Oligonucleotide primers specific for babR or 16S rRNA were used to amplify the target genes by PCR, and quantification of the amplified DNA fragments was performed using SYBR green incorporation. The values represent the relative abundances of specific mRNAs, with the level of mRNA in parental strain 2308 designated 1.000.
To test if Hfq is modulating the amount BabR protein at the level of transcription or translation, the transcription of babR in the B. abortus hfq mutant was assessed (Fig. 2C and D). Similar to the virB-lacZ transcriptional fusion, a plasmid-borne babR-lacZ transcriptional fusion containing the region from −72 to +93 relative to the babR transcriptional start site was constructed in pMR15, and the β-galactosidase activity of Brucella strains carrying this fusion was measured. There was no significant difference between the β-galactosidase activities produced by parental strain 2308 and the hfq mutant strain (Fig. 2C). Unlike strains harboring the babR-lacZ translational fusion, strains harboring the babR-lacZ transcriptional fusion did not exhibit any color differential when grown on agar containing X-Gal (Fig. 2B). As an independent approach to assessing the link between Hfq and babR transcription, real-time RT-PCR was used to assess the abundances of babR mRNA in the different B. abortus strains, and it was determined that babR mRNA levels are not significantly decreased in the hfq mutant compared to those in wild-type strain 2308 (Fig. 2D). Taken together, these data suggest that the positive link between Hfq and BabR translation is direct.
Definition of the babR promoter and the 5′ UTR of the babR mRNA.
In order to regulate gene expression at the posttranscriptional level, Hfq must bind to mRNAs, and, in some instances, this binding occurs at the 5′ UTR of the target mRNA (45). Therefore, it was important to define the promoter region of babR in order to further characterize the Hfq-mediated regulation of babR expression (Fig. 3). The babR gene (BAB1_0190) in B. abortus 2308 is found on chromosome 1. The genes in this locus include metH (B12-dependent methionine synthase; BAB1_0188), hyp (hypothetical protein; BAB1_0189), babR (LuxR-type transcriptional regulator; BAB1_0190), and atf (aminotransferase; BAB1_0191) (Fig. 3A). The transcriptional start site for the babR mRNA was determined by primer extension analysis and confirmed by 5′ RACE analysis, and these techniques revealed the presence of a 119-nucleotide 5′ UTR upstream of the babR coding region in the babR transcript (Fig. 3B). The predicted RNA secondary structure of the babR 5′ UTR and the first five codons of the coding sequence was determined using mfold (http://mfold.rna.albany.edu/?q=mfold), and interestingly, this program predicted only two very similar secondary structures (Fig. 3C). The calculated free-energy (ΔG) values for these structures were −22.44 and −24.20 kcal/mol, indicating that the formation of these predicted structures is highly favorable.
Truncation of the babR 5′ UTR alleviates the requirement of Hfq for babR expression.
The direct binding of Hfq to the babR 5′ UTR is evidence for the involvement of Hfq in the modulation of babR expression at the posttranscriptional level. In order define the region of the babR 5′ UTR responsible for the effect of Hfq on expression, the first 20 nucleotides of the babR 5′ UTR were deleted, and a babR-lacZ translational fusion with this truncated 5′ UTR was constructed (Fig. 4A). The truncated fusion, as well as the full-length UTR fusion, was transformed into parental B. abortus strain 2308 and isogenic hfq mutant strain B8, and β-galactosidase activity was measured (Fig. 4B). Similar to previous data (Fig. 2A), the β-galactosidase activity produced by the strains harboring the full-length babR-lacZ translational fusion was significantly decreased in the hfq mutant compared to that in strain 2308; however, there was no significant difference between the strains carrying the truncated babR 5′ UTR fusion. As shown in Fig. 2B, parental strain 2308 produces blue colonies when grown on agar containing the β-galactosidase indicator X-Gal, and the hfq mutant produced white colonies when grown on the same medium. On the other hand, both parental strain 2308 and the hfq mutant strain harboring the truncated babR 5′ UTR-lacZ fusion produced blue colonies (Fig. 4B). Altogether, these data show that the first 20 nucleotides of the babR 5′ UTR are required for the effect of Hfq on the babR-lacZ translational fusion.
Fig 4.

Truncation of the babR 5′ UTR alleviates the requirement for Hfq. (A) Schematic of the babR-lacZ translational fusions used in this experiment. The full-length fusion construct pC3008 (also used in Fig. 2) contains the babR promoter region and the entire babR 5′ UTR cloned in frame with lacZ. The truncated fusion construct pC3014 (Table 2) contains the babR promoter region and a truncated babR 5′ UTR cloned in frame with lacZ. (B) β-Galactosidase activities of strains carrying the full-length and truncated babR 5′ UTR-lacZ translational fusions. B. abortus 2308 and the isogenic hfq mutant harboring pC3008 or pC3014 were growth in brucella broth, and β-galactosidase activity was measured. The asterisk indicates a statistically significant difference between the β-galactosidase activities of parental strain 2308 and the hfq mutant strain (P < 0.01). To the right of the graph is an image of Brucella stains carrying pC3008 or pC3014 grown on tryptic soy agar containing X-Gal. ND, no statistically significant difference.
In vitro, bacterial Hfq proteins will bind to target mRNAs in the absence of a cognate sRNA, and in this study, Brucella Hfq was tested for the ability to bind the 5′ UTR of the babR mRNA. A recombinant version of B. abortus 2308 Hfq was expressed and purified, and EMSAs were used to test for interactions of Hfq with the in vitro-transcribed babR 5′ UTR and a mutated form of the babR 5′ UTR that is missing the first 20 nucleotides (Fig. 5). Hfq bound to the full-length babR 5′ UTR in a concentration-dependent manner, and while Hfq also bound the mutated babR 5′ UTR, higher concentrations of Hfq were required for binding of the mutated 5′ UTR than for that of the full-length babR 5′ UTR These data indicate that Hfq binds directly to the babR mRNA and furthermore that the first 20 nucleotides of the babR 5′ UTR are required for maximal binding by Hfq. Therefore, these interactions are likely important for the requirement of Hfq in mediating wild-type BabR protein levels. From the babR-lacZ fusion data (Fig. 4) and the babR 5′ UTR/Hfq EMSA data (Fig. 5), it is concluded that Hfq functions at the posttranscriptional level to modulate the expression of babR in B. abortus by binding directly to the 5′ UTR of the babR mRNA, and moreover, the first 20 nucleotides of the babR 5′ UTR are required for this regulatory event.
Fig 5.

Hfq binds the 5′ UTR of the babR transcript. The full-length 5′ UTR of the babR mRNA and a mutated version of the babR 5′ UTR (−20 nucleotides from the 5′ end) were transcribed in vitro and radiolabeled by [α-32P]UTP incorporation. An EMSA was performed using the labeled in vitro-transcribed 5′ UTRs and purified recombinant B. abortus Hfq. Increasing concentrations of rHfq were incubated with the labeled RNA probe, and the binding reaction products were resolved in 5% native polyacrylamide gels and visualized by autoradiography.
BabR is a repressor of virB expression and binds to the virB promoter.
It was originally reported that BabR (also called BlxR) has a positive influence on the virB promoter (32), but more recently, another study concluded that BabR is a repressor of virB transcription (49). Both studies were performed using B. melitensis 16M, and the differences in the results obtained could be attributed to differences in experimental and growth conditions. For this study, it was important to clearly define the effect of BabR on virB transcription in B. abortus 2308 in order to establish whether or not it plays a role in the genetic circuitry between Hfq and VirB1 protein levels. Therefore, an isogenic babR mutant of B. abortus 2308 was constructed and the virB1 transcription levels in this strain were assessed (Fig. 6).
Fig 6.

BabR is a repressor of virB expression. (A) The virB-lacZ transcriptional fusion construct pC3003 (Table 2) was transformed into B. abortus 2308, the babR isogenic mutant (babR), and the babR mutant strain expressing babR from a plasmid (babR/Com), and following cultivation in low-pH (pH 4.5) defined medium, β-galactosidase activity was assessed. The asterisk denotes a statistically significant difference between the β-galactosidase activities of parental strain 2308 and the babR mutant strain (P < 0.01). (B) Real-time RT-PCR analysis of virB1 transcript levels. B. abortus 2308 strains were grown in low-pH (pH 4.5) defined medium, and total RNA was isolated. Oligonucleotide primers specific for virB1 or 16S rRNA were used to amplify the target genes by PCR, and quantification of the amplified DNA fragments was performed using SYBR green incorporation. The values represent the relative abundances of specific mRNAs, with the level of mRNA in parental strain 2308 designated 1.000.
The same virB-lacZ transcriptional fusion used previously in Fig. 1 was transformed into the babR mutant strain and the mutant strain expressing babR from a plasmid, and β-galactosidase activity was measured (Fig. 6A). The activity of this fusion was significantly increased in the babR mutant compared to that in wild-type strain 2308, and β-galactosidase was returned to wild-type levels by complementation of babR in the mutant. Real-time RT-PCR analysis of relative virB1 mRNA levels demonstrated a similar trend (Fig. 6B). The relative abundance of virB1 mRNA was approximately 1.9-fold higher in the babR mutant than in parental strain 2308. Altogether, these results confirm the role of BabR as a regulator of virB transcription, and these data also indicate that BabR acts as a repressor of virB transcription in B. abortus 2308.
The role of BabR as a transcriptional regulator of the virB operon was previously determined by other laboratories, and these groups employed targeted mutagenesis of babR and subsequent experiments with virB promoter fusions and/or microarray analyses (32, 49). However, no studies have addressed the ability of BabR to bind directly to the virB promoter region, and therefore, it was not known if BabR directly or indirectly regulates virB transcription. In this study, a recombinant version of BabR was expressed and purified, and binding of BabR to the virB promoter region was tested by EMSA (Fig. 7). A DNA fragment corresponding to the first 150 bp directly upstream of the virB1 start codon was used in these experiments, and BabR bound this fragment of DNA in a concentration-dependent manner. Importantly, the binding of BabR to the PvirB DNA was specific, as indicated by the ability of excess unlabeled PvirB DNA to abolish binding, while a nonspecific fragment of DNA (the coding region of BAB2_0612) was not able to compete with PvirB DNA for binding to BabR. From the EMSA analyses, it was concluded that BabR directly binds to the virB promoter in order to repress transcription.
Fig 7.

BabR binds the virB1 promoter. An EMSA was employed to test the binding of purified rBabR to the virB promoter region. A DNA probe containing the first 150 bp of the virB upstream region was amplified by PCR, and the fragment was radiolabeled with [γ-32P]ATP. Increasing concentrations of rBabR were incubated with the labeled DNA, and in some binding reaction mixtures, unlabeled specific (PvirB DNA) and nonspecific (BAB2_0612 coding sequence DNA) competitor DNA fragments were included as controls. The binding reaction products were resolved in 6% native polyacrylamide gels and visualized by autoradiography.
DISCUSSION
The Brucella virB locus has been the subject of numerous studies, and several transcriptional regulators have been identified that influence virB expression (31). These regulators include the histidine utilization regulator HutC (41) and the two-component regulator BvrRS (22). The stringent response (p)ppGpp synthetase/hydrolase protein Rsh has also been linked to virB expression in Brucella sp. (8). Additionally, mutational analyses of 88 transcriptional regulators in B. melitensis 16M revealed five additional transcriptional activators (ArsR6, AraC2, AraC8, DeoR1, and GntR4) of virB expression (16). The LuxR-type regulators VjbR (6, 50) and BabR (also called BlxR) (32, 50) have also been reported to influence virB expression. In the present study, we have shown that Hfq, most likely in conjunction with an sRNA, is also involved in the regulation of virB expression. From the data collected (Fig. 1 and 2), it was hypothesized that Hfq functions via an intermediate transcriptional regulator to modulate virB expression. This hypothesis was based on the fact that in other bacteria, Hfq has been shown to alter the levels of transcriptional regulators, which, in turn, leads to the altered transcription of genes targeted by these regulators (17, 18). Subsequent experiments determined that BabR, a known transcriptional regulator of virB, requires Hfq for normal expression. Therefore, a working hypothesis was developed for this regulatory circuit in which Hfq and a small RNA are needed for the efficient translation of BabR, which, in turn, leads to regulation of virB expression.
The present work confirmed a regulatory link between BabR and virB, and it was established that BabR is a virB repressor in B. abortus 2308 (Fig. 6). Previously, the proper expression of virB had been linked to BabR, as mutation of babR was reported to lead to changes in virB expression (32, 49), and these studies utilized promoter fusions to reporter genes and quantitative RT-PCR to draw these conclusions. The ability of BabR to directly bind to DNA to exert its regulatory effects was not previously reported, but here it is shown that BabR binds to the promoter region of virB (Fig. 7). Thus, we demonstrated a direct and specific interaction between BabR and the virB promoter. BabR is a member of the LuxR superfamily, which consists of transcriptional regulators that are involved in the regulation of quorum-sensing systems, and these regulators often respond to small-molecule signals, such as homoserine lactones (HSL), that alter the capacity of the proteins to bind target promoters in DNA (26). Interestingly, addition of the Brucella quorum-sensing molecule C12-HSL (47) to the EMSA reaction mixtures did not affect the binding of BabR to the virB promoter (data not shown). This supports results reported by Uzureau et al. showing that BabR-mediated regulation of the virB system is independent of the C12-HSL quorum-sensing molecule (49). By DNase I footprinting analyses, the specific DNA sequence in the virB promoter that is bound by another Brucella LuxR-type regulator, VjbR, has been identified (2). In order to compare the binding sites of the two Brucella LuxR-type regulators, the BabR binding box in the virB promoter was sought. Unfortunately, repeated attempts using DNase I footprint analysis failed to identify the specific sequence bound by BabR. Nonetheless, BabR binds directly to the virB promoter, but how and under what conditions VjbR and BabR cooperate to regulate virB expression remain to be determined.
Having determined that Hfq is required for the proper translation of BabR, we next sought to better define the link between Hfq and BabR. Using EMSAs, it was shown that Brucella Hfq binds the babR 5′ UTR with a high degree of affinity (Fig. 5). Additionally, the first 20 nucleotides of the babR 5′ UTR are required in order for Hfq to mediate its effect on babR expression (Fig. 4). The 5′ UTR of the babR mRNA is predicted to form a highly stable secondary structure with hairpins that occlude the RBS (Fig. 3), and Brucella Hfq binds this 5′ UTR with high affinity (Fig. 5). Even though the ability of Hfq to serve as a modulator of gene expression is usually through its capacity to bring together target mRNAs with sRNAs (15), it is possible that binding of Hfq alone (without an sRNA) to the babR 5′ UTR can alleviate the secondary structure of the 5′ UTR and allow the proper translation of BabR. Indeed, a babR-lacZ translational fusion is completely unable to be activated in the B. abortus hfq mutant, but expression of hfq from a plasmid restores the production of β-galactosidase in this mutant (Fig. 2A and B). There is evidence from work with E. coli that Hfq can alter gene expression in the absence of an sRNA (10, 53), and therefore, the possibility that Hfq functions without an sRNA to regulate babR expression cannot be excluded. While the present study identified BabR as a target for Hfq in B. abortus 2308, the identity of, or requirement for, an sRNA in this regulatory event was not described. It is likely that Hfq works in concert with an sRNA in order to exert its regulatory effects on the babR mRNA, and our laboratory is currently employing several approaches to identify such an sRNA molecule. While Hfq is essential for babR expression, these experiments do not confirm or exclude the requirement of an sRNA for proper babR expression. Rather, it can only be concluded that Hfq is absolutely required for the expression of babR.
The role of Hfq in the virulence of Brucella is clear and well documented (33, 35), but this requirement of Hfq for wild-type virulence has remained undefined mechanistically. The present study sheds some light on this subject, as we have shown that Hfq aids in the proper expression of the virB locus, which is an important genetic determinant of Brucella virulence (11, 19, 27, 40, 55). It was determined that both the transcription and the translation of virB are affected by Hfq (Fig. 1), and these findings led to the hypothesis that Hfq indirectly alters VirB1 protein levels by controlling the expression of a virB transcriptional regulator. However, from these data, the possibility cannot be excluded that Hfq also acts directly on the virB mRNA to affect the expression of the virB system, and furthermore, it is possible that Hfq is regulating virB expression both directly and indirectly (Fig. 8). Additionally, the present study was carried out using B. abortus 2308, where virB is constitutively expressed at a low level, and can be further induced in a low-pH environment (40). Other Brucella spp., such as B. melitensis and B. suis, do not exhibit constitutive virB expression, and thus, how Hfq is involved in virB expression in other Brucella spp. is not known. Nonetheless, studies are ongoing in our laboratory to determine if Hfq directly regulates VirB1 protein levels by acting at the level of the virB mRNA, as well as to clearly define the indirect regulatory relationship between Hfq and virB expression.
Fig 8.
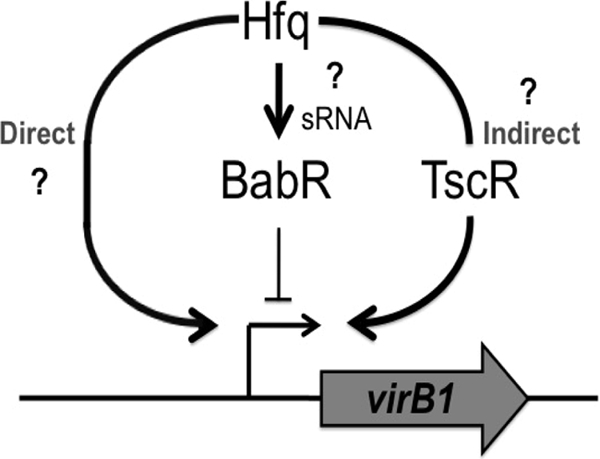
Model of Hfq-mediated regulation of babR and virB1. There is a clear relationship between Hfq and wild-type levels of VirB1 in B. abortus 2308. It was shown in this study that Hfq positively regulates the production of BabR, a known virB1 transcriptional regulator; however, BabR is a repressor of virB1 transcription. Therefore, BabR is not the sought-after genetic link between Hfq and virB1 expression. Two alternative means of virB1 regulation by Hfq may be taking place. Hfq may be directly altering the levels of VirB1 protein by binding to the virB1 mRNA and altering translation (Direct). Alternatively, Hfq may be affecting the levels of a transcriptional regulator protein (TscR), and altered levels of this regulator would lead to increased levels of virB1 mRNA, resulting in increased levels of VirB1 protein (Indirect).
It is important to note that the data presented here indicate that BabR is not the sought-after genetic link between Hfq and virB expression. It was initially hypothesized that BabR served as this link; however, it was determined that BabR is a repressor of virB transcription in B. abortus 2308 (Fig. 6). Hfq positively influences BabR production (Fig. 2), and therefore, Hfq-mediated increases in BabR levels should result in decreased virB expression. Given that Hfq also elicits a positive effect on virB expression (Fig. 1), BabR cannot serve as the direct connection between Hfq and virB expression. Thus, it is concluded that Hfq coordinates the expression of virB in B. abortus 2308 through both BabR-dependent and BabR-independent mechanisms. This seems likely, given that mutations in either hfq or virB result in the attenuation of Brucella strains, but mutation of babR is not linked to Brucella virulence (32). As discussed earlier, the possibility exists that Hfq is functioning at multiple levels, both directly and indirectly, to maximally modulate the expression of the virB locus (Fig. 8). In conclusion, Hfq independently mediates the proper expression of the LuxR-type transcriptional regulator BabR and the VirB type IV secretion system in B. abortus, but the sRNA(s) involved in these regulatory events remains to be elucidated.
ACKNOWLEDGMENTS
We thank Ivan Ndamukong for technical assistance with real-time RT-PCR.
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases to R.M.R. (AI48499).
Footnotes
Published ahead of print 21 October 2011
REFERENCES
- 1. Aiba H. 2007. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10:134–139 [DOI] [PubMed] [Google Scholar]
- 2. Arocena GM, Sieira R, Comerci DJ, Ugalde RA. 2010. Identification of the quorum-sensing target DNA sequence and N-acyl homoserine lactone responsiveness of the Brucella abortus virB promoter. J. Bacteriol. 192:3434–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bellaire BH, Elzer PH, Baldwin CL, Roop RM., II 2003. Production of the siderophore 2,3-dihydroxybenzoic acid is required for wild-type growth of Brucella abortus in the presence of erythritol under low-iron conditions in vitro. Infect. Immun. 71:2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Celli J, Grovel JP. 2004. Organelle robbery: Brucella interactions with the endoplasmic reticulum. Curr. Opin. Microbiol. 7:93–97 [DOI] [PubMed] [Google Scholar]
- 5. Comerci DJ, Martínez-Lorenzo MJ, Sieira R, Grovel JP, Ugalde RA. 2001. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell. Microbiol. 3:159–168 [DOI] [PubMed] [Google Scholar]
- 6. Delrue RM, et al. 2005. A quorum-sensing regulator controls expression of both the type IV secretion system and the flagellar apparatus of Brucella melitensis. Cell. Microbiol. 7:1151–1161 [DOI] [PubMed] [Google Scholar]
- 7. Ding Y, Davis BM, Waldor MK. 2004. Hfq is essential for Vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol. 53:345–354 [DOI] [PubMed] [Google Scholar]
- 8. Dozot M, et al. 2006. The stringent response mediator Rsh is required for Brucella melitensis and Brucella suis virulence, and for expression of the type IV secretion system virB. Cell. Microbiol. 8:1791–1802 [DOI] [PubMed] [Google Scholar]
- 9. Fantappiè L, et al. 2009. The RNA chaperone Hfq is involved in stress response and virulence in Neisseria meningitidis and is a pleiotropic regulator of protein expression. Infect. Immun. 77:1842–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Folichon M, et al. 2003. The poly(A) binding protein Hfq protects RNA from RNase E and exoribonucleolytic degradation. Nucleic Acids Res. 31:7302–7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foulongne V, Bourg G, Cazevieille C, Michaux-Charachon S, O'Callaghan D. 2000. Identification of Brucella suis genes affecting intracellular survival in an in vitro human macrophage infection model by signature-tagged transposon mutagenesis. Infect. Immun. 68:1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franco MP, Mulder M, Gilman RH, Smits HL. 2007. Human brucellosis. Lancet Infect. Dis. 7:775–786 [DOI] [PubMed] [Google Scholar]
- 13. Fröhlich KS, Vogel J. 2009. Activation of gene expression by small RNA. Curr. Opin. Microbiol. 12:674–682 [DOI] [PubMed] [Google Scholar]
- 14. Gee JM, et al. 2005. The Brucella abortus Cu/Zn superoxide dismutase is required for optimal resistance to oxidative killing by murine macrophages and wild-type virulence in experimentally infected mice. Infect. Immun. 73:2873–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gottesman S. 2004. The small RNA regulators of Escherichia coli: roles and mechanisms. Annu. Rev. Microbiol. 58:303–328 [DOI] [PubMed] [Google Scholar]
- 16. Haine V, et al. 2005. Systematic targeted mutagenesis of Brucella melitensis 16M reveals a major role for GntR regulators in the control of virulence. Infect. Immun. 73:5578–5586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hansen AM, Kaper JB. 2009. Hfq affects the expression of the LEE pathogenicity island in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 73:446–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holmqvist E, et al. 2010. Two antisense RNAs target the transcriptional regulator CsgD to inhibit curli synthesis. EMBO J. 29:1840–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hong PC, Tsolis RM, Ficht TA. 2000. Identification of genes required for chronic persistence of Brucella abortus in mice. Infect. Immun. 68:4102–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovach ME, et al. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
- 21. Kulakov YK, Guigue-Talet PG, Ramuz MR, O'Callaghan D. 1997. Response of Brucella suis 1330 and B. canis RM6/66 to growth at acid pH and induction of an adaptive acid tolerance response. Res. Microbiol. 148:145–151 [DOI] [PubMed] [Google Scholar]
- 22. Martínez-Núñez C, et al. 2010. The two-component system BvrR/BvrS regulates the expression of the type IV secretion system VirB in Brucella abortus. J. Bacteriol. 192:5603–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meibom KL, et al. 2009. Hfq, a novel pleiotropic regulator of virulence-associated genes in Francisella tularensis. Infect. Immun. 77:1866–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller JH. 1972. Experiments in molecular genetics, p 352–355 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 25. Møller T, et al. 2002. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol. Cell 9:23–30 [DOI] [PubMed] [Google Scholar]
- 26. Nasser W, Reverchon S. 2007. New insights into the regulatory mechanisms of the LuxR family of quorum sensing regulators. Anal. Bioanal. Chem. 387:381–390 [DOI] [PubMed] [Google Scholar]
- 27. O'Callaghan D, et al. 1999. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Plt type IV secretion systems is essential for intracellular survival of Brucella suis. Mol. Microbiol. 33:1210–1220 [DOI] [PubMed] [Google Scholar]
- 28. Pappas G, N. Akritidis, M. Bosilkovski, Tsianos E. Brucellosis 2005. Brucellosis. N. Engl. J. Med. 352:2325–2336 [DOI] [PubMed] [Google Scholar]
- 29. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. 2006. The new global map of human brucellosis. Lancet Infect. Dis. 6:91–99 [DOI] [PubMed] [Google Scholar]
- 30. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:2002–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rambow-Larsen AA, Petersen EM, Gourley CR, Splitter GA. 2009. Brucella regulators: self-control in a hostile environment. Trends Microbiol. 17:371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rambow-Larsen AA, Rajashekara G, Petersen E, Splitter G. 2008. Putative quorum-sensing regulator BlxR of Brucella melitensis regulates virulence factors including the type IV secretion system and flagella. J. Bacteriol. 190:3274–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robertson GT, Roop RM., II 1999. The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol. Microbiol. 34:690–700 [DOI] [PubMed] [Google Scholar]
- 34. Robertson GT, et al. 2000. The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J. Bacteriol. 182:3482–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roop RM, II, et al. 2000. Virulence of Brucella melitensis hfq, katE and bacA mutants in pregnant goats, abstr 93, p 89 In Brucellosis 2000. 53rd Annual Brucellosis Research Conference, Nimes, France [Google Scholar]
- 36. Roop RM, II, Gaines JM, Anderson ES, Caswell CC, Martin DW. 2009. Survival of the fittest: how Brucella strains adapt to their intracellular niche in the host. Med. Microbiol. Immunol. 198:221–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sauter C, Basquin J, Suck D. 2003. Sm-like proteins in eubacteria: the crystal structure of the Hfq protein from Escherichia coli. Nucleic Acid Res. 31:4091–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schweizer HP, Hoang TT. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22 [DOI] [PubMed] [Google Scholar]
- 39. Seleem MN, Boyle SM, Sriranganathan N. 2008. Brucella: a pathogen without classic virulence genes. Vet. Microbiol. 129:1–14 [DOI] [PubMed] [Google Scholar]
- 40. Sieira R, Comerci DJ, Sánchez DO, Ugalde RA. 2000. A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence intracellular multiplication. J. Bacteriol. 182:4849–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sieira R, Arocena GM, Bukata L, Comerci DJ, Ugalde RA. 2010. Metabolic control of virulence genes in Brucella abortus: HutC coordinates virB expression and the histidine utilization pathway by direct binding to both promoters. J. Bacteriol. 192:217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simonsen KT, et al. 2011. A role for the RNA chaperone Hfq in controlling adherent-invasive Escherichia coli colonization and virulence. PLoS One 6:e16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sittka A, Pfeiffer V, Tedin K, Vogel J. 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 63:193–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sonnleitner E, et al. 2003. Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb. Pathog. 35:217–228 [DOI] [PubMed] [Google Scholar]
- 45. Soper TJ, Woodson SA. 2008. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA 14:1907–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Spratt BG, Hedge PJ, te Heesen S, Edelman A, Broome-Smith JK. 1986. Kanamycin-resistant vectors that are analogues of plasmids pUC8, pUC9, pEMBL8 and pEMBL9. Gene 41:337–342 [DOI] [PubMed] [Google Scholar]
- 47. Taminiau B, et al. 2002. Identification of a quorum-sensing signal molecule in the facultative intracellular pathogen Brucella melitensis. Infect. Immun. 70:3004–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tiedeman AA, Smith JM. 1988. lacZY gene fusion cassettes with KanR resistance. Nucleic Acids Res. 16:3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Uzureau S, et al. 2010. Global analysis of quorum sensing targets in the intracellular pathogen Brucella melitensis 16 M. J. Proteome Res. 9:3200–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uzureau S, et al. 2007. Mutations of the quorum sensing-dependent regulator VjbR lead to drastic surface modifications in Brucella melitensis. J. Bacteriol. 189:6035–6047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Valderas MW, et al. 2005. Role of HdeA in acid resistance and virulence in Brucella abortus 2308. Vet. Microbiol. 107:307–312 [DOI] [PubMed] [Google Scholar]
- 52. Valentin-Hansen P, Eriksen M, Udesen C. 2004. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol. Microbiol. 51:1525–1533 [DOI] [PubMed] [Google Scholar]
- 53. Vytvytska O, Moll I, Kaberdin VR, von Gabain A, Bläsi U. 2000. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev. 14:1109–1118 [PMC free article] [PubMed] [Google Scholar]
- 54. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 55. Zygmunt MS, Hagius SD, Walker JV, Elzer PH. 2006. Identification of Brucella melitensis 16M genes required for bacterial survival in the caprine host. Microbes Infect. 8:2849–2854 [DOI] [PubMed] [Google Scholar]