

Background: Tideglusib is a GSK-3 inhibitor currently undergoing clinical trials for Alzheimer disease and progressive supranuclear palsy.
Results: Removal of unbound compound does not recover the enzyme activity, and the dissociation rate constant is close to zero. The protein shows a low turnover rate in neurons.
Conclusion: Tideglusib is an irreversible inhibitor of GSK-3β.
Significance: The irreversibility and the long enzyme half-life may possess interesting pharmacodynamic implications.
Keywords: Alzheimer Disease, Drug Discovery, Enzyme Inhibitors, Enzyme Kinetics, Glycogen Synthase Kinase 3, Irreversible Inhibitors, NP-12, NP031112, PSP, Tideglusib
Abstract
Tideglusib is a GSK-3 inhibitor currently in phase II clinical trials for the treatment of Alzheimer disease and progressive supranuclear palsy. Sustained oral administration of the compound to a variety of animal models decreases Tau hyperphosphorylation, lowers brain amyloid plaque load, improves learning and memory, and prevents neuronal loss. We report here that tideglusib inhibits GSK-3β irreversibly, as demonstrated by the lack of recovery in enzyme function after the unbound drug has been removed from the reaction medium and the fact that its dissociation rate constant is non-significantly different from zero. Such irreversibility may explain the non-competitive inhibition pattern with respect to ATP shown by tideglusib and perhaps other structurally related compounds. The replacement of Cys-199 by an Ala residue in the enzyme seems to increase the dissociation rate, although the drug retains its inhibitory activity with decreased potency and long residence time. In addition, tideglusib failed to inhibit a series of kinases that contain a Cys homologous to Cys-199 in their active site, suggesting that its inhibition of GSK-3β obeys to a specific mechanism and is not a consequence of nonspecific reactivity. Results obtained with [35S]tideglusib do not support unequivocally the existence of a covalent bond between the drug and GSK-3β. The irreversibility of the inhibition and the very low protein turnover rate observed for the enzyme are particularly relevant from a pharmacological perspective and could have significant implications on its therapeutic potential.
Introduction
Alzheimer disease (AD)3 is the most prevalent form of dementia. Around 26 million people worldwide suffer from this withering disorder according to the World Health Organization, and it is believed that this figure will grow to reach nearly 80 million cases by 2050 (1). There currently is no effective treatment that delays the onset or slows the progression of AD. Despite extensive research efforts over the past decades to identify the precise cause of the disease and to understand the mechanisms leading to this pathological state, they still remain elusive, although significant progress has been achieved and many aspects of the biology of AD have been unveiled.
The “amyloid hypothesis” has become the leading hypothesis to explain AD pathophysiology. It suggests that the accumulation of the amyloid β peptide is the primary factor that triggers a cascade of pathogenic events leading to alterations in Tau protein, synaptic dysfunction, and neuronal death (2). Despite recent clinical failures of several drug candidates targeted towards the key steps of the amyloid cascade, the critical role of amyloid in the pathogenesis of AD is still widely accepted, and many efforts are currently being directed to establish how the oligomers formed in the initial steps of amyloid aggregation affect the neurodegenerative process characteristic of this condition and influence the pernicious events that characterize the pathology (3–5).
As a consequence, drug discovery efforts against AD over the past two decades have primarily focused on targets defined by the amyloid cascade hypothesis, so far with disappointing results. These failures underscore the need for novel therapeutic approaches to successfully halt or reverse the pathology and cognitive decline in AD. By contrast, strategies focused on the alterations suffered by Tau protein have received relatively little attention until recently despite the fact that the presence of extensive Tau pathology is central to the disease. In this context, glycogen synthase kinase-3β has recently been proposed as a link between the two major pathological pathways in AD, amyloid and Tau (6–8), leading to the “GSK-3 hypothesis of AD” (9). Thus, GSK-3β inhibition has emerged as one of the most promising therapeutic strategies in AD.
Originally discovered because of its role in the regulation of glucose metabolism, GSK-3β appears to be a cellular hub, integrating several signaling systems. The regulation of its activity occurs by complex mechanisms that are each dependent upon specific signaling pathways (reviewed in Ref. 10). GSK-3β has been identified as the main kinase responsible for the hyperphosphorylation of Tau, leading to the formation of neurofibrillary tangles in AD brains (11–13), and its expression seems to be up-regulated in the hippocampus of AD patients (14–16). Consequently, the enzyme has been recognized as a relevant player not only in the pathogenesis of AD but in other tauopathies as well (10).
Besides having been identified as the major Tau protein kinase, GSK-3β also mediates Aβ neurotoxicity, plays an essential role in synaptic plasticity and memory, might be involved in Aβ formation, and has an important role in inflammation and neuronal death, all key features of AD neuropathology (reviewed in Ref. 17). Indeed, GSK-3β is believed to induce neuronal mortality by prompting the degradation of β-catenin, hence avoiding the transcription of genes involved in cell survival, and also by promoting apoptosis (18, 19). Furthermore, GSK-3β impairs axonal transport through a mechanism triggered by Aβ oligomers and mediated by NMDA receptors (20). GSK-3β has also been confirmed as a key player in the process of memory impairment, one of the characteristic symptoms of AD; it negatively affects neuronal plasticity by promoting the inactivation of cAMP-response element-binding protein, a transcription factor involved in synaptic plasticity. GSK-3β also contributes to the dysfunction of APC (adenomatous polyposis coli gene product) and CRMP2 (collapsin response mediator protein-2), two structural proteins implicated in synaptic remodeling (21, 22). In addition, recent results demonstrate that GSK-3β is directly involved in the long term potentiation inhibition caused by Aβ oligomers (23), and several reports point to a genetic association of the GSK3 gene with the risk of AD either by itself (24, 25) or synergistically with Tau (26) or Cdk5 (cyclin-dependent kinase 5) (27).
Therefore, the discovery of small molecule inhibitors of GSK-3 has attracted significant attention both as a therapeutic agent and as a means to understand the molecular basis of AD and other tauopathies. A significant effort has been made in the past few years to synthesize highly selective, potent GSK-3 inhibitors. Some of them have shown in vivo efficacy in various animal models of AD (28, 29), and a couple of them have advanced to clinical testing (30). Among them, the thiadiazolidindiones (TDZDs) were the first non-ATP competitive inhibitors of GSK-3 that were reported (31). Members of this family have been tested in a number of animal models of AD. Remarkably, sustained oral administration of tideglusib (NP-12, NP031112; chemical structure shown in supplemental Fig. 1) to amyloid precursor protein/Tau transgenic mice led to an improvement in cognitive and behavioral deficits; a significant decrease in the levels of Tau phosphorylation, amyloid deposition and plaque-associated astrocytic proliferation and reduced neuronal loss (32). To the best of our knowledge, tideglusib is the only GSK-3 inhibitor currently in clinical development for the treatment of AD; it is presently being tested in phase II clinical trials for both AD and progressive supranuclear palsy (33).
Despite the successful results obtained for tideglusib in vivo, no data have been reported so far on the in vitro activity of the drug. Indeed, the only published data for compounds of the TDZD series show modest potencies on GSK-3β inhibition, with IC50 values in the micromolar range (31, 34), but little was known about the mechanism of such inhibition, an aspect that is key to understanding and interpreting the effects of the drug as well as guiding the design of novel derivatives with altered features. Therefore, we have undertaken enzymological studies on the inhibition of GSK-3β by tideglusib, and this paper describes the results obtained, which show an irreversible inhibition of the enzyme. The potential pharmacodynamic implications of this mechanism are discussed accordingly.
EXPERIMENTAL PROCEDURES
Tideglusib was prepared in house at the Medicinal Chemistry Department. [35S]Tideglusib (2.28 GBq/mmol on the day the experiment was run) was prepared by Huntingdon Life Sciences (Huntingdon, UK). N-terminal His6-tagged human recombinant GSK-3β expressed by baculovirus in infected Spodoptera frugiperda Sf21 cells was purchased from Millipore (Billerica, MA). Sephadex G-25 (PD-10 columns) was from GE Healthcare. Z'-LYTETM reagents, expression vectors, and mouse monoclonal anti-GSK-3α/β antibody were from Invitrogen. β-Tubulin antibody was from Abcam (Cambridge, MA). HRP-conjugated polyclonal rabbit anti-mouse immunoglobulins were from Dako (Glostrup, Denmark) Alsterpaullone and hypothemycin were from Calbiochem. CT99021 was from Selleck (Houston, TX). All other reagents were of the highest purity available from Sigma unless otherwise stated.
Generation of Mutant GSK-3β
The mutant version of GSK-3β C199A was generated by using the QuikChange multisite-directed mutagenesis kit (Stratagene, Santa Clara, CA) using a human GSK-3β cDNA from a commercial cDNA library in pcDNA4/HisMaxB and a suitable primer (5′-TATTAAAACTCGCTGACTTTGGAAGTG-3′) prepared in house. Constructs were transformed into Escherichia coli JM109 competent cells, and plasmid DNA was extracted with the Wizard® Miniprep kit (Promega, Madison, WI), the mutation being confirmed by DNA sequence analysis. Large scale expression and purification was done at Protein Alternatives S.L. (Tres Cantos, Spain). The plasmid DNA with the mutation was subsequently engineered into pFastBac-Dual with a His6 tag, and MAX Efficiency® DH10BacTM competent cells (Invitrogen) were used to produce recombinant baculovirus molecules. Recombinant bacmid was then transfected into baculovirus-infected S. frugiperda Sf9 insect cells using Cellfectin® II reagent (Invitrogen) following the manufacturer's instructions, resulting in the production of infectious recombinant baculovirus particles. The protein was expressed at 27 °C in a Wave Bioreactor system (GE Healthcare) using the transfected insect cells with a multiplicity of infection of 1. After 72 h, the cells were harvested, and extracts were prepared by sonication with 0.5% Triton X-100 in His-binding buffer (20 mm Tris-HCl, 0.5 m NaCl, 40 mm imidazole, pH 8.0). After removing cellular debris by centrifugation, the protein was purified from the cleared supernatant by immobilized metal ion affinity chromatography on a HiTrapTM column using an ÄKTATM system (GE Healthcare) with a 40–500 mm imidazole gradient in the same buffer over 10 column volumes. Purity and identity of the obtained protein was confirmed by SDS-PAGE and peptide mass fingerprinting by MALDI-TOF/TOF. Final material was dialyzed into 20 mm Tris-HCl, pH 7.4, 500 mm NaCl, 0.5 mm EDTA, 0.5 mm PMSF, 25% glycerol and finally aliquoted, frozen in an ethanol-dry ice bath, and stored at −80 °C.
Enzymatic Assays
The activity of GSK-3β (both wild-type and C199A versions) was measured with the FRET-based Z′-LYTETM technology from Invitrogen using as peptide substrate the so-called “Ser/Thr-9,” a fluorescein- and coumarin-double-labeled 11-mer based on the sequence of human glycogen synthase I containing Ser-641. The assay was carried out at 25 °C according to the manufacturer's instructions in a final volume of 10 μl in 384-well low volume round bottom black plates (Corning Glass) with 50 mm Hepes, pH 7.5, 10 mm MgCl2, 1 mm EGTA, and 0.01% Brij-35 as assay buffer. Enzyme concentration ranged from 2 to 5 nm as determined by active-site titration with the well known inhibitor CT99021 (35, 36). Unless otherwise stated, ATP and peptide concentrations were 12.5 and 2 μm, respectively, corresponding to their respective previously determined Km values. Typically, the assays were run for 1 h in the presence or absence of compounds in a final DMSO concentration of 1%, and samples were processed according to the manufacturer's instructions. When time courses were evaluated, the reactions were stopped at the intended times and processed as above. The final readout (emission ratio between coumarin and fluorescein) was obtained in an EnVision® Xcite plate reader (PerkinElmer Life Sciences), and the resulting values were converted to the amount of product formed using a standard curve with phosphorylated and intact peptides. When concentration responses of the compounds were evaluated, their potencies were determined as the negative logarithm of the IC50 in molar units (i.e. the pIC50) as the experiments were carried out by performing serial dilutions, and therefore the range of concentrations was evenly distributed in a logarithmic rather than a linear scale. pIC50 values were calculated by fitting the resulting data to Equation 1, using the nonlinear regression function of Prism® 5.0 (GraphPad Software Inc., La Jolla, CA).
Equation 1 is a modified version of the classic isotherm equation, where Min and Max are the lower and higher asymptotes of the resulting sigmoid curve, [I] is the concentration of inhibitor in molar units, and n is the Hill coefficient. Percentage inhibition was calculated from the ratio of the enzyme activity in the presence and absence of compound. The mean pIC50 values from duplicate experiments were finally calculated, and these mean values were converted to IC50 values by taking the antilog. Likewise, the range of IC50 values defined by both duplicates was calculated by taking the antilog of the pIC50 range; the resulting value corresponds to the factor that must multiply or divide each IC50 to define such a range.
When double titrations of ATP and tideglusib were performed, the assay was carried out as described above for 15 min (after having proved that linearity was kept during that time and a reasonably good approximation to initial rates was obtained), varying both ATP and tideglusib concentrations while keeping the concentration of peptide substrate fixed. Data analysis was performed by fitting the experimental data to the appropriate equations for competitive, uncompetitive, and non-competitive inhibition. The best results (as judged by F-test) were obtained when fitting to the non-competitive model described by Equation 2,
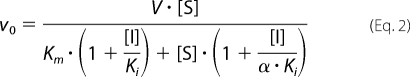 |
where v0 is the initial rate, α is the parameter showing how the binding of the substrate and the inhibitor are mutually affected, Ki is the inhibition constant, Km is the Michaelis constant for ATP, V is the maximum velocity, and [I] and [S] are tideglusib and ATP concentrations, respectively.
Filtration Experiments
The reversibility of GSK-3β inhibition was tested by filtration through Amicon Ultra-0.5 centrifugal filter devices (Millipore) with a cut-off of 30 kDa followed by dilution of the concentrated retentate. Samples of 20 nm GSK-3β in the assay buffer described above were incubated with 1% DMSO or with a 1 μm concentration of the tested compounds in 1% DMSO for 30 min and then filtered by centrifugation according to the manufacturer's instructions. The retentate sample was diluted 20-fold with assay buffer to recover the original volume. This procedure was repeated twice (i.e. total dilution was 400-fold), and finally, the diluted retentate was used to measure the GSK-3β activity as described above.
Binding Experiments with Radiolabeled Tideglusib
To further investigate the interaction of tideglusib with GSK-3β, binding studies with radioactive compound were performed. [35S]tideglusib (207 Bq/nmol) at 55 μm was incubated with 5 μm GSK-3β for 1 h at 25 °C in 315 μl of 50 mm Tris-HCl, pH 7.5, containing 150 mm NaCl and 0.1 mm EGTA. The incubation was extended for another 30 min after having added 35 μl of the same buffer with or without 100 mm DTE. Samples were then processed in three different ways. First, an aliquot of 125 μl of each sample was mixed with 375 μl of 8 m GdnHCl in H2O and heated at 80 °C for 5 min. A second aliquot of 125 μl was diluted up to 500 μl with H2O and left at room temperature for 5 min. In both cases, the free drug was removed afterwards by gel filtration through Sephadex G-25, and the amount of bound drug was determined by liquid scintillation counting on a 1450-MicroBeta TriLux counter (PerkinElmer Life Sciences). Finally, a third 40-μl aliquot of each original sample was mixed with 10 μl of denaturing electrophoresis sample buffer without reducing agents, and 35 μl of this mixture was loaded onto a 10% polyacrylamide gel and subjected to SDS-PAGE (again in the absence of reducing agents except for the DTE already included in the corresponding sample), followed by fluorography of the dried gel.
Protein Turnover Rate Experiments
To determine the turnover rate of GSK-3β, cortical neurons were treated with the protein synthesis inhibitor cycloheximide, and the total GSK-3β concentration was measured at the indicated times by immunoblotting. Cortical neurons from CD-1 mouse embryos of 18-day gestation were obtained and maintained following procedures already described (37). After 4 days in vitro, the cells were treated with 50 μm cycloheximide, and at the intended time, they were lysed with buffer containing 10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EDTA, 2 mm Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% sodium deoxycholate, 1 mm PMSF, and the CompleteTM protease inhibitor mixture (Roche Applied Science). The lysates were centrifuged at 13,000 × g for 3 min at 4 °C, resuspended in denaturing electrophoresis sample buffer, submitted to electrophoresis in a 10% SDS-polyacrylamide gel, and transferred to Hybond ECL nitrocellulose membranes (GE Healthcare). Membranes were then blocked with 10% nonfat milk and incubated overnight with the mouse monoclonal anti-GSK-3α/β antibody (1:1000 dilution) and β-tubulin antibody (1:500) as the loading control. Finally, they were treated with the corresponding HRP-conjugated anti-mouse immunoglobulin, and the immunoreactive proteins were visualized with an enhanced chemiluminescence detection system (GE Healthcare). The relative levels of GSK-3β were quantified by densitometry of the scanned images of the immunoblot. The half-life of the enzyme was calculated by fitting the time course of GSK-3β disappearance to a single exponential decay equation. Cell viability was monitored in parallel (LDH cell cytotoxicity kit from Roche Applied Science) to ensure that cell survival was above 90%.
Evaluation of Inhibition on Kinase Panel
The inhibitory activities of tideglusib and hypothemycin on a panel of selected kinases were evaluated in the Invitrogen European Screening Center (Paisley, UK). Compounds were tested in duplicate at a single concentration of 10 μm on a group of selected kinases, the enzymatic activity of which was measured using the Z′-LYTETM technology at ATP and peptide concentrations around their Km values, except for MEK1, MEK2, p38α, and JNK1, for which ATP was at 100 μm. In a few cases where it was not possible to monitor the activity of the kinases (MEK3, NIK, TAK1-TAB1, MNK2, NLK, and ZAK) the ability of the compounds to displace the binding of fluorescent analogues of known ATP-competitive inhibitors was measured using the time-resolved FRET-based LanthaScreenTM technology. Details on the nature of the kinases tested and the average of the results obtained in each case are presented in supplemental Table 1.
RESULTS
The synthesis of TDZDs was first reported in 2002 (31). In that paper, the compounds were described to inhibit GSK-3β in a non-competitive mode with respect to ATP. We therefore first checked if, as expected, tideglusib also shares this property. To that end, double ATP and inhibitor titrations were run while keeping the peptide substrate concentration fixed. Fig. 1 shows that the experimental results fit well to a non-competitive inhibition model (termed “mixed” by some authors (38) and “non-competitive” by others (39)) with a Ki of 60 ± 7 nm and an α value of 22 ± 5. The high value for this latter parameter suggests a preponderance of the competitive component of the inhibition. Nonetheless, it must be noticed that such inhibition pattern is only meaningful from a mechanistic perspective if the compound was reversible, because the equations defining this type of inhibition, to which the experimental data have been fitted, were deduced on the basis of a rapid, reversible binding of the inhibitor to the enzyme. Indeed, irreversible inhibitors often display a non-competitive pattern when double titrations are performed. Consequently, we decided to test the reversibility of the inhibition by running filtration experiments. The aim of such studies was to investigate if the removal of the unbound compound promoted the dissociation of the enzyme-inhibitor complex and led to inhibition fading. As observed in Fig. 2, the inhibition caused by 1 μm tideglusib (∼20 times over its calculated Ki) remained after the free, unbound drug had been removed by two consecutive filtration and dilution cycles, causing a 400-fold dilution of the small molecular weight material. In the same experiment, the kinase activity was fully recovered when the enzyme was treated with its known reversible inhibitor SB-415286 at 1 μm (30 times over its reported Ki) (40). These results strongly suggest that the inhibition caused by tideglusib on GSK-3β was irreversible or at least behaved as irreversible within the time frame of the experiment (1 h of postdilution incubation with buffer followed by a 1-h enzymatic reaction), meaning that the off-rate of the drug is low enough to avoid the recovery of the enzyme activity within this 2-h period. Accordingly, it is reasonable to expect that slow inhibition kinetics must be governing the process, and, therefore, commonly used efficacy parameters, such as IC50, will depend on the enzyme-drug incubation period. This fact is clearly shown in Fig. 3A, where a significant shift in the dose-response curve was observed as a consequence of increasing the incubation time between enzyme and inhibitor by 1 h, the shift leading to a 20-fold decrease in the IC50 value. Such time-dependent inhibition is not an exclusive feature of tideglusib but rather a compound class property, because it is also noticeable in other structurally related TDZDs covering a wide (3 orders of magnitude) range of potencies; as observed in the correlation plot displayed in Fig. 3B, all of the experimental points fall below the y = x line, meaning that the potencies were significantly higher (i.e. the IC50 values were lower) when a 1-h preincubation step was included.
FIGURE 1.
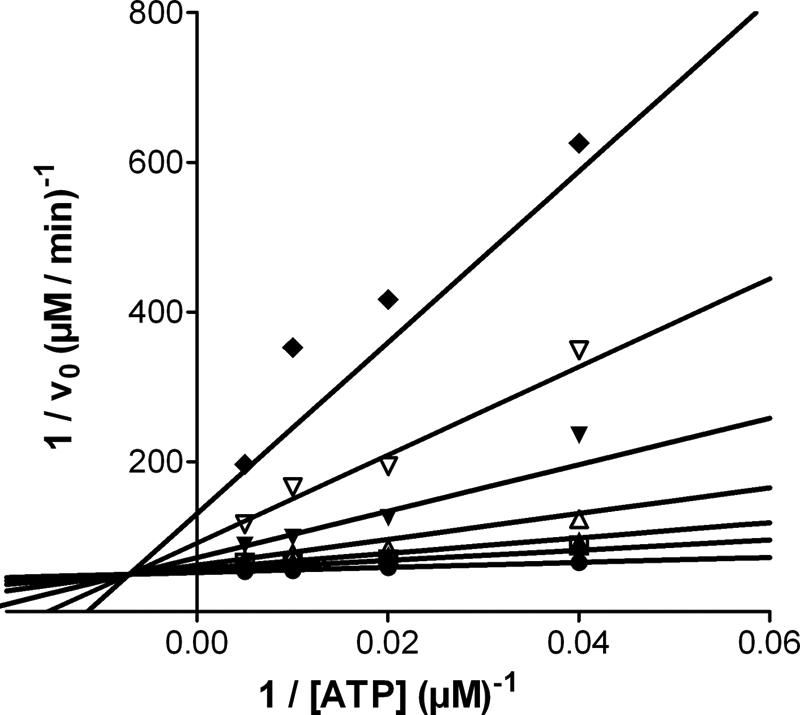
Double reciprocal plots of the inhibition caused by tideglusib on GSK-3β. The experiment was carried out as described under “Experimental Procedures” at a fixed non-saturating concentration of substrate peptide (2 μm) and at varying ATP concentrations. Rates were calculated from the amount of phosphopeptide produced after 15 min and are expressed in μm product/min, whereas ATP concentration is in μm. The lines correspond to the double-reciprocal transformation of the curves obtained by nonlinear regression fitting of the experimental data to Equation 2. Tideglusib concentrations were 0 (●), 0.063 (■), 0.125 (▴), 0.25 (△), 0.5 (▾), 1 (▿), and 2 μm (♦).
FIGURE 2.
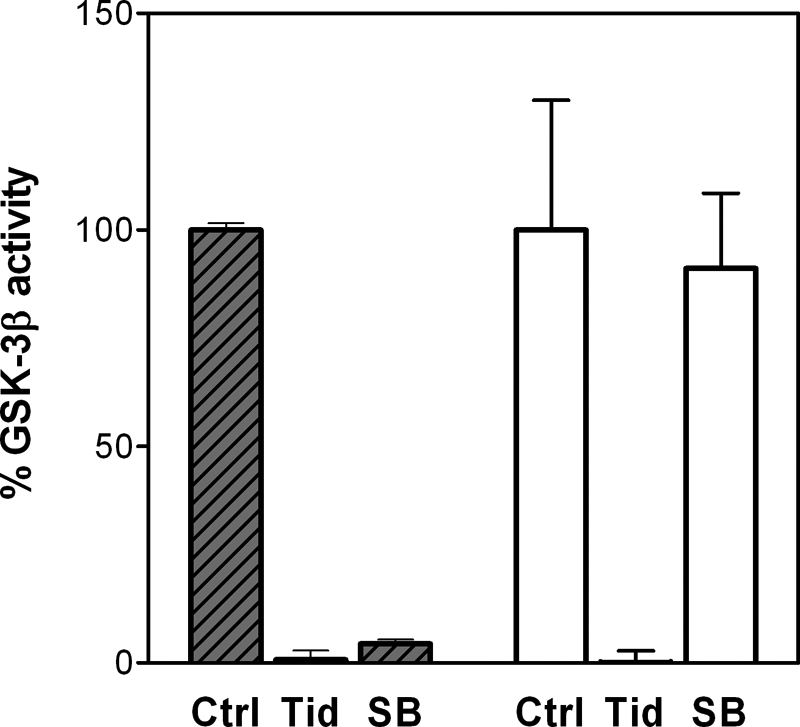
Activity of samples from the filtration-dilution process. The values on the vertical axis are expressed as percentage of GSK-3β activity with respect to the control sample before the process was started. Data represent the average of three independent samples measured in duplicate, with error bars denoting S.D. Shadowed bars, samples before the process. Clear bars, samples after two filtration-dilution processes leading to a 400-fold dilution of the drugs. Ctrl, control, untreated GSK-3β; Tid, GSK-3β treated with 1 μm tideglusib; SB, GSK-3β treated with 1 μm SB-415286.
FIGURE 3.

Tideglusib and other TDZDs as time-dependent inhibitors. A, concentration-response curves of tideglusib on GSK-3β. The experimental conditions are described under “Experimental Procedures,” including (●) or not including (○) a 1-h preincubation of the enzyme with the compound. Data were fitted to Equation 1 to calculate the IC50 values, which resulted in a value of 5 nm for the preincubation case and 105 nm for the non preincubation one. The data presented here correspond to the average of triplicate determinations ± S.E. B, effect of preincubation on the potencies of TDZDs. IC50 values were calculated for several TDZD derivatives as before, with or without a 1-h preincubation. Each compound is represented by a dot; tideglusib is denoted with a filled star. The dashed line denoting identity (y = x) is presented for visual reference.
The slow inhibition kinetics rendered by tideglusib enables the determination of the microscopic kinetic constants defining the binding of the compound to GSK-3β. This type of behavior can be widely described by the mechanism shown in Scheme 1 (described in Ref. 41), which assumes a two-step process, including a first recognition step (i.e. the formation of the enzyme-inhibitor complex) followed by a second step that leads to a different form of this complex (denoted as E*-I in Scheme 1) (e.g. by enzyme isomerization or conformational rearrangement).
SCHEME 1.

Mechanism of two-step inhibitor binding.
Equation 3 describes the kinetics of product formation by an enzyme inhibited through such a mechanism at a given inhibitor concentration, as described by Morrison and Walsh (41).
where v0 and vs represent the initial and steady-state velocities of product formation while the enzyme is being inhibited, and kobs is the apparent rate constant to approach the inhibition equilibrium at a given inhibitor concentration. Such a concentration dictates the value of kobs according to Equation 4 (assuming that k2 ≫ k3; i.e. the first equilibrium is reached rapidly),
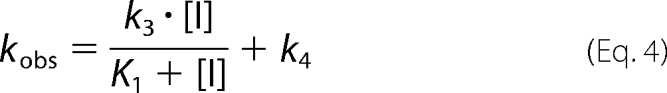 |
where K1 is the dissociation constant of the first E-I complex (i.e. the quotient k2/k1). Therefore, a secondary plot of kobs versus inhibitor concentration would yield a hyperbola allowing the determination of k4 (from the y intercept), k3 (from the upper asymptote after subtracting k4), and K1 (from the abscissa corresponding to the midpoint between the y intercept and the upper asymptote of the hyperbola). Thus, to gain further insight into the kinetics of tideglusib inhibition on GSK-3β, we obtained progress curves in the presence of several concentrations of the compound, and the experimental data were fitted to Equation 3, from which kobs values were derived and plotted against tideglusib concentration. As shown in Fig. 4, the experimental data nicely fitted to Equation 3, and the secondary plot delivered a straight line (inset, R2 = 0.998) instead of a hyperbola. This result points to a large K1 value so that tideglusib concentrations used in the experiment were far from those leading to the limiting value of kobs (i.e. K1 ≫ [I] in Equation 4). In such a case, the intermediate E-I is kinetically insignificant relative to the formation of E*-I, and the whole process behaves as if it was composed on one single step yielding this latter complex (41–43). Consequently, the value of the slope of this line corresponds to k3/K1, which is the pseudo-second-order rate constant leading to the formation of E*:I (44). Such a value ± S.E. from the linear fitting is 1.16 ± 0.04 × 103 m−1 s−1, a relatively small figure that fits well with the assumption made above about the high K1 figure (see “Discussion”). On the other hand, k4 was obtained from the intercept and calculated to be 2.15 ± 2.16 × 10−5 s−1, a value not significantly different from zero as inferred from two observations; the S.E. value is similar in magnitude to the deduced value for the parameter, and the linear fitting obtained when the line was forced to pass through the origin yielded a similar S.D. of the residuals (1.654 × 10−3 versus 1.652 × 10−3), indicating that the goodness of the fit was equally acceptable. Therefore, k4 can be considered zero for practical purposes, suggesting that there is no dissociation of the E*-I complex, which means that the inhibition of GSK-3β caused by tideglusib is functionally irreversible, a conclusion consistent with the results obtained from the filtration experiments.
FIGURE 4.
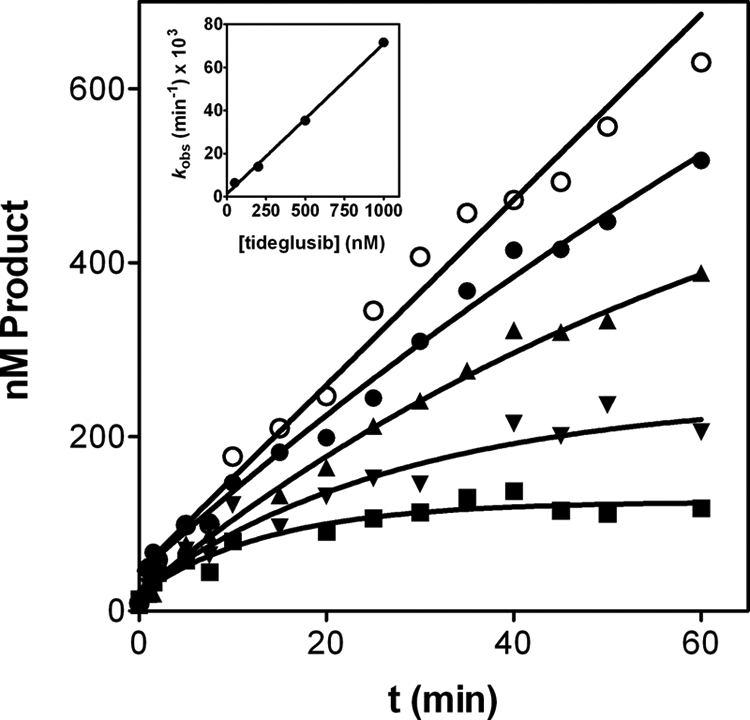
Time course of tideglusib inhibition on GSK-3β. Progress curves were obtained with 2 nm enzyme and tideglusib at 0 (○) 50 (●), 200 (▴), 500 (▾), and 1000 nm (■). Experimental data were fitted to Equation 3; the lines represent the best fit yielding the corresponding kobs values that were plotted versus tideglusib concentration (inset) and fitted to a straight line with R2 = 0.998. Data are representative of two independent experiments performed in duplicate.
It is very common to confound irreversible inhibition with covalent binding as the majority of irreversible inhibitors covalently modify their target enzymes. However, both terms must be differentiated, and the scientific literature actually provides several examples of irreversible inhibitors not acting through a covalent mechanism, such as aryl methyl sulfonyls and sulfonamides for COX-2 (45), as well as common drugs like allopurinol for xanthine oxidase or acyclovir for HSV-1 DNA polymerase (see Ref. 46 for a detailed review). Nonetheless, the results obtained in classical molecular interaction potential mapping studies led some authors to suggest that TDZDs may bind covalently to GSK-3β through its Cys-199 residue (34). Consequently, we believed it appropriate to investigate whether this hypothesis was true and whether such covalent binding took place and was responsible for the irreversible inhibition of GSK-3β caused by tideglusib.
Hypothemycin, a fungus-derived polyketide, is a resorcylic acid lactone that contains a cis-enone in the macrocycle. This molecule has been reported to inhibit several kinases. This cis-enone is an effective Michael acceptor of nucleophiles, such as Cys groups. Schirmer et al. (47) completed a detailed bioinformatics analysis on the kinases inhibited by hypothemycin and found that many of them included a conserved Cys residue in the ATP-binding site, whereas such a residue was absent in the active site of all of the kinases unaffected by this compound. In their analysis, they identified a total of 46 kinases in the human kinome containing this Cys residue, which is adjacent to the conserved Asp involved in Mg2+ binding. GSK-3β was among these kinases, and the relevant Cys residue was Cys-199. We therefore decided to evaluate the activity of tideglusib in a panel of kinases, including as many as possible of those 46 that contain the conserved Cys, comparing the inhibitory profiles of tideglusib and hypothemycin. A panel of 68 kinases (including 26 of those 46) was selected on the basis of their structural and functional diversity in an effort to include a representative sample of the entire human kinome. The evaluation was carried out for both compounds at a fixed concentration of 10 μm, and the results obtained were compared in the correlation plot displayed in Fig. 5, where the line denoting identity (y = x) is shown for visual reference. As observed, the inhibitory profiles of the compounds are completely dissimilar, and only a few dots are placed near the identity line. Moreover, this lack of correlation is even more pronounced if the analysis focuses only on the kinases with the conserved Cys residue, because most of the dots corresponding to these kinases appear in the upper part of the graph, meaning that the extent of the inhibition caused by hypothemycin on these kinases is larger than that caused by tideglusib. This disparity and the fact that only 10 of the 26 “Cys-conserved” kinases were significantly inhibited (more than 50%) by 10 μm tideglusib suggest that, even if the drug was acting on GSK-3β by modifying a Cys residue in the active site, the process should involve specific interactions that accounted for the differential effects observed between tideglusib and hypothemycin, and it was not simply the result of a reactive, non-selective chemical modification.
FIGURE 5.
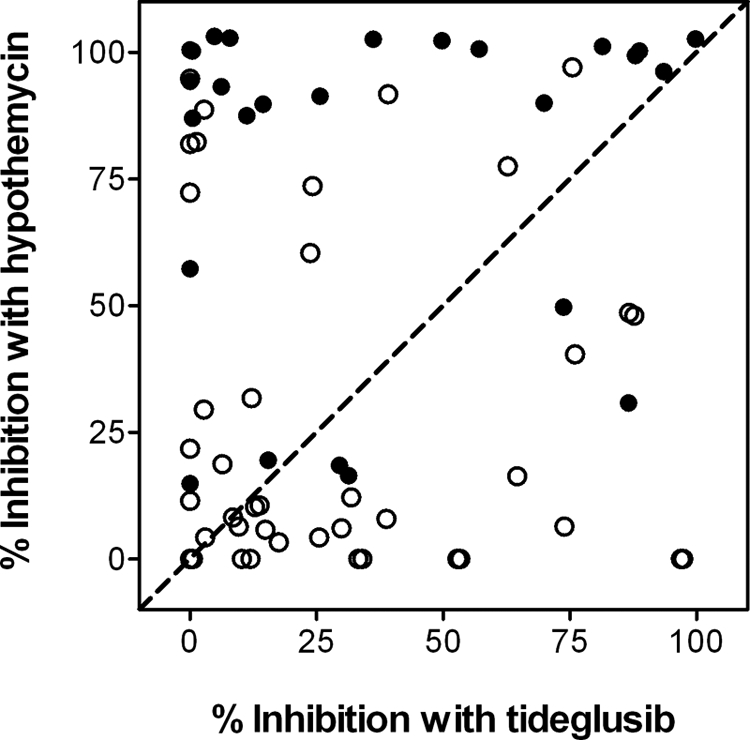
Inhibition caused by hypothemycin and tideglusib on a panel of selected human recombinant kinases. As detailed under “Experimental Procedures,” both compounds were tested in duplicate at a single concentration of 10 μm. Each dot in the plot corresponds to the average of the effect caused on a single kinase. ●, kinases containing a conserved Cys in their active site; ○, kinases not containing such residue. The dashed line denoting identity (y = x) is presented for visual reference.
To gain further insight into the potential role of Cys-199 on the inhibition caused by tideglusib, a C199A mutant version of human recombinant GSK-3β was prepared, and the effect of the drug was again compared with that of hypothemycin. Because hypothemycin belongs to a compound class known to inhibit with slow kinetics (48), the effect of a 1-h preincubation step was also studied. Alsterpaullone, a commonly used, well known reversible inhibitor of the enzyme that acts through a rapid equilibrium process (49), was used as a control. The results, summarized in Table 1, show that the three compounds inhibited the wild-type enzyme with different potencies, the effect of alsterpaullone being independent of the preincubation step, as expected. However, hypothemycin failed to inhibit the mutant enzyme even when a preincubation step was included, whereas tideglusib showed a modest inhibition, which became potent (IC50 = 60 nm) when the C199A enzyme and the drug were preincubated for 1 h. Alsterpaullone showed identical results with the wild-type and the mutant enzymes, demonstrating that its mechanism of inhibition is independent of the presence of the Cys-199 residue.
TABLE 1.
IC50 values (expressed in nm) for GSK-3β inhibition with or without a 1-h preincubation of the enzyme with each compound
Data correspond to the mean of two independent determinations; the mean was calculated from the logarithm values as described under “Experimental Procedures.” The values shown in parentheses correspond to the factor that must multiply or divide the IC50 to define the range corresponding to the duplicate determinations.
| Inhibitor | GSK-3 β WT |
GSK-3 β C199A |
||
|---|---|---|---|---|
| No preincubation | 1-h preincubation | No preincubation | 1-h preincubation | |
| Tideglusib | 105 (1.02) | 5 (1.02) | 7,000 (1.95) | 60 (1.02) |
| Hypothemycin | 14,000 (1.02) | 210 (1.15) | >100,000 | >100,000 |
| Alsterpaullone | 45 (1.12) | 40 (1.10) | 15 (1.15) | 50 (1.10) |
This result highlights the differences between the mechanisms by which hypothemycin and tideglusib inhibit GSK-3β. Cys-199 is essential for the inhibitory effect of the former, but the role of this residue on the inhibition caused by tideglusib is less clear. Indeed, although replacing Cys-199 impaired notably the inhibitory effect of tideglusib, such an effect was not fully abolished. Moreover, the compound demonstrated a significant potency on the mutant enzyme if enough time was provided for the interaction to proceed. Bearing this in mind, kinetic studies were performed with tideglusib and the mutant enzyme in order to compare the results obtained with the wild-type version. Progress curves in the presence of several concentrations of tideglusib were obtained (Fig. 6), and the experimental data were processed as above. As was the case with the wild-type enzyme, the kobs displayed a linear dependence on the concentration of tideglusib (Fig. 6, inset) rendering a k3/K1 value of 1.91 ± 0.15 × 102 m−1 s−1 and a k4 of 8.8 ± 0.8 × 10−5 s−1. It is noticeable that the C199A mutation led to a decrease in the k3/K1 value of 1 order of magnitude with respect to that of the wild-type enzyme (see Table 2), suggesting that Cys-199 replacement decelerated the process of E*-I formation. The most relevant information was provided by the dissociation constant k4, which is 4-fold higher and, more remarkably, significantly different from zero. Still, tideglusib showed a long residence time in the mutant enzyme (t½ = 2.2 h as deduced from k4), but the fact that k4 was distinct from zero suggests that this GSK-3β mutation turned tideglusib from an irreversible to a longstanding reversible inhibitor, and this might be consistent with the formation of a covalent bond between the drug and Cys-199. However, the long half-life and the reduced but still significant potency (Ki* around 500 nm) suggests that the Cys residue is not totally essential for the inhibitory activity of the compound.
FIGURE 6.
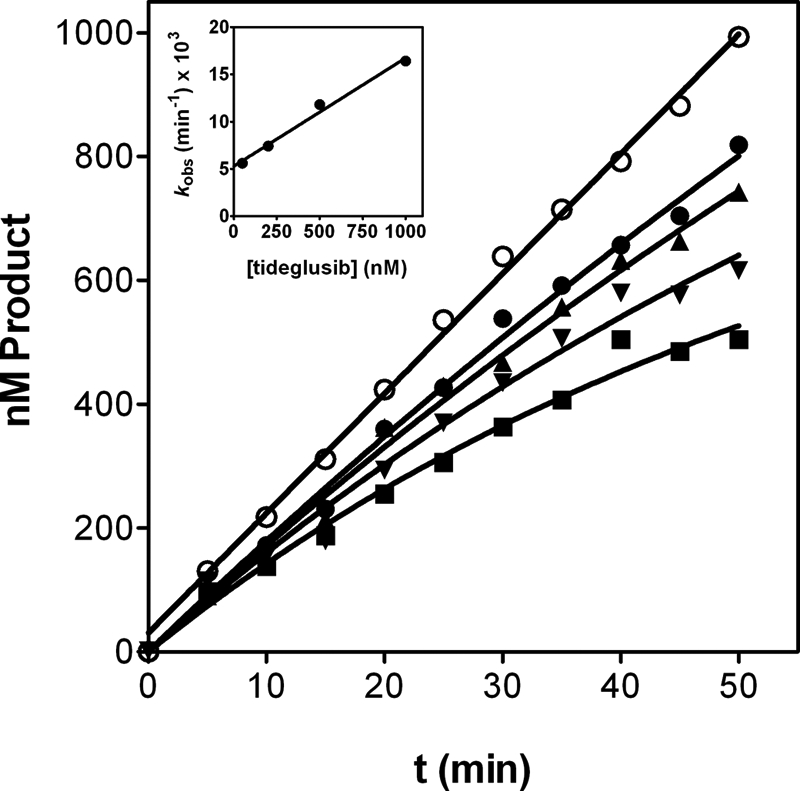
Time course of tideglusib inhibition on GSK-3β C199A. Progress curves were obtained with 5 nm enzyme and tideglusib at 0 (○), 50 (●), 200 (▴), 500 (▾), and 1000 nm (■). Experimental data were fitted to Equation 3. The lines represent the best fit yielding the corresponding kobs values that were plotted versus tideglusib concentration (inset) and fitted to a straight line with R2 = 0.988. Data are representative of two independent experiments performed in duplicate.
TABLE 2.
Summary of kinetic parameters for the inhibition caused by tideglusib on GSK-3β WT and C199A
Errors correspond to the S.E. deduced from the linear fitting to Equation 4. t½ was deduced from k4, whereas Ki* has been approximated as the quotient between k4 and k3/K1 assuming k4 ≪ k3. Neither t½ nor Ki* have been calculated for WT GSK-3β, given the large k4 error.
| GSK-3β version | k3/K1 | k4 | t½ | Ki* |
|---|---|---|---|---|
| m−1s−1 | s−1 | h | nm | |
| WT | 1.16 ± 0.04 × 103 | 2.15 ± 2.16 × 10−5 | ||
| C199A | 1.91 ± 0.15 × 102 | 8.8 ± 0.8 × 10−5 | 2.2 ± 0.2 | 461 ± 78 |
To shed light on the mode of interaction of tideglusib with GSK-3β, binding experiments with radiolabeled [35S]tideglusib were performed. The aim of these experiments was to investigate whether the compound remained bound to the enzyme after having subjected the complex to denaturing conditions. Given that the possible interaction with Cys-199 might involve the formation of a disulfide bond with the sulfur atom of the TDZD ring, the effect of a reducing agent (DTE) was also explored. GSK-3β and [35S]tideglusib were mixed and incubated at room temperature before adding buffer with or without DTE. The samples then followed a double-pronged strategy designed to obtain both qualitative and quantitative information of the same approach; while a small aliquot was submitted to electrophoresis under denaturing conditions followed by fluorography, the remaining part of each sample was either treated with a chaotropic agent (6 m GdnHCl at 80 °C for 5 min) or left untreated before removing the unbound drug by gel filtration chromatography on Sephadex G-25 and measuring the remaining radioactivity by liquid scintillation counting. As can be observed in Fig. 7A, only the DTE-untreated samples rendered a very faint band, hardly visible to the naked eye, which could be detected in the fluorography at a position corresponding to the molecular mass of GSK-3β (∼47 kDa) only after very long exposure times (8 days). Although the presence of a band could suggest covalent modification, such a very weak response with respect to the amount of GSK-3β loaded on the gel (126 pmol) and the specific activity of the radiolabel (207 Bq/nmol) prevents us from obtaining clear conclusions from this result and rather points to a possible artifactual nature. Furthermore, the quantitative analysis after gel filtration (Fig. 7B) shows that whereas the sample treated under non-denaturing conditions presented significant levels of tideglusib binding (0.47 mol/mol of GSK-3β), these levels were notably reduced when the sample was denatured with GdnHCl (0.14 mol/mol of GSK-3β) and were fully abolished when DTE was included in the treatment. In principle, the levels of bound drug in the GdnHCl-treated sample should have been either unaffected or completely suppressed with respect to the untreated sample, depending on whether the binding was covalent or not, respectively. Still, the observed residual bound levels might be consistent with non-covalent binding, assuming that the protein has suffered an incomplete denaturation, but the fact that the levels were so decreased is harder to explain if the binding was covalent. Likewise, the possibility of a covalent modification of the protein that eventually progresses to a final adduct in which the sulfur atom of tideglusib (i.e. the one being isotopically labeled) is lost cannot be excluded. Therefore, although the covalent binding between tideglusib and GSK-3β cannot be completely ruled out, its presence is not unequivocally supported by the currently available evidence.
FIGURE 7.

Binding experiments with [35S]tideglusib. A, fluorography of samples prepared and processed as described under “Experimental Procedures,” obtained after 8 days of exposure. Samples were loaded in duplicate lanes, leaving empty lanes among them (denoted with dashes) to avoid cross-lane contamination issues. Lane 1, 14C-labeled molecular weight markers (355 Bq loaded in 3 μl). Lanes 2 and 3, samples not treated with DTE. Lanes 4 and 5, DTE-treated samples. B, amount of bound drug after gel filtration on Sephadex G-25. Samples were processed as described under “Experimental Procedures”; the stoichiometry of the binding was calculated from the amount of radioactivity and the protein recovered from the column as determined by absorbance at 280 nm. Error bars, S.D.
The functionally irreversible inhibition of GSK-3β by tideglusib makes this compound a potent pharmacological tool. To further understand the physiological consequences of this fact, the protein turnover rate of the enzyme was monitored in primary cultures of mouse cortical neurons. Initial approaches to that goal with classical pulse and chase experiments did not deliver any interpretable result, because the incubation of cells with [35S]Met/Cys in Met- and Cys-free medium for up to 3 h failed to label enough endogenous GSK-3β. Longer incubation times could not be sustained due to toxic effects on the cell culture. This result by itself may suggest a slow turnover for this protein and led us to take a different approach monitoring the enzyme degradation after halting protein synthesis with cycloheximide (Fig. 8), a methodology that, despite some caveats (see “Discussion”), has previously rendered results similar to those obtained by pulse and chase (50, 51). The experiment could be extended up to 48 h before significant cell toxicity occurred, and the exponential decay observed enabled us to estimate a t½ of 41 ± 4 h for GSK-3β, despite the fact that about 40% of the enzyme levels at t = 0 was still present after 48 h of cycloheximide treatment. The inclusion of 1 and 10 μm tideglusib in the culture medium did not significantly change this result (data not shown). As suggested above, such a long half-life might explain the failure of the pulse and chase approach and is consistent with results reported by other authors using a similar procedure in rat cerebellar granule neurons (52) and in HEK-293 cells (53). A similar result was also obtained when the study was performed by our group with human neuroblastoma SH-SY5Y cells as well as with other non-neuronal cell lines, such as HepG2 and Jurkat cells (data not shown). This long half-life and the functionally irreversible inhibition caused by tideglusib point to a longstanding effect of the compound and may imply pharmacodynamic consequences of potential clinical application.
FIGURE 8.
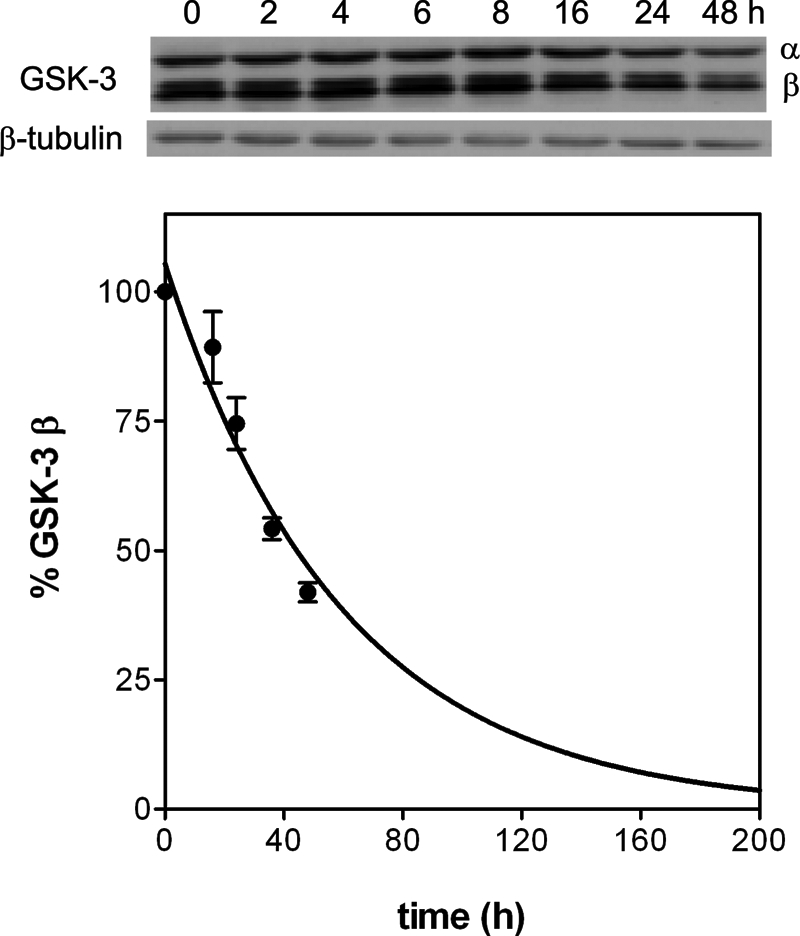
Half-life of GSK-3β in mouse cortical neurons. Cortical neurons at 4-days in vitro were treated with 50 μm cycloheximide and at the intended times were processed as described under “Experimental Procedures.” The intensity of the bands observed in the immunoblots (above) was used to quantify the amount of GSK-3β after normalizing with the amount of β-tubulin. The line corresponds to the nonlinear regression fitting of the experimental points to a single exponential decay equation yielding a decay rate of 0.017 ± 0.02 h−1, which corresponds to a half-life of 41 ± 4 h.
DISCUSSION
Our data demonstrate that tideglusib is an irreversible inhibitor of GSK-3β. This was evidenced by the lack of recovery of enzymatic activity once the unbound drug was removed, but also by the extremely low value of the dissociation rate constant k4, which was not significantly different from zero. Such irreversibility may explain the apparent non-competitive pattern with respect to ATP (54) that has been obtained in double titrations and had also been previously reported for other TDZDs (31, 34). Thus, it is not possible to derive a straightforward conclusion from such an inhibitory pattern (e.g. interpreting that the inhibitor and ATP bind to different enzyme forms and maybe at different sites). It is interesting to observe that the α value deduced from the fitting of the experimental data to the non-competitive equation was significantly higher than 1 (α = 22 ± 5), pointing to a predominantly competitive component.
Noticeably, when Cys-199 was replaced by Ala, the value of k4, although small, became significantly different from zero, denoting the possible existence of a slow dissociation step for the complex formed with this mutant enzyme. Although this fact might be consistent with the hypothesis of a covalent binding to Cys-199, none of the currently available experimental evidence supports this possibility unequivocally. The fluorography obtained after SDS-PAGE of preincubated mixtures of [35S]tideglusib and GSK-3β revealed very weak bands only for the DTE-untreated samples, but the faint intensity of these bands is not consistent with the amount of protein loaded and the specific activity of the radiolabeled drug. The quantification of the binding after removing the unbound drug by gel filtration showed a significant decrease in the levels of bound [35S]tideglusib after denaturation with 6 m GdnHCl, a result that would support the non-covalent nature of the interaction. Moreover, the low levels of binding inferred from the small amount of drug that remains stuck to the denatured sample are not consistent with the high levels of inhibition exerted by tideglusib on GSK-3β, these latter being more in agreement with the binding levels observed for the untreated sample. On the other hand, the complete absence of bound drug in the DTE-treated samples may be a reflection of the instability of TDZDs in the presence of thiolic agents at high concentrations, which cannot be prevented even if the drug is bound to GSK-3β. In any case, the significant but partial suppression of bound tideglusib in the GdnHCl-treated sample compared with the total elimination in the DTE-treated ones can probably be explained by incomplete denaturation of the former. Alternatively, the formation of a covalent complex that evolves and finally yields a modified protein that does not include the sulfur atom of tideglusib (hence not being radioactive) cannot be ruled out. Notably, alternative approaches, such as tryptic digestion followed by MALDI-TOF analysis and peptide mass fingerprinting, have not revealed any difference between the GSK-3β-tideglusib sample and GSK-3β alone (data not shown), suggesting that either there is no covalent modification, or it is too subtle to be detected by the methodologies used. In any case, the kinetic data have shown that, regardless of the existence of a covalent bond, other critical interactions between tideglusib and GSK-3β, not involving Cys-199, must take place, as suggested by two important findings: first, the ability of the drug to inhibit the C199A mutant enzyme with both moderate affinity (Ki* ∼500 nm) and long residence time (t½ = 2.2 h), and, second, the lack of effect on other kinases that include a Cys homologous to Cys-199 in their active site, suggesting that such a residue is not essential for the effect of the drug. These results support the nature of tideglusib as a specific inhibitor and differentiate it from other more reactive molecules, which may eventually lead to the same form of inactivated enzyme.
The kinetics of inhibition by tideglusib showed a linear dependence of the rate constant of approach to equilibrium, kobs, with inhibitor concentration. This suggests that, under our experimental conditions, the intermediate complex E-I (see Scheme 1) was kinetically insignificant relative to the rate of inactivation leading to E*:I and precluded us from determining the values of k3 and K1. In a classic article about slow binding inhibition, Morrison and Walsh (41) presented a comprehensive table with the relevant kinetic parameters for many inhibitors of this kind, including drugs like methotrexate and trimethoprim. It is noticeable that all of the tabulated values for k3 fell in a narrower band than those for K1: 2–3 orders of magnitude versus 6. This fact explains the negative linear correlation between the pseudo-second-order rate constant k3/K1 and the equilibrium constant K1 shown in Fig. 9 (R = 0.96, p < 0.0001). Such a correlation may enable us to obtain a gross estimate of K1 (hence also k3) from the experimental value of k3/K1; for the interaction of WT GSK-3β and tideglusib, this approximation suggests that K1 may be on the order of 10−4 m, and therefore k3 may be in the range of 10−1 s−1. The high value for K1 would be consistent with the poor kinetic significance of the E-I complex and would explain the linear dependence of kobs with tideglusib because the tested concentrations of the inhibitor were not higher than 1 μm, (i.e. K1 ≫ [I] in Equation 4, as mentioned under “Results”). This may indicate a relatively weak initial binding of the compound to the enzyme, which is strengthened during the subsequent step, leading to the formation of the irreversible complex with a pseudo-second-order rate constant of 1.16 ± 0.04 × 103 m−1 s−1.
FIGURE 9.
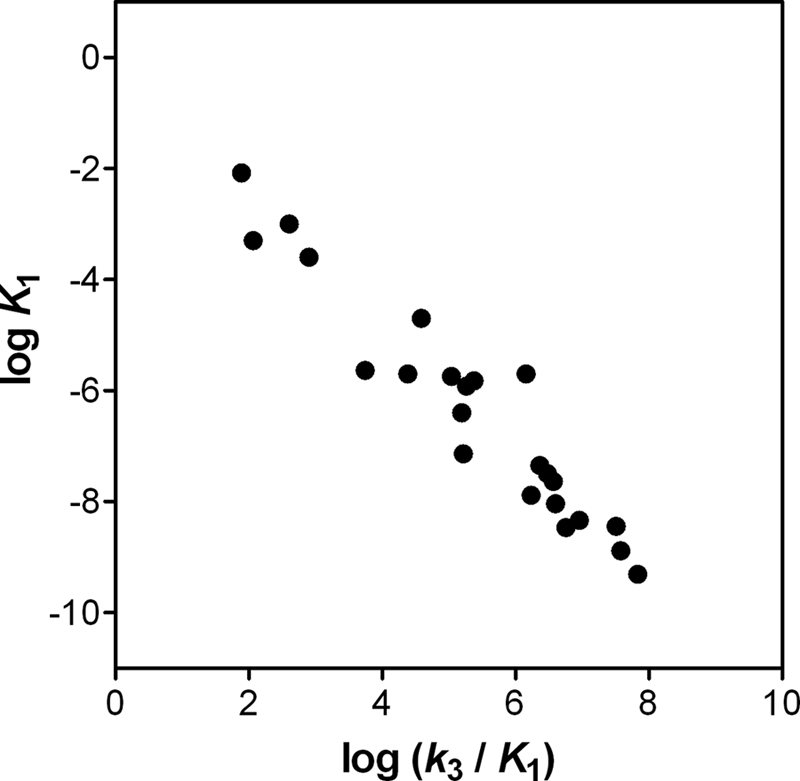
Correlation between K1 and k3/K1 values reported in Ref. 41. The values correspond to 23 enzyme-inhibitor pairs reported in Ref. 41.
Enzyme inhibitors acting through covalent binding to their targets or being functionally irreversible have usually been dismissed when found in drug discovery programs, often due to concerns about safety liabilities. However, their use in the clinic has a long tradition of success, and several voices have recently defended their use (55). Indeed, a variety of approaches have been followed by different groups to design either irreversible or covalently bound kinase inhibitors with therapeutic potential as a strategy to overcome drug resistance (56) or to improve the selectivity of the inhibition (57). Although the currently available data do not prove unequivocally the covalent binding of tideglusib to GSK-3β, the irreversible inhibition it causes becomes more relevant when considering the slow turnover rate of GSK-3β in neuronal cells. Nevertheless, the impossibility of running a classical pulse and chase experiment has precluded us from obtaining an accurate value for the enzyme half-life, and two main caveats must therefore be considered when interpreting this result. First, because protein synthesis was not synchronized in our experiments, the population of GSK-3β being degraded was a mixture of aged molecules and recently synthesized ones, so the observed decay rate might have been accelerated by the influence of the former, and the half-life obtained might consequently have been underestimated; hence, it must be considered as a sort of lowest value. Second, because protein synthesis has been halted with cycloheximide, it may be simplistic to consider that the protein degradation machinery was unaffected, and therefore the validity of the result can be questioned. Nonetheless, besides the fact that this procedure is common and widely used, the long half-life estimated here for GSK-3β is consistent with values described by other authors (although using the same approach) in different cell lines and in primary neurons and is also consistent with the most likely explanation for the failure of the pulse and chase strategy. On the basis of this reasoning, we believe that the GSK-3β half-life figure we have determined is valid enough at least to propose that this is a long lived protein.
In summary, our results demonstrate that tideglusib is an irreversible inhibitor of GSK-3β, thus explaining the non-competitive inhibition pattern with respect to ATP that has also been observed and was previously reported for other TDZDs. Although the evidence presented here does not seem to indicate a covalent modification of the enzyme, the functional irreversibility of the inhibition combined with the low turnover rate determined for GSK-3β may extend the duration of the pharmacological effect caused by this drug in a way that can be exploited to maximize its therapeutic potential.
Supplementary Material
Acknowledgments
We thank colleagues from the Chemistry Department for providing tideglusib; Susana Morales for the generation of mutant plasmids; Juan Ignacio Imbaud, José Ramón Gutiérrez-Pulgar, and María del Carmen García-Hernández (Protein Alternatives S.L.) for help in producing mutant recombinant GSK-3β; and Félix Hernández (Centro de Biología Molecular Severo Ochoa) for helpful discussions.
This work was supported by European Commission Grant EU-FP7 (Neuro.GSK3, Project 223276) and CDTI Grant IDI-20110884. All of the authors are employees of Noscira, the owner of tideglusib.

This article contains supplemental Fig. 1 and Table 1.
- AD
- Alzheimer disease
- Aβ
- amyloid β peptide
- DTE
- dithioerythritol
- GSK-3β
- glycogen synthase kinase-3β
- TDZD
- thiadiazolidindione
- GdnHCl
- guanidine hydrochloride.
REFERENCES
- 1. Alzheimer's Association (2011) 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 7, 208–244 [DOI] [PubMed] [Google Scholar]
- 2. Hardy J. (2006) A hundred years of Alzheimer's disease research. Neuron 52, 3–13 [DOI] [PubMed] [Google Scholar]
- 3. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 4. Moreno H., Yu E., Pigino G., Hernandez A. I., Kim N., Moreira J. E., Sugimori M., Llinás R. R. (2009) Synaptic transmission block by presynaptic injection of oligomeric amyloid β. Proc. Natl. Acad. Sci. U.S.A. 106, 5901–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ittner L. M., Ke Y. D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B. C., Christie M. J., Napier I. A., Eckert A., Staufenbiel M., Hardeman E., Götz J. (2010) Dendritic function of Tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell 142, 387–397 [DOI] [PubMed] [Google Scholar]
- 6. Muyllaert D., Kremer A., Jaworski T., Borghgraef P., Devijver H., Croes S., Dewachter I., Van Leuven F. (2008) Glycogen synthase kinase-3β, or a link between amyloid and Tau pathology? Genes Brain Behav. 7, 57–66 [DOI] [PubMed] [Google Scholar]
- 7. Hernández F., Gómez de Barreda E., Fuster-Matanzo A., Lucas J. J., Avila J. (2010) GSK3: a possible link between β amyloid peptide and Tau protein. Exp. Neurol. 223, 322–325 [DOI] [PubMed] [Google Scholar]
- 8. Ittner L. M., Götz J. (2011) Amyloid-β and Tau–a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72 [DOI] [PubMed] [Google Scholar]
- 9. Hooper C., Killick R., Lovestone S. (2008) The GSK3 hypothesis of Alzheimer's disease. J. Neurochem. 104, 1433–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Medina M., Wandosell F. (2011) Deconstructing GSK-3: The fine regulation of its activity. Int. J. Alz. Dis. 2011, 479249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanger D. P., Hughes K., Woodgett J. R., Brion J. P., Anderton B. H. (1992) Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of Tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62 [DOI] [PubMed] [Google Scholar]
- 12. Ishiguro K., Shiratsuchi A., Sato S., Omori A., Arioka M., Kobayashi S., Uchida T., Imahori K. (1993) Glycogen synthase kinase 3 β is identical to Tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 325, 167–172 [DOI] [PubMed] [Google Scholar]
- 13. Muñoz-Montaño J. R., Moreno F. J., Avila J., Diaz-Nido J. (1997) Lithium inhibits Alzheimer's disease-like Tau protein phosphorylation in neurons. FEBS Lett. 411, 183–188 [DOI] [PubMed] [Google Scholar]
- 14. Blalock E. M., Geddes J. W., Chen K. C., Porter N. M., Markesbery W. R., Landfield P. W. (2004) Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pei J. J., Tanaka T., Tung Y. C., Braak E., Iqbal K., Grundke-Iqbal I. (1997) Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 56, 70–78 [DOI] [PubMed] [Google Scholar]
- 16. Leroy K., Boutajangout A., Authelet M., Woodgett J. R., Anderton B. H., Brion J. P. (2002) The active form of glycogen synthase kinase-3β is associated with granulovacuolar degeneration in neurons in Alzheimer's disease. Acta Neuropathol. 103, 91–99 [DOI] [PubMed] [Google Scholar]
- 17. Medina M., Avila J. (2011) The role of glycogen synthase kinase-3 (GSK-3) in Alzheimer's disease in Alzheimer's Disease Pathogenesis: Core Concepts, Shifting Paradigms, and Therapeutic Targets, pp. 197–222, Intech Open Access Publisher, Rijeka, Croatia [Google Scholar]
- 18. Turenne G. A., Price B. D. (2001) Glycogen synthase kinase3 β phosphorylates serine 33 of p53 and activates p53's transcriptional activity. BMC Cell Biol. 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., She H., Mao Z. (2009) Phosphorylation of neuronal survival factor MEF2D by glycogen synthase kinase 3beta in neuronal apoptosis. J. Biol. Chem. 284, 32619–32626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grimes C. A., Jope R. S. (2001) CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. 78, 1219–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A., Kaibuchi K. (2005) GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149 [DOI] [PubMed] [Google Scholar]
- 22. Zumbrunn J., Kinoshita K., Hyman A. A., Näthke I. S. (2001) Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 β phosphorylation. Curr. Biol. 11, 44–49 [DOI] [PubMed] [Google Scholar]
- 23. Jo J., Whitcomb D. J., Olsen K. M., Kerrigan T. L., Lo S. C., Bru-Mercier G., Dickinson B., Scullion S., Sheng M., Collingridge G., Cho K. (2011) Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 14, 545–547 [DOI] [PubMed] [Google Scholar]
- 24. Mateo I., Infante J., Llorca J., Rodríguez E., Berciano J., Combarros O. (2006) Association between glycogen synthase kinase-3β genetic polymorphism and late-onset Alzheimer's disease. Dement. Geriatr. Cogn. Disord. 21, 228–232 [DOI] [PubMed] [Google Scholar]
- 25. Schaffer B. A., Bertram L., Miller B. L., Mullin K., Weintraub S., Johnson N., Bigio E. H., Mesulam M., Wiedau-Pazos M., Jackson G. R., Cummings J. L., Cantor R. M., Levey A. I., Tanzi R. E., Geschwind D. H. (2008) Association of GSK3B with Alzheimer disease and frontotemporal dementia. Arch. Neurol. 65, 1368–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwok J. B., Loy C. T., Hamilton G., Lau E., Hallupp M., Williams J., Owen M. J., Broe G. A., Tang N., Lam L., Powell J. F., Lovestone S., Schofield P. R. (2008) Glycogen synthase kinase-3β and Tau genes interact in Alzheimer's disease. Ann. Neurol. 64, 446–454 [DOI] [PubMed] [Google Scholar]
- 27. Mateo I., Vázquez-Higuera J. L., Sánchez-Juan P., Rodríguez-Rodríguez E., Infante J., García-Gorostiaga I., Berciano J., Combarros O. (2009) Epistasis between Tau phosphorylation regulating genes (CDK5R1 and GSK-3β) and Alzheimer's disease risk. Acta Neurol. Scand. 120, 130–133 [DOI] [PubMed] [Google Scholar]
- 28. Gentles R. G., Hua S., Dubowchik G. M. (2009) Recent advances in the discovery of GSK-3 inhibitors and a perspective on their utility for the treatment of Alzheimer's disease. Annu. Rep. Med. Chem. 44, 3–26 [Google Scholar]
- 29. Medina M., Avila J. (2010) Glycogen synthase kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer's disease. Curr. Pharm. Des. 16, 2790–2798 [DOI] [PubMed] [Google Scholar]
- 30. Medina M., Castro A. (2008) Glycogen synthase kinase-3 (GSK-3) inhibitors reach the clinic. Curr. Opin. Drug Discov. Devel. 11, 533–543 [PubMed] [Google Scholar]
- 31. Martinez A., Alonso M., Castro A., Pérez C., Moreno F. J. (2002) First non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J. Med. Chem. 45, 1292–1299 [DOI] [PubMed] [Google Scholar]
- 32. Serenó L., Coma M., Rodríguez M., Sánchez-Ferrer P., Sánchez M. B., Gich I., Agulló J. M., Pérez M., Avila J., Guardia-Laguarta C., Clarimón J., Lleó A., Gómez-Isla T. (2009) A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 35, 359–367 [DOI] [PubMed] [Google Scholar]
- 33. del Ser T. (2010) Phase IIA clinical trial on Alzheimer's Disease with NP-12, a GSK-3 inhibitor. Alzheimers and Dement. 6, S147 [Google Scholar]
- 34. Martinez A., Alonso M., Castro A., Dorronsoro I., Gelpí J. L., Luque F. J., Pérez C., Moreno F. J. (2005) SAR and 3D-QSAR studies on thiadiazolidinone derivatives: exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J. Med. Chem. 48, 7103–7112 [DOI] [PubMed] [Google Scholar]
- 35. Ring D. B., Johnson K. W., Henriksen E. J., Nuss J. M., Goff D., Kinnick T. R., Ma S. T., Reeder J. W., Samuels I., Slabiak T., Wagman A. S., Hammond M. E., Harrison S. D. (2003) Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 52: 588–595 [DOI] [PubMed] [Google Scholar]
- 36. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gimenez-Cassina A., Lim F., Cerrato T., Palomo G. M., Diaz-Nido J. (2009) Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase-3. J. Biol. Chem. 284, 3001–3011 [DOI] [PubMed] [Google Scholar]
- 38. Segel I. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, 1st Ed., pp. 100–160, John Wiley & Sons, Inc; Hoboken, NJ [Google Scholar]
- 39. Cook P. F., Cleland W. W. (2007) Enzyme Kinetics and Mechanism, 1st Ed., Garland Science, New York, NY [Google Scholar]
- 40. Coghlan M. P., Culbert A. A., Cross D. A., Corcoran S. L., Yates J. W., Pearce N. J., Rausch O. L., Murphy G. J., Carter P. S., Roxbee Cox L., Mills D., Brown M. J., Haigh D., Ward R. W., Smith D. G., Murray K. J., Reith A. D., Holder J. C. (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7, 793–803 [DOI] [PubMed] [Google Scholar]
- 41. Morrison J. F., Walsh C. T. (1988) The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301 [DOI] [PubMed] [Google Scholar]
- 42. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., pp. 318–349, Wiley-VCH, New York [Google Scholar]
- 43. Marcinkeviciene J., Luo Y., Graciani N. R., Combs A. P., Copeland R. A. (2001) Mechanism of Inhibition of beta-site amyloid precursor protein-cleaving enzyme (BACE) by a statine-based peptide. J. Biol. Chem. 276, 23790–23794 [DOI] [PubMed] [Google Scholar]
- 44. Fersht A. (1998) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, 1st Ed., W.H. Freeman, New York [Google Scholar]
- 45. Copeland R. A., Williams J. M., Giannaras J., Nurnberg S., Covington M., Pinto D., Pick S., Trzaskos J. M. (1994) Mechanism of selective inhibition of the inducible isoform of prostaglandin G/H synthase. Proc. Natl. Acad. Sci. U.S.A. 91, 11202–11206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robertson J. G. (2005) Mechanistic basis of enzyme-targeted drugs. Biochemistry 44, 5561–5571 [DOI] [PubMed] [Google Scholar]
- 47. Schirmer A., Kennedy J., Murli S., Reid R., Santi D. V. (2006) Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl. Acad. Sci. U.S.A. 103, 4234–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao A., Lee S. H., Mojena M., Jenkins R. G., Patrick D. R., Huber H. E., Goetz M. A., Hensens O. D., Zink D. L., Vilella D., Dombrowski A. W., Lingham R. B., Huang L. (1999) Resorcylic acid lactones: naturally occurring potent and selective inhibitors of MEK. J. Antibiot. 52, 1086–1094 [DOI] [PubMed] [Google Scholar]
- 49. Leost M., Schultz C., Link A., Wu Y. Z., Biernat J., Mandelkow E. M., Bibb J. A., Snyder G. L., Greengard P., Zaharevitz D. W., Gussio R., Senderowicz A. M., Sausville E. A., Kunick C., Meijer L. (2000) Paullones are potent inhibitors of glycogen synthase kinase-3β and cyclin-dependent kinase 5/p25. Eur. J. Biochem. 267, 5983–5994 [DOI] [PubMed] [Google Scholar]
- 50. Patrick G. N., Zhou P., Kwon Y. T., Howley P. M., Tsai L. H. (1998) p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J. Biol. Chem. 273, 24057–24064 [DOI] [PubMed] [Google Scholar]
- 51. Zhou P. (2004) Determining protein half-lives. Methods Mol. Biol. 284, 67–77 [DOI] [PubMed] [Google Scholar]
- 52. Hongisto V., Vainio J. C., Thompson R., Courtney M. J., Coffey E. T. (2008) The Wnt pool of glycogen synthase kinase 3β is critical for trophic-deprivation-induced neuronal death. Mol. Cell Biol. 28, 1515–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cole A., Frame S., Cohen P. (2004) Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 377, 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cornish-Bowden A. (2004) Fundamentals of Enzyme Kinetics, 3rd Ed., pp. 113–144, Portland Press, Oxford, UK [Google Scholar]
- 55. Singh J., Petter R. C., Baillie T. A., Whitty A. (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317 [DOI] [PubMed] [Google Scholar]
- 56. Klüter S., Simard J. R., Rode H. B., Grütter C., Pawar V., Raaijmakers H. C., Barf T. A., Rabiller M., van Otterlo W. A., Rauh D. (2010) Characterization of irreversible kinase inhibitors by directly detecting covalent bond formation: a tool for dissecting kinase drug resistance. ChemBioChem 11, 2557–2566 [DOI] [PubMed] [Google Scholar]
- 57. Cohen M. S., Zhang C., Shokat K. M., Taunton J. (2005) Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308, 1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.