

Abstract
Lytic granules in cytotoxic lymphocytes, which include T cells and natural killer (NK) cells, are secretory lysosomes that release their content upon fusion with the plasma membrane (PM), a process known as degranulation. Although vesicle exocytosis has been extensively studied in endocrine and neuronal cells, much less is known about the fusion of lytic granules in cytotoxic lymphocytes. Here, we used total internal reflection fluorescence microscopy to examine lytic granules labeled with fluorescently tagged Fas ligand (FasL) in the NK cell line NKL stimulated with phorbol ester and ionomycin and in primary NK cells activated by physiological receptor–ligand interactions. Two fusion modes were observed: complete fusion, characterized by loss of granule content and rapid diffusion of FasL at the PM; and incomplete fusion, characterized by transient fusion pore opening and retention of FasL at the fusion site. The pH-sensitive green fluorescence protein (pHluorin) fused to the lumenal domain of FasL was used to visualize fusion pore opening with a time resolution of 30 ms. Upon incomplete fusion, pHluorin emission lasted several seconds in the absence of noticeable diffusion. Thus, we conclude that lytic granules in NK cells undergo both complete and incomplete fusion with the PM, and propose that incomplete fusion may promote efficient recycling of lytic granule membrane after the release of cytotoxic effector molecules.
Keywords: degranulation, fusion, human, lytic granules, natural killer cells
Target cell killing by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells requires exocytosis of the content of lytic granules (LGs),1, 2, 3 which store effector molecules, such as granzymes, perforin and Fas ligand (FasL, also known as CD95-L and CD178). LG fuse with the plasma membrane (PM), following CTL and NK cell activation, and release cytolytic effector molecules that kill susceptible target cells.4 Several molecules that play an important role in the fusion of LG with the PM of cytotoxic lymphocytes have been identified.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 Munc13-4 is essential in the priming of LG docked at the PM,18 and Rab27a is required for the delivery of LG to and/or their retention at the PM.19, 20 Defects in Munc18-2, myosin IIA or syntaxin-11 in NK cells result in LG that polarize toward the immunological synapse, but are unable to release their content.21, 22, 23, 24, 25 Imaging studies have shown that polarized LG are released by CTL and NK cells at a defined secretory domain within the immunological synapse;26, 27 however, how LG fuse with the PM in cytotoxic lymphocytes is unclear.
Although molecules that regulate exocytosis of granules could vary in different cell types, increasing evidence shows that LG may share a similar mechanism with granules in endocrine cells and synaptic vesicles in neuronal cells for their fusion with the PM.11, 12, 28 Incomplete fusion of synaptic vesicles at the frog neuromuscular junction has been described more than four decades ago by electron microscopy.29 Extensive evidence from imaging and electrophysiological techniques suggests that both complete and incomplete fusion occur in endocrine cells and several different neural synapses, including the calyx of held synapse,30 neuromuscular junction31, 32 and cultured hippocampal synapse.33
To test whether complete fusion or incomplete fusion of LG with the PM occurs in cytotoxic lymphocytes, we used the human NK cell line NKL, as well as primary NK cells from human peripheral blood, as a model to study the fusion modes of LG during degranulation. Here, we labeled LG with DsRed-FasL-pH-sensitive green fluorescence protein (GFP) (pHluorin). FasL, a type II transmembrane protein that belongs to the tumor necrosis factor family,34 contributes to cytotoxicity mediated by CTL and NK cells.35, 36, 37 FasL is present in the LG membrane of CTL and NK cells.4 pHluorin is a pH-sensitive variant of the GFP and has been used to monitor vesicle fusion in endocrine and neuronal cells as a fusion protein with the integral vesicular protein-associated membrane protein 2 (refs 38, 39 and 40). We attached pHluorin to the C-terminal, lumenal portion of FasL and DsRed to the N-terminal, cytosolic tail of FasL. By specifically labeling LG with different fluorescent proteins, and by using total internal reflection fluorescence (TIRF) microscopy (also called evanescent wave microscopy), we show that NK cells use two distinct modes for exocytosis of LG: complete fusion, where granule content is lost and FasL diffuses rapidly at the PM; and incomplete fusion, whereby formation of a transient fusion pore at the PM is accompanied by retention of FasL.
Results
Complete fusion of LG visualized with GFP-FasL
Complete fusion is generally accepted as the major pathway of exocytotic release of neuropeptides and hormones in neuronal and endocrine cells.32, 33 To determine if complete fusion occurs during exocytosis of LG, NKL cells were transiently transfected with GFP-FasL (GFP fused to the N terminus of FasL). GFP-FasL-positive compartments overlapped with perforin-containing granules, as shown by labeling of fixed cells with anti-perforin antibodies (data not shown). Transfected NKL cells were plated on poly--lysine-coated glass coverslips and imaged by TIRF. As shown with the membrane dye DiIC16, NKL cells formed stable contacts with poly--lysine-coated coverslips, as seen by TIRF microscopy (data not shown). To test whether phorbol 12-myristate 13-acetate (PMA) and ionomycin induced degranulation, the appearance of lysosome-associated membrane protein 1 (LAMP-1, also known as CD107a) at the cell surface was monitored with a directly labeled Fab of CD107a monoclonal antibody and imaged by TIRF microscopy, as described.27, 41 Approximately 15 min after stimulation, NKL cells acquired surface LAMP-1 staining, which appeared in dynamic and dispersed clusters, and accumulated over time (Supplementary Figure S1 and Supplementary Movie S1). Very little LAMP-1 staining, as shown in Supplementary Figure S1A, was observed without PMA and ionomycin stimulation. Treatment of NKL cells with PMA and ionomycin increased the maximum fluorescence intensity (arbitrary units, AU) within the evanescent field from 28.4±3.2 in the absence of stimulation (control, n=9 cells) to 363.8±88.7 (PMA and ionomycin treatment, n=9 cells) (Supplementary Figure S1B).
When NKL cells expressing GFP-FasL were stimulated with PMA and ionomycin, as shown in Supplementary Movie S2, LG approached the PM, appeared to dock and rapidly fused with the PM, releasing a bright fluorescence cloud that diffused at the PM, consistent with complete fusion. When an LG entered the TIRF evanescent field (TIRF imaging plane), an increase of the total fluorescence intensity of GFP-FasL was observed before fusion with the PM (Figure 1a). The width of the single fluorescence intensity, as indicated by the full-width at half-maximum (FWHM) of the fitted Gaussian function, increased exponentially, whereas the total intensity increased in a roughly linear manner, remained at a plateau and then rapidly decreased as GFP-FasL diffused at the PM (Figures 1b and c, and Supplementary Movie S2). The fusion event resulted in the dispersion of the fluorescent signal radiating outwardly from the point of LG contact (Supplementary Movie S2). Therefore, complete fusion of LG was observed after PMA and ionomycin stimulation.
Figure 1.

Complete fusion of a GFP-FasL-positive LG with the PM. (a) Selected frames from a time-lapsed live imaging are shown. The scale bar is 0.75 μm. (b) Diagram showing a possible pathway of LG fusion with the PM. The evanescent field is illustrated by the light blue bar. The membrane of LG labeled by GFP-FasL is illustrated by green circle. (c) The relative total intensity and FWHM of single vesicle were plotted over time. The vertical dashed lines indicate the possible distinct phase of the fusion process. Images representative of at least four fusion events observed from 20 cells in three independent experiments.
NKL cells were transfected with the LG cargo protein granzyme B fused to DsRed and examined after PMA and ionomycin stimulation. TIRF microscopy images revealed the appearance of DsRed-labeled LG: the DsRed signal remained steady for a short time (usually ∼500 ms) and disappeared within ∼100 ms (Supplementary Figure S2). This sudden drop of DsRed signal was faster than what could be expected from the retrieval of a partially fused LG into the cytoplasm (5–30 s, our own observations) or the movement of an LG out of the evanescent field without fusion (3–16 s, our own observations). Taken together, our data with GFP-FasL and granzyme B-DsRed indicate that complete fusion of LG occurs in NKL cells.
Visualization of incomplete fusion by double labeling of LG membrane and cargo protein
It would be difficult to identify incomplete fusion events using GFP-FasL-labeled LG alone, as it would not be distinguishable from docking followed by release from the PM without fusion. Therefore, to test whether incomplete fusion occurs, NKL cells were co-transfected with GFP-FasL (LG membrane protein) and granzyme B-DsRed (LG cargo protein). As expected, granzyme B-DsRed and GFP-FasL fluorescence overlapped, indicating that these tagged molecules were targeted to the same compartments (Supplementary Figure S3). Moreover, stably transfected GFP-FasL also colocalized with perforin-containing lytic granules (Supplementary Figure S4), which suggested that FasL, perforin and granzyme B represented a single pool of lytic granules in NKL cells. Two groups of LG were observed after stimulation with PMA and ionomycin. Thirty out of 55 LG (from five cells) stopped moving during the 1-min observation period. This group of LG was docked at the PM without fusion. The second group of LG (25 out of 55) showed active movement and fluctuations in fluorescence intensity of both DsRed and GFP during the 1-min observation period. A typical sample of LG fusing to the PM (six separate fusion events from 20 cells) is shown in Supplementary Movie S3. Approach toward the PM was followed by a short period of attachment (docking and priming), fusion and perpendicular movement away from the PM (Supplementary Movie S3). We first classified and analyzed this group. Figure 2a (granzyme B-DsRed) and Figure 2b (GFP-FasL) show an example of an exocytic event. Figure 2c depicts a diagram showing a possible pathway of incomplete LG fusion with the PM. As an LG appeared in the TIRF evanescent field, we observed a concurrent increase in fluorescence intensity in both the green and red channel (Figures 2a, b and d). Figures 2d, e and f show the occurrence over time of both the green and red images in the upper traces, which are the total fluorescence profiles, FWHM and relative Z-position (see Methods) of one single LG at different time points, respectively.
Figure 2.

Incomplete fusion of an LG labeled with GFP-FasL and granzyme B-DsRed. (a, b) Individual fusion event of an LG double labeled by granzyme B-DsRed (a) and GFP-FasL (b), and imaged simultaneously with dual view on 605/50 and 525/50 channels, respectively. The scale bar is 0.75 μm. (c) Diagram showing a possible pathway of LG fusion with the PM. The evanescent field is illustrated by green bar. The membrane and content of LG are illustrated by green and red colors, respectively. (d) Time course of total intensity of GFP-FasL- (right, green) and granzyme B-DsRed- (left, red) positive single LG. (e) Corresponding with panel d, the FWHM of single vesicle was shown in GFP (right, green) and DsRed (left, red). (f) The plots of relative vertical z-position of LG in GFP (right, green) and DsRed (left, red) was shown as a function time, respectively. The number ‘0' in the relative z-position indicates the closest proximity to the PM. (g) Comparison of diffusion rate of GFP-FasL (green) and granzyme B-DsRed (red). A single exponential fit (dashed line) to the fluorescence decay after fusion reveals time constants (τ) of 472.77 ms for GFP-FasL and 133.23 ms for granzyme B-DsRed. The vertical dashed lines indicate six distinct phase of LG fusion, according to changes in fluorescence intensity and relative Z-position. Images are representative of at least six fusion events observed from 20 cells in three independent experiments.
We dissected the exocytosis into six different periods. In period 1, the LG enters the evanescent field, which is indicated by a synchronous increase of total DsRed and GFP intensity (Figure 2d), FWHM (Figure 2e) and a synchronous decrease of relative Z-position (relative axial position of LG) to the PM (Figure 2f). Decreased Z-position (perpendicular to the PM) indicated that the LG was approaching the coverslips. In period 2, the LG was tethered to the PM for 400 ms (docking and priming), which is indicated by a relatively constant total intensity, FWHM and relative Z-position. In period 3, the LG fused with the PM, which is indicated by a maximum in total intensity and FWHM (Figures 2d and e) and closest proximity to the PM (Figure 2f). In period 4, the subsequent decrease of total intensity in this phase was probably due to release of the contents of LG or to gradual movement of the LG out of the evanescent field. In period 5, which lasts about 1 s, the Z-position, FWHM and total intensity are relatively stable. In period 6, the LG escaped from the evanescent field as indicated by a simultaneous decrease of total intensity and relative Z-position. The peak intensity (see Methods) can serve as an effective marker to monitor the diffusion of granule membrane protein.42 To further characterize period 4, we measured the diffusion rate (τ) difference between the GFP-FasL and granzyme B-DsRed fluorescence by fitting the peak fluorescence intensity to a mono-exponential function (see Methods) (Figure 2g). We observed that the decay of membrane protein-FasL-GFP was considerably slower (τ=472.77 ms) than the released soluble granzyme B (τ=133.23 ms) upon LG fusion with the PM. These data indicate that incomplete fusion of LG occurs in NKL cells.
Use of DsRed-FasL-pHluorin to analyze incomplete fusion of LG
We developed a more sensitive tool to validate the existence of incomplete fusion events. A tripartite DsRed-FasL-pHluorin fusion protein was generated and transfected into NKL cells. To test whether DsRed-FasL-pHluorin was targeted properly to LG, fixed and permeabilized cells were stained with perforin antibody. DsRed-FasL-pHluorin colocalized with perforin (Figure 3a). As expected, owing to the acidic lumen of LG,43, 44 pHluorin emission was very low in live cells. The addition of ammonium chloride to raise the pH in LG resulted in a strong pHluorin signal (Figure 3b and Supplementary Movie S4).
Figure 3.

Colocalization of DsRed-FasL-pHluorin with perforin in NKL cells. (a) Confocal image of a paraformaldehyde-fixed NKL cell showing the distribution of DsRed, pHluorin and perforin. Human NKL cells were attached on the poly-coated slides for 10 min at 37 °C, fixed, permeabilized and stained with mouse monoclonal antibody IgG2b against human perforin and followed by goat anti-mouse IgG2b secondary antibody conjugated with Alexa Fluor 647 dye. The scale bar is 5.0 μm. (b) Confocal image of DsRed-FasL-pHluorin in a live cell after the addition of 500 m ammonium chloride (pH 7.4). The images are representative of at least 50 cells in two independent experiments. The scale bar is 5.0 μm.
Figure 4a depicts how the fluorescence intensity of pHluorin increases upon fusion of LG with the PM. In NKL cells expressing DsRed-FasL-pHluorin and stimulated with PMA and ionomycin, DsRed fluorescent spots appeared in the TIRF evanescent field and preceded the appearance of pHluorin signal by a time interval that varied from ∼30 ms to several seconds (∼6–8 s). After a first period of DsRed fluorescence signal, pHluorin fluorescent spots appeared without any apparent diffusion of either DsRed or pHluorin fluorescence, by dual-color TIRF imaging (Figure 4b and Supplementary Movie S5). After the appearance of pHluorin, indicative of fusion pore opening, a decrease in total intensity of DsRed and pHluorin was observed (Figure 4c). The duration of DsRed and pHluorin signals persisted for about 30 s and was observed in four separate fusion events (31.5±5.9 s), as shown in Figure 4c. In a separate group of four fusion events, the dwelling time at the PM of DsRed and pHluorin was much shorter (4.9±0.46 s) (Figures 4d and e). In one particular cell, we were able to observe both a short and a longer dwelling time of the incompletely fused granule (data not shown), which indicates that this variability is determined by granule-intrinsic rather than cell-intrinsic properties.
Figure 4.

Incomplete fusion of an LG labeled with DsRed-FasL-pHluorin. (a) This diagram shows that the fluorescence intensity of pHluorin increases upon LG fusion with the PM. (b) Selected frames are shown from time-lapsed live imaging for a single LG. Times indicated are relative to the onset of fusion (appearance of pHluorin fluorescence, shown by an arrow on both pHluorin and DsRed channels). Dual-color images were acquired simultaneously by TIRF microscopy equipped with GFP/DsRed dual-view microimager. The scale bar is 1.5 μm. (c) Time course of relative pHluorin and DsRed fluorescence intensity were plotted from approaching/docking (appearance of DsRed) to vesicle escaping from evanescent field (disappearance of DsRed). The vertical dashed lines indicate the distinct phases of LG according to the change of DsRed and pHluorin fluorescence intensity. Images are representative of at least three fusion events observed from 11 cells in two independent experiments. (d) Selected frames are shown from a time-lapsed imaging of a single LG during fusion. The pHluorin fluorescent spots indicating single LG (top panel) and the corresponding position of DsRed signals (bottom panel) are shown as a function of time. Dual-color images were acquired simultaneously by TIRF microscopy equipped with GFP/DsRed dual-view microimager. The scale bar is 1.0 μm. (e) Time course of mean fluorescence intensity of pHluorin (left, green) and DsRed fluorescence (right, red) intensity were plotted, respectively. Images representative of at least four fusion events observed from 11 cells in two independent experiments.
In agreement with our data using GFP-FasL, diffusion of FasL at the PM, indicative of complete fusion, was also observed for some of the LG in DsRed-FasL-pHluorin-expressing NKL cells (Supplementary Figure S5 and Supplementary Movie S6).
The experiments described so far were carried out after stimulation of the cell line NKL with phorbol ester and ionomycin, a combination that induces strong calcium mobilization.45 This experimental system raised two concerns. First, stimulation of NK cells through activation receptors may result in a different usage of complete and partial fusion during degranulation. Second, the cell line NKL may not have the same properties as primary NK cells. To address both of these concerns, primary NK cells, freshly isolated from human peripheral blood, were nucleofected with DsRed-FasL-pHluorin. Despite very low transfection efficiency, we were able to image resting NK cells expressing DsRed-FasL-pHluorin. DsRed-FasL-pHluorin-positive compartments overlapped with perforin-containing granules, as shown by labeling of fixed cells with anti-perforin antibodies (data not shown).
Transfected primary NK cells were deposited on coverslips to which the Fc fragment of human IgG1 (a ligand for activation receptor CD16) and intercellular adhesion molecule-1 (ICAM-1, a ligand for LFA-1) had been attached. (Stimulation of primary human NK cells through CD16 and LFA-1 results in polarized degranulation.46) As a control, transfected primary NK cells were attached to poly--lysine-coated coverslips. Images were acquired at 33 frames per second. There was no detectable pHluorin fluorescent signal at the position corresponding to DsRed fluorescence, under simultaneous dual-color TIRF imaging, in unstimulated control cells, indicating that no fusion pore opening took place (Supplementary Figure S6). The number of individual fusion events observed with and without stimulation with physiological ligands of activation receptors was determined. No fusion event was observed in 13 unstimulated NK cells, and 12 fusion events were observed in eight NK cells after stimulation by physiological ligands Fc and ICAM-1. Each cell was imaged for 1 min at 2000 frames per min. Despite the lack of degranulation in unstimulated cells, a total of 65 granules have been detected at the PM. (Some lytic granules are at the PM of resting NK cells.27) Not a single one underwent fusion, as shown by a steady signal of DsRed in the absence of a pHluorin signal (Supplementary Figure S6). The unfused granule eventually left the TIRF field, as shown by the gradual loss of DsRed signal at the end of the recording (Supplementary Figure S6). We conclude that the granule fusion events observed are the result of ligand-induced stimulation.
After addition to lipid bilayers carrying ICAM-1 and IgG1 Fc and image acquisition at 33 frames per second, different types of fusion events were observed (Figure 5). Upon complete fusion, the pHluorin signal appeared, remained steady for ∼60 ms and disappeared within ∼100 ms (Figure 5a). The width of the single fluorescence intensity, as indicated by the FWHM of the fitted Gaussian function, increased rapidly before the loss of signal, which indicated that FasL diffused at the PM (Figure 5b). Upon incomplete fusion, similar to NKL cells, two different kinetics of DsRed and pHluorin fluorescence signal over time in primary NK cells upon LG fusion were observed. In a group of six fusion events from eight cells, the dwelling time at the PM of DsRed and pHluorin was about 5 s (4.55±1.19 s). A representative sample is shown in Figure 5c. In a separate group of three incomplete fusion events from eight cells, both DsRed and pHluorin signals remained steady for about 23 s (23.66±4.07 s). In the example shown in Figure 5d, there was no apparent diffusion of DsRed and pHluorin. Therefore, LG displayed incomplete fusion in primary NK cells activated by physiological receptor–ligand interactions.
Figure 5.

Fusion of LG labeled with DsRed-FasL-pHluorin in primary NK cells. (a) Selected frames are shown from a time-lapsed imaging of a single LG during fusion. Times indicated are relative to the onset of fusion (appearance of pHluorin fluorescence). Images are representative of three fusion events observed from eight cells in two independent experiments. The scale bar is 2.0 μm. (b) Three-dimensional plot images of four different time points (0, 30, 60 and 90 ms) from (a) are shown to illustrate the change of FWHM as a function of time. The pixel-based intensity values are plotted from a range of 900–980 counts. The intensity of images is shown as a color-coded scale. Each plot is scaled by its respective minimum and maximum intensity values. Note the maximum of FWHM at time point of 90 ms, which indicates the diffusion of FasL on the PM. (c) and (d) Two representative fusion events displaying either a short (c) or a long (d) dwelling time of pHluorin and DsRed signals are shown. Selected frames are shown from a time-lapsed live imaging of single LG during fusion with the PM. The pHluorin fluorescent spots indicating fusion pore opening (top panel) and the corresponding position of DsRed signals (bottom panel) are shown as a function of time. Dual-color images were acquired simultaneously by TIRF microscopy equipped with GFP/DsRed dual-view microimager. Images representative of nine fusion events observed from eight cells in two independent experiments. The scale bars are 2.0 μm.
Discussion
CTLs and NK cells kill target cells by polarized release of the content of LG at immunological synapses.2, 5 To date, the precise steps of LG fusion in cytotoxic lymphocytes such as CTL and NK cells have not been imaged in live cells. In this study, we describe the development of tools to examine LG fusion events at the PM. By specifically labeling LG with fluorescently tagged proteins, we observed that LG use two distinct modes—complete and incomplete fusion—to fuse with the PM. We have generated a triple fusion protein, consisting of DsRed-FasL-pHluorin, as a novel approach to image LG fusion events. pHluorin does not emit fluorescence at acidic pH.38 Upon fusion with the PM, the pHluorin signal increases owing to exposure to the neutral extracellular pH, thereby providing a specific and very rapid readout for exocytosis. At the same time, DsRed in the cytosolic tail provides a marker to monitor lateral or perpendicular movement of LG at the PM.
Multiple steps are required for the fusion of LG with the PM,5 including transport of LG to the PM, tethering to the PM, and priming and fusion with the PM. By labeling the lumen of LG with granzyme B-DsRed and the LG membrane with GFP-FasL, we could dissect distinct steps in LG fusion with the PM. During transport, as shown in Figure 2, the fact that the relative Z-position decreased, whereas the peak intensity and total intensity increased, suggested that the LG approached the PM. During a brief tethering phase, there was no detectable movement of LG perpendicular to the PM (that is, no change in Z-position). This period is similar to the docking phase for vesicles in neurons and endocrine cells.42, 47 We also observed a small and short plateau of peak intensity and total intensity in both GFP-FasL and granzyme B-DsRed signals. Furthermore, the FWHM remained constant, which indicated that GFP-FasL and granzyme B-DsRed were not diffusing into the PM during the tethering phase. In addition, the relative Z-position of the LG gradually approached the position where the evanescent field was greatest. At that point, the LG is probably docked at the PM. In the fusion phase, the total intensity, peak intensity and FWHM reached their maximal value. In the following phase, the exponential decay of the DsRed peak fluorescence signal and the exponential, but partial, decay of GFP peak fluorescence intensity could result from gradual movement out of the evanescent field or by release of LG cargo protein into extracellular space and diffusion of some of LG membrane protein FasL on the PM. The rate of decay of granzyme B-DsRed signal was greater than that of GFP-FasL, implying that granzyme B-DsRed was indeed released. The remaining GFP and DsRed fluorescence signals were stable for about a second, before their synchronous decline and disappearance, suggesting movement out of the evanescent field. The partial fusion mode of LG in NKL cells described here is reminiscent of ‘kiss and run' exocytosis in synaptic vesicle release.33, 48, 49, 50
To obtain stronger evidence for an incomplete fusion mode, a DsRed-FasL-pHluorin fusion protein was expressed in NKL cells. In this case, appearance of pHluorin fluorescence corresponded to fusion pore opening and neutralization of the pH. We observed that pHluorin and DsRed signals could persist on the same LG for up to 30 s after fusion (in four out of eight incomplete fusion events), which is reminiscent of the ‘kiss and stay' pathway in neurotransmitter release.51 We also observed shorter concurrent appearance of pHluorin and DsRed fluorescence signals (∼5 s) upon incomplete fusion. In some cases, vesicles did not appear to pause for docking and priming before fusion, which is reminiscent of ‘Crash fusion' in embryonic chromaffin cells.52 Overall, our results suggest that small fusion pores may form, which hinder diffusion of vesicle membrane protein on the PM, as observed recently during exocytosis of post-Golgi vesicles.53
The complete and incomplete modes of fusion occur also in primary NK cells receiving physiological activation signals, as we were able to observe them in resting NK cells transfected with DsRed-FasL-pHluorin, and stimulated by ligands of CD16 and LFA-1. Incomplete fusion events in NK cells are most likely not due to weak activation signals, as they were observed in NKL cells stimulated with PMA and ionomycin, a stimulus known to induce maximum calcium mobilization.45 Furthermore, they occurred also in resting, primary NK cells stimulated through CD16, which is known to induce strong degranulation in primary NK cells.46 Incomplete fusion events, with limited LG membrane protein diffusion on the PM, may explain the stable clusters of exocytosed LAMP-1 molecules observed on live degranulating NK cells in the absence of granule polarization.27
The existence of incomplete fusion of synaptic vesicles at neurological synapses is generally accepted.33 Cytotoxic lymphocytes, including CTL and NK cells, may share similar molecular mechanism for vesicle fusion with neuronal cells.28, 54, 55 Incomplete fusion could be a useful mechanism for rapid and efficient recycling of LG membrane in cytotoxic lymphocytes.27 Ultimately, it will be interesting to discover how fusion modes vary in NK cells under different physiological stimulation conditions.
Methods
NK cells
The human NK cell line NKL was cultured in RPMI medium 1640 (Gibco, Grand Island, NY, USA) containing 10% fetal calf serum, 1% -glutamine, 1% sodium pyruvate and 200 U ml−1 recombinant interleukin (IL)-2 (National Cancer Institute-FCRDCM, Frederick, MD, USA). Primary NK cells were isolated from human peripheral blood by negative selection with an NK isolation kit (Stemcell Technologies Inc., Vancouver, BC, Canada) and were >99% CD3− CD56+. Freshly isolated NK cells were resuspended in Iscove's modified Dulbecco's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% human serum (Valley Biomedical, Winchester, VA, USA) without IL-2, and were used within 2 days.
Reagents
Degranulation by NKL cells was triggered with 100 n PKC activator PMA (Sigma-Aldrich, St Louis, MO, USA) and 3 m calcium ionophore ionomycin (Sigma-Aldrich). Human IgG1 Fc was fused to a 6-histidine amino-acid tag and purified from the supernatant of transfected 293T cells over ProBond Nickel-chelating Resin (Invitrogen), as described.27 Human ICAM-1 protein (R&D Systems, Minneapolis, MN, USA) and IgG1 Fc were coated on coverslips in 100 m sodium bicarbonate (pH 9.2) at 10 μg ml−1 at 4 °C overnight. Fetal bovine serum (5%) in culture medium was used to block nonspecific binding, as described.56 The following monoclonal antibodies were used: anti-CD107a (clone H4A3; BD Bioscience, San Diego, CA, USA) and anti-perforin (clone δG9; Pierce Chemical Co., Rockford, IL, USA).
DNA construct of pHluorin-FasL-DsRed and DsRed-granzyme B
The plasmid for human GFP-FasL fusion protein (a gift from G Griffiths, Cambridge, UK) has been described.4 To generate a granzyme B-DsRed fusion protein, total RNA was extracted from NK92 cells with RNeasy Mini Kit (Qiagen, GmbH, Hilden, Germany), and a human granzyme B cDNA was generated by reverse transcription-polymerase chain reaction (PCR) with the forward primer 5′-AAGGCCTCTGTCGACATGCAACCAATCCTGCTTCTG-3′ and reverse primer 5′-AGAATTCGCAAGCTTGTAGCGTTTCATGGTTTTCTT-3′. The PCR product was digested with SalI and HindIII and inserted into pDsRed-Monomer-N in-Fusion Ready vector (Clontech Laboratories, Mountain View, CA, USA).
The pHluorin-FasL-DsRed fusion protein was constructed as follows: human FasL was amplified by PCR from the GFP-FasL cDNA using the forward primer 5′-TGCAGTCGACGGTACCATGCAGCAG-3′, which includes a SalI site, and the reverse primer 5′-ATATGGATCCTGCTGCGGCCGCGAGCTTATATAAGCCGAAAAACGTCTG-3′, which includes NotI and BamHI sites. The PCR product was cloned into TOPO TA Cloning vector (Invitrogen) and verified by sequencing. The SalI–BamHI insert was cloned into SalI–BamHI-digested DsRed-monomer-C1 vector (Clontech Laboratories). This resulted in the insertion of a 17-amino-acid long linker between DsRed and FasL, identical to the one in GFP-FasL. The pHluorin coding sequence was amplified from a vesicle-associated membrane protein 2-pHluorin plasmid (gift from G Miesenböck, Yale University, New Haven, CT, USA)38 using the forward primer 5′-ATATGCGGCCGCCAGCGGCGGAAGCGGCGGGACCGG-3′, which included a NotI site, and the reverse primer 5′-ATATGGATCCTCTAGATTAACCGGTTTTGTATAGTTCATC-3′, which included a BamHI site. The pHluorin PCR product was cloned into the TOPO TA Cloning vector and verified by sequencing. The NotI–BamHI fragment was inserted into NotI–BamHI-digested DsRed-FasL vector. The boundary between FasL and pHluorin includes three alanines encoded by the NotI site and the original 9-amino-acid-long, serine-glycine-rich linker from the vesicle-associated membrane protein 2-pHluorin fusion construct.
Transfection of NK cells
NKL cells were transfected with Amaxa nucleofection technology (Amaxa, Cologne, Germany). Briefly, 2 × 106 NKL cells and 4.0 μg DNA construct were resuspended in solution nucleofector kit V and transfected using O-17 program. After transfection, cells were transferred into six-well plates at 37 °C. Images were acquired 12–24 h after transfection. Human primary NK cells were transfected with Amaxa nucleofection technology. Briefly, 3.0 × 106 NK cells and 6.0 μg DNA were resuspended in human NK cells nucleofector solution and transfected using U-001 program. After transfection, cells were transferred into pre-warmed opTimizer CTS T-cell expansion SFM (Invitrogen) containing 1 U ml−1 rIL-2 (Roche, Basel, Switzerland) and 10 ng ml−1 recombinant IL-15 (PeproTech Inc., Rocky Hill, NJ, USA). NK cells were cultured <12 h with low-dose IL-15 as a survival factor.57
Fixation and permeabilization of NKL cells
NKL cells were fixed with 4% freshly prepared paraformaldehyde for 15–30 min at room temperature (RT), washed with phosphate-buffered saline (PBS) for three times. Cells were permeabilized in 0.5% Triton X-100 and 10% normal donkey serum in PBS for 30 min at RT. Cells were stained with anti-perforin monoclonal antibody mouse IgG2b (clone δG9; Pierce Chemical Co.) for 60 min at RT. The primary antibody was diluted with 0.05% Triton X-100 and 3% normal donkey serum in PBS (1:333 dilution). After three washes in PBS, cells were incubated for 1 h at RT with appropriate secondary antibodies in 0.05% Triton X-100 and 3% normal donkey serum in PBS. Secondary antibody used was Alexa Fluor 647-conjugated goat anti-mouse IgG2b (1:1000 dilution) (Molecular Probes, Eugene, OR, USA).
Confocal microscopy
Confocal images were collected on a Zeiss LSM510 Meta Confocal microscope using a plan apochromat × 63/1.4 oil-immersion objective. GFP-FasL and pHluorin were excited by 488 nm excitation wavelength (argon/krypton) and collected at the emission wavelength of 530 nm. DsRed was excited at 543 or 488 nm excitation wavelength (helium/neon) and collected at 620 nm emission wavelength. Differential interference contrast images were collected simultaneously with the fluorescent images. Multi-track acquisition mode was used to avoid crosstalk between the different fluorophores.
TIRF microscopy
TIRF images were acquired using an Olympus inverted IX-81 microscope equipped with a Xenon-arc lamp as light source, shutter and filter wheels equipped with appropriate excitation and emission filters, ASI MS-2000 controller (Applied Scientific Instrumentation Inc., Eugene, OR, USA) motorized stage for xyz movements, electron-multiplier charge-coupled devices (Photometrics Casade II:512, Roper Scientific Inc., Tucson, AZ, USA), Olympus TIRF module and lasers launched in a single mode fiber via an acoustic-optical tunable filter (NEOS Technologies, Melbourne, FL, USA). The 100 × 1.45 NA TIRF objective from Zeiss (Zeiss, ∂ plan-Fluar × 100/1.45 oil) was used for TIRF experiments. TIRF illumination was provided by the 488 nm line of an argon laser (Laser Physics, Salt Lake City, UT, USA). The hardware on the microscope was controlled by the Metamorph software (Molecular Devices, Downingtown, PA, USA).
Image acquisition and analysis
Images were acquired using the electron-multiplier charge-coupled devices and analyzed with Image Pro Plus 6.1 software (Media Cybernetics, Sliver Spring, MD, USA). The 488 nm laser line was selected using acoustic-optical tunable filter for simultaneous illumination of pHluorin and DsRed using a GFP/DsRed dual-view microimager (Optical Insights, Tucson, AZ, USA). The total intensity of a single vesicle was computed by integrating the background-subtracted intensity over a circle (approximately 1.0 μm diameter) that centered on the selected vesicle. To obtain the peak intensity, we fitted the radial intensity distribution I(r) of LG with a nonlinear Levenberg–Marquardt routine to the Gaussian:42
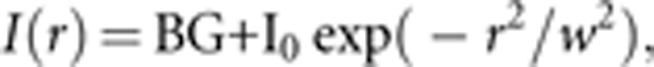 |
where r is the distance of each pixel to the center of mass. The fitting parameters are I0, w and BG, where I0 is the peak intensity, BG is the background intensity and w is the measure of the width (the Gauss width). The relative movement in the Z-position (ΔZn) from time point (n−1) to (n) can be calculated from the following formula:58 ΔZn=−d ln (Fn−Fn−1), where Fn and Fn−1 are background corrected fluorescence at time point (n) and (n−1) and d is the penetration depth. The penetration depth of the evanescent field was calculated as ∼87 nm, assuming a cell refractive index of 1.37. The penetration depth was calculated according to the equation: d=λ/4π [(NAi^2−n2^2)]1/2, in which λ is the wavelength of light and NAi=n1 sin α is the numerical aperture of incidence; n2 is the refractive index of the cell (typically n2=1.37). The single of LG was characterized by its total intensity, FWHM, relative Z-position and peak intensity. In some experiments, to illustrate the three-dimensional appearance of single fluorescent spot, we fitted the single fluorescent spot with a two-dimensional Gaussian function, as described.27, 59 The results of this fit yielded the integrated intensity, background and FWHM.
Acknowledgments
We thank G Griffiths (University of Cambridge) for the GFP-FasL plasmid, G Miesenböck (Yale University) for the vesicle-associated membrane protein 2-pHluorin plasmid, and J Brzostowski and P Tolar (NIAID-NIH, USA) for advice with imaging. We also thank the NIH Fellows Editorial Board for editorial assistance. This work has been supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Disease, and National Heart, Lung, and Blood Institute.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Immunology and Cell Biology website (http://www.nature.com/icb)
Supplementary Material
References
- Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Page LJ, Griffiths GM. Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients. Traffic. 2000;1:435–444. doi: 10.1034/j.1600-0854.2000.010508.x. [DOI] [PubMed] [Google Scholar]
- Bossi G, Griffiths GM. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat Med. 1999;5:90–96. doi: 10.1038/4779. [DOI] [PubMed] [Google Scholar]
- Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8:713–725. doi: 10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey TM, Meade JL, Hewitt EW. Organelle proteomics: identification of the exocytic machinery associated with the natural killer cell secretory lysosome. Mol Cell Proteomics. 2007;6:767–780. doi: 10.1074/mcp.M600365-MCP200. [DOI] [PubMed] [Google Scholar]
- de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10:568–579. doi: 10.1038/nri2803. [DOI] [PubMed] [Google Scholar]
- Dressel R, Elsner L, Novota P, Kanwar N, Fischer von Mollard G. The exocytosis of lytic granules is impaired in Vti1b- or Vamp8-deficient CTL leading to a reduced cytotoxic activity following antigen-specific activation. J Immunol. 2010;185:1005–1014. doi: 10.4049/jimmunol.1000770. [DOI] [PubMed] [Google Scholar]
- Loo LS, Hwang LA, Ong YM, Tay HS, Wang CC, Hong W. A role for endobrevin/VAMP8 in CTL lytic granule exocytosis. Eur J Immunol. 2009;39:3520–3528. doi: 10.1002/eji.200939378. [DOI] [PubMed] [Google Scholar]
- Arneson LN, Segovis CM, Gomez TS, Schoon RA, Dick CJ, Lou Z, et al. Dynamin 2 regulates granule exocytosis during NK cell-mediated cytotoxicity. J Immunol. 2008;181:6995–7001. doi: 10.4049/jimmunol.181.10.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Palacios M, Odemuyiwa SO, Coughlin JJ, Garofoli D, Ewen C, Davidson CE, et al. Vesicle-associated membrane protein 7 (VAMP-7) is essential for target cell killing in a natural killer cell line. Biochem Biophys Res Commun. 2008;366:617–623. doi: 10.1016/j.bbrc.2007.11.079. [DOI] [PubMed] [Google Scholar]
- Menager MM, Menasche G, Romao M, Knapnougel P, Ho CH, Garfa M, et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol. 2007;8:257–267. doi: 10.1038/ni1431. [DOI] [PubMed] [Google Scholar]
- Pores-Fernando AT, Gaur S, Doyon MY, Zweifach A. Calcineurin-dependent lytic granule exocytosis in NK-92 natural killer cells. Cell Immunol. 2009;254:105–109. doi: 10.1016/j.cellimm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pores-Fernando AT, Ranaghan MY, Zweifach A. No specific subcellular localization of protein kinase C is required for cytotoxic T cell granule exocytosis. J Biol Chem. 2009;284:25107–25115. doi: 10.1074/jbc.M109.011866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pores-Fernando AT, Zweifach A. Calcium influx and signaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol Rev. 2009;231:160–173. doi: 10.1111/j.1600-065X.2009.00809.x. [DOI] [PubMed] [Google Scholar]
- Ma JS, Haydar TF, Radoja S. Protein kinase C delta localizes to secretory lysosomes in CD8+ CTL and directly mediates TCR signals leading to granule exocytosis-mediated cytotoxicity. J Immunol. 2008;181:4716–4722. doi: 10.4049/jimmunol.181.7.4716. [DOI] [PubMed] [Google Scholar]
- Herz J, Pardo J, Kashkar H, Schramm M, Kuzmenkina E, Bos E, et al. Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary T lymphocytes. Nat Immunol. 2009;10:761–768. doi: 10.1038/ni.1757. [DOI] [PubMed] [Google Scholar]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol. 2001;152:825–834. doi: 10.1083/jcb.152.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Meckel T, Long EO. Distinct role of rab27a in granule movement at the plasma membrane and in the cytosol of NK cells. PLoS One. 2010;5:e12870. doi: 10.1371/journal.pone.0012870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119:3765–3773. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andzelm MM, Chen X, Krzewski K, Orange JS, Strominger JL. Myosin IIA is required for cytolytic granule exocytosis in human NK cells. J Exp Med. 2007;204:2285–2291. doi: 10.1084/jem.20071143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn KB, Rak GD, Maru SY, Demers K, Difeo A, Martignetti JA, et al. Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. J Immunol. 2009;182:6969–6984. doi: 10.4049/jimmunol.0804337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–1915. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneson LN, Brickshawana A, Segovis CM, Schoon RA, Dick CJ, Leibson PJ. Cutting edge: syntaxin 11 regulates lymphocyte-mediated secretion and cytotoxicity. J Immunol. 2007;179:3397–3401. doi: 10.4049/jimmunol.179.6.3397. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751–761. doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- Liu D, Bryceson YT, Meckel T, Vasiliver-Shamis G, Dustin ML, Long EO. Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity. 2009;31:99–109. doi: 10.1016/j.immuni.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Cytotoxic T. Lymphocyte exocytosis: bring on the SNAREs! Trends Cell Biol. 2005;15:644–650. doi: 10.1016/j.tcb.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP, Mauro A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1973;57:499–524. doi: 10.1083/jcb.57.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wu XS, Mohan R, Wu LG. Two modes of fusion pore opening revealed by cell-attached recordings at a synapse. Nature. 2006;444:102–105. doi: 10.1038/nature05250. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Curran M, Cohen FS, Brodwick M. Simultaneous electrical and optical measurements show that membrane fusion precedes secretory granule swelling during exocytosis of beige mouse mast cells. Proc Natl Acad Sci USA. 1987;84:1585–1589. doi: 10.1073/pnas.84.6.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wu LG. The debate on the kiss-and-run fusion at synapses. Trends Neurosci. 2007;30:447–455. doi: 10.1016/j.tins.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Cretney E, Street SE, Smyth MJ. TNF contributes to the immunopathology of perforin/Fas ligand double deficiency. Immunol Cell Biol. 2002;80:436–440. doi: 10.1046/j.1440-1711.2002.01108.x. [DOI] [PubMed] [Google Scholar]
- Smyth MJ. Fas ligand-mediated bystander lysis of syngeneic cells in response to an allogeneic stimulus. J Immunol. 1997;158:5765–5772. [PubMed] [Google Scholar]
- Rouvier E, Luciani MF, Golstein P. Fas involvement in Ca(2+)-independent T cell-mediated cytotoxicity. J Exp Med. 1993;177:195–200. doi: 10.1084/jem.177.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Presynaptic imaging techniques. Curr Opin Neurobiol. 2001;11:544–549. doi: 10.1016/s0959-4388(00)00247-6. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- Beal AM, Anikeeva N, Varma R, Cameron TO, Norris PJ, Dustin ML, et al. Protein kinase C theta regulates stability of the peripheral adhesion ring junction and contributes to the sensitivity of target cell lysis by CTL. J Immunol. 2008;181:4815–4824. doi: 10.4049/jimmunol.181.7.4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoranzer J, Goulian M, Axelrod D, Simon SM. Imaging constitutive exocytosis with total internal reflection fluorescence microscopy. J Cell Biol. 2000;149:23–32. doi: 10.1083/jcb.149.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Xu L, Yang F, Li D, Gong F, Xu T. Rapid biogenesis and sensitization of secretory lysosomes in NK cells mediated by target-cell recognition. Proc Natl Acad Sci USA. 2005;102:123–127. doi: 10.1073/pnas.0405737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, Hester S, Lapham CK, Argon Y. The lytic granules of natural killer cells are dual-function organelles combining secretory and pre-lysosomal compartments. J Cell Biol. 1990;111:2327–2340. doi: 10.1083/jcb.111.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundt M, Schmidt RE. The glycosylphosphatidylinositol-linked Fc gamma receptor III represents the dominant receptor structure for immune complex activation of neutrophils. Eur J Immunol. 1992;22:811–816. doi: 10.1002/eji.1830220327. [DOI] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. 2005;202:1001–1012. doi: 10.1084/jem.20051143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A, et al. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 2007;5:47–57. doi: 10.1016/j.cmet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Smith SM, Renden R, von Gersdorff H. Synaptic vesicle endocytosis: fast and slow modes of membrane retrieval. Trends Neurosci. 2008;31:559–568. doi: 10.1016/j.tins.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO, Jahn R. Kiss-and-run, collapse and ‘readily retrievable' vesicles. Traffic. 2007;8:1137–1144. doi: 10.1111/j.1600-0854.2007.00614.x. [DOI] [PubMed] [Google Scholar]
- Harata NC, Aravanis AM, Tsien RW. Kiss-and-run and full-collapse fusion as modes of exo-endocytosis in neurosecretion. J Neurochem. 2006;97:1546–1570. doi: 10.1111/j.1471-4159.2006.03987.x. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Verhage M, Sorensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Rivera VM, Simon SM. Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell. 2009;137:1308–1319. doi: 10.1016/j.cell.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths GM, Tsun A, Stinchcombe JC. The immunological synapse: a focal point for endocytosis and exocytosis. J Cell Biol. 2010;189:399–406. doi: 10.1083/jcb.201002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross CC, Brzostowski JA, Liu D, Long EO. Tethering of intercellular adhesion molecule on target cells is required for LFA-1-dependent NK cell adhesion and granule polarization. J Immunol. 2010;185:2918–2926. doi: 10.4049/jimmunol.1000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100:3633–3638. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- Oheim M, Stuhmer W. Tracking chromaffin granules on their way through the actin cortex. Eur Biophys J. 2000;29:67–89. doi: 10.1007/s002490050253. [DOI] [PubMed] [Google Scholar]
- Holtzer L, Meckel T, Schmidt T. Nanometric three-dimensional tracking of individual quantum dots in cells. Appl Phys Lett. 2007;90:053902. doi: 10.1063/1.2437066. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.