

Abstract
The Mycobacterium tuberculosis prrA-prrB (Rv0903c-Rv0902c) two-component regulatory system is expressed during intracellular growth in human macrophages and is required for early intracellular multiplication in murine macrophages, suggesting its importance in establishing infection. To better understand the function of the prrA-prrB two-component system, we defined the transcriptional characteristics of the prrA and prrB genes during exponential and stationary growth and upon exposure to different environmental stresses and attempted to generate a prrA-prrB deletion mutant. The prrA and prrB genes constitute an operon and are cotranscribed during logarithmic growth, with transcriptional levels decreasing in stationary phase and during hypoxia. Despite the transcriptional differences, PrrA protein levels remained relatively stable throughout growth and in hypoxia. Under conditions of nitrogen limitation, prrAB transcription was induced, while acidic pH stress and carbon starvation did not significantly alter transcript levels. Deletion of the prrAB operon on the chromosome of M. tuberculosis H37Rv occurred only in the presence of an episomal copy of the prrAB genes, indicating that this two-component system is essential for viability. Characterization of the prrAB locus in M. tuberculosis Mt21D3, a previously described prrA transposon mutant, revealed that this strain is not a true prrA knockout mutant. Rather, Tn5367 transposon insertion into the prrA promoter only decreased prrA and prrB transcription and PrrA levels in Mt21D3 compared to those in the parental Mt103 clinical strain. These data provide the first report describing the essentiality of the M. tuberculosis prrAB two-component system and reveal insights into its potential role in mycobacterial growth and metabolism.
INTRODUCTION
Tuberculosis continues to be a global health emergency. According to 2009 WHO statistics, 9.4 million new cases of tuberculosis were diagnosed and 1.7 million people died from the disease (equivalent to 4,700 deaths each day) (29). This global emergency is further exacerbated by multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis strains that are resistant to our best antibiotics and thus difficult to treat (14). A hallmark in the life cycle of M. tuberculosis is its intracellular residence within the human macrophage. To ensure its intracellular survival, M. tuberculosis must adapt to the host environment by appropriately regulating the expression of genes involved in virulence and metabolism. Understanding how M. tuberculosis regulatory systems coordinate complex adaptations is critical to deciphering the ongoing interactions that govern establishment and progression of tuberculosis disease.
Like most bacteria, M. tuberculosis uses two-component systems to execute transcriptional reprogramming in response to changing environments. Of the 11 paired two-component systems, two orphan histidine kinase genes, and six orphan response regulator genes (15), at least three M. tuberculosis response regulators (phoP, devR, and mprA) have been implicated in mycobacterial virulence (6, 19, 23) and persistence (30, 32). However, thus far, only the M. tuberculosis mtrA response regulator gene has been deemed essential for viability (31). Although mtrA and mtrB (histidine kinase gene) are genetically linked on the chromosome, mtrB was not essential for M. tuberculosis growth in vitro (31).
The prrAB two-component regulatory system has been shown to be expressed during growth in human macrophages and is required for early intracellular multiplication (7, 12, 15). Moreover, the prrAB regulatory system is one of only four two-component systems conserved in all mycobacterial species (28), thus strongly suggesting its fundamental importance in mycobacteria. Based on these data and observations, we hypothesized that the prrAB two-component system was critical for M. tuberculosis survival or virulence within the host. In this study, we have begun delineating prrAB expression characteristics and have determined that the prrAB signal transduction system is essential for M. tuberculosis viability and may play a role in regulating gene expression during nitrogen-limiting conditions.
MATERIALS AND METHODS
Bacterial media and culture conditions.
Escherichia coli JM109 was grown in Luria-Bertani (LB) broth or on LB agar plates at 37°C with hygromycin (Hyg) (150 μg/ml) added as necessary. M. tuberculosis H37Rv cultures were grown at 37°C in Middlebrook 7H9 basal liquid medium (Difco) supplemented with 10% ADS (0.5% bovine serum albumin fraction V, 0.2% dextrose, 140 mM NaCl) enrichment and 0.05% Tween 80 (herein described as supplemented Middlebrook 7H9) with or without 0.2% glycerol and Hyg (100 μg/ml) when required. M. tuberculosis H37Rv was cultivated on Middlebrook 7H9, 7H10, or 7H11 agar (Difco) supplemented with 10% ADS enrichment with or without 0.2% glycerol and various concentrations of Hyg (described below). M. tuberculosis Mt103 and Mt21D3 strains were grown similarly to H37Rv, with kanamycin (Km) (25 μg/ml) included for Mt21D3 growth. Mycobacterium smegmatis mc2155 was grown at 37°C in Middlebrook 7H9 liquid medium or LB broth supplemented with 0.5% Tween 80 or on LB agar plates. During M. tuberculosis H37Rv prrAB mutagenesis attempts, additional medium supplements, varied post-mycobacteriophage infection outgrowth times, and modifications of Middlebrook 7H9, 7H10, or 7H11 agar with various concentrations of Hyg were as follows: (i) medium supplements, Casamino Acids (0.2% or 0.4%), Casitone (0.1%), and tryptophan (20 or 40 μg/ml); (ii) outgrowth times, 24 h and 72 h; (iii) Hyg concentrations, 50, 75, 100, 125, or 150 μg/ml.
Exposure of M. tuberculosis to environmental stresses.
M. tuberculosis H37Rv cultures (optical density at 600 nm [OD600] of ∼0.2 to 0.4) were harvested, washed with Middlebrook 7H9 basal medium (minus ADS supplement), and resuspended in stress-specific medium. For acid stress, the cultures were resuspended in Middlebrook 7H9-Tween 80-ADS medium with the pH adjusted to 5.5. For carbon stress, cultures were resuspended in Middlebook 7H9-Tween 80-albumin-NaCl medium (without dextrose or glycerol). To generate nitrogen-limiting conditions, the Amon et al. (2) model, whereby cells are incubated in Middlebrook 7H9-Tween 80-ADS medium supplemented with 200 μM l-methionine S-sulfoximine (MSX), was employed. Treated and untreated control cultures were grown aerobically for 4 h and 4 days, followed by RNA isolation as described below. For hypoxia, M. tuberculosis cultures were grown in Dubos-Tween 80-albumin broth to an OD600 of ∼0.6 and subjected to hypoxia in standing, sealed tubes for 24 and 48 h as described previously (24).
RNA isolation.
RNA isolation was performed using TRI Reagent solution (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer's protocol and as described previously (20). Briefly, cells were lysed mechanically using a FastPrep bead beater (Qbiogene/MP Biomedicals) with three pulses of 30 s each at 6,500 rpm in a mixture of 0.1 mm zirconium-silica beads and 1 ml of TRI Reagent solution in a 2-ml sterile screw-cap tube. The RNA in the aqueous phase was extracted with chloroform, precipitated using isopropanol, and treated with RNase-free Turbo DNase (Applied Biosystems/Ambion) for 1 h at 37°C to remove any DNA contamination. The DNase-treated RNA was then determined to be free of DNA contamination by confirming the lack of 16S rRNA gene amplification. The RNA concentration was determined using a NanoDrop 1000 spectrophotometer (NanoDrop/Thermo Scientific), and the quality of the RNA was assessed using Experion, an automated capillary electrophoresis system (Bio-Rad Laboratories).
qRT-PCR.
For quantitative reverse transcription-PCR (qRT-PCR), 100 ng of RNA was converted to cDNA by reverse transcription using the iScript cDNA synthesis kit (Bio-Rad Laboratories). To further verify the absence of contaminating genomic DNA, RNA samples were concurrently analyzed using RT-PCR without reverse transcriptase. The cDNA was diluted (1:10) and used as a template for quantitative PCR (qPCR) using gene-specific primers and SYBR green dye. Optimization of the PCR amplification parameters was performed using iQ SYBR green supermix (Bio-Rad Laboratories). Standard curve generation and melting-curve analyses were performed as described previously (20). For each qPCR experiment, the calculated threshold cycle (CT) was normalized to the CT of the internal 16S rRNA control (amplified from the same samples) before calculating the fold change between the test and control samples.
Northern analyses.
Antisense prrA and prrB RNA digoxigenin (DIG)-labeled probes were prepared using the DIG Northern starter kit (Roche, Indianapolis, IN), following the manufacturer's protocol. One microgram of total M. tuberculosis H37Rv RNA was isolated from logarithmic-phase cells, separated via a 1% agarose-glyoxyl gel, and transferred to a BrightStar membrane (Applied Biosystems/Ambion) using NorthernMax kit reagents (Applied Biosystems/Ambion). The transferred RNA was hybridized with the gene-specific, denatured DIG-labeled RNA probe and detected using the NorthernMax protocol (Applied Biosystems/Ambion). To ensure complete stripping of the DIG-labeled probe before subsequent hybridizations, membranes were incubated twice in a 50% formamide solution containing 50 mM Tris-HCl, pH 7.5, and 5% SDS at 80°C for 60 min and then subjected to autoradiography.
Mycobacterial protein extracts.
M. tuberculosis cells were washed twice in wash buffer (phosphate-buffered saline [PBS] buffer containing protease inhibitor cocktail [Roche], 1 mM dithiothreitol [DTT], 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) and lysed in wash buffer or two-dimensional (2D)-rehydration buffer (Bio-Rad Laboratories) with 0.1-mm-diameter zirconia/silica beads (Biospec Products) in a Fast Prep homogenizer. Lysed cells were centrifuged at 12,000 rpm at 4°C for 10 min, and the supernatant containing the cell-free protein extract was collected. Protein concentrations were determined using the RC-DC protein assay kit (Bio-Rad Laboratories) with bovine serum albumin as a standard.
PrrA antibody generation and Western analyses.
The recombinant 6×-His-tagged PrrA protein was produced in E. coli and purified as previously described (17). The purified PrrA protein (∼100 μg) was emulsified in Freund's incomplete adjuvant and used to immunize two New Zealand White rabbits by subcutaneous injection. Rabbits were given booster injections of PrrA antigen at 4-week intervals, and antisera were collected after at least two booster injections. After protein quantitation, the samples were resolved via SDS-PAGE, transferred onto a nitrocellulose membrane, and stained with Ponceau S stain to ensure equivalent amounts of transferred protein. Immunoblotting was performed using rabbit anti-PrrA rabbit polyclonal antiserum (1:20,000) and detected with the Supersignal West Dura chemiluminescent substrate (Thermo Scientific). Quantitations were performed using the ImageJ 1.44 densitometry software program (NIH).
Generation of a ΔprrAB::hyg thermosensitive mycobacteriophage and mutagenesis procedures.
The ΔprrAB::hyg allele was constructed by amplifying the flanking regions of the prrAB genes and cloning the fragments into pYUB854 (4) on either side of the hyg resistance cassette. The 1,000-bp region upstream of the prrAB genes was amplified using the primers SH81 (5′-CCCAAGCTTGATGCTGGATCGTGCGTTGAC-3′) and SH82 (5′-CTAGCTAGCTTTGCCTGATTACCGTCCAGC-3′), which contain HindIII and NheI sites (underlined) at their respective 5′ termini. The 989-bp region downstream of the prrAB genes was amplified using the primers SH83 (5′-TGCTCTAGAGGCACCGCGTCGCTGGAGAA-3′) and SH84 (5′-CGGGGTACCACCGGCAGCGAAGGTATCAA-3′), which contain XbaI and KpnI sites (underlined) at their respective 5′ termini. The PCR products were cloned into pYUB854 (4), flanking the hyg cassette, to create pSH270. The ligation mixture of PacI-digested pSH270 and PacI-digested concatamerized phAE87 DNA was packaged using Gigapack III packaging extracts (Stratagene) and transduced into E. coli HB101. Phasmid DNA was prepared from pooled Hyg-resistant (Hygr) transductants, digested with appropriate restriction enzymes to verify the presence of the desired insert, and electroporated into M. smegmatis mc2155. Transformants were plated and incubated at 30°C for the generation of mycobacteriophage plaques. A high-titer mycobacteriophage stock was generated from a confirmed temperature-sensitive phage (phSH270) and was used to infect M. tuberculosis H37Rv as previously described (4, 16).
The prrAB-complementing plasmid, pSH439, consists of 125 bp upstream of prrA, the prrAB coding regions, and 15 bp downstream of the prrB stop codon cloned into pMV306, a site-specific (att) integrating mycobacterial vector (26). Confirmed integration of pSH439 into M. tuberculosis H37Rv generated M. tuberculosis STS16, which harbored an ectopic prrAB locus. The phSH270 ΔprrAB::hyg thermosensitive mycobacteriophage was used to infect M. tuberculosis STS16 as previously described (4, 16), and Hyg- and Km-resistant colonies were screened by PCR or Southern blot analysis for deletion of the wild-type prrAB locus. Two confirmed prrAB-complemented ΔprrAB::hyg deletion mutants were isolated and designated M. tuberculosis STS22 and STS23.
Southern blot analyses.
Genomic DNA was isolated via enzymatic lysis from M. tuberculosis cells harvested in the mid-logarithmic phase of growth, digested with the appropriate restriction enzymes, separated by agarose gel electrophoresis, and transferred to a positively charged nylon membrane. Transferred DNAs were then hybridized with the gene-specific DIG-labeled probes and detected using anti-DIG-alkaline phosphatase antibody and the CDP-Star chemiluminescent reagent as recommended by the manufacturer (Roche). Membranes were then exposed to X-ray film at room temperature.
Statistical analyses.
Statistical analyses were performed using the Prism 5 software program (GraphPad Software, San Diego, CA) and were calculated using a Student t test or analysis of variance (ANOVA). A P value of <0.05 was considered statistically significant.
RESULTS
Characterization of prrA and prrB transcription during in vitro growth.
Since the prrAB two-component system is evolutionarily conserved among all mycobacterial genomes and plays a role in early stages of M. tuberculosis infection, we investigated its physiological characteristics. Compared to 16S rRNA levels, transcription of prrA and prrB is generally low throughout exponential growth of M. tuberculosis H37Rv in vitro, with transcriptional levels of prrA consistently more abundant than those of prrB (Fig. 1A). Transcription of prrA and prrB was greatest during early logarithmic growth and was slightly decreased as the culture progressed into late exponential growth (Fig. 1A). These prrA and prrB transcriptional trends during exponential growth were similar during M. tuberculosis H37Rv growth in the absence of glycerol (data not shown).
Fig 1.

prrA and prrB transcription and PrrA levels during in vitro growth. (A) Quantitation of prrA and prrB transcripts during M. tuberculosis H37Rv in vitro exponential growth in supplemented Middlebrook 7H9 broth with glycerol. The optical density at 600 nm of the cultures is indicated by the solid line and scaled on the right y axis. (B) qRT-PCR comparison of prrA and prrB expression in logarithmic and stationary phases of M. tuberculosis growth in supplemented Middlebrook 7H9 broth without glycerol. Normalized expression of prrA and prrB with respect to 16S rRNA expression is presented as the mean ± SD of data from three independent experiments. The decrease in prrA and prrB expression during stationary phase is presented relative to the logarithmic-phase expression, which is set at 1.0. ∗∗∗, P < 0.0001. (C) Western blot analyses of PrrA in total M. tuberculosis H37Rv protein (20 μg) collected from logarithmic (lane 1) and stationary (lane 2) growth phases and detected using anti-PrrA antibodies. Lane 3 represents the purified His-tagged PrrA protein (40 ng). (D) Northern analysis of M. tuberculosis H37Rv with prrA and prrB single-stranded RNA probes. RNA samples were collected from exponential-phase cultures of M. tuberculosis H37Rv grown in Middlebrook 7H9 broth with glycerol. (E) Diagram of the prrA-prrB region of the chromosome, drawn to scale and showing the prrA and prrB gene lengths.
The PrrA protein is stable in stationary phase.
To investigate prrA and prrB transcriptional trends throughout growth, we compared prrA and prrB levels during M. tuberculosis H37Rv logarithmic (3 days) and stationary (30 days) phases of growth. After 30 days of growth in supplemented Middlebrook 7H9 without glycerol, representing stationary phase, prrA and prrB transcription was decreased by 60% compared to expression during logarithmic growth (3 days) (Fig. 1B). However, Western blot analysis of total protein isolated at the above-specified times, followed by densitometry quantitation, revealed that production of PrrA in stationary-phase cultures was slightly decreased, by ∼20%, from that in logarithmic-phase cultures (Fig. 1C and Fig. 2C, lanes 4 and 5). The levels of PrrA in stationary phase did not correlate with the decreased transcript level (Fig. 1B), indicating that the PrrA protein is relatively stable in stationary phase.
Fig 2.

M. tuberculosis prrA and prrB are differentially transcribed and translated upon exposure to in vitro environmental stress conditions. (A) qRT-PCR comparison of prrA and prrB expression upon M. tuberculosis H37Rv exposure to low pH (pH 5.5), carbon starvation, or nitrogen-limiting environments. The fold change in prrA and prrB expression in H37Rv grown under environmental stress conditions with respect to H37Rv grown in Middlebrook 7H9-Tween-ADS medium (untreated control) (baseline expression set to 1.0) is presented as the mean ± SD of data from three independent experiments. ∗∗, prrA P value = 0.0013 or prrB P value = 0.0032. d, days. (B) qRT-PCR analysis of prrA and prrB expression, comparing logarithmic growth and 24- or 48-h exposure to hypoxic conditions. Normalized expression of prrA and prrB with respect to 16S rRNA expression is presented as the mean ± SD of data from three independent experiments. The decrease in prrAB transcript levels in hypoxia is presented relative to logarithmic-phase expression, which is set at 1.0. ∗∗∗, prrA P value = 0.0002 by ANOVA or prrB P value < 0.0001 by ANOVA. (C) Western blot analysis of PrrA during M. tuberculosis H37Rv logarithmic growth, stationary phase, and hypoxia. Total protein (40 μg) was separated via SDS-PAGE and detected using polyclonal anti-PrrA antibodies with purified His-tagged PrrA protein (40 ng) serving as a positive control. Lanes: 1, aerobic growth control; 2, hypoxia, 24 h; 3, hypoxia, 48 h; 4, logarithmic growth, 3 days; 5, stationary phase, 30 days; 6, purified PrrA protein. (D) Western blot analysis of PrrA during M. tuberculosis H37Rv nitrogen limitation, logarithmic growth, stationary phase, and hypoxia. Total protein (40 μg) was separated via SDS-PAGE and detected using polyclonal anti-PrrA antibodies, with the purified His-tagged PrrA protein (20 ng) serving as a positive control. Lanes: 1, MSX exposure, 4 h; 2, MSX exposure, 4 days; 3, purified PrrA protein.
prrA and prrB are cotranscribed in an operon.
To determine if the transcriptional differences were due to the presence of independent prrA and prrB expression, we used Northern blot analyses to establish the length of the prrA and prrB transcripts. Northern analyses using DIG-labeled prrA and prrB single-stranded RNA probes revealed the presence of a single transcript of approximately 2.1 kb (Fig. 1D). Repeated hybridizations did not reveal the presence of smaller transcripts. Therefore, we concluded that in M. tuberculosis H37Rv, prrA and prrB, like most genetically linked two-component systems, are cotranscribed under the growth conditions tested in an apparent operon with a 10-bp intergenic region separating prrA and prrB (Fig. 1E).
prrA and prrB transcription is induced under nitrogen limitation and repressed in hypoxia.
To begin characterizing environmental conditions that could stimulate PrrAB activity, we analyzed prrA and prrB transcript levels in M. tuberculosis H37Rv upon short (4 h) and prolonged (4 days) exposure to nitrogen-limiting conditions, acid stress, carbon starvation, and hypoxia. Nitrogen limitation was induced by exposure to MSX, which is known to inhibit M. tuberculosis glutamine synthetase GlnA1, a crucial enzyme in nitrogen metabolism and virulence (13, 21, 27). Based on qRT-PCR analyses, prrA and prrB were induced 7- and 5-fold, respectively, when M. tuberculosis was incubated for 4 days under nitrogen-limiting conditions (Fig. 2A). No induction was observed in MSX-treated samples at 4 h postexposure. prrA and prrB transcriptional changes upon exposure to a low-pH environment and during carbon starvation were not statistically significant (Fig. 2A). Incubation of M. tuberculosis under hypoxic conditions for 24 h or 48 h resulted in significantly decreased transcription of both prrA and prrB (Fig. 2B). However, despite prrA transcriptional changes (Fig. 2B), PrrA levels were similar and stable in hypoxia (Fig. 2C, lanes 2 and 3) compared to those of the aerobic control culture (Fig. 2C, lane 1). Most importantly, comparison of MSX-treated cultures revealed that after 4 days of MSX exposure, PrrA levels were increased by 89% compared to results with 4 h of MSX exposure (Fig. 2D), thus corroborating the induction observed at the transcription level (Fig. 2A). Together, these results suggest that the PrrAB two-component system is capable of responding to environmental changes and may be required in stress-induced metabolic and physiological adaptation of M. tuberculosis, specifically adaptation to nitrogen limitation.
prrAB is an essential two-component system.
Since the prrAB system is significantly induced under nitrogen-limiting conditions (Fig. 2A), expressed after 48 h of intracellular growth in human macrophages, and required for early intracellular multiplication in murine bone marrow-derived macrophages (7, 12, 15), we attempted to delete the M. tuberculosis prrAB two-component system using the thermosensitive mycobacteriophage mutagenesis strategy, in which a hygromycin resistance cassette replaced the entire prrA open reading frame (ORF) and 99% of the prrB ORF. A total of 11 independent M. tuberculosis H37Rv ΔprrAB::hyg thermosensitive mycobacteriophage infections were performed, yielding 1,325 Hyg-resistant (Hygr) CFU, of which 231 independent cultures were grown and screened by PCR or Southern blot analysis for the presence of the ΔprrAB::hyg mutation. After screening, none had undergone allelic exchange to yield prrAB deletion mutants. In an attempt to recover an M. tuberculosis ΔprrAB::hyg mutant, we also supplemented either Middlebrook 7H9, 7H10, or 7H11 agar with various concentrations of Hyg, Casamino Acids, Casitone, and/or tryptophan and varied post-mycobacteriophage infection outgrowth times (see Materials and Methods). The inability to delete the prrAB genes from M. tuberculosis after 11 successive, independent phage mutagenesis attempts, while incorporating various supplementation and outgrowth approaches, strongly suggested that the prrAB two-component system is essential for viability under the described experimental conditions. To confirm the requirement of prrAB for viability, a second copy of prrAB was introduced into the M. tuberculosis H37Rv chromosome, generating M. tuberculosis STS16. After a single M. tuberculosis STS16 mycobacteriophage transduction with the phSH270 ΔprrAB::hyg thermosensitive mycobacteriophage, 2 of 18 clones (designated STS22 and STS23), or 11%, harbored a mutated wild-type prrAB locus (Fig. 3). Thus, the presence of an ectopic prrAB locus permitted the disruption of the chromosomal prrAB genes and confirmed the essentiality of this regulatory system.
Fig 3.
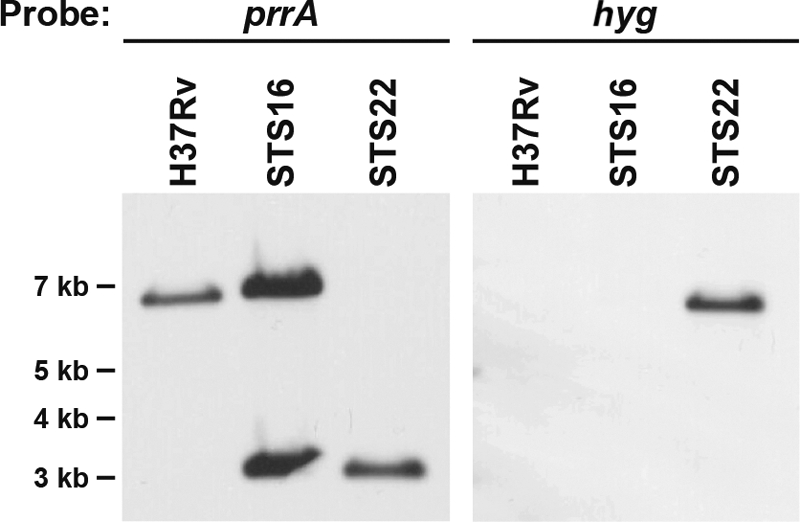
Southern blot analysis of M. tuberculosis chromosomal DNA from wild-type H37Rv, STS16 (H37Rv::prrAB), and STS22 (prrAB-complemented ΔprrAB::hyg deletion mutant), confirming generation of a wild-type ΔprrAB::hyg mutation in the presence of an ectopic copy of prrAB. Genomic DNA was digested with HpaI and SpeI and hybridized with the prrA gene and the hygromycin (hyg) resistance cassette. Expected sizes of the hybridized DNA fragments: wild-type prrA, 6,823 bp; episomal copy of prrA, 3,048 bp; hyg, 6,730 bp. Molecular size markers (in kilobases) are shown on the left.
Comparative genomics of the prrAB operon in H37Rv, Mt103, and Mt21D3.
Given our results that the prrAB system is essential in H37Rv and that a prrA transposon mutant in the M. tuberculosis Mt103 clinical strain has been described (7), we wanted to investigate if different M. tuberculosis strains had differential requirements for the PrrAB regulatory system. In order to verify the Mt103 and Mt21D3 strains and to compare the sequence with M. tuberculosis H37Rv, we amplified and sequenced the intergenic regions upstream of prrA and the prrA and prrB ORFs in M. tuberculosis Mt21D3 and Mt103. Our sequencing results indicated that Tn5367 inserted 14 bp upstream of the prrA start codon in Mt21D3, which is slightly further upstream than previously reported (5 bp upstream of the prrA ORF) (7). Compared to H37Rv, there were three nucleotide differences that existed in the Mt103 prrA and prrB genes—prrA C221 to T221, prrB C281 to T281, and prrB C848 to T848—leading to three amino acid changes, PrrA T74 to M74, PrrB S94 to F94, and PrrB T283 to I283, respectively. No frame shifts or deletions were detected when the Mt103 prrA and prrB sequences were compared with the H37Rv prrA and prrB sequences, revealing 711-bp and 1,341-bp ORFs, respectively, in both strains (Fig. 1E). Southern blot analyses, using prrA and aph (Km) probes, revealed hybridization patterns identical to those in the Ewann et al. (7) study and further confirmed the validity of the Mt103 and Mt21D3 strains (Fig. 4A).
Fig 4.

Characterization of prrA from M. tuberculosis Mt103 wild-type and Mt21D3 prrA transposon mutant strains. (A) Genomic DNA from Mt103 and Mt21D3 was digested with BamHI, which is present within Tn5367 (7), and hybridized with the prrA gene or the Km resistance (aph) cassette. Based on BamHI sites located within Tn5367 and flanking the prrA and prrB genes (7), the expected sizes of the hybridized DNA fragments are as follows: prrA (Mt103), 7,801 bp; prrA with Tn5367 inserted in the promoter (Mt21D3), 6,607 bp; and aph (Mt21D3), 4,292 bp. Molecular size markers (in kilobases) are shown on the left. (B) qRT-PCR analysis of prrA and prrB expression in Mt103 and Mt21D3. Results were normalized with respect to 16S rRNA expression and are shown as expression differences from results for the Mt103 control sample, which were assigned a value of 1. The data represent the means ± SD of data from three independent experiments. ∗, prrA P value = 0.0378; ∗∗, prrB P value = 0.0037. (C) Western blot analysis of whole-cell lysates from M. tuberculosis Mt103 (1×, 25 μg) and Mt21D3 (1×, 25 μg; 2×, 50 μg) strains probed with polyclonal anti-PrrA antibody, with the purified His-tagged PrrA protein (40 ng) serving as a control.
prrAB transcription and PrrA protein expression in the wild-type Mt103 and Mt21D3 prrA transposon mutant strains.
Due to our failed attempts in generating a ΔprrAB::hyg knockout mutant, we characterized prrAB expression in the M. tuberculosis Mt21D3 prrA transposon mutant. Although it was previously reported that prrA was transiently required for early intracellular multiplication in mouse bone marrow-derived macrophages (7), the authors did not report transcriptional analysis of prrA and prrB transcripts in the Mt21D3 prrA transposon mutant. Based on qRT-PCR analyses, prrA and prrB transcript levels in Mt21D3 were 30% and 42%, respectively, less than those in wild-type Mt103 (Fig. 4B). These results established that the Tn5367 insertion upstream of prrA in Mt21D3 did not generate a true prrA-deficient mutant but rather reduced prrA and prrB transcription. Moreover, the decrease in prrB expression as a result of the transposon insertion is indicative of cotranscription of prrAB genes in Mt103 and is similar to the transcriptional results obtained with H37Rv. In accordance with the qRT-PCR results, production of the PrrA protein is also greatly diminished in Mt21D3 compared to that in Mt103 (Fig. 4C). Densitometry analysis revealed that PrrA expression was 71% less in Mt21D3 than in wild-type Mt103 (Fig. 4C). These PrrA expression differences in Mt21D3 are significant considering that PrrA levels in H37Rv were relatively stable regardless of the growth phase or hypoxic conditions (Fig. 2C). Further investigations into defining the PrrAB regulon and determining the role of the PrrAB system in M. tuberculosis stress adaptation will be critical in assessing the basis of prrAB essentiality.
DISCUSSION
M. tuberculosis two-component regulatory systems are key facilitators of transcriptional adaptation of mycobacteria in response to dynamic and subtle changes in the environment during culture and during infection. M. tuberculosis has 11 genetically linked two-component system genes and eight unpaired (or orphaned) histidine kinase or response regulator genes (15) that orchestrate and coordinate adaptive strategies required for survival. The conservation of the PrrAB regulatory system in all mycobacterial species (28) infers that PrrAB has been evolutionarily selected to play an important role in mycobacterial biology, yet we know nothing about its function. The M. tuberculosis PrrB histidine kinase and PrrA response regulator represent a functional, cognate two-component signal transduction circuit (22). Autophosphorylation of the PrrB histidine kinase domain was readily demonstrated, and subsequent transphosphorylation of PrrB was rapid and highly specific (8, 22). Here we describe transcriptional analysis of the prrAB system and, most important, provide evidence that the PrrAB system is essential for mycobacterial viability. We should note that an identifiable transposon site hybridization (TraSH) signal in the prrB gene suggests that the histidine kinase gene can sustain a mutation (25); however, attempts to independently delete the prrB gene have not yet been pursued. The induction of prrAB under conditions of nitrogen limitation potentially links its function to regulation of nitrogen metabolism.
With this study, we have determined that M. tuberculosis harbors not only one but two response regulators, MtrA and PrrA, that are essential for viability. The previously determined essential mtrA response regulator gene is transcribed during M. tuberculosis growth in macrophages (15, 31), and its overexpression prevents M. tuberculosis multiplication in macrophages and mouse lungs and spleens (9). Interestingly, crystal structures of the PrrA and MtrA response regulators show remarkable similarities (10, 22). Both PrrA and MtrA have N-terminal phosphorylation receiver domains with a classic α/β fold and a C-terminal effector domain with a winged helix DNA-binding motif (10, 22). Of all crystallized response regulators described to date, the domain orientation of MtrA is most similar to that of PrrA, with respective inactive domain orientations providing a barrier to activation (3, 10).
Extensive structural investigations have classified response regulators into two categories: (i) readily phosphorylatable (e.g., Thermotoga maritima DrrD and E. coli PhoB) and (ii) poorly phosphorylatable (e.g., M. tuberculosis PrrA and MtrA) (3). Response regulators that are poorly phosphorylated exhibit significant interfaces that exist between the N-terminal phosphorylation receiver domain and the C-terminal DNA-binding domain, thus occluding phosphorylation, stabilizing the inactive conformation of the receiver domain, and sterically inhibiting the helix domain from binding DNA (3, 11). While these substantial interdomain interactions prohibit phosphorylation via small molecule phospho donors (e.g., acetyl phosphate), PrrA domain interactions do not inhibit phosphorylation via its cognate PrrB histidine kinase (3). Structural evidence indicates that unphosphorylated PrrA will not bind DNA (3, 11). However, in vitro studies have demonstrated that unphosphorylated, recombinant PrrA binds the prrAB promoter region and autoregulates expression (8). Additionally, in support of the reported structural properties, PrrA phosphorylation enhanced DNA binding (8). Taken altogether, these results reveal dynamic, inherent regulatory control of PrrA response regulator function and that maintenance of environmental signal specificity is crucial for PrrAB activation and DNA binding.
Previously, Ewann et al. (7) reported that a prrA transposon mutant of the M. tuberculosis Mt103 clinical strain, designated Mt21D3, harbors a Tn5367 transposon inserted 5 bp upstream of the predicted prrA start codon. While intracellular growth of Mt21D3 in mouse bone marrow-derived macrophages was impaired during the first 6 days of infection, with 10-fold decreases in CFU compared to levels of the wild-type strain, Mt103, growth recovery of the Mt21D3 mutant was similar to that of Mt103 at day 9 (7). Moreover, transcription of a prrA-gfp transcriptional fusion in Mycobacterium bovis BCG-infected mouse bone marrow-derived macrophages revealed a 2-fold increase in fluorescence intensity over background levels during the first 3 days of infection (7). After 9 days of intracellular multiplication, prrA-gfp expression returned to background levels, indicating transient expression of prrA during early phases of intracellular growth (7). These data correlate with our previous reports demonstrating that prrA is expressed after 48 h of M. tuberculosis growth in human peripheral blood monocyte-derived macrophages (12, 15).
In light of our results revealing the essentiality of the PrrAB two-component system in M. tuberculosis H37Rv, it was imperative to evaluate its requirement in other M. tuberculosis strains. Our results from sequence comparisons between the wild-type M. tuberculosis H37Rv and Mt103 strains revealed one and two nucleotide differences in prrA and prrB, respectively. We also determined that the Tn5367 transposon inserted 14 bp upstream of prrA in Mt21D3, rather than 5 bp as described previously (7). Furthermore, the Tn5367 insertion did not knock out prrA expression completely in M. tuberculosis Mt21D3 but rather decreased prrA and prrB expression. Accordingly, rather than the absence of the PrrA protein in the Mt21D3 mutant, we observed the presence of the PrrA protein, albeit at levels that were considerably diminished compared to those in the wild-type Mt103 strain. Since prrA is expressed during M. tuberculosis intracellular growth (12, 15), diminished prrA and prrB levels in Mt21D3 likely led to the detected intracellular growth deficiencies (7). While it is possible that PrrA is transiently required during early infection events, we cannot exclude the possibility that the observed loss of phenotype at later time points in Mt21D3 occurred as a result of residual PrrA activity in the mutant. Based on our determinations that the prrAB two-component system is essential for viability, the Mt21D3 strain could serve as a suitable prrA knockdown expression strain for future studies. Given the potential role of prrA in M. tuberculosis intracellular adaptation, its expression during intracellular growth in human macrophages (12, 15), and its requirement for early intracellular multiplication in murine macrophages (7), further investigations into the basis of prrAB essentiality are warranted.
The determination that prrA and prrB represent a polycistronic operon is not unexpected, since genetically linked two-component system genes are commonly cotranscribed. Comparative analysis of prrA and prrB transcripts at different growth phases showed maximal levels during exponential growth, with greatly diminished expression in stationary phase. The discordance of prrA transcript levels with protein levels in actively replicating exponential-phase cultures versus slow-growing stationary phase brings forth the possibility that the PrrA protein is stable and involved in transcriptional regulation during stationary phase. Although much work needs to be done to validate this hypothesis, significant induction of prrAB transcripts and PrrA protein levels during nitrogen limitation provides valuable insights into PrrA function. Nitrogen-limiting conditions are typically induced either by limiting the nitrogen source in the culture medium or by blocking assimilation/utilization of the nitrogen source. In a previous report, Amon et al. (2) demonstrated that exogenous addition of MSX to M. smegmatis cultures in Middlebrook 7H9 medium induced a cellular nitrogen starvation response at the transcriptional level. MSX inactivates glutamine synthetase (GS) and consequently blocks nitrogen metabolism and impairs ammonia assimilation (2, 18, 21). Interestingly, another key function associated with GS is the synthesis of poly-l-glutamate/glutamine, a virulence-associated M. tuberculosis cell wall component. Inhibition of GS via MSX showed correlation with the decreased synthesis of poly-l-glutamate/glutamine in the M. tuberculosis cell wall (13). Using the model for nitrogen limitation described by Amon et al. (2), we observed that MSX treatment resulted in a dramatic increase in the transcription of prrA and prrB, suggesting that this two-component system regulates genes that respond to nitrogen availability or modulates acclimation to nitrogen limitation. Considering the essentiality of the PrrAB system for M. tuberculosis viability, the link between PrrAB regulation, GS activity, and cell wall structure prompts further investigation. It should be noted that MSX-treated cultures analyzed in this study maintained glucose as a carbon source. It is known that nitrogen-starved cells that have access to a carbon source (in this case, glucose) exhibit substantial metabolic activity compared to cells subjected to carbon and nitrogen starvation or carbon starvation alone (1); hence, it is plausible that PrrA may also be involved in carbon-derived metabolism.
The global response to nitrogen starvation has been previously studied in M. tuberculosis as a model mimicking persistence (5). In this 6-week nitrogen deprivation model, wherein cells exhibit little or no replication, a low respiration rate, and no loss of viability, the stringent response is induced while aerobic respiration, translation, cell division, and lipid biosynthesis are decreased (5). Although prrA was not identified in the Betts et al. (5) study, the method of mediating a nitrogen starvation environment was different from the nitrogen-limiting conditions that we used in this study. Given the complex nitrogen deprivation response and its potential link to persistence mechanisms, it will be pertinent to investigate the underlying function of the essential PrrA response regulator during nitrogen-limiting conditions and its influence on M. tuberculosis viability, pathogenesis, and persistence.
ACKNOWLEDGMENTS
We thank Robin Treuer for performing the initial qRT-PCR experiments and Northern analyses and Lourdes Arteaga-Cortés for assistance with cloning, sequencing, and Western analyses. We gratefully acknowledge Mary Jackson and Brigitte Gicquel for providing the M. tuberculosis Mt103 and Mt21D3 strains and Shaleen Korch for assistance with the environmental stress studies.
This research was supported by grant AI46428 to Josephine E. Clark-Curtiss from the NIH National Institute of Allergy and Infectious Diseases and by ASU faculty research funds to Shelley E. Haydel.
Footnotes
Published ahead of print 11 November 2011
REFERENCES
- 1. Albers E, Larsson C, Andlid T, Walsh MC, Gustafsson L. 2007. Effect of nutrient starvation on the cellular composition and metabolic capacity of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 73: 4839–4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amon J, et al. 2008. Nitrogen control in Mycobacterium smegmatis: nitrogen-dependent expression of ammonium transport and assimilation proteins depends on the OmpR-type regulator GlnR. J. Bacteriol. 190: 7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. 2010. Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 285: 32325–32335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bardarov S, et al. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148: 3007–3017 [DOI] [PubMed] [Google Scholar]
- 5. Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43: 717–731 [DOI] [PubMed] [Google Scholar]
- 6. Converse PJ, et al. 2009. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77: 1230–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ewann F, et al. 2002. Transient requirement of the PrrA-PrrB two-component system for early intracellular multiplication of Mycobacterium tuberculosis. Infect. Immun. 70: 2256–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ewann F, Locht C, Supply P. 2004. Intracellular autoregulation of the Mycobacterium tuberculosis PrrA response regulator. Microbiology 150: 241–246 [DOI] [PubMed] [Google Scholar]
- 9. Fol M, et al. 2006. Modulation of Mycobacterium tuberculosis proliferation by MtrA, an essential two-component response regulator. Mol. Microbiol. 60: 643–657 [DOI] [PubMed] [Google Scholar]
- 10. Friedland N, et al. 2007. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry 46: 6733–6743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao R, Mack TR, Stock AM. 2007. Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem. Sci. 32: 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graham JE, Clark-Curtiss JE. 1999. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. U. S. A. 96: 11554–11559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harth G, Horwitz MA. 1999. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets. J. Exp. Med. 189: 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haydel SE. 2010. Extensively drug-resistant tuberculosis: a sign of the times and an impetus for antimicrobial discovery. Pharmaceuticals (Basel) 3: 2268–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haydel SE, Clark-Curtiss JE. 2004. Global expression analysis of two-component system regulator genes during Mycobacterium tuberculosis growth in human macrophages. FEMS Microbiol. Lett. 236: 341–347 [DOI] [PubMed] [Google Scholar]
- 16. Haydel SE, Clark-Curtiss JE. 2006. The Mycobacterium tuberculosis TrcR response regulator represses transcription of the intracellularly expressed Rv1057 gene, encoding a seven-bladed β-propeller. J. Bacteriol. 188: 150–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haydel SE, Dunlap NE, Benjamin WH., Jr 1999. In vitro evidence of two-component system phosphorylation between the Mycobacterium tuberculosis TrcR/TrcS proteins. Microb. Pathog. 26: 195–206 [DOI] [PubMed] [Google Scholar]
- 18. Khan A, Akhtar S, Ahmad JN, Sarkar D. 2008. Presence of a functional nitrate assimilation pathway in Mycobacterium smegmatis. Microb. Pathog. 44: 71–77 [DOI] [PubMed] [Google Scholar]
- 19. Malhotra V, et al. 2004. Disruption of response regulator gene, devR, leads to attenuation in virulence of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 231: 237–245 [DOI] [PubMed] [Google Scholar]
- 20. Malhotra V, Tyagi JS, Clark-Curtiss JE. 2009. DevR-mediated adaptive response in Mycobacterium tuberculosis H37Ra: links to asparagine metabolism. Tuberculosis 89: 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manning JM, Moore S, Rowe WB, Meister A. 1969. Identification of L-methionine S-sulfoximine as the diastereoisomer of L-methionine SR-sulfoximine that inhibits glutamine synthetase. Biochemistry 8: 2681–2685 [DOI] [PubMed] [Google Scholar]
- 22. Nowak E, Panjikar S, Konarev P, Svergun DI, Tucker PA. 2006. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J. Biol. Chem. 281: 9659–9666 [DOI] [PubMed] [Google Scholar]
- 23. Pérez E, et al. 2001. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 41: 179–187 [DOI] [PubMed] [Google Scholar]
- 24. Saini DK, et al. 2004. DevR-DevS is a bona fide two-component system of Mycobacterium tuberculosis that is hypoxia-responsive in the absence of the DNA-binding domain of DevR. Microbiology 150: 865–875 [DOI] [PubMed] [Google Scholar]
- 25. Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48: 77–84 [DOI] [PubMed] [Google Scholar]
- 26. Stover CK, et al. 1991. New use of BCG for recombinant vaccines. Nature 351: 456–460 [DOI] [PubMed] [Google Scholar]
- 27. Tullius MV, Harth G, Horwitz MA. 2003. Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect. Immun. 71: 3927–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tyagi JS, Sharma D. 2004. Signal transduction systems of mycobacteria with special reference to M. tuberculosis. Curr. Sci. (Bangalore) 86: 93–102 [Google Scholar]
- 29. World Health Organization 2010. Global tuberculosis control fact sheet. World Health Organization, Geneva, Switzerland [Google Scholar]
- 30. Zahrt T, Deretic V. 2001. Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc. Natl. Acad. Sci. U. S. A. 98: 12706–12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zahrt TA, Deretic V. 2000. An essential two-component signal transduction system in Mycobacterium tuberculosis. J. Bacteriol. 182: 3832–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zahrt TC, Wozniak C, Jones D, Trevett A. 2003. Functional analysis of the Mycobacterium tuberculosis MprAB two-component signal transduction system. Infect. Immun. 71: 6962–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]