

Background: Alternative splicing diversifies calcium channel structure to change channel properties.
Results: Extensive C-terminal alternative splicing generates channels differing in activation potential and voltage- and calcium-dependent inactivation properties.
Conclusion: Diversification of channel function through altered structure is fine-tuned by alternative splicing.
Significance: CaV1.4 C-terminal splice variations recapitulate some aspects of native photoreceptor calcium currents.
Keywords: Alternative Splicing, Calcium Channels, Ion Channels, Patch Clamp Electrophysiology, RNA Splicing, CaV1.4, Calcium-dependent Inactivation, Current Density, L-type Calcium Channels, Retina
Abstract
The CaV1.4 voltage-gated calcium channel is predominantly expressed in the retina, and mutations to this channel have been associated with human congenital stationary night blindness type-2. The L-type CaV1.4 channel displays distinct properties such as absence of calcium-dependent inactivation (CDI) and slow voltage-dependent inactivation (VDI) due to the presence of an autoinhibitory domain (inhibitor of CDI) in the distal C terminus. We hypothesized that native CaV1.4 is subjected to extensive alternative splicing, much like the other voltage-gated calcium channels, and employed the transcript scanning method to identify alternatively spliced exons within the CaV1.4 transcripts isolated from the human retina. In total, we identified 19 alternative splice variations, of which 16 variations have not been previously reported. Characterization of the C terminus alternatively spliced exons using whole-cell patch clamp electrophysiology revealed a splice variant that exhibits robust CDI. This splice variant arose from the splicing of a novel alternate exon (43*) that can be found in 13.6% of the full-length transcripts screened. Inclusion of exon 43* inserts a stop codon that truncates half the C terminus. The CaV1.4 43* channel exhibited robust CDI, a larger current density, a hyperpolarized shift in activation potential by ∼10 mV, and a slower VDI. Through deletional experiments, we showed that the inhibitor of CDI was responsible for modulating channel activation and VDI, in addition to CDI. Calcium currents in the photoreceptors were observed to exhibit CDI and are more negatively activated as compared with currents elicited from heterologously expressed full-length CaV1.4. Naturally occurring alternative splice variants may in part contribute to the properties of the native CaV1.4 channels.
Introduction
CaV1.4 is a member of the L-type family of voltage-gated calcium channels (LTCC)2 that are predominantly expressed in the rod photoreceptor and retina bipolar synapses. Mutations in the CACNA1F gene encoding CaV1.4 channels have been associated with congenital stationary night blindness type-2 (CSNB2), a condition characterized by various visual impairments in addition to night blindness (1, 2). Electroretinography of CSNB2 patients suggests that CaV1.4 mediates neurotransmitter release at the synapse (3).
The electrophysiological properties of heterologously expressed CaV1.4 included a slow voltage-dependent inactivation (VDI) and a unique absence of calcium-dependent inactivation (CDI) (4–6). CDI is a property well documented in the other LTCCs as follows: CaV1.2, CaV1.3, and recently, CaV1.1 channels (7). In the CaV1.4 pore-forming α1-subunit, an autoinhibitory domain (ICDI) was recently identified that resides in the distal end of the C terminus, and it profoundly blunts CDI by interacting with the calcium-sensing and inactivation-coupling machinery in the proximal C terminus (8–11). The electrophysiological properties of CaV1.4 channels are supportive of its physiological role as glutamate release at the photoreceptor synapse occurs in a sustained and graded manner. However, the electrophysiological properties characterized using heterologously expressed CaV1.4 channels differed from the properties of the native Ca2+ currents measured at the rod photoreceptors and bipolar cells (12–14). The heterologously expressed channels activated at membrane potentials that were ∼30 mV more positive compared with the rod Ca2+ currents. In addition, Ca2+ currents measured in rod photoreceptor and bipolar synapses do exhibit CDI (15–17). These discrepancies suggest that the endogenous CaV1.4 channels could be modulated to activate at more hyperpolarized potentials as well as exhibit CDI. This may occur by mechanisms including cytoplasmic Ca2+ protein buffers or post-transcriptional modifications such as alternative splicing. Interaction with a calcium-binding protein CaBP4 was shown to negatively shift the voltage of activation by 10 mV (18). Additional shifts in activation potential and modulations to other electrophysiological properties may be brought about by other mechanisms. Indeed, in other LTCCs like CaV1.2 and CaV1.3, alternative splicing has been found to modulate their biophysical and pharmacological characteristics (19–21). Alternative splicing in voltage-gated calcium channels is extensive throughout the length of calcium channel coding region. In CaV1.2, at least 12 exon loci in the gene are alternatively spliced with up to 12 different variations of splicing occurring in one locus (compiled in Ref. 22). Other examples include seven alternatively spliced loci in CaV2.1 and six alternatively spliced loci in CaV3.1 (23, 24). However, so far, only three alternatively spliced loci have been described for CaV1.4 (25).
Here, we systematically screened for alternatively spliced exons in CaV1.4 transcripts from the human retina using the “transcript-scanning method” (23, 24, 26) and showed that CaV1.4 is extensively alternatively spliced. We characterized the alternatively spliced exons in the C-terminal region and uncovered a population of splice variants that can activate within the photoreceptor operating potentials as well as exhibit CDI. Other electrophysiological properties that were altered include voltage-dependent inactivation (VDI) and current density.
EXPERIMENTAL PROCEDURES
Reference Sequences
The genomic sequence for CaV1.4 was from GenBankTM accession number AJ006216. The reference CaV1.4 cDNA sequence was from GenBankTM accession number AF201304. This sequence was considered as wild type (WT) in this work. CaV1.4 exon positions were determined by aligning the cDNA sequence against the genomic sequence (using the MegAlign module of the Lasergene® software suite, DNASTAR), and the exons were number in order from 1 to 48. The reference amino acid sequence used was from GenBankTM accession number NP005174. The reference CaV1.2 cDNA sequence used was from GenBankTM accession number NM000719.
Nomenclature for Describing Alternatively Spliced Exon Variant
Various suffixes and a prefix applied to an exon number are used to describe the type of alternative splicing that occurred at that exon locus (supplemental Fig. 6). Δ denotes a cassette exon that was skipped in the course of alternative splicing. a or d denote the changes to exon length at the acceptor or donor site, respectively. + or − indicates if the exon is lengthened or shortened, respectively. i suffix denotes retained intron, and x denotes mutually exclusive exons. * denotes a novel exon (not found in public database).
Transcript-scanning Method
To determine the different alternatively spliced exons in the CaV1.4 gene, we employed the transcript strategy previously described by Mittman and co-workers (24, 26) and Soong et al. (23). In this method, we first designed PCR primer pairs that span at least two exons or four splice boundaries along the length of the CaV1.4 gene (supplemental Fig. 1). Sufficient pairs of primers were made such that amplicons form overlapping segments along the entire CaV1.4 sequence. The primers, designed using Oligo Primer Analysis Software (Molecular Biology Insights), are given in supplemental Table 7. PCR was performed using human retina Marathon®-Ready cDNA (Clontech; catalog no. 7449-1, lot no. 0120657 and 3060598) as template and TaqDNA polymerase (Promega). Each pair of primers produced amplicons of varying sizes, corresponding to different alternative splice variants (or nonspecific products), visualized as multiple bands on an agarose gel (supplemental Fig. 1). Each band was extracted and ligated into pGEM®-T Easy vector and transformed into DH10B Escherichia coli. For every band cloned, 8–30 positive transformants (indicated by blue/white colony selection) were picked and further PCR screened using specific primers. Colonies yielding different sized PCR products were expanded and the plasmid DNAs extracted for DNA sequencing. The DNA sequences were analyzed by comparison with the CaV1.4 genomic and cDNA sequences to identify the type of alternative splicing that had occurred and to determine the exact location of the alternative exon-intron splice junctions as well as their adherence to the “gt … ag” rule. To transcript scan exon 1, we made use of the Marathon® adaptors that were ligated to the ends of Marathon®-ready cDNAs. The adaptor primer was provided by the manufacturer, and this was paired with a reverse primer residing on exon 2 or exon 3. To scan exon 48, we used a forward primer from exon 47 or 46 paired with an oligo(dT) that annealed to the poly(A) tail. As one primer in each pair was nonspecific (i.e. adaptor primer, oligo(dT)), the PCRs yielded diffused and multiple bands when separated on agarose gel. These were extracted, cloned, and rigorously screened in the same manner as described above.
Construction of Cloned Full-length CaV1.4 Library
Primers that reside in the 5′- and 3′-untranslated region (UTR) of the CaV1.4 gene were employed for long PCR amplification using human retina cDNA as template and the Elongase® Enzyme Mix (Invitrogen), a polymerase mixture having proofreading function and capable of long PCRs. The resultant ∼6-kb amplicons were subcloned into pCR®-XL-TOPO® vector (Invitrogen) and transformed into DH10B bacteria. Positive transformants were then picked and grown in 96-well plates. Random clones were picked and verified by restriction digest profiling as well as DNA sequencing.
Screening Full-length Library to Determine Abundance of Splice Variants
Pairs of primers were selected that flank exons shown by transcript scanning to have alternatively spliced variants. The clones in the full-length library were then screened using each selected pair of primers by PCR. The clones that produced PCR products and migrated with the expected sizes (WT or alternative splicing) on the agarose gel were counted. Clones were picked at random for sequencing to verify the validity of the screen. The quantity of alternatively spliced exon variants was expressed as a percentage of the total number of clones counted (supplemental Fig. 3).
Generation of Expression Constructs for Electrophysiology Characterization
Ch-WT is a chimera consisting of a CaV1.2 backbone and the entire C terminus from CaV1.4 cloned in a mammalian expression vector. The CaV1.2 fragment was excised using HindIII and BclI restriction sites from the α1C77WT clone (kindly provided by Dr. Roger D. Zühlke, University of Bern, Switzerland). The CaV1.4 C-terminal fragment was PCR-amplified from the human CaV1.4-pCDNA3.1 clone (kindly given by Dr. J. E. McRory, University of British Columbia, Canada), with the addition of a NotI site after the stop codon. The PCR also introduced a silent mutation to remove the second of the two BclI sites. The CaV1.2 fragment was ligated to CaV1.4 at the remaining BclI site, within a region of sequence identity in the IVS6 domain. The final chimeric construct was assembled in the pCDNA3 vector (Invitrogen) between HindIII and NotI sites.
Subsequent constructs containing the CaV1.4 C terminus alternatively spliced exons were generated by appending the C terminus of Ch-WT. DNA fragments of CaV1.4 C terminus that contained either Δ37, 42d+, 43*, or 45a− splicing were obtained by PCR or restriction digests from CaV1.4 clones in the full-length library. These were substituted into Ch-WT using various restriction sites: Δ37, NsiI/NotI; 42d+, SbfI/SbfI; 43* and 45a−, BamHI/XhoI.
For the deletion constructs, different fragments of the CaV1.4 C terminus were first PCR-amplified with the insertion of stop codon to terminate at His-1718, Arg-1755, Ala-1797, Gln-1835, and Leu-1878 for the clones Ch-e43, Ch-e44, Ch-e45, Ch-e46, and Ch-ΔICDI, respectively. These were substituted into Ch-WT using BamHI and XbaI sites.
The ICDI-mCherry construct consists of the ICDI domain of CaV1.4 fused in-frame to a red fluorescent protein, mCherry. ICDI was PCR-amplified from CaV1.4-pcDNA3.1 template with the addition of Kozak sequence and start codon. An XbaI restriction site replaced the endogenous stop codon. The mCherry fragment was amplified from the pRSET-B mCherry clone (gift from Dr. Roger Y. Tsien, University of California, San Diego) with the addition of an XbaI site at the 5′ end and a stop codon at the 3′ end. These fragments were assembled in a pCDNA3 vector between EcoRI and NotI sites.
The mutant construct Ch-P2A1 contains two mutations P1766A and P1767A, and Ch-P2A2 contains two mutations P1773A and P1774A. CaV1.4 C-terminal fragments containing either pair of mutations were first generated using overlap-extension PCR (27) and then substituted into Ch-WT using BamHI and XbaI sites. The integrity of all constructs was validated by DNA sequencing.
Transient Expression of Calcium Channels in HEK-293 Cells
HEK-293 cells were co-transfected with 1.75 μg of α (Ch-WT or variants), 1.25 μg each of β2a and α2δ-subunits (kind gifts of Dr. Terry P. Snutch, University of British Columbia, Canada), and 0.2 μg of T-antigen plasmids using the calcium phosphate method. The cells were then incubated for at least 36 h in a water-saturated 5% CO2 incubator at 37 °C before whole-cell patch clamp recordings were made.
Whole-cell Patch Clamp Electrophysiology
Whole-cell patch clamp recordings were performed on transfected cells between 36 and 72 h after transfection. The external bath solution was composed of the following (in mm): 10 HEPES, 140 tetraethylammonium methanesulfonate, 5 BaCl2 or CaCl2, pH 7.4, 300–310 mosm (adjusted with glucose). The pipette internal solution contained (in mm): 138 Cs-MeSO3, 5 CsCl, 0.5 EGTA, 10 HEPES, 1 MgCl2, 2 mg/ml Mg-ATP, pH 7.3, 290–300 mosm (adjusted with glucose). The junction potential was determined to be −11 mV, and the voltages reported here are uncorrected for junction potential. True voltages may be calculated by subtracting 11 mV from the reported values. Whole-cell currents were obtained under voltage clamp with either the Axopatch 200A or Axopatch 200B amplifier, operated using pClamp software (Molecular Devices). The signals were filtered at 1–5 kHz and sampled at 5–50 kHz. Series resistance was typically 1.2–1.8 megohms after 80% compensation. A P/4 protocol was applied on line to subtract leak and capacitive transients.
To assess the current-voltage (I-V) relationship of the channels, transfected cells were depolarized to a family of test potentials of −60 to 60 mV, in steps of 10-mV increments, from a holding potential of −90 mV. The peak current evoked by each voltage was normalized to the maximal current obtained for each cell recorded and fitted with Equation 1,
where Gmax is the maximum conductance; Erev is the reversible potential; V½, act is the half-activation potential, and kact is the slope.
For steady-state activation, we analyzed the tail currents (G) obtained at the end of a short depolarizing pulses. Cells were held at −90 mV before depolarizing to a family of voltages ranging from −60 to 100 mV, in steps of 10-mV increments, for 20 ms. Following that, a repolarization to −50 mV for 10 ms evokes the tail currents that are measured. The peak of each tail current was normalized to the maximum obtained for each cell recording and fitted with dual Boltzmann Equation 2,
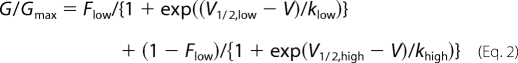 |
where G is the tail current and Gmax is the maximum tail current; Flow is the fraction of the low threshold component; V½, low, V½, high, klow, and khigh are the half-activation potentials and slope factors for the low and high threshold components, respectively, and V½, act may be calculated when G = 0.5 Gmax.
Steady-state inactivation properties were determined by comparing the test current obtained after a long depolarizing pulse to an initial pre-pulse. For the −90-mV holding potential, a pre-pulse current was evoked by stepping to 10 mV for 30 ms. This was followed by a family of 15-s-long depolarizing pulses ranging from −120 to 20 mV. Next, a test pulse current was evoked by stepping the cell to 10 mV for 100 ms. The peak current evoked by each test pulse was divided by the peak current evoked by the pre-pulse to obtain the normalized current. The values were fitted with a single Boltzmann Equation 3,
where Irelative is the normalized current; V½,inact is the potential for half-inactivation, and kinact is the slope value.
The strength of CDI exhibited by the channel is expressed by the f value (28). To determine the f value, first the current amplitude that remained at a given time point (i.e. 30, 50, 100, 20, and 300 ms) after depolarization was measured and normalized against the peak current to obtain the residual current (i.e. r30 and r300 for 30 and 300 ms, respectively). Next, the f value was calculated by subtracting the residual IBa with the residual ICa. An f value of 0 indicates no CDI, whereas the maximum f value of 1 indicates complete CDI. For comparison between channels, we compared the f values obtained from current evoked by depolarization to 10 mV. The presence of CDI may also be seen in the profile of residual ICa evoked by a family of voltages. When plotted against voltage, the “U”-shaped dependence of residual ICa values on voltage is a tell-tale trait of CDI.
For VDI, cells were placed in the external solution with Ba2+ as the charge carrier and depolarized to Vmax, from a holding potential of −90 mV. As time elapsed, the current evoked by Vmax decayed, and the amplitude was measured at different time points. These were subtracted from the peak current evoked to obtain the inactivated current. This was then expressed as a percentage of the peak current (percentage inactivation).
Fractional recovery from inactivation was determined using a two-pulse protocol and following a 2-s depolarizing pre-pulse to Vmax (determined from the I-V properties of the transfected cell), and a test pulse was applied after a certain time period, ΔT, has elapsed. The peak IBa measured during the test pulse was expressed as a fraction of peak IBa obtained at the beginning of the pre-pulse to obtain the fraction of recovery. This was plotted against ΔT. The curve was obtained by fitting the values with a double exponential Equation 4,
where Y is the fraction of recovery, Af and As are the maximum values of the fast and slow component, and τf and τs are their time constants, respectively.
Data and Statistical Analyses
All values presented are means ± S.E. Electrophysiology data were analyzed using Clampfit software (Molecular Devices). Data values were exported to Microsoft® Office Excel® software (Microsoft Corp.) for calculations. Data plotting, curve fitting, and statistical analyses were performed using GraphPad Prism® software (GraphPad Software, Inc.). Statistical significance of the difference between means was determined using Student's t test or one-way analysis of variance. Statistical significance was accepted at p < 0.05.
RESULTS
Transcript Scanning of CaV1.4 from Human Retinal cDNAs
To determine the different alternatively spliced exons in the CaV1.4 gene, we employed the transcript scanning strategy that has been previously reported (23, 24, 26). As the CaV1.4 transcripts are predominantly expressed in the retina (1, 2, 4), we used human retina cDNA in PCRs to amplify across all the splice junctions of the CaV1.4 gene; and we generated at least 15 overlapping PCR amplicons (supplemental Fig. 1). Bacterial colonies obtained from plasmids subcloned with each of the 15 PCR amplicons were screened, and the individual clones harboring different size PCR products were subject to automated DNA sequencing (supplemental Fig. 2). The identity and mechanism of the alternative splicing for each splice locus were identified by inspection of the exon-intron boundaries and determined by analysis of the DNA sequences in comparison with the CaV1.4 genomic sequence (Fig. 2; GenBank accession number AJ006216).
FIGURE 2.

Alternative splice exons in CaV1. 4. The left column lists (in boldface) the alternatively spliced exons found in CaV1.4 transcripts. The nomenclature used to denote the type of splicing is described in supplemental Fig. 7. The percentage occurrences of each alternative splice form in full-length CaV1.4 transcripts are given in parentheses, together with the number of clones analyzed. A description on the mode of splicing follows, with the consequent alterations to the translation reading frame as well as amino acid changes. The splice scheme in the middle column illustrates the mechanism of splicing in terms of the exact splice junction used by the wild type (WT) and alternative modes of splicing. Dotted boxes indicate the extended boundaries of alternative exons and the exon cassettes. Italicized are the canonical gt … ag sites found almost invariably on the intronic side of splice junctions. Underlined are stop codons. The right column lists the channel structures that are affected by the alternative splicing.
Transcript scanning of CaV1.4 from human retina cDNA revealed that 16 exons are alternatively spliced, of which some may undergo more than one mode of alternative splicing with some adjacent exons alternatively spliced in tandem (Fig. 2); this results in 19 different exon variants. These include cassette exons that can be fully excluded by alternative splicing and alternate use of splice donor/acceptor junctions that resulted in lengthening or shortening of the exons and mutually exclusive exons. Two of the cassette exons are novel and have not been predicted in the public database. In all cases, the canonical “gt … ag ” splice junctions were preserved at the intron-exon junctions of the alternative splice sites (47). Not all alternative splicings were productive as some resulted in frameshifts that gave rise to premature stop codons. The relative positions of the alternatively spliced loci along the entire length of the CaV1.4 open-reading frame are illustrated in Fig. 1. Alternative splicing at the C terminus produced four splice variants (Δ37, 42d+, 43*, and 45a−) that contained different lengths of the C-terminal tail. The Δ37 splice variant is truncated the entire C terminus, although in contrast the use of an alternative donor site in 42d+ adds an additional 13 amino acids to the region downstream of the IQ motif. The inclusion of the novel alternative exon 43* adds a novel 19-amino acid peptide past exon 42 but contains a stop codon that resulted in premature termination of the C terminus with a truncation of the last 257 amino acids. Conversely, 21 amino acids are removed in the 45a− variant by the use of an alternative acceptor site with no frameshift of downstream codons. This occurs in a region of the C terminus that has poor conservation among LTCCs (supplemental Fig. 3). Notably, the splice variants 16a− and 17a+1, Δ16 and 17a+2, and Δ17 and 18a, are pairs of adjacent alternatively spliced exons that when spliced in tandem gave rise to transcripts coding for functional CaV1.4 channels. Here, alternative splicing of the first exon generated a frameshift that was corrected by the alternative splicing in the second exon. Had alternative splicing occurred independently in one locus frameshift would result in a premature stop codon. This mode of splicing is interesting and unusual because it manages to alter the gross peptide sequence without relying on a different set of nucleotide sequences.
FIGURE 1.

Schematic illustration of CaV1.4 alternative splicing in channel structure. The four repeat domain structure of the voltage-gated calcium channel is represented here and annotated with the relative exon positions, alternatively spliced loci, and important functional domains. Exon boundaries are indicated by thin lines. Exons are numbered in order from 1 to 48. The α interaction domain (binding site for the β-subunit), EF-hand, IQ motif, and the inhibitor of calcium-dependent inactivation domain (ICDI) are denoted using shaded ovals. Alternatively spliced exons are labeled in parentheses, and the relative positions in the channel schematic is darkened. The exact site and mode of alternative splicing is detailed in Fig. 2, and the nomenclature used to denote the alternatively spliced exons is given in supplemental Fig. 7.
We also transcript-scanned the bracketing exons (i.e. exon 1 and exon 48) by using Marathon®-Ready human retinal cDNA (Clontech) (29) as template in PCRs that contained a primer that targets the sequence in the Marathon® adapter and a CaV1.4-specific primer. Based on the results from the PCR screen, coupled with DNA sequencing of clones, we found no additional alternate exons to exon 1 or exon 48.
Abundance of Each Alternative Splice Locus within CaV1.4 Transcripts
Proteomic variations generated by alternative splicing may alter the electrophysiological properties of CaV1.4 channels. The relative abundance of each CaV1.4 splice variant would no doubt influence the overall property of the native CaV1.4 currents. To determine the levels of occurrence of the splice variants, we first PCR-amplified and cloned full-length CaV1.4 cDNAs from human retina to obtain a pool of full-length CaV1.4 clones. Using primer pairs that target alternatively spliced loci, we characterized the full-length CaV1.4 clones by PCR screening to identify the combinatorial splicing pattern of each clone. Clones producing PCR product size alterations that tallied with the expected changes were counted and expressed as a percentage of the total population (Fig. 2, left column). Random clones were selected for DNA sequencing to validate the PCR screen. The most abundant splice variant we have found was 16a− (61.7%). Here, the alternative use of an alternative splice acceptor site at exon 16 resulted in a frameshift with a consequent stop codon at IIS6 domain. This variant may give rise to the formation of a hemi-channel. The most abundant productive splice variant we detected was Δ32 (17.9%). The skipping of exon 32 shortens the IVS3-S4 linker by 7-amino acid residues. Shortening of the IVS3-S4 linker by alternative splicing may be a common mechanism for diversity among LTCC. In CaV1.1, alternative splicing at this locus can include 10% of transcripts in adult muscle and more than 66% in regenerating muscle (30). In CaV1.2, alternative splicing of exons encoding IVS3 and the IVS3-S4 linker could produce up to 12 variations (22). Although the amino acid content of the IVS3-S4 linker varies greatly between the four LTCC, the length of the linker, however, may be important in modulating channel activation (22).
Electrophysiological Characterizations of CaV1.4 C-terminal Splice Variants
Alternatively splicing at four loci produced splice variants Δ37, 42d+, 43*, and 45a− that have various lengths of the C terminus. Reports from previous investigations into the biophysical properties of full-length CaV1.4 channels have shown that these channels expressed relatively small currents (about three times lower than Cav1.2 (5)), despite the use of high barium concentration in the external solution (15–30 mm), and tend to have a low transfection efficiency (22% (6)). To screen, in a more efficacious manner, the C-terminal splice variants for distinct electrophysiological properties, we chose the chimera approach. We generated a chimeric reference construct (Ch-WT) switching the full-length C terminus of human CaV1.4 into the human CaV1.2 channel backbone (Fig. 3A). The CaV1.2 was selected because it is a well characterized L-type subtype and has an almost identical I-V relationship to CaV1.4 (5). Similar chimeras have been successfully used in a robust manner to investigate the effects of the CaV1.4 C terminus on channel properties and the mechanisms involved (9–11).
FIGURE 3.

Current-voltage relationship of CaV1. 2–1.4 chimera wild type and alternatively spliced variants. A, schematic representation of chimera constructs. The channel backbone consists of CaV1.2 (white box), whereas the cytosolic tail consists of CaV1.4 wild type or alternatively spliced variants, Δ37, 42d+, 43*, and 45a− (black). The stop codons for Δ37 and 43* are indicated by white and black filled circles, respectively. Amino acid positions are numbered according to the CaV1.4 sequence, GenBankTM accession number NP005174. B, voltage protocol for determining current-voltage (I-V) relationship. 400-ms depolarizations to potentials ranging, in 10-mV increments, from −60 to 70 mV (holding potential −90 mV). C, representative IBa (gray) and ICa (black) traces during a 400-ms depolarization to 10 mV. The IBa and ICa traces were scaled to enable comparison between the two profiles. Current scale bars were drawn for both IBa (gray) and ICa (black). The time scales for each IBa and ICa pair are the same. D–G, normalized I-V plots for IBa of WT, 42d+, 43*, and 45a− constructs, respectively. The curves were fitted with the equation described under “Experimental Procedures.” In parentheses are the number of cells recorded. The WT curve was redrawn in each graph for comparison. H–K are the same as D–G but for ICa.
The entire cytosolic tail is truncated in the Δ37 splice variant, and this channel may be nonfunctional. However, because a CSNB2 CaV1.4 mutant with no cytosolic tail was previously reported to mediate current (4), we attempted to characterize this splice variant as well, but we were not able to evoke any current in the Δ37 chimeric channel.
43* Splice Variant Inactivated More Rapidly in Ca2+ and Activated in a More Hyperpolarized Potential
To assess the current-voltage (I-V) relationship of the splice variants, transfected cells were depolarized to a family of test potentials from −60 to 70 mV, in steps of 10-mV increments, from a holding potential of −90 mV (Fig. 3B). The current trace profile for Ch-43* displayed a much slower inactivating IBa compared with Ch-WT or the other two functional C-terminal splice variants (Fig. 3C, gray traces), but it showed an early and more pronounced inactivation of ICa (Fig. 3C, black traces). These inactivation properties of the channels in Ba2+ compared with Ca2+ will be further analyzed below.
Ch-43* also activated at a more negative potential with a hyperpolarized shift in voltage for half-maximal activation, V½, act, by 21.0 mV in Ba2+ (supplemental Table 1) and 19.4 mV in Ca2+ (supplemental Table 2) compared with Ch-WT (Fig. 3, F and J; gray trace; both p < 0.001; unpaired t test). Such changes in channel activation are slight and less apparent in Ch-42d+ and Ch-45a−. The V½, act of the Ch-42d+ hyperpolarized shift by 2.6 mV in Ba2+ and 2.0 mV in Ca2+ (p < 0.001; unpaired t test), whereas the V½, act of Ch-45a-depolarized-shift by 1.2 mV in Ba2+ and 0.5 mV in Ca2+ (p < 0.001; unpaired t test). We then appended the CaV1.4 full-length channel with the 43* exon and found that in a similar manner, exon 43* mediated a pronounced hyperpolarized shift in activation (12.5 mV; p < 0.0001; unpaired t test) in the native channel (Fig. 4B). Also, CaV1.4 4.3* mediated a rapidly inactivating ICa, whereas the WT displayed little inactivation (Fig. 4A).
FIGURE 4.

Current-voltage relationship of CaV1.4 wild type and 43* splice variant. A, representative IBa (gray) and ICa (black) traces during a 400-ms depolarization to 10 mV. B, normalized I-V plots for IBa of CaV1.4 WT and 43*. The curves were fitted with the equation described under “Experimental Procedures.” In parentheses are the number of cells recorded.
43* Splice Variant Exhibited a Negative Shift in Window Current
We have shown that exon 43* caused a pronounced shift of the I-V relationship in the hyperpolarized direction, indicating a more negatively activating channel. To enable a more accurate assessment of the voltage activation of these channels, we analyzed the tail currents (G) obtained at the end of short depolarizing pulses to various potentials (Fig. 5A). Also, to determine the inactivation properties of the channels under steady-state conditions, transfected cells were held at various potentials for 15 s, and currents evoked before and after each inactivating pulse were compared (Fig. 5B).
FIGURE 5.

Activation and steady-state inactivation properties of wild type and 43* splice variant channels. A, voltage protocol used for determining activation properties and an exemplar current trace (WT). Tail currents were measured after 20 ms depolarization to potentials ranging from −60 to 100 mV, in 10-mV steps. B, voltage protocol used for determining steady-state inactivation, SSI, properties, and an exemplar current trace (WT). Currents evoked at 10-mV test potentials were measure before and after a 15-s depolarization to potentials ranging from −120 to 20 mV, in 10-mV steps. A holding potential of −90 mV was maintained for all experiments. C and D, normalized plots for activation and SSI for chimeric channels Ch-WT and Ch-43*, respectively. For SSI, peak currents obtained after the 15-s inactivating pulse were normalized to that obtained before inactivation and plotted against voltage. The curves are fits with the Boltzmann relationship. For the activation plots, the peak of the tail currents (G) were normalized against the largest peak and plotted against voltage. The curves were fitted using the equation given under “Experimental Procedures.” The number of cells recorded are given in parentheses. E and F, same as C and D but for native channels CaV1.4 WT and CaV1.4 43*, respectively.
Compared with Ch-WT, the voltage of half-maximal activation in Ch-43* was shifted by −29.9 mV (Fig. 5C; V½, act, Ch-WT, 21.9 ± 5.0 mV, n = 11; Ch-43*, −8.1 ± 2.8 mV, n = 16; p < 0.0001, unpaired t test). There was no change in the lower part of the slope klow; however, an increase was detected in khigh, although the difference was not statistically significant due to large error. Likewise, exon 43* mediated a hyperpolarized shift in V½, act of the native channel by 7.9 mV (Fig. 5E; V½, act, CaV1.4 WT, −18.1 ± 1.8 mV, n = 12; CaV1.4 43*, −26.7 ± 1.1 mV, n = 11; p < 0.001, unpaired t test), with no significant changes in the slope values. Alternative splicing of exon 43* in the chimera resulted only in a minor shift in the voltage for half-maximal steady-state inactivation (Fig. 5D; V½, inact, Ch-WT, −45.1 ± 0.2, n = 11; Ch-43*, −43.8 ± 0.1 mV, n = 8; p < 0.0001, unpaired t test) but gave rise to a significantly steeper SSI slope by decreasing the kinact by 5.7 (kinact, Ch-WT, 10.8 ± 0.1; 43*, 5.1 ± 0.1; p < 0.0001, unpaired t test). In the native CaV1.4 channel, both WT and 43* displayed similar SSI at more negative membrane potentials. However, beyond −40 mV, CaV1.4 43* could not be further inactivated (Fig. 5F; V½, act, CaV1.4 WT, −36.5 ± 1.0 mV, n = 16; CaV1.4 43*, −44.5 ± 1.8 mV, n = 5, p < 0.001, unpaired t test. kinact, CaV1.4 WT, 9.5 ± 0.9 mV, n = 16; CaV1.4 43*, 7.1 ± 1.5 mV, n = 5, p = 0.17, unpaired t test). Taken together, exon 43* supported a window current that is more hyperpolarized than WT (supplemental Fig. 4A) but with a reduced SSI at higher potentials.
CDI Is Restored by 43* Splicing
Rapid inactivation of activated channels due to Ca2+ is a phenomenon displayed by many high voltage activated channels. Interaction of Ca2+-bound calmodulin with the IQ domain triggers CDI; however, in the CaV1.4 channel the ICDI domain at the distal end of the C terminus interacts with the calcium-sensing apparatus, consisting of the EF-hand and IQ motif, to profoundly blunt CDI (8–11). From the profiles of the exemplar traces in Figs. 3C and 4A, it was clear that the 43* variant still displayed robust CDI. To quantify the degree of CDI exhibited by the channels, the fractions of current that remained at a given time point after depolarization (residual current; i.e. r30 and r300 for 30 and 300 ms, respectively; Fig. 6, A–D) to different voltages were determined. The difference between the residual current of Ba2+ and Ca2+ is a measure of CDI strength (f value). Here, we calculated the f value obtained at 10 mV (f10; Fig. 6, E and F). In addition, because the channels exhibit a fair amount of calcium-independent inactivation (VDI), we also used the ratiometric approach to calculate CDI. Here, “netCDI” is obtained by taking the ratio of the remaining current of Ca2+ to Ba2+ mV (netCDI10; Fig. 4, I and J). This method complements the classical f value measurements by avoiding the underestimation of CDI strength in the presence of strong VDI (31–33).
FIGURE 6.

Calcium-dependent inactivation and voltage-dependent inactivation of current through WT and 43* spliced variant channels. A and C, fraction of peak current, Ipeak, that remained at 30-ms upon depolarization to the indicated voltages, r30. The difference between the remaining current for IBa and ICa, f value, indicates the strength of calcium-dependent inactivation. The curves are visual fits of the values plotted to facilitate comparison. Number of cells recorded are given in parentheses. B and D, fraction of remaining current at 300-ms of depolarization, r300, otherwise same as A and C. E and F, f values obtained at 10 mV, f10, for r30 and r300, respectively. netCDI is the ratio between the remaining currents for IBa and ICa. G, percentage of IBa inactivation during depolarization to Vmax in the time course indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (compared with WT; unpaired t test).
For the Ch-WT channel, the r30 for Ca2+ was slightly lower than for Ba2+ (Fig. 6A). This became more evident with residual currents measured at a longer depolarization time of 300 ms (Fig. 6B), indicating a small degree of inactivation by Ca2+ (f10, 30 ms, 0.05 ± 0.03; 300 ms, 0.18 ± 0.06. netCDI10, 30 ms, 0.06 ± 0.03; 300 ms, 0.33 ± 0.14). In the native WT channel, however, this is less pronounced (Fig. 6E; f10, 30 ms, 0.02 ± 0.02; netCDI10, 0.02 ± 0.02) and even barely detectable at 300 ms (Fig. 6F; f10, 0.00 ± 0.03; netCDI10, 0.00 ± 0.03).
In contrast, 43* led to a gross increase in CDI in both the chimera and native channels at 300 ms (Fig. 6, E and F; f10, 0.63 ± 0.03 and 0.46 ± 0.03, respectively; netCDI10, 0.79 ± 0.11 and 0.48 ± 0.03, respectively). Even as early as 30 ms, CDI was already very robust with an increase of at least 8-fold in Ch-43* (f10, 0.47 ± 0.03; netCDI10, 0.48 ± 0.07) and >13-fold in the native spliced variant (f10, 0.27 ± 0.02; netCDI10, 0.28 ± 0.02). The residual current of 43* channels in Ca2+ exhibits a U-shaped dependence on voltage (Fig. 6, C and D) that is distinctive of CDI.
VDI Is Suppressed by 43* Splicing
An appreciable amount of slow inactivation in the absence of Ca2+ is apparent in the current trace profiles of Ch-WT (Fig. 3C) and, to a lesser degree, CaV1.4 WT (Fig. 4). We next queried whether VDI may be modulated by alternative splicing in the C terminus. Fig. 6G shows the percentage inactivation of peak IBa, evoked at Vmax, as it decays over various time points. Inactivation of IBa through Ch-WT increased steadily over time; displaying 66.0 ± 5.1% inactivation at 300 ms after depolarization and reaching 85.3 ± 3.6% by the end of 1 s. Truncation of the distal portions of the C terminus by 43* splicing resulted in a channel that inactivated in Ba2+ more slowly compared with Ch-WT. At 300 ms, IBa inactivation for Ch-43* (21.9 ± 3.2%) was about 3-fold slower than Ch-WT (p < 0.0001, unpaired t test), and at 1 s the inactivation of Ch-43* was half that of WT (Ch-WT, 85.3 ± 3.6%; Ch-43* 45.7 ± 4.3%; p < 0.0001, unpaired t test). The native CaV1.4 WT displayed a much lower VDI compared with the chimera, exhibiting only 14.3 ± 2.5% inactivation at 1 s. The presence of exon 43*, however, still reduced VDI of the native channel by half (7.1 ± 1.6%).
43* Splicing Mediated an Increase in Current Density
Previous reports of LTCC with C-terminal deletions had described gross increase in current density (Cav1.1 (34), Cav1.2 (35), and Cav1.2 (36)). We therefore examined the currents mediated by the 43* channels. Appending exon 43* to either the chimera or native channel both led to a significant increase in current density (Fig. 7, A and B). The increase in current density, when evoked at 60 mV, was 4.3 times in the chimera (Ch-WT, 35.6 ± 6.7 pA/pF; Ch-43*, 153.2 ± 27.1 pA/pF; p < 0.01, unpaired t test) and 2.1 times larger in the native channel when evoked at 0 mV (CaV1.4 WT, 25.5 ± 4.4 pA/pF; CaV1.4 43*, 53.2 ± 11.7 pA/pF; p < 0.01, unpaired t test).
FIGURE 7.

Current density of IBa through WT and 43* spliced variant channels and gating charge. A and B, peak of tail currents measured at the end of short depolarizing pulses evoked at different potentials were normalized against the membrane capacitance (Cm) of the recorded cell to obtain the current density. The number of cells recorded are shown in parentheses. C, gating charge was calculated by taking the integral of the current transient (gray areas, inset), at the onset, of the depolarizing pulse to the reversal potential of the ionic current. Shown here are representative gating and ionic tail current traces evoked at reversal potential. D, average gating charge for CaV1.4 WT (n = 14) and CaV1.4 43* (n = 21) normalized against membrane capacitance. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (compared with WT; unpaired t test).
Current density is a function of the number of channels expressed on the plasma membrane, the channel's unitary conductance, and open probability. We next measured the gating charge of CaV1.4 WT and 43* channels. As gating charge arises from the voltage-driven movement of charged residues within the activating channel structure, this gives an index of the quantity of channels expressed on the cell membrane (Fig. 7C). CaV1.4 43* displayed a gating charge that was more than 6-fold smaller than the WT (Fig. 7D; WT, 1.28 ± 0.14 femtocoulombs/pF; 43*, 0.19 ± 0.08 femtocoulombs/pF), indicating that the larger current density it exhibits is likely due to a larger single channel conductance or open probability or both. Single channel analyses would be required to determine these two properties.
Determinants for Activation and Inactivation in CaV1.4 C Terminus
In the 43* splice variant, the distal portion of the C terminus is truncated. This results in the loss of the ICDI, a domain that has been previously demonstrated to be responsible for the suppression of CDI in the CaV1.4 channels. Removal of a short length of this ICDI had also been shown to slow VDI (10). It is conceivable that the robust CDI and slow VDI in Ch-43* can be fully attributed to the lack of the ICDI domain. However, as the ICDI is composed of only the last 100 amino acids of the C terminus, we did not rule out the possible participation of the segment between exon 43 and ICDI in channel activation and inactivation.
First, we established whether the changes to channel properties by 43* splicing can be fully reproduced by deleting only the ICDI or whether it requires a greater length of the C terminus to be deleted. We next co-expressed a peptide containing the ICDI together with the Ch-43* to determine whether the ICDI itself can reverse the effects of 43* alternative splicing (i.e. suppression of CDI as well as increased VDI). To these ends, another two chimeric channels were constructed. These consisted of the CaV1.2 backbone and truncated C termini of the CaV1.4, one terminating after exon 43 and another terminating just before the ICDI (labeled Ch-e43 and Ch-ΔICDI, respectively; Fig. 7A). The ICDI peptide was also cloned into a mammalian expression vector and fused with an mCherry fluorescent protein (37). These CaVα1 clones were each co-expressed, in HEK-293 cells, with the auxiliary subunits and with either the mCherry-fused ICDI peptide (ICDI-mCherry) or mCherry alone as a control. Only cells that were found to express both green and red fluorescence were selected for whole-cell electrophysiological recordings (supplemental Fig. 6).
Hyperpolarized I-V Shift by 43* Splicing Was Due to Loss of ICDI
Analysis of the I-V relationship of Ch-ΔICDI showed that deletion of the ICDI from the full-length Ch-WT enabled a hyperpolarized shift reminiscent of that mediated by the 43* splicing (compare between Figs. 8D, gray curve, and 3F). This implies that the I-V shift shown by 43* was primarily due to the loss of the ICDI domain. Co-expression of Ch-ΔICDI with ICDI-mCherry reversed the shift with a 16.3- and 25.4-mV more positive V½, act in Ba2+ and Ca2+, respectively (supplemental Tables 4 and 5, p < 0.001, unpaired t test). The slope of activation, kact, was also decreased by 3.3 and 3.7 in Ba2+ and Ca2+, respectively (supplemental Tables 4 and 5, p < 0.001, unpaired t test). In the control experiments, co-expression of mCherry with the CaVα and auxiliary subunits failed to mediate the I-V shifts. Hence, these data demonstrated that the ICDI domain modulated the activation kinetics of the channel.
FIGURE 8.

Current-voltage relationship of Ch-43* and deletion constructs Ch-e43 and Ch-ΔICDI, co-expressed with ICDI-containing peptide. A, schematic representation of the various constructs. The channel backbone consists of CaV1.2 (white box), whereas the cytosolic tail consists of CaV1.4 containing exon 43* (Ch-43*) or deleted at amino acid positions 1718 (Ch-e43) or 1878 (Ch-ΔICDI). A co-expression construct consisting of a 100-amino acid peptide encompassing the ICDI fused to the mCherry fluorescent protein (ICDI-mCherry). The stop codon for 43* is indicated by a black filled circle. Amino acid positions are numbered according to the CaV1.4 sequence, GenBankTM accession number NP005174. B–D, normalized I-V plots for IBa of Ch-43*, Ch-e43, and Ch-ΔICDI co-expressed with either ICDI-mCherry (black) or mCherry (gray). The curves were fitted with the equation described under “Experimental Procedures.” In parentheses are the numbers of cells recorded. E–G, same as B–D but for ICa.
Whole-cell electrophysiological recordings of Ch-e43 showed that it displayed an I-V relationship similar to Ch-43* or Ch-ΔICDI (Fig. 8, B–G). Similarly, co-expression with ICDI-mCherry led to depolarized shifts in V½, act of 15.4 and 17.1 mV in Ba2+ and Ca2+, respectively, as well as a decrease in kact by 4.0 and 3.0 in Ba2+ and Ca2+, respectively (supplemental Tables 4 and 5).
Co-expression of Ch-43* with ICDI-mCherry also triggered a right shift in the I-V curve (Fig. 8, B and E). The presence of mCherry did not affect the channel properties as the I-V curve for the control was similar to that of Ch-43* in the previous experiments (Fig. 3, F and J). The depolarized shift in V½, act effected by ICDI-mCherry was 16.8 mV in Ba2+ and 19.3 mV in Ca2+ (supplemental Tables 4 and 5; p < 0.001, unpaired t test). The magnitude of V½, act shift in Ca2+ reflected a complete reversal of the hyperpolarized shift caused by the 43* splicing. Similarly, co-expression of Ch-43* with ICDI-mCherry restored kact to values similar to Ch-WT (supplemental Tables 1 and 2).
Co-expression with ICDI-mCherry Suppressed CDI and Increased VDI in Ch-43*
The representative traces shown in Fig. 9, A–F, reflect the current profiles obtained when cells in the co-expression studies were subjected to the square-pulse protocol illustrated in Fig. 3B. For Ch-43*, Ch-e43 and Ch-ΔICDI co-expressed with the mCherry control of the current profile matched that of Ch-43*, whereby ICa rapidly decayed, and IBa displayed only modest inactivation (Fig. 9, A, C, and E). However when co-expressed with ICDI-mCherry, the fast inactivation of ICa was much retarded, and in the case of Ch-e43 and Ch-ΔICDI, ICa decayed as slowly as IBa (Fig. 9, B, D, and F).
FIGURE 9.

Current traces, CDI, and VDI of Ch-43* and deletion constructs in co-expression experiments. Channel constructs were co-expressed with either ICDI-mCherry or mCherry (labeled on the top) as described earlier. A–F, representative IBa (gray) and ICa (black) traces during a 400-ms depolarization to 10 mV. G–L, residual IBa (gray) and ICa (black) for the corresponding co-expression experiments above, measured at 30- and 300-ms as labeled on the axes. M and N, strength of CDI (f-value and netCDI) calculated at 10 mV for r30 and r300, respectively. O, percentage of IBa inactivation during depolarization to Vmax in the time course indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (compared as indicated; unpaired t test).
Ch-43*, Ch-e43, and Ch-ΔICDI when co-expressed with mCherry control, exhibited a residual ICa with a U-shaped dependence on voltage (Fig. 9, G, I, and K) that is indicative of CDI. This U-shape residual ICa was lost when the channels were co-expressed with ICDI-mCherry and the residual ICa attained profiles similar to residual IBa when plotted against voltage (Fig. 9, H, J, and L).
In Fig. 9M, the calculated f10 as well as netCDI showed that ICDI-mCherry completely eliminated CDI at 30 ms. At 300 ms (Fig. 9N), co-expression with ICDI-mCherry suppressed f10 from 0.54 ± 0.12 to 0.05 ± 0.15 in Ch-43*, from 0.62 ± 0.08 to 0.04 ± 0.19 in Ch-e43, and from 0.57 ± 0.13 to 0.08 ± 0.16 in Ch-ΔICDI. Calculated values of netCDI also indicated similar degrees of CDI abolishment. Here, ICDI-mCherry suppressed CDI to levels lower than that measured in Ch-WT. This is perhaps due to the greater abundance of the overexpressed ICDI-mCherry as well as the increased mobility that ICDI has when expressed as a separate peptide.
Co-expression with ICDI-mCherry enhanced VDI in Ch-43*, Ch-e43, and Ch-ΔICDI. In the absence of ICDI-mCherry, Ch-43* exhibited slow IBa inactivation with 1.5 ± 0.5% at 30 ms after activation and only 17.3 ± 3.3% by 300 ms (Fig. 9O). Ch-e43 and Ch-ΔICDI also displayed similarly slow VDI (300 ms, Ch-e43, 18.0 ± 2.6%; Ch-ΔICDI, 22.4 ± 5.6%; p > 0.1, unpaired t test). When co-expressed with ICDI-mCherry, the inactivation of IBa through Ch-43* was increased to 7.9 ± 1.5% at 30 ms and 47.3 ± 3.0% at 300 ms (p < 0.001, unpaired t test). Similarly, for Ch-e43 and Ch-ΔICDI, the presence of ICDI-mCherry increased IBa inactivation at 300 ms to 37.3 ± 3.9 and 37.4 ± 3.4% (p < 0.001, unpaired t test), respectively. However, for Ch-ΔICDI, the increase in VDI due to the presence of ICDI-mCherry was only significant at 300 ms, although an average increase was already apparent at 200 ms. Unlike Ch-e43, significant VDI increase due to ICDI-mCherry could be detected from 50 ms onwards. This “delayed” response displayed by Ch-ΔICDI may be attributed to the additional sequences of the C terminus that Ch-ΔICDI has compared with Ch-e43 and Ch-43*.
Taken together, the data so far demonstrated that the hyperpolarized shift in I-V relationship, the restoration of CDI, as well as slow VDI conferred by 43* splicing was predominantly due to the absence of the ICDI domain rather than other portions of the C terminus. This is because the construct with only the ICDI deleted, Ch-ΔICDI, displayed similar activation and inactivation properties as Ch-43*. Besides, further deletion of the remaining length of C terminus between the ICDI and exon 43 did not significantly alter these properties. Moreover, co-expression with a peptide containing the ICDI was able to restore Ch-WT-like characteristics to all three constructs; however, VDI in Ch-ΔICDI required a longer depolarization to respond. Therefore, it seems that the ICDI is a multifunction domain capable of modulating activation and inactivation properties of the channel.
DISCUSSION
Voltage-gated calcium channels are extensively and alternatively spliced, and such post-transcriptional modifications diversify the biophysical properties of the channel. In our work, we identified 19 alternative splice variants of CaV1.4 in the human retina, 16 of which are completely novel. Importantly, we showed that alternative splicing can restore CDI to the CaV1.4 channel that was previously reported to be CDI-insensitive (4, 6). Also, the splice variant 43* exhibited a substantial hyperpolarized shift in activation as well as an increase in current density. We had also characterized the recovery of each C-terminal splice variant from inactivation in the chimeric channels, and we showed that 43* dramatically slowed recovery, whereas 45a− slowed recovery only at the early phase. The involvement of proline residues within proline-rich exon 45 was also implicated. However, because voltage inactivation was much reduced in the native channel, we were unable to recapitulate these effects here. Nonetheless, the figures for the recovery experiments are given in supplemental Figs. 8 and 9.
Although in previous studies artificially engineered truncations of the C terminus of CaV1.4 have determined that removal of ICDI resulted in disinhibition of CDI (9, 10), we now showed that this process may be natively regulated by alternative splicing. This is found in the 43* variant that introduces a termination signal into the C terminus, 137 residues from the IQ motif. The placement of the stop codon by the novel exon cassette is strategic because while maintaining the integrity of the calcium-sensing apparatus, it also generates a channel with a twice larger current density than the original. This increase in current density was not seen in another C-terminal truncated CaV1.4 (L1591X mutant) (10)), whereby the entire length of the C terminus immediately after the IQ was removed. These data suggested the importance of the additional sequences retained by the 43* splicing. In support of this, an appreciable degree of conservation across the LTCC isoforms is found in the proximal part of this region (supplemental Fig. 3). In CaV1.1 and -1.2, truncation of the C terminus after this region also gave rise to increased current density (Cav1.1 (34), Cav1.2 (35), and Cav1.2 (36)). In addition, a segment of this region (PCRD, supplemental Fig. 3) was also defined here to be the binding target of a distal C-terminal autoinhibitory domain (DCRD) that is responsible for repressing current density. Sequences homologous to the DCRD may also be found within the ICDI of CaV1.4. This raises an important question of whether the PCRD and DCRD homologues of CaV1.4 interact. This is especially so in the context of abolishment of CDI by ICDI because the requirement for the “A” region, encompassing the PCRD, for ICDI efficacy have been reflected in several previous works (9–11). Investigating this interaction may mold our understanding on the two mechanisms that have been described for ICDI (8, 11).
The increased current density in CaV1.4 43* may be due to one or a combination of three causes as follows: an increase in expression of the channel on the plasma membrane, an increase in unitary conductance, and an increase in open probability. We have shown that this was not due to an increased membrane expression. On the contrary, CaV1.4 43* membrane expression was six times lower than WT channels. In the CaV1.2 truncated channel, there was neither an increase in channel expression, as shown by gating charge analyses, nor an increase in single channel conductance (35). In CaV1.3, there was no increase in expression density detected either (38). For CaV1.4, it remains to be seen, with single channel analyses, if 43* splicing could alter unitary conductance and open probability.
The 43* splicing also resulted in a hyperpolarized shift in activation, and this may appear to conflict with some of the previous reports on other C-terminal truncated LTCCs. However, such left shifts were observed in C-terminal truncated LTCCs co-expressed either with β2a or β3 (CaV1.2 (36), CaV1.3 (38), and CaV1.1 (39)) and not seen when co-expressed with β1b (CaV1.3 (40) and CaV1.2 (41)).
Our data may be used to provide insights on certain discrepancies between the currents obtained when recording from the rod photoreceptors versus the current characteristics of cloned CaV1.4 heterologously expressed in cells. Cloned CaV1.4 channels characterized in a heterologous expression system are activated by membrane depolarization to potentials that are more positive compared with the rod photoreceptor dark resting membrane potential of approximately −40 mV (17, 42–44). This posed a problem because rod photoreceptors hyperpolarize upon activation by light, apparently implying that in the normal operating range of the rod photoreceptor membrane potential CaV1.4 channels are minimally activated or not at all. Also, when native calcium currents in the rod photoreceptor were measured, they could be evoked at ∼30 mV more hyperpolarized potentials than that obtained from cultured cells expressing cloned full-length CaV1.4 channels (12–14). It is possible that the native currents resulted from the presence of other calcium channel isoforms (i.e. T-type). Taken together, these findings seemingly excluded CaV1.4 from the role of inducing neurotransmission at the photoreceptor synapse (45). However given its localization together with its association with CSNB2 etiology (1, 2, 13), CaV1.4 activity should play an important role in normal rod photoreceptor function. The possibility of channel modulation by intracellular binding factors was thus explored, and it was found (18) that interaction with CaBP4 was able to shift the activation of the CaV1.4 channel by −10 mV, therefore pushing the channel into the operating range of the rod photoreceptor. In this study, we showed that post-transcriptional modification of the CaV1.4 channel by alternative splicing provided another means to shift channel activation potential. Alternative splicing generating the novel exon 43* mediated an ∼10 mV more hyperpolarized activated Ca2+ current.
Although 43* variants comprised only 13.6% of total CaV1.4 transcripts expressed in the human retina, it exhibited a 2-fold larger in current density. Therefore, the current contributed by 43* variants may make up a larger proportion of total endogenous CaV1.4 calcium current in the retina.
Another discrepancy between cloned CaV1.4 and native currents is the presence of CDI in the latter. This was detected in Ca2+ currents measured from rod photoreceptors and retinal bipolar synapses (15–17). We likewise propose that the CDI observed here may have been contributed by alternatively spliced variants of CaV1.4 like those containing exon 43* that exhibits robust CDI. The main difference between the CDI exhibited by the 43* splicing and the CDI shown in these reports is that the native CDI occurred with slower kinetics. In addition to differences in experimental setup (i.e. levels of charged carriers used, strength of Ca2+ buffering, etc.), the conditions present under the more physiological setting may modulate the rate and degree of CDI. For example, in CaV1.2 and CaV1.3, the presence of calcium-binding proteins was shown to reduce CDI strength (46). Also, CDI measured from rods and bipolar cells had different inactivation time constants (15), and CDI was stronger, and the rate of recovery from inactivation was pronouncedly slower in rods present in retinal slices than in isolated rod cells (16).
Supplementary Material
Acknowledgments
We sincerely thank the following people for the invaluable gifts of molecular clones: Dr. Terry P. Snutch (University of British Columbia, Canada) for β2a and α2δ clones; Dr. Roger D. Zühlke (University of Bern, Switzerland) for the CaV1.2 α1 clone (α1C77WT pBluescript); Dr. John E. McRory (University of British Columbia, Canada) for the CaV1.4 α1 clone (CaV1.4 pcDNA3.1), and Dr. Roger Y. Tsien (University of California, San Diego) for the mCherry clone (pRSET-B mCherry).

This article contains supplemental Figs. 1–9 and Tables 1–7.
- LTCC
- L-type calcium channel
- CDI
- calcium-dependent inactivation
- CSNB2
- congenital stationary night blindness type-2
- ICDI
- inhibitor of calcium-dependent inactivation
- I-V
- current-voltage
- SSI
- steady-state inactivation
- VDI
- voltage-dependent inactivation
- pF
- picofarad.
REFERENCES
- 1. Strom T. M., Nyakatura G., Apfelstedt-Sylla E., Hellebrand H., Lorenz B., Weber B. H., Wutz K., Gutwillinger N., Rüther K., Drescher B., Sauer C., Zrenner E., Meitinger T., Rosenthal A., Meindl A. (1998) An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 19, 260–263 [DOI] [PubMed] [Google Scholar]
- 2. Bech-Hansen N. T., Naylor M. J., Maybaum T. A., Pearce W. G., Koop B., Fishman G. A., Mets M., Musarella M. A., Boycott K. M. (1998) Loss-of-function mutations in a calcium-channel α1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 19, 264–267 [DOI] [PubMed] [Google Scholar]
- 3. Miyake Y., Yagasaki K., Horiguchi M., Kawase Y., Kanda T. (1986) Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 104, 1013–1020 [DOI] [PubMed] [Google Scholar]
- 4. McRory J. E., Hamid J., Doering C. J., Garcia E., Parker R., Hamming K., Chen L., Hildebrand M., Beedle A. M., Feldcamp L., Zamponi G. W., Snutch T. P. (2004) The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J. Neurosci. 24, 1707–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baumann L., Gerstner A., Zong X., Biel M., Wahl-Schott C. (2004) Functional characterization of the L-type Ca2+ channel Cav1.4α1 from mouse retina. Invest. Ophthalmol. Vis. Sci. 45, 708–713 [DOI] [PubMed] [Google Scholar]
- 6. Koschak A., Reimer D., Walter D., Hoda J. C., Heinzle T., Grabner M., Striessnig J. (2003) Cav1.4α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J. Neurosci. 23, 6041–6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stroffekova K. (2008) Ca2+/CaM-dependent inactivation of the skeletal muscle L-type Ca2+ channel (Cav1.1). Pflugers Arch. 455, 873–884 [DOI] [PubMed] [Google Scholar]
- 8. Griessmeier K., Cuny H., Rötzer K., Griesbeck O., Harz H., Biel M., Wahl-Schott C. (2009) Calmodulin is a functional regulator of Cav1.4 L-type Ca2+ channels. J. Biol. Chem. 284, 29809–29816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wahl-Schott C., Baumann L., Cuny H., Eckert C., Griessmeier K., Biel M. (2006) Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc. Natl. Acad. Sci. U.S.A. 103, 15657–15662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh A., Hamedinger D., Hoda J. C., Gebhart M., Koschak A., Romanin C., Striessnig J. (2006) C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat. Neurosci. 9, 1108–1116 [DOI] [PubMed] [Google Scholar]
- 11. Liu X., Yang P. S., Yang W., Yue D. T. (2010) Enzyme-inhibitor-like tuning of Ca(2+) channel connectivity with calmodulin. Nature 463, 968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thoreson W. B., Tranchina D., Witkovsky P. (2003) Kinetics of synaptic transfer from rods and cones to horizontal cells in the salamander retina. Neuroscience 122, 785–798 [DOI] [PubMed] [Google Scholar]
- 13. Morgans C. W., Bayley P. R., Oesch N. W., Ren G., Akileswaran L., Taylor W. R. (2005) Photoreceptor calcium channels: insight from night blindness. Vis. Neurosci. 22, 561–568 [DOI] [PubMed] [Google Scholar]
- 14. Cia D., Bordais A., Varela C., Forster V., Sahel J. A., Rendon A., Picaud S. (2005) Voltage-gated channels and calcium homeostasis in mammalian rod photoreceptors. J. Neurophysiol. 93, 1468–1475 [DOI] [PubMed] [Google Scholar]
- 15. von Gersdorff H., Matthews G. (1996) Calcium-dependent inactivation of calcium current in synaptic terminals of retinal bipolar neurons. J. Neurosci. 16, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rabl K., Thoreson W. B. (2002) Calcium-dependent inactivation and depletion of synaptic cleft calcium ions combine to regulate rod calcium currents under physiological conditions. Eur. J. Neurosci. 16, 2070–2077 [DOI] [PubMed] [Google Scholar]
- 17. Corey D. P., Dubinsky J. M., Schwartz E. A. (1984) The calcium current in inner segments of rods from the salamander (Ambystoma tigrinum) retina. J. Physiol. 354, 557–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haeseleer F., Imanishi Y., Maeda T., Possin D. E., Maeda A., Lee A., Rieke F., Palczewski K. (2004) Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat. Neurosci. 7, 1079–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao P., Yu D., Li G., Yong T. F., Soon J. L., Chua Y. L., Soong T. W. (2007) A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 282, 35133–35142 [DOI] [PubMed] [Google Scholar]
- 20. Shen Y., Yu D., Hiel H., Liao P., Yue D. T., Fuchs P. A., Soong T. W. (2006) Alternative splicing of the Ca(v)1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J. Neurosci. 26, 10690–10699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao P., Yong T. F., Liang M. C., Yue D. T., Soong T. W. (2005) Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc. Res. 68, 197–203 [DOI] [PubMed] [Google Scholar]
- 22. Tang Z. Z., Liang M. C., Lu S., Yu D., Yu C. Y., Yue D. T., Soong T. W. (2004) Transcript scanning reveals novel and extensive splice variations in human L-type voltage-gated calcium channel, Cav1.2α1 subunit. J. Biol. Chem. 279, 44335–44343 [DOI] [PubMed] [Google Scholar]
- 23. Soong T. W., DeMaria C. D., Alvania R. S., Zweifel L. S., Liang M. C., Mittman S., Agnew W. S., Yue D. T. (2002) Systematic identification of splice variants in human P/Q-type channel α1(2.1) subunits: implications for current density and Ca2+-dependent inactivation. J. Neurosci. 22, 10142–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mittman S., Guo J., Agnew W. S. (1999) Structure and alternative splicing of the gene encoding α1G, a human brain T calcium channel α1 subunit. Neurosci. Lett. 274, 143–146 [DOI] [PubMed] [Google Scholar]
- 25. Boycott K. M., Maybaum T. A., Naylor M. J., Weleber R. G., Robitaille J., Miyake Y., Bergen A. A., Pierpont M. E., Pearce W. G., Bech-Hansen N. T. (2001) A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Hum. Genet. 108, 91–97 [DOI] [PubMed] [Google Scholar]
- 26. Mittman S., Guo J., Emerick M. C., Agnew W. S. (1999) Structure and alternative splicing of the gene encoding α1I, a human brain T calcium channel α1 subunit. Neurosci. Lett. 269, 121–124 [DOI] [PubMed] [Google Scholar]
- 27. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 28. Mori M. X., Erickson M. G., Yue D. T. (2004) Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science 304, 432–435 [DOI] [PubMed] [Google Scholar]
- 29. Chenchik A., Diachenko L., Moqadam F., Tarabykin V., Lukyanov S., Siebert P. D. (1996) Full-length cDNA cloning and determination of mRNA 5′ and 3′ ends by amplification of adaptor-ligated cDNA. BioTechniques 21, 526–534 [DOI] [PubMed] [Google Scholar]
- 30. Jurkat-Rott K., Lehmann-Horn F. (2004) The impact of splice isoforms on voltage-gated calcium channel α1 subunits. J. Physiol. 554, 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barrett C. F., Tsien R. W. (2008) The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc. Natl. Acad. Sci. U.S.A. 105, 2157–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tadross M. R., Dick I. E., Yue D. T. (2008) Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Findeisen F., Minor D. L., Jr. (2009) Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J. Gen. Physiol. 133, 327–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morrill J. A., Cannon S. C. (2000) COOH-terminal truncated α(1S) subunits conduct current better than full-length dihydropyridine receptors. J. Gen. Physiol. 116, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wei X., Neely A., Lacerda A. E., Olcese R., Stefani E., Perez-Reyes E., Birnbaumer L. (1994) Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J. Biol. Chem. 269, 1635–1640 [PubMed] [Google Scholar]
- 36. Gao T., Cuadra A. E., Ma H., Bunemann M., Gerhardstein B. L., Cheng T., Eick R. T., Hosey M. M. (2001) C-terminal fragments of the α1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated α1C subunits. J. Biol. Chem. 276, 21089–21097 [DOI] [PubMed] [Google Scholar]
- 37. Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., Tsien R. Y. (2004) Improved monomeric red, orange, and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 [DOI] [PubMed] [Google Scholar]
- 38. Singh A., Gebhart M., Fritsch R., Sinnegger-Brauns M. J., Poggiani C., Hoda J. C., Engel J., Romanin C., Striessnig J., Koschak A. (2008) Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ren D., Hall L. M. (1997) Functional expression and characterization of skeletal muscle dihydropyridine receptors in Xenopus oocytes. J. Biol. Chem. 272, 22393–22396 [DOI] [PubMed] [Google Scholar]
- 40. Xu W., Lipscombe D. (2001) Neuronal Ca(V)1.3α(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hulme J. T., Yarov-Yarovoy V., Lin T. W., Scheuer T., Catterall W. A. (2006) Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J. Physiol. 576, 87–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Witkovsky P., Schmitz Y., Akopian A., Krizaj D., Tranchina D. (1997) Gain of rod to horizontal cell synaptic transfer: relation to glutamate release and a dihydropyridine-sensitive calcium current. J. Neurosci. 17, 7297–7306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moriondo A., Pelucchi B., Rispoli G. (2001) Calcium-activated potassium current clamps the dark potential of vertebrate rods. Eur. J. Neurosci. 14, 19–26 [DOI] [PubMed] [Google Scholar]
- 44. Schneeweis D. M., Schnapf J. L. (2000) Noise and light adaptation in rods of the macaque monkey. Vis. Neurosci. 17, 659–666 [DOI] [PubMed] [Google Scholar]
- 45. Thoreson W. B. (2007) Kinetics of synaptic transmission at ribbon synapses of rods and cones. Mol. Neurobiol. 36, 205–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cui G., Meyer A. C., Calin-Jageman I., Neef J., Haeseleer F., Moser T., Lee A. (2007) Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. J. Physiol. 585, 791–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sharp P. A., Burge C. B. (1997) Classification of introns: U2-type or U12-type. Cell 91, 875–879 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.