

Abstract
N-Myristoyltransferase (NMT) represents a promising drug target for human African trypanosomiasis (HAT), which is caused by the parasitic protozoa Trypanosoma brucei. We report the optimization of a high throughput screening hit (1) to give a lead molecule DDD85646 (63), which has potent activity against the enzyme (IC50 = 2 nM) and T. brucei (EC50 = 2 nM) in culture. The compound has good oral pharmacokinetics and cures rodent models of peripheral HAT infection. This compound provides an excellent tool for validation of T. brucei NMT as a drug target for HAT as well as a valuable lead for further optimization.
Introduction
Human African trypanosomiasis (HAT) or sleeping sickness is caused by two subspecies of the protozoan parasite Trypanosoma brucei (T. brucei gambiense and T. b. rhodesiense), which are transmitted by the bite of an infected tsetse fly. The disease, which is fatal unless treated, has two stages: an initial peripheral infection during which the parasites are found in the bloodstream and gives rise to nonspecific symptoms; a second stage when parasites enter the central nervous system, causing the classic symptoms of HAT, eventually leading to coma and death. Currently there are five treatments available, although none of them are satisfactory because of toxicity, treatment failures, and the requirement for parenteral administration, which is inappropriate in a rural African setting.1
The enzyme N-myristoyltransferase (NMT) represents a promising drug target, since its essentiality has been demonstrated in many organisms. Moreover, in T. brucei, RNAi knockdown of NMT has been shown to be lethal in cell culture and to abrogate infectivity in a mouse model of infection.2,3
NMT catalyzes the co-translational transfer of myristate from myristoyl-CoA to the N-terminal glycine of a subset of eukaryotic proteins, a modification that has been implicated in subcellular targeting to membrane locations and/or activation and stabilization of the substrate protein (Figure 1).4 While there is an incomplete knowledge of the targets of NMT in T. brucei and their subsequent downstream effects, bioinformatic analysis suggests there are in excess of 60 potential substrates,2 two of which (ADP-ribosylation factor-1 protein (ARF-1) and ADP-ribosylation factor-like protein (ARL-1)) have been characterized and shown to be essential for bloodstream parasite viability.5 Therefore, inhibition of the enzyme would be expected to have pleiotropic effects through its potential to affect multiple pathways.
Figure 1.

Format and performance of the HTS assay: (A) % inhibition and IC50 values determined using a scintillation proximity assay;2(B) frequency histogram representing % inhibition values for the HTS. Hits are designated as those compounds that displayed a % inhibition equal to or greater than 3 standard deviation units above the mean (40% inhibition). (C) Comparison of replicate % inhibition values for the 352 primary screen hits.
The enzyme operates via a Bi-Bi mechanism in which binding of myristoyl-CoA induces a conformational rearrangement that reveals the peptide binding site.2 The myristate group is then transferred in a nucleophilic addition–elimination reaction, which is followed by sequential release of CoA followed by the myristoylated protein. Several industrial research groups have targeted NMT from the yeast Candida albicans, initially developing peptidomimetic inhibitors,6−10 which had limited cellular activity. More recently promising small molecule inhibitors, based around benzofuran11−14 and benzothiazole15,16 cores, have been discovered.2 Development of these compounds as antifungal agents has not been continued presumably because of the inability to derive compounds with broad-spectrum antifungal activity.
We have previously reported biological studies that demonstrate the validity of T. brucei NMT (TbNMT) as a druggable target for HAT, using the prototypic TbNMT inhibitor DDD85646 (63).17,18 In this paper, we describe the medicinal chemistry that led to the discovery of this key inhibitor.
Results and Discussion
Hit Discovery
TbNMT entered our discovery portfolio for HAT following a positive assessment of its potential as a drug target, according to our published criteria.19 When the project commenced, there was a clinical need for safe oral drugs active in the first acute stage of the disease, in addition to the most favored profile of activity in both stages of HAT. Consequently, there was not an absolute requirement for compounds to access the CNS, relaxing some of the criteria required for compound progression, for example, polar surface area (PSA). Screening of a selection of antifungal compounds against TbNMT (a repositioning or piggy-back approach4) by Smith and colleagues at the University of York, U.K., failed to identify substantially potent or druglike compounds.20 Similarly we did not observe significant inhibition of TbNMT with the potential benzofuran leads RO-09-460913 and RO-09-487913 (see Supporting Information), or related analogues, originally developed by Roche against the fungal species C. albicans. Subsequently, comparison of X-ray crystal structures has revealed that there are substantial differences between the C. albicans NMT structure and that of NMT from Leishmania major. The T. brucei NMT enzyme (for which there is no structure to date) is most closely related to that of the L. major enzyme (74% sequence identity overall, 94% sequence identity within the active site).
In the absence of any reported tractable chemical start points, we opted to carry out a high-throughput screen (HTS) using our 63362-compound diversity library.21 A convenient homogeneous scintillation–proximity based assay in 384-well plate format was used to screen compounds, which employed TbNMT, tritiated myristoyl-CoA, the biotinylated peptide CAP5.522 (sequence GCGGSKVKPQPPQAK), and streptavidin coated polyvinyltoluene scintillation beads (Figure 1). Compounds were initially screened in singlicate at 30 μM, and the screen performance was excellent, with a mean (±SD) Z′ = 0.79 ± 0.05 (n = 210 plates). Compounds showing greater than 50% inhibition were reconfirmed in duplicate, and then potency was determined via 10-point half-log dilution curves (30 μM to 1.5 nM). Hit compounds shown to nonspecifically inhibit the assay signal were eliminated from further evaluation. Key compounds were also screened against human NMT-1 (HsNMT-1) and against the bloodstream form of the T. b. brucei (BSF427, VSG118) parasite in culture. Inhibition of proliferation of human MRC5 cells was used as an initial toxicity counterscreen (see Experimental Section).
Optimization of the Pyrazole Sulfonamides
Of the compounds confirmed as validated hits by testing of resynthesized material, we opted to focus our attention on compound 1 (Figure 2), the most potent example of a series of nine N-pyrazole arylsulfonamides identified by the HTS. The compound had potencies of 1.9 and 22 μM against TbNMT and HsNMT-1, respectively, demonstrating encouraging selectivity over the human isoform while having promising ligand efficiency (LE = 0.34).23 Moreover, 1 had moderate activity against the parasite in vitro (EC50 = 21 μM). Testing of 1 against varying concentrations of substrate peptide indicated that it is competitive for substrate binding and hence binds to the peptide binding site. Thus, changing the concentration of CAP5.5 from 0.5 to 16 μM caused the IC50 for TbNMT to change from 1.0 to 4.3 μM, as previously described.17
Figure 2.

Structure and biological activity of compound 1.
In the absence of any structural information for TbNMT to guide rational inhibitor design, we adopted a pragmatic chemistry-driven approach through which we aimed to (i) understand the importance for binding of existing parts of the inhibitor with a view to optimizing these and (ii) identify appropriate positions from which to extend the molecule and seek further ligand–protein interactions that improve affinity. A systematic program was therefore carried out to define the key pharmacophoric features of 1. A common requirement for high potency in previously reported NMT inhibitors is a basic functionality that interacts electrostatically with the C-terminal carboxylic acid of NMT, in the region of the active site normally occupied by the amino group of the peptide substrate’s terminal glycine. Although the binding mode of 1 was unknown from the outset, we anticipated that incorporation of a basic center in an appropriate location would significantly enhance potency of the hits, as experienced by previous NMT inhibitor programs.7−14,16,17,24
Investigation of SAR at the Pyrazole Group
Initially we investigated changes around the pyrazole of 1 (Table 1). Compounds were prepared by reaction of an amine with 3-methoxy-2,4,5-trimethylbenzenesulfonyl chloride. The pyrazole N-substituent could be modified by using a standard alkylation reaction on the symmetrical intermediate (2) (Scheme 1).
Table 1. Activities of Pyrazoles and Pyrazole Replacements against TbNMTa.

IC50 values are shown as mean values of two or more determinations. Standard deviation is typically within 2-fold from the IC50.
Scheme 1.

Reagents: (i) NH2R, pyridine; (ii) R1Br, Cs2CO3, DMF.
Removal of the N-methyl substituent of 1 (2) and replacement of the N-methyl substituent with a larger n-propyl (3) or isopropyl (4) group significantly reduced activity. A number of potential aromatic replacements of the pyrazole, including imidazopyridine (7), pyridine (8), substituted pyridine (9), isoxazole (10 and 11), phenyl (12), and benzyl (13) together with addition of a methylene between the sulfonamide and the pyrazole (14), were investigated. The only replacements that gave some potency were the 2-methylpyridyl derivative 9 (IC50 = 10 μM) and the isoxazole derivative 10 (IC50 = 19 μM), although these were both less potent than the original N-methylpyrazole 1 (IC50 = 1.9 μM). In combination, these results implied that the pyrazole N2 nitrogen was interacting with the enzyme as a hydrogen bond acceptor, since removal of it (e.g., 12) or increasing steric bulk around it (3 and 4) caused marked reductions in potency. Only isoelectronic heterocycles 9 and 10, with a similarly located hydrogen bonding acceptor capability, retained activity. Therefore, the trimethylpyrazole moiety of 1 was retained in further studies.
Investigation of SAR at the Sulfonamide Group
A series of analogues were made to investigate changes at the sulfonamide linker of compound 15 (Table 2). These were prepared as described in Scheme 2. In general, amides and sulfonamides were prepared by reaction of the appropriate amine with a benzoyl chloride or sulfonyl chloride, respectively.
Table 2. Activities of Sulfonamide Replacements against TbNMTa.

IC50 values are shown as mean values of two or more determinations. Standard deviation is typically within 2-fold from the IC50.
Scheme 2.

Reagents: (i) 4-bromobenzenesulfonyl chloride, pyridine, DCM; (ii) MeI, Cs2CO3, DMF; (iii) 4-bromobenzoyl chloride, pyridine, DCM; (iv) isobutyraldehyde, AcOH, NaBH(OAc)3, DCM, 0 °C → room temp; (v) 4-bromobenzyl bromide, K2CO3, KI, DMF; (vi) diisopropyl azodicarboxylate, polymer supported PPh3, pyridine, DCM; (vii) SOCl2, chloroform; (viii) sodium 4-bromobenzene sulfinate, H2O, acetone.
Replacement of the sulfonamide of 15 with an amide (17) as well as reversing the sulfonamide (19) and amide (24) led to a loss of activity. This suggests that either both sulfonamide oxygen atoms are involved in binding, or the sulfonamide causes a conformational effect important for locating the pyrazole ring relative to the aromatic system. It is known from crystallographic studies that N-substituted sulfonamides prefer to adopt a “kinked” conformation with substituents on the same side of the S–N bond.25 Therefore, amide 17 was N-substituted (18) with the aim of forcing the amide into a similar cis-conformation, but this compound was also inactive. Similarly, using an amine (21) or ether (22) as a linker led to a reduction in activity, presumably due to their greater flexibility and/or loss of hydrogen bond acceptor capability. Addition of an extra methylene (23) caused a loss of activity. Only the sulfone replacement (20) showed some activity (IC50 = 42 μM), although this was less than the corresponding sulfonamide (15).
The sulfonamide could be N-methylated with little effect on activity (16), suggesting that the sulfonamide NH group is not a participant in a significant interaction with TbNMT. Further substitution of the sulfonamide nitrogen of a related compound (29) with a variety of simple alkyl groups, while not unduly detrimental, did not significantly enhance activity (see Table S1 in the Supporting Information), with potencies observed in the range of 0.5 μM (R = methyl) to 9.3 μM (R = CH2C≡CCH3).
Investigation of SAR at the Aryl Group
An investigation of diverse substitutions of the arylsulfonyl ring was carried out. Compounds were prepared using parallel synthesis techniques (Scheme 3). We initially prepared 60 analogues (see Table S2 in the Supporting Information); key compounds are shown in Table 3.
Scheme 3.

Reagents: (i) pyridine, DCM.
Table 3. Activities of Aryl Replacements against TbNMTa.

IC50 values are shown as mean values of two or more determinations. Standard deviation is typically within 2-fold from the IC50.
In general, we found SAR around the ring to be comparatively flat, with even relatively sterically demanding substitutions in ortho-, meta-, and particularly para-positions tolerated, albeit without significant enhancements in potency. Given the largely inconsequential effect on potency of groups around the aromatic ring and the ability to tolerate even large substituents (e.g., 31, IC50 = 1.9 μM), we speculated that these substituents occupy a “sequence insensitive” region of the active site (i.e., the region that accommodates residues 2–4 of the substrate peptide), which is known to be capable of binding a variety of residues but to favor hydrophobic residues.26,27Given the approximately 10-fold improvement in activity obtained with the 2,6-dichloro substitution of 28 (IC50 = 1.0 μM) compared with 25 (IC50 = 12 μM), we continued to search for improvements in activity using this framework and utilized compound 29 in a Suzuki reaction for the preparation of a small speculative library of biaryl derivatives (see Table S3 in the Supporting Information). Of the 14 analogues prepared, only 36 (IC50 = 0.9 μM) had activity comparable to that of 29.
Additional Protein–Ligand Interactions from an Aminopyridine Scaffold
Since relatively bulky substitution was best tolerated in the para-position of the sulfonamide aromatic system (e.g., 36), we thought it most appropriate to seek additional protein–ligand interactions by probing from this position. The aminopyridyl moiety did not appear to be detrimental to binding per se (e.g., 31, IC50 = 1.9 μM); therefore, an additional exploratory library of 44 compounds was produced using a nucleophilic aromatic substitution reaction between chloropyridine 37 and a diverse set of commercially available amines (Scheme 4). Selected compounds are shown in Table 4 (full data can be seen in Table S4 in the Supporting Information).
Scheme 4.

Reagents: (i) pyridine, DCM; (ii) HNR1R2, EtOH, 130–155 °C.
Table 4. Activities of Aryl Replacements against TbNMT and HsNMT-1a.

IC50 values are shown as mean values of two or more determinations. Standard deviation is typically within 2-fold from the IC50. n.d. = not determined.
Again, a variety of substituents were tolerated, but with flat SAR, with activity generally in the low micromolar range. However, two compounds were particularly noteworthy because of the striking difference in potency despite their structural similarity. Compounds 41 (IC50 = 38 μM) and 42 (IC50 = 0.14 μM) differ only in having a morpholine or a methylpiperazine at the terminus of an ethyl linkage. We attributed the greater potency of 42 to its potential to form an electrostatic interaction via the protonatable terminal amine, since this approximately 100-fold improvement in activity is consistent with formation of a ligand–protein salt bridge, typically worth 10–400 kJ/mol. This interaction would not be possible via the morpholine of 41. We subsequently demonstrated through surrogate crystallographic studies in LmNMT that this is the most probable explanation for the increase in activity against TbNMT (see Structural Studies). Furthermore, replacement of the basic terminal nitrogen of 42 with a methylene (i.e., 44, IC50 = 14 μM) gave a large drop in activity, consistent with our reasoning. Given that enzyme kinetic studies using 1 indicated that this series binds in the peptide binding site, we speculated that 42 forms a salt bridge with the carboxyl group of the terminal residue of TbNMT (Val446), a mechanistically vital residue for myristoyl transfer. Moreover, this is an interaction that is found with all currently known inhibitors of NMT that have very high potency.6−16
An important objective of target-based discovery programmes is to achieve compounds with high selectivity against relevant human isoforms in order to minimize the risk of target-driven toxicity. While there is a high degree of sequence similarity between TbNMT and HsNMT-1 and -2 (41% identity, 69% similarity vs HsNMT-1; 44% identity, 68% similarity vs HsNMT-2) particularly at the active site (83% identity, 90% similarity vs both isoforms), fortuitously key compounds demonstrate upward of 60-fold selectivity against HsNMT-1 (e.g., 42).
A number of interesting observations were made when we further investigated a variety of analogues with pendent amines (Table 4). Differences in pKa (calculated using ACDlabs software, version 11.02) might account for the greater potency of the piperidine analogue 47 (IC50 = 0.03 μM; pKa = 9.6), which is 4-fold more active than the analogous piperazine 42 (IC50 = 0.14 μM; pKa = 7.9), and the lower potency of imidazole 50 (IC50 = 1.3 μM; pKa = 7.1). We were somewhat surprised that imidazole proved to be a poor substitute for an alkylamine, since this finding is in contrast to that of Searle.7 The des-methylpiperazine 43 (IC50 = 1.0 μM; pKa = 9.3), which we would expect to be more basic and therefore more potent, was actually around 8-fold less active than 42 potentially because of loss of a hydrophobic interaction at the terminal methyl group. Moreover, the loss of this methyl group causes a reduction in selectivity against HsNMT-1 compared to 42 (16-fold vs more than 60-fold). The spacer length to the amine could be varied; compare the two-carbon linker of 42 (IC50 = 0.14 μM) with the three-carbon linker of 49 (IC50 = 0.11 μM). However, the linker length in compound 46 (IC50 = 2.1 μM) would appear to be too short to optimally form the key interaction with the terminal nitrogen. There were indications that the orientation of the amine has an effect on activity (for example, compare conformationally constrained analogues 51, IC50 = 0.11 μM, 52, IC50 = 0.03 μM, and 40, IC50 = 0.36 μM) in compounds where the amines are constrained in slightly different orientations. Whereas methylation of the terminal nitrogen appears to be beneficial for activity, higher substitution, i.e., with isopropyl (45), phenyl (48), benzyl (53) or tert-butyloxycarbonyl groups (54), was not tolerated.
Effect of Conformational Restraint on the Activity of 42
Having identified a key protein–ligand electrostatic interaction in compounds typified by the prototype 42, we continued to explore the concept of using conformational restraint to orient the amine in an optimal manner to increase potency. We chose to revisit the biaryl system of 36 as a suitable scaffold from which to attach an amine moiety because it conveyed the dual benefits of an unambiguous conformation combined with the potency-enhancing effect of the 2,6-dichlorophenyl moiety. A series of compounds was examined where a protonatable nitrogen was attached in various positions around the distal aryl ring (Table 5). Biaryl compounds 56, 59, 60, and 63 could be prepared using a Suzuki cross-coupling reaction of commercially available arylboronic acids or esters with 29, as the key step (Scheme 5). Alternatively, 29 could be converted to the corresponding pinacolboronic ester by cross-coupling with bispinacolatodiboron, allowing Suzuki reaction in the reverse sense with the appropriate commercially available aryl bromide. Benzylamine compounds 57, 58, 61, and 62 were prepared by reductive amination of benzaldehyde 55.
Table 5. Activities of Constrained Analogues against TbNMT, HsNMT-1, and T. bruceia.

IC50 values are shown as mean values of two or more determinations. Standard deviation is typically within 2-fold from the IC50.
Scheme 5.

Reagents: (i) B(OH)2R or B(OR)2R, K3PO4, DMF, H2O, Pd(dppf)Cl2.DCM; (ii) bis-pinacolatodiboron, K3PO4, THF, Pd(PPh3)4; (iii) RBr, K3PO4, DMF, H2O, Pd(PPh3)4; (iv) HNR2, NaBH(OAc)3, CHCl3.
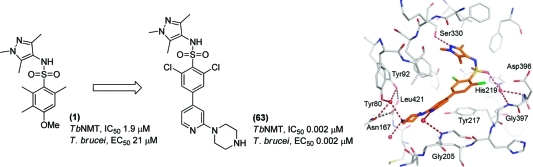
It is clear from these variations that substituents in the meta-position consistently gave significantly higher potency than those in the para-position; for example, compare 57 (IC50 = 1.2 μM) with 61 (IC50 = 0.005 μM); 58 (IC50 = 11 μM) with 62 (IC50 = 0.02 μM); and 59 (IC50 = 0.29 μM) with 63 (IC50 = 0.002 μM). A notable feature of compound 63, the most potent of these constrained molecules, is that it also potently inhibits the human isoform of NMT (TbNMT IC50 = 0.002 μM vs HsNMT-1 IC50 = 0.003 μM). In addition, DDD85646 also significantly inhibits the N-myristoyltransferase of other species, including Leishmania major (LmNMT IC50 = 0.002 μM) and Trypanosoma cruzi (TcNMT IC50 = 0.003 μM), which suggests that this compound has potential utility as a lead for other infectious diseases.
Pharmacology of 63
The intravenous (3 mg/kg) and oral (10 mg/kg) pharmacokinetics of 63 have been assessed in the female NMRI mouse, the species and strain used in the acute animal model of HAT. Compound 63 has low blood clearance (Clb = 6 mL min–1 kg–1) and low volume of distribution (Vd = 0.4 L/kg) with moderate half-life (t1/2 = 1.2 h) and oral bioavailability (F = 19%). Compound 63 (DDD85646) as the hydrochloride salt has high aqueous solubility (>15 mg/mL) and an appreciable unbound fraction in mouse and human plasma (11% and 18%, respectively).17
The pharmacokinetic properties of 63 therefore have enabled twice a day oral dosing, at a tolerated level, to achieve high exposure of free drug relative to the EC90 for parasite proliferation, making it a useful tool compound for validating the target in the animal model of acute HAT. In this model, the mice are infected with parasites and then treated with compound 3 days later for 4 days. After treatment mice are monitored for levels of parasitemia. Cure is defined as mice showing no signs of parasitemia 30 days after infection. The Kaplan–Meier survival plot for female NMRI mice (n = 5 per dose group) after infection with T. b. brucei s427 (inoculum 1 × 104 parasites) following oral treatment with 63 has been described previously.17 The minimal orally efficacious dose was found to be 12.5 mg/kg b.i.d. for 4 days. In contrast, the minimal orally efficacious dose in the more refractory but clinically relevant T. b. rhodesiense model of HAT was 50 mg/kg b.i.d. for 4 days.17 This reduced sensitivity in vivo is not due to reduced sensitivity of the compound in vitro but may be a result of the known precedent for this species to occupy privileged sites in vivo.17
Unfortunately, 63 has restricted brain penetration (brain/blood ratio of <0.1), consistent with its physicochemical properties (PSA = 92, MW = 495). Moreover, 63 also appears to be a weak P-glycoprotein substrate as evidenced by the Caco-2 efflux ratio (5.4) and the 3-fold increase in brain/blood ratio observed in rat when the P-glycoprotein inhibitor GF12091828 is co-administered, which at least partially explains the lack of CNS penetration. The low concentration of 63 measured in brain might simply represent residual compound present in the intravascular volume. That said, given its high potency, 63 was progressed to a stage 2 HAT efficacy study at the maximum tolerated dose (100 mg/kg po b.i.d.) to investigate whether efficacy could be observed. Female NMRI mice (n = 5 per dose group) were infected with T. b. brucei GVR35 (inoculum 1 × 104 parasites), and treatment commenced with 63 at 100 mg/kg po b.i.d. on day 21 postinfection for 5 days. No difference in survival time from that of vehicle control animals was observed. In contrast, melarsoprol (20 mg/kg ip once daily for 5 days), as positive control, was fully curative (100% survival to 180 days postinfection).
In terms of developability, the hepatic microsomal intrinsic clearance in rat (0.5 mL min–1 g–1) and human (1.2 mL min–1 g–1) is similar to that observed in mouse (0.6 mL min–1 g–1), suggesting that blood clearance in these species should not be too dissimilar from that observed in mouse. Compound 63 does not have a significant hERG liability (IC50 = 28 μM; automated patch clamp) and no significant drug–drug interaction liability (≤10% inhibition at 1 μM across the five major human isoforms CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) and therefore represents an exciting druggable lead for further development of a stage 1 compound for treatment of HAT.
Structural Studies
Attempts to crystallize the TbNMT have been unsuccessful to date. However, during the course of this project, we were able to obtain a structure for Leishmania major NMT (LmNMT), which we have used as a surrogate model for TbNMT. To further understand and confirm the SAR, we have carried out a retrospective structural analysis of selected ligands. This shows that 1, 29, 42, 59, and 63 occupy the substrate binding groove of LmNMT (Figure 3A). The pyrazole moieties superimpose well, occupying a pocket formed predominantly by the side chains of Val81, Asp83, Phe88, Phe90, Phe232, Ser330, Leu341, and Tyr345 that bind residues 4 and 5 of the peptide substrate (Figure 3B). This pocket has not been exploited in previously reported series of NMT inhibitors developed against the C. albicans and Saccharomyces cerevisiae enzymes because this pocket does not exist in these two species. Instead of a serine, there is a phenylalanine that occupies the same space as the pyrazole group of our ligands. In LmNMT the pyrazole nitrogen lone pair forms a hydrogen bond with the side chain hydroxyl group of Ser330. The pyrazole N-methyl group interacts with the side chains of Val81 and the aromatic ring of Phe90. As seen from the SAR, substituents larger than methyl at this nitrogen are not tolerated because of the steric constraints exerted by this pocket. The methyl in the 3-position of the pyrazole is packed against the aromatic group of Phe88 and the side chain of Leu341. Subsequent SAR studies (to be reported in due course) have shown that this interaction provides an important contribution to binding.
Figure 3.

Structural analysis of the pyrazole sulfonamide inhibitors of NMT. Panel A shows the binding mode of screening hit 1 bound to LmNMT (gold C atoms) overlaid with the peptide GLYASKLA bound to ScNMT (PDB code 1IID, green C atoms). Panels B–F show the binding modes of ligands (gold C atoms) bound to LmNMT (gray C atoms). Key water molecules are shown as red spheres and hydrogen bonds as dashed lines. Key residues are labeled for clarity. Panel B shows the binding mode of 1 forming canonical interactions formed by pyrazole sulfonamide moieties. Panel C shows the binding mode of 42 extending down toward the C-terminus, picking up water mediated interactions close to the terminal carboxylate. Panel D shows in detail the hydrogen bonding interactions of the conserved hydration site around the C-terminus and the interaction with the methylpiperazine of 42. Panel E shows the binding mode of the highly potent molecule 63 (PDB code 2WSA). Panel F shows the binding mode of the para-substituted molecule 59 (gold C atoms) overlaid with the binding mode of the meta-substituted 63 (purple C atoms, partially transparent). This illustrates the strained conformation of 59 and the altered hydrogen bonding pattern of the piperazine moiety.
The sulfonamide moiety of 1, 59, and 63 hydrogen-bonds through a highly coordinated water molecule to the side chain of His219 and the backbone amides of Asp396 and Gly397. This water molecule mimics the side chain hydroxyl of a Ser/Thr residue, an important feature of the consensus sequence at position 5 of the substrate peptide. There is a degree of flexibility in the position and orientation of the sulfonamide group; in 29 this moiety sits 0.8 Å closer to the Asn376-His219 pair and the water mediated hydrogen bonds are retained (Supporting Information, Table S6). In 42 the hydrogen bonding pattern of the sulfonamide is altered with the water bridged interaction with His219 being replaced by a direct contact between the sulfonamide oxygen and the side chain of Asn376. This tendency for the sulfonamide to occupy various positions makes it difficult to rationalize the exact importance of its binding contribution, and we suggest that this group may indeed only be important for optimal disposition of the pyrazole relative to the central aromatic. The benzene rings of 1, 29, 59, and 63 do not form directional interactions with the protein, though they stack above the side chain of Tyr217, which lines the floor of the peptide binding site. Hydrophobic packing is increased further by the two chlorine atoms of 63, explaining their beneficial effect on potency. In 42, the pyridyl nitrogen of this core ring potentially forms a water bridged interaction with the side chains of Tyr345 and Tyr217 with the linker nitrogen atom forming a water bridged interaction with the backbone carbonyl of His398.
As previously observed, the presence of a basic functionality near the C-terminal carboxylate results in large gains in potency. However, in contrast to the Roche benzofuran that interacts directly with the carboxylate group (PDB code 1IYL), the series described here hydrogen-bond indirectly to the C-terminal carboxylate via a network of water molecules (Figure 3C and Figure 3D). The residues that form this hydration site are all fully conserved throughout all NMTs. The alkyl chain of 42 extends down the peptide binding groove, allowing the methylated nitrogen of the piperazine to hydrogen bond with a water network formed by the side chains of Tyr92 and Asn167. An additional hydrogen bond is formed between the water molecule and the phenolic hydroxyl of Tyr80. The most potent inhibitor 63 presents the terminal piperazine nitrogen 2.8 Å from a tightly bound water molecule, and an additional hydrogen bond is made with another water network close to Met420. An additional water mediated hydrogen bond is formed between the pyridyl nitrogen atom and the amide of Gly205 (Figure 3E).
Comparison of the binding modes of 63 and its para-substituted counterpart 59 provides some insight into the large potency difference between the two molecules, despite retaining the pyrazole, sulfonamide, and to some extent the piperazine interactions (Figure 3F). The piperazine moiety of 63 forms two strong hydrogen bonds with excellent geometry to the solvent network around the C-terminal carboxylate. In the case of 59 two potential hydrogen bonds can be fulfilled, first to the well conserved water molecule described for 42 and to the side chain carbonyl of Asn167; however, the geometry of these interactions is suboptimal with the stronger of the two hydrogen bonds formed directly to the asparagine side chain. The meta-substitution of 63 allows the molecule to access a greater range of spatial orientations as opposed to the linear para-substitution of 59 which results in a strained conformation within the active site, suggesting that entropic constraints play a significant role in binding. The reduced enzyme activity of the para-substituted molecules may be attributed to a combination of the steric constraint and the suboptimal hydrogen bonding network around the C-terminal carboxylate.
Conclusion
Chemistry-driven optimization of a screening hit has resulted in the discovery of pyrazolesulfonamide DDD85646 (63), a highly potent, trypanocidal, orally active prototype inhibitor of TbNMT, which cures rodents of systemic infection with T. b. brucei and T. b. rhodesiense strains. As previously reported, a number of biological studies have already established that 63 acts on target:17 (i) overexpression of TbNMT in T. brucei(5-fold) led to a reduced sensitivity to 63 (8-fold); (ii) incubation of the parasites with 63 prevented incorporation of radiolabeled myristoyl-CoA into proteins; (iii) more generally over the series, there was a good correlation between inhibition of the enzyme and inhibition of parasite growth in culture. The precise mechanism by which inhibition of NMT causes its antiparasitic effects is not known. However, we suggest that a pleiotropic effect, caused by the inhibition of myristoylation of multiple important substrates, accounts for the effectiveness of 63 in cell culture and in vivo.
Although during this program we did not have structural information to guide inhibitor design, we are presently able to rationalize SAR using the structural information gained from surrogate crystallography in LmNMT. The majority of residues lining the active site are conserved between the T. brucei and human NMT enzymes, which may explain why many of the inhibitors including 63 show little selectivity. In the absence of a structure of TbNMT, we are unable to delineate useful differences between HsNMT and TbNMT that could inform the design of selective inhibitors. However, compounds within this series, such as 42, can possess a promising level of selectivity.
We have discovered a useful prototype inhibitor of TbNMT that is trypanocidal, has good oral pharmacokinetics, and is effective in acute models of disease, thus allowing us to successfully validate TbNMT as a target for drug discovery. The limitations of 63, its inability to cross the blood–brain barrier and lack of selectivity against HsNMT, are clearly factors that need to be addressed. Work is currently ongoing to optimize the selectivity and pharmacokinetic properties of compounds within this series to identify potential clinical candidates that can deliver an efficacious free concentration in the brain at a well-tolerated dose.
Experimental Section
General Experimental Information
Chemicals and solvents were purchased from Aldrich Chemical Co., Fluka, ABCR, VWR, Acros, Fisher Chemicals, and Alfa Aesar and were used as received unless otherwise stated. Air- and moisture-sensitive reactions were carried out under an inert atmosphere of argon in oven-dried glassware. Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (layer 0.20 mm silica gel 60 with fluorescent indicator UV254, from Merck). Developed plates were air-dried and analyzed under a UV lamp (UV 254/365 nm). Flash column chromatography was performed using prepacked silica gel cartridges (230–400 mesh, 40–63 μm, from SiliCycle) using a Teledyne ISCO Combiflash Companion or Combiflash Retrieve. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance II 500 spectrometer (1H at 500.1 MHz, 13C at 125.8 MHz) or a Bruker DPX300 spectrometer (1H at 300.1 MHz). Chemical shifts (δ) are expressed in ppm recorded using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), multiplet (m), broad (br), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz. LC–MS analyses were performed with either an Agilent HPLC 1100 series connected to a Bruker Daltonics MicrOTOF or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole spectrometer, where both instruments were connected to an Agilent diode array detector. LC–MS chromatographic separations were conducted with a Waters Xbridge C18 column, 50 mm × 2.1 mm, 3.5 μm particle size; mobile phase, water/acetonitrile + 0.1% HCOOH, or water/acetonitrile + 0.1% NH3; linear gradient from 80:20 to 5:95 over 3.5 min and then held for 1.5 min; flow rate of 0.5 mL min–1. All assay compounds had a measured purity of ≥95% (by TIC and UV) as determined using this analytical LC–MS system. High resolution electrospray measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer. Microwave-assisted chemistry was performed using a Biotage initiator microwave synthesizer. Compounds 25, 26, 33, and 34 were purchased from Enamine as solids. Purity (>97%) and molecular mass were confirmed by HPLC and high resolution mass spectrometry.
Prototypical Procedure for Preparation of a Sulfonamide from an Amine and a Sulfonyl Chloride
4-Methoxy-2,3,6-trimethyl-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)benzenesulfonamide (1)
4-Methoxy-2,3,6-trimethylbenzenesulfonyl chloride (500 mg, 2.0 mmol) was added portionwise to a stirred solution of 4-amino-1,3,5-trimethyl-1H-pyrazole (250 mg, 2.0 mmol) in pyridine (10.0 mL) at room temperature. The mixture was stirred for 24 h and then concentrated to dryness in vacuo. The resulting residue was diluted with DCM and washed with saturated aqueous NaHCO3. The organic phase was separated, dried (MgSO4), filtered, and concentrated to dryness in vacuo. Trituration from Et2O and collection by vacuum filtration gave the title compound 1 as a fine off-white solid (380 mg, 1.13 mmol, 57%). 1H NMR (500 MHz, DMSO-d6): δ 8.87 (s, 1H), 6.67 (s, 1H), 3.48 (s, 3H), 3.28 (s, 3H), 2.33 (s, 3H), 2.21 (s, 3H), 2.01 (s, 3H), 1.77 (s, 3H), 1.54 (s, 3H). HRMS (m/z): [MH+] calcd for C16H24N3SO3, 338.1533; found 338.1533.
4-Bromo-2,6-dichloro-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)benzenesulfonamide (29)17
Compound 29 was prepared from 4-bromo-2,6-dichlorobenzenesulfonyl chloride (5.0 g, 15.4 mmol) and 4-amino-1,3,5-trimethyl-1H-pyrazole (1.93 g, 15.4 mmol) in pyridine (35.0 mL) according to the method of 1, to give the title compound 29 as an off-white solid (5.64 g, 13.7 mmol, 89%). 1H NMR (300 MHz, DMSO-d6): δ 9.75 (s, 1H), 8.00 (s, 2H), 3.57 (s, 3H), 1.93 (s, 3H), 1.72 (s, 3H). HRMS (m/z): [MH+] calcd for C12H13N3SO2Cl2Br, 411.9283; found 411.9282.
6-Chloropyridine-3-sulfonic Acid (1,3,5-Trimethyl-1H-pyrazol-4-yl)amide (37)
37 was prepared from 6-chloropyridine-3-sulfonyl chloride (4.8 g, 22.7 mmol) and 4-amino-1,3,5-trimethyl-1H-pyrazole (2.84 g, 22.7 mmol) in pyridine (35.0 mL) according to the method of 1, to give the title compound 37 as an off-white solid (5.13 g, 17.1 mmol, 75%). 1H NMR (500 MHz, DMSO-d6): δ 9.51 (s, 1H), 8.59 (d, J = 2.3 Hz, 1H), 8.03 (dd, J = 7.6 Hz, 2.3 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 3.58 (s, 3H), 1.84 (s, 3H), 1.63 (s, 3H). HRMS (m/z): [MH+] calcd for C11H14N4SO2Cl, 301.0521; found 301.0523.
Prototypical Procedure for N-Alkylation of Compound 2
N-(1-Propyl-3,5-dimethyl-1H-pyrazol-4-yl)-4-methoxy-2,3,6-trimethylbenzenesulfonamide (3)
A solution of N-(3,5-dimethyl-1H-pyrazol-4-yl)-4-methoxy-2,3,6-trimethylbenzenesulfonamide 2 (100 mg, 0.31 mmol), cesium carbonate (202 mg, 0.62 mmol), and 1-bromopropane (76 mg, 0.62 mmol) in DMF (10.0 mL) was heated to 80 °C for 1 h in a microwave. The mixture was partitioned between ethyl acetate (25 mL) and brine (25 mL), dried (MgSO4), filtered, and concentrated to dryness in vacuo. The resulting residue was purified by column chromatography (SiO2, 1:1 ethyl acetate/hexane) to give the title compound as an off-white solid (27 mg, 0.07 mmol, 24%). 1H NMR (500 MHz, DMSO-d6): δ 8.67 (s, 1H), 6.45 (s, 1H), 4.46 (t, J = 6.9 Hz, 2H), 3.78 (s, 3H), 2.58 (s, 3H), 2.36 (s, 3H), 2.16 (s, 3H), 2.04 (s, 3H), 1.98 (m, 2H), 1.83 (s, 3H), 1.21 (t, J = 6.9 Hz, 3H). HRMS (m/z): [MH+] calcd for C18H28N3SO3, 366.1846; found 366.1847.
Prototypical Procedure for Preparation of a 2-Aminopyridine by SNAr Reaction of 2-Chloropyridine (37) with an Alkylamine
6-[2-(4-Methylpiperazin-1-yl)ethylamino]pyridine-3-sulfonic Acid (1,3,5-Trimethyl-1H-pyrazol-4-yl)amide (42)
6-Chloro-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)pyridine-3-sulfonamide (37) (225 mg, 0.75 mmol) and 4-(2-aminoethyl)methylpiperazine (215 mg, 1.5 mmol) in ethanol (2.0 mL) were heated at 155 °C for 1 h by microwave in a sealed vessel. Dilution with DCM (25 mL), washing with saturated aqueous sodium hydrogen carbonate solution (2 × 5 mL), drying (MgSO4), and concentration in vacuo gave a residual oil which was subjected to chromatography (SiO2, 10:90 MeOH/EtOAc) to give the title compound as an off-white powder (198 mg, 0.49 mmol, 65%). 1H NMR (300 MHz, DMSO-d6): δ 8.77 (s, 1H), 8.07 (d, J = 2.2 Hz, 1H), 7.45 (dd, J = 2.2 Hz, 8.9 Hz, 1H), 7.28 (s br, 1H), 6.54 (d, J = 8.9 Hz, 1H), 3.57 (s, 3H), 3.44–3.39 (m, 2H), 2.41 (t, J = 6.1 Hz, 2H), 2.41 (s br, 4H), 2.36–2.31 (s br, 4H and 3H), 2.16 (s, 3H), 1.89 (s, 3H), 1.67 (s, 3H). HRMS (m/z): [MH+] calcd for C18H30N7SO2, 408.2176; found 408.2185.
Prototypical Procedure for the Suzuki Reaction between an Aryl Bromide and a Boronic Acid/Boronate Ester
2,6-Dichloro-4-(2-piperazin-1-ylpyridin-4-yl)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)benzenesulfonamide (63)17
A deoxygenated solution of 4-bromo-2,6-dichloro-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)benzenesulfonamide (29) (13.84 g, 33.3 mmol), 2-(1-piperazinyl)pyridine-4-boronic acid pinacol ester (11.57 g, 40.0 mmol), tribasic potassium phosphate (9.73 g, 44.0 mmol), and Pd(PPh3)4 (1.50 g, 0.96 mmol) in DMF (200 mL) and water (40 mL) in a round-bottomed flask under argon was heated at 120 °C for 1 h. The reaction mixture was then concentrated in vacuo, diluted with DCM (400 mL), washed with saturated aqueous ammonia solution (2 × 100 mL), dried (MgSO4), and concentrated in vacuo. The residual solid was triturated from Et2O and collected by filtration to give a solid which was recrystallized from EtOAc to give the title compound 63 as an off-white powder (15.22 g, 30.7 mmol, 92%). 1H NMR (500 MHz, DMSO-d6): δ 9.79 (s, 1H), 8.25 (d, J = 5.9 Hz, 1H), 8.20 (s, 2H), 7.61 (s, 1H), 7.40 (d, J = 5.9 Hz, 1H), 4.08 (s br, 4H), 3.63 (s, 3H), 3.28 (s br, 4H), 2.00 (s, 3H), 1.77 (s, 3H). 13C NMR (125 MHz, DMSO-d6): 147.5, 147.3, 143.8, 137.4, 136.3, 135.2, 129.8, 111.8, 111.7, 109.1, 108.9, 42.7, 42.0, 36.2, 10.4. HRMS (m/z): [MH+] calcd for C21H25N6SO2Cl2, 495.1131; found 495.1124.
Enzyme Inhibition Assay
N-Myristoyltransferase is an enzyme that catalyzes the addition of myristic acid from myristoyl-CoA to the N-terminal glycine residue of numerous substrate proteins and peptides with the subsequent release of coenzyme A. 3H-labeled myristoyl-CoA (GE Healthcare) can be used in the reaction to transfer 3H-myristic acid to a biotinylated substrate peptide (GCGGSKVKPQPPQAK(biotin)-amide, Pepceuticals Inc.). The reaction can be measured by the subsequent binding of the labeled peptide to streptavidin-coated scintillation proximity assay (SPA) beads (GE Healthcare) and monitoring of β-particle excitation of the embedded scintillant (Figure 1).
Measurement of the ability of compounds to inhibit the N-myristoyltransferase enzyme(s) of human (HsNMT-1 and HsNMT-2) and kinetoplast (T. brucei, T. cruzi, and L. major) species was performed using a modification of the scintillation proximity assay platform described previously by Panethymitaki et al.20 as follows: Compounds were solubilized in DMSO at a top concentration of 10 mM and serially diluted in half log steps to achieve a range of final assay concentrations of 100 μM to 1.5 nM. Compound at each concentration was added to white 384-well plates in a volume of 0.5 μL. N-Myristoyltransferase enzyme (HsNMT-1, HsNMT-2, TcNMT, TbNMT, or LmNMT), dissolved to a working concentration of 10 nM in assay buffer (30 mM Tris-HCl, pH 7.4, 0.5 mM EGTA, 0.5 mM EDTA, 1.25 mM DTT, 0.1% Triton X-100), was then added to columns 1–11 and 13–23 of the plates in a volume of 20 μL. To columns 12 and 24, 20 μL of assay buffer was added to provide a no enzyme control. Following a 5 min incubation at room temperature the substrates (GCGGSKVKPQPPQAK(biotin)-amide and myristoyl-CoA), dissolved in assay buffer, were added to all wells in a volume of 20 μL to start the reaction. The final concentrations of peptide and 3H-myristoyl-CoA were 0.5 μM and 125 nM, respectively, and the specific activity of the radiolabel was 8 Ci/mmol. Plates were then incubated at room temperature for up to 50 min (dependent upon the period of linearity for the different enzyme species) before SPA beads, suspended to 1 mg/mL in a stop solution (200 mM phosphoric acid/NaOH, pH 4, 750 mM MgCl2), were added in a volume of 40 μL. Plates were then read on a TopCount microplate luminometer and data analyzed by calculating the percentage inhibition compared to the maximum and minimum assay controls. Concentration effect curves were fitted using nonlinear regression using XLFit 4.2, and IC50 values were determined.
Cell Viability Assay
Measurement of the ability of the compounds to inhibit human (MRC5, human lung fibroblast cells) and trypanosome (T. b. brucei, BSF427, VSG118) cell growth was performed using a modification of the cell viability assay previously described by Raz et al.29 Compounds were dissolved in DMSO at a top concentration of 10 mM and serially diluted in half log steps to achieve a range of final assay concentrations of 50 μM to 0.5 nM. Compound at each concentration (200-fold final) was added to clear 96-well tissue culture plates in a volume of 1 μL. Then 2000 cells per well in relevant growth medium (HMI-9T for T. brucei, a modification of HMI-9 as described by Hurumi et al.,30 where 0.2 mM 2-mercaptoethanol was replaced with 0.056 mM thiolglycerol, and MEM with 10% FBS for MRC5) were then added to columns 1–11 of the plates in a volume of 199 μL. To column 12, 200 μL of medium was added to provide a no cells control. Plates were then incubated at 37 °C in an atmosphere of 5% CO2 for 69 h, before the addition of 20 μL of 500 μM rezasurin solution, and a further incubation period of 4 h. Plates were then read on a BioTek flx800 fluorescent plate reader, and percentage inhibition was compared to the maximum and minimum assay controls. Concentration effect curves were fitted using nonlinear regression using XLFit 4.2 and EC50 values determined.
Crystallography
The expression construct for 6xHis-TEV site, LmNMT (5-421), was obtained from the Structural Genomics Consortium (SGC), Toronto, Canada. The protein was expressed in E. coli Rosetta (DE3) using autoinduction medium. Cells were harvested by centrifugation and resuspended in 50 mM HEPES, pH 7.5, 0.5 M NaCl, 5 mM imidazole, 5% glycerol (plus DNase, lysozyme, and a protease inhibitor cocktail) and lysed by passage through a constant cell disruptor (Constant Cell Systems). Chromatography steps were carried out using an AKTA system (GE Healthcare). The lysate was cleared by centrifugation (50K rcf 30 min) and loaded onto a 5 mL HisTRAP crude column (GE Healthcare), and target protein was eluted by a 5–250 mM imidazole gradient. Fractions containing LmNMT were pooled, desalted, and applied to a 6 mL Resource-Q column pre-equilibrated with 10 mM HEPES, pH 7.5. LmNMT was eluted by 0–500 mM NaCl gradient, and fractions were analyzed by SDS–PAGE with fractions greater than 95% purity concentrated to 9 mg/mL for crystallization.
Crystals of LmNMT with myristoyl-CoA were obtained by incubating the protein with 1 mM myristoyl-CoA for 1 h at 4 °C prior to crystallization. Protein crystals were obtained via the hanging drop vapor diffusion method by mixing 2 μL of protein solution mixed with 2 μL of reservoir solution that consisted of 24–30% PEG1500, 0.2 M NaCl, and 0.1 M Na cacodylate, pH 5.6. Rod shaped crystals appeared after 2–4 days at 20 °C.
Protein–ligand complexes were obtained by soaking the crystals for 16 h in mother liquor derived cryoprotectant (25% PEG1500, 0.2 M NaCl, 0.1 M Na cacodylate, pH 5.6, 20% glycerol) + 15 mM ligand, prepared from a stock concentration of 0.2 M in DMSO. Crystals were flash frozen in liquid nitrogen and stored before data measurement. X-ray diffraction data were measured at the ESRF using beamlines ID-29 and ID23-2 and the data reduced using the HKL suite.31 Molecular replacement was carried out using MOLREP32 with the binary LmNMT + myristoyl-CoA structure (PDB code 3H5Z) used as a search model. The models were refined using REFMAC5,33 and manual alteration was carried out using COOT34 with ligand coordinate and topology files created with PRODRG.35 Coordinates and associated diffraction data for complexes of LmNMT + 1, 29, 42, 62, and 59 have been deposited in the Protein Data Bank (PDB) with accession codes 4A2Z, 4A30, 4A31, 4A32, and 4A33, respectively.
Acknowledgments
Funding for this work was provided by the Wellcome Trust (Grants WT077705, WT083481, WT085622). The authors thank Gina McKay for performing HRMS analyses and for assistance with performing other NMR and MS analyses, Daniel James for data management, Bhavya Rao for performing parasite assays, Dr. Gian-Fillipo Ruda for synthesis of RO-09-4879, and Dr. Stephen Patterson for critical evaluation of the manuscript.
Glossary
Abbreviations Used
- NMT
N-myristoyltransferase
- HAT
human African trypanosomiasis
- ARF-1
ADP-ribosylation factor-1 protein
- ARL-1
ADP-ribosylation factor-like protein
- HTS
high throughput screen
- CAP5.5
cytoskeleton associated protein 5.5
- SPA
scintillation proximity assay
- PSA
polar surface area
- NMRI
Naval Medical Research Institute
- CYP
cytochrome P450
- hERG
human ether-a-go-go-related gene
- PDB
Protein Data Bank
Supporting Information Available
Synthetic details for all compounds, additional compound biological data (Tables S1–S4), and X-ray crystallographic data (Tables S5 and S6). This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Jacobs R. T.; Nare B.; Phillips M. A. State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 2011, 11, 1255–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer P. W.; Tate E. W.; Leatherbarrow R. J.; Holder A. A.; Smith D. F.; Brown K. A. N-Myristoyltransferase: a prospective drug target for protozoan parasites. ChemMedChem 2008, 3, 402–408. [DOI] [PubMed] [Google Scholar]
- Price H. P.; Guther M. L. S.; Ferguson M. A. J.; Smith D. F. Myristoyl-CoA:protein N-myristoyltransferase depletion in trypanosomes causes avirulence and endocytic defects. Mol. Biochem. Parasitol. 2010, 169, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb M. H.; Van Voorhis W. C.; Buckner F. S.; Yokoyama K.; Eastman R.; Carpenter E. P.; Panethymitaki C.; Brown K. A.; Smith D. F. Protein farnesyl and N-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol. Biochem. Parasitol. 2003, 126, 155–163. [DOI] [PubMed] [Google Scholar]
- Price H. P.; Goulding D.; Smith D. F. ARL1 has an essential role in Trypanosoma brucei. Biochem. Soc. Trans. 2005, 33, 643–645. [DOI] [PubMed] [Google Scholar]
- Brown D. L.; Devadas B.; Lu H. F.; Nagarajan S.; Zupec M. E.; Freeman S. K.; McWherter C. A.; Getman D. P.; Sikorski J. A. Replacements for lysine in l-seryl-l-lysyl dipeptide amide inhibitors of Candida albicans myristoyl-CoA:protein N-myristoyltransferase. Bioorg. Med. Chem. Lett. 1997, 7, 379–382. [Google Scholar]
- Devadas B.; Freeman S. K.; Zupec M. E.; Lu H. F.; Nagarajan S. R.; Kishore N. S.; Lodge J. K.; Kuneman D. W.; McWherter C. A.; Vinjamoori D. V.; Getman D. P.; Gordon J. I.; Sikorski J. A. Design and synthesis of novel imidazole-substituted dipeptide amides as potent and selective inhibitors of Candida albicans myristoylCoA:protein N-myristoyltransferase and identification of related tripeptide inhibitors with mechanism-based antifungal activity. J. Med. Chem. 1997, 40, 2609–2625. [DOI] [PubMed] [Google Scholar]
- Devadas B.; Zupec M. E.; Freeman S. K.; Brown D. L.; Nagarajan S.; Sikorski J. A.; McWherter C. A.; Getman D. P.; Gordon J. I. Design and syntheses of potent and selective dipeptide inhibitors of Candida albicans myristoyl-CoA:protein N-myristoyltransferase. J. Med. Chem. 1995, 38, 1837–1840. [DOI] [PubMed] [Google Scholar]
- Nagarajan S. R.; Devadas B.; Zupec M. E.; Freeman S. K.; Brown D. L.; Lu H. F.; Mehta P. P.; Kishore N. S.; McWherter C. A.; Getman D. P.; Gordon J. I.; Sikorski J. A. Conformationally constrained [p-(omega-aminoalkyl)phenacetyl]-l-seryl-l-lysyl dipeptide amides as potent peptidomimetic inhibitors of Candida albicans and human myristoyl-CoA:protein N-myristoyl transferase. J. Med. Chem. 1997, 40, 1422–1438. [DOI] [PubMed] [Google Scholar]
- Sikorski J. A.; Devadas B.; Zupec M. E.; Freeman S. K.; Brown D. L.; Lu H. F.; Nagarajan S.; Mehta P. P.; Wade A. C.; Kishore N. S.; Bryant M. L.; Getman D. P.; McWherter C. A.; Gordon J. I. Selective peptidic and peptidomimetic inhibitors of Candida albicans myristoylCoA:protein N-myristoyltransferase: a new approach to antifungal therapy. Biopolymers 1997, 43, 43–71. [DOI] [PubMed] [Google Scholar]
- Ebiike H.; Masubuchi M.; Liu P. L.; Kawasaki K.; Morikami K.; Sogabe S.; Hayase M.; Fujii T.; Sakata K.; Shindoh H.; Shiratori Y.; Aoki Y.; Ohtsuka T.; Shimma N. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 2. Bioorg. Med. Chem. Lett. 2002, 12, 607–610. [DOI] [PubMed] [Google Scholar]
- Kawasaki K.; Masubuchi M.; Morikami K.; Sogabe S.; Aoyama T.; Ebiike H.; Niizuma S.; Hayase M.; Fujii T.; Sakata K.; Shindoh H.; Shiratori Y.; Aoki Y.; Ohtsuka T.; Shimma N. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 3. Bioorg. Med. Chem. Lett. 2003, 13, 87–91. [DOI] [PubMed] [Google Scholar]
- Masubuchi M.; Ebiike H.; Kawasaki E.; Sogabe S.; Morikami K.; Shiratori Y.; Tsujii S.; Fujii T.; Sakata K.; Hayase M.; Shindoh H.; Aoki Y.; Ohtsuka T.; Shimma N. Synthesis and biological activities of benzofuran antifungal agents targeting fungal N-myristoyltransferase. Bioorg. Med. Chem. 2003, 11, 4463–4478. [DOI] [PubMed] [Google Scholar]
- Masubuchi M.; Kawasaki K.; Ebiike H.; Ikeda Y.; Tsujii S.; Sogabe S.; Fujii T.; Sakata K.; Shiratori Y.; Aoki Y.; Ohtsuka T.; Shimma N. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N-myristoyltransferase. Part 1. Bioorg. Med. Chem. Lett. 2001, 11, 1833–1837. [DOI] [PubMed] [Google Scholar]
- Ebara S.; Naito H.; Nakazawa K.; Ishii F.; Nakamura M. FTR1335 is a novel synthetic inhibitor of Candida albicansN-myristoyltransferase with fungicidal activity. Biol. Pharm. Bull. 2005, 28, 591–595. [DOI] [PubMed] [Google Scholar]
- Yamazaki K.; Kaneko Y.; Suwa K.; Ebara S.; Nakazawa K.; Yasuno K. Synthesis of potent and selective inhibitors of Candida albicansN-myristoyltransferase based on the benzothiazole structure. Bioorg. Med. Chem. 2005, 13, 2509–2522. [DOI] [PubMed] [Google Scholar]
- Frearson J. A.; Brand S.; McElroy S. P.; Cleghorn L. A. T.; Smid O.; Stojanovski L.; Price H. P.; Guther M. L. S.; Torrie L. S.; Robinson D. A.; Hallyburton I.; Mpamhanga C. P.; Brannigan J. A.; Wilkinson A. J.; Hodgkinson M.; Hui R.; Qiu W.; Raimi O. G.; Van Aalten D. M. F.; Brenk R.; Gilbert I. H.; Read K. D.; Fairlamb A. H.; Ferguson M. A. J.; Smith D. F.; Wyatt P. G. N-Myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand S.; Wyatt P.; Thompson S.; Smith V.; Bayliss T.; Harrison J.; Norcross N.; Cleghorn L.; Gilbert I.; Brenk R.. N-Myristoyl Transferase Inhibitors. WO2010026365, 2010.
- Frearson J. A.; Wyatt P. G.; Gilbert I. H.; Fairlamb A. H. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007, 23, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panethymitaki C.; Bowyer P. W.; Price H. P.; Leatherbarrow R. J.; Brown K. A.; Smith D. F. Characterization and selective inhibition of myristoyl-CoA:protein N-myristoyltransferase from Trypanosoma brucei and Leishmania major. Biochem. J. 2006, 396, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenk R.; Schipani A.; James D.; Krasowski A.; Gilbert I. H.; Frearson J. A.; Wyatt P. G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Fowler C.; Ersfeld K.; Gull K. CAP5.5, a life-cycle-regulated, cytoskeleton-associated protein is a member of a novel family of calpain-related proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 2001, 116, 25–34. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Ebara S.; Naito H.; Nakazawa K.; Ishii F.; Nakamura M. FTR1335 is a novel synthetic inhibitor of Candida albicansN-myristoyltransferase with fungicidal activity. Biol. Pharm. Bull. 2005, 28, 591–595. [DOI] [PubMed] [Google Scholar]
- Parkin A.; Collins A.; Gilmore C. J.; Wilson C. C. Using small molecule crystal structure data to obtain information about sulfonamide conformation. Acta Crystallogr. 2008, B64, 66–71. [DOI] [PubMed] [Google Scholar]
- McWherter C. A.; Rocque W. J.; Zupec M. E.; Freeman S. K.; Brown D. L.; Devadas B.; Getman D. P.; Sikorski J. A.; Gordon J. I. Scanning alanine mutagenesis and de-peptidization of a Candida albicans myristoyl-CoA:protein N-myristolytransferase octapeptide substrate reveals three elements critical for molecular recognition. J. Biol. Chem. 1997, 272, 11874–11880. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S.; Eisenhaber B.; Eisenhaber F. N-Terminal N-myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences. J. Mol. Biol. 2002, 317, 523–540. [DOI] [PubMed] [Google Scholar]
- Polli J. W.; Jarrett J. L.; Studenberg S. D.; Humphreys J. E.; Dennis S. W.; Brouwer K. R.; Woolley J. L. Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor. Pharm. Res. 1999, 16, 1206–1212. [DOI] [PubMed] [Google Scholar]
- Raz B.; Iten M.; Grether-Buhler Y.; Kaminski R.; Brun R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T. b. rhodesiense and T. b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [DOI] [PubMed] [Google Scholar]
- Hirumi H.; Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum-protein without feeder cell-layers. J. Parasitol. 1989, 75, 985–989. [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D 2000, 56, 1622–1624. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.