

Abstract
A major advance in the microscopic study of cells and tissues is the introduction of photoactivatable fluorescent proteins which can specifically mark proteins of interest within a living cell. Fluorescent proteins are now available that allow a pool of molecules to be “turned on” by photoactivation. This unit discusses technical aspects for the general use of photoactivatable fluorescent proteins and introduces some specific applications in the concluding remarks.
Keywords: photoactivatable, fluorescent protein, microscopy
Introduction
Cell biologists rely on the many advances in microscopy methods to study cells and tissues. A major advance is the introduction of optical highlighter fluorescent proteins, which can specifically mark proteins of interest within a living cell. Optical highlighter fluorescent proteins allow a pool of molecules to be “turned on” by photoactivation, photoconversion, and photoswitching. Here, selected members of this class of fluorescent proteins will be introduced, technical aspects for the general use of these fluorescent proteins will be outlined and some specific applications will be discussed.
Background
Optical highlighter fluorescent proteins differ from normal fluorescent proteins in that they exhibit little or no fluorescence at certain excitation and emission wavelengths prior to photoactivation. However, they do retain many of the usual advantages associated with fluorescent proteins. The term “certain wavelengths” is used to avoid confusion about some optical highlighters that are highly fluorescent in their native inactivated states; this fluorescence arises from a different spectral region than that of the protein in its photoactivated state. Also confusing at times is the classification of these molecules. Here, they will be designated photoactivatable (fluorescence changes from off to on), photoconvertible (fluorescence shifts from one wavelength to another), and photoswitchable (fluorescence can be reversibly and repeatedly turned on and off). Proteins from several marine species have been reported, along with several variants and approaches for their use as photoactivatable fluorescent proteins ((Lukyanov et al., 2005; Day and Davidson, 2009; Lippincott-Schwartz and Patterson, 2009); Table 1). For the purpose of a photoactivation protocol, these molecules are categorized in this unit on the basis of their spectral characteristics. Descriptions of their other characteristics that might be of interest for other types of experiments, as well as evidence for the mechanism for photoconversion, are discussed elsewhere (Lukyanov et al., 2005; Day and Davidson, 2009; Lippincott-Schwartz and Patterson, 2009).
Table 1.
| Protein | Activation wavelength (nm)a |
Stateb | Excitation wavelength (nm) |
Emission wavelength (nm) |
Contrast (fold increase) | Reference |
|---|---|---|---|---|---|---|
| PA-GFP | 405 | Pre | 400 | 515 | 70 | Patterson and Lippincott-Schwartz (2002) |
| Post | 504 | 517 | ||||
| PS-CFP | 405 | Pre | 402 | 468 | 300 green 1500 green-to-cyan ratio | Chudakov et al. (2004) |
| Post | 490 | 511 | ||||
| PS-CFP2 | 405 | Pre | 400 | 470 | See supplier's Web site | |
| Post | 490 | 511 | ||||
| PAmRFP1-1 | 405 | Post | 578 | 605 | 70 | Verkhusha and Sorkin (2005) |
| PAmCherry1 | 405 | Post | 564 | 595 | Subach et al. (2009) | |
| PATagRFP | 405 | Post | 562 | 595 | Subach et al. (2010) | |
| Dronpac | 490, 405 | Pre | 503 | 518 | Ando et al. (2004) | |
| Dronpa-2 | 490, 405 | ON | 486 | 513 | Ando et al. (2007) | |
| Dronpa-3 | 490, 405 | ON | 487 | 514 | Ando et al. (2007) | |
| rsFastlime | 490, 405 | ON | 496 | 518 | 67 | Stiel et al. (2007) |
| Padron | 490, 405 | ON | 503 | 522 | 143 | Andresen et al. (2008) |
| bsDronpa | 490, 405 | ON | 460 | 504 | 17 | Andresen et al. (2008) |
| rsCherry | 470, 561 | ON | 572 | 610 | 6.7 | Stiel et al. (2008) |
| rsCherryRev | 470, 561 | ON | 572 | 608 | 20 | Stiel et al. (2008) |
| KFP1 | 532 | Post | 580 | 600 | Chudakov et al. (2003) | |
| Kaede | 405 | Pre | 508 | 518 | 2000 red-to-green ratio | Ando et al. (2002) |
| Post | 572 | 582 | ||||
| Kikume Green-Red (KikGR) | 405 | Pre | 507 | 517 | 2000 red-to-green ratio | Tsutsui et al. (2005) |
| Post | 583 | 593 | ||||
| mKikGR | 405 | Pre | 505 | 515 | Habuchi et al. (2008) | |
| Post | 580 | 591 | ||||
| EosFP | 405 | Pre | 506 | 516 | Wiedenmann et al. (2004) | |
| Post | 571 | 581 | ||||
| mEosFP | 405 | Pre | 505 | 516 | Wiedenmann et al. (2004) | |
| Post | 573 | 584 | ||||
| IrisFP | 405 photoconversion | Pre | 488 | 516 | Adam et al. (2008) | |
| 488 or 561 photoswitching | Post | 551 | 580 | |||
| Dendra | 405 or 488 | Pre | 486 | 505 | 4500 red-to-green ratio | Gurskaya et al. (2006) |
| Post | 558 | 575 | ||||
| Dendra2 | 405 or 488 | Pre | 486 | 505 | See supplier's Web site | |
| Post | 558 | 575 |
The wavelength range for activation can be broad for each protein. The wavelengths listed here are suggestions based on commonly available laser lines.
The wavelengths to the right of Pre represent the major peaks prior to photoactivation. The wavelengths to the right of Post represent the major peaks after photoactivation; absence of Pre wavelengths indicates that the protein is not initially fluorescent (values not published).
Dronpa displays green fluorescence with excitation at 503 nm and emission at 518 nm. Excitation at 490 nm results in loss of absorbance at 503 nm. Upon irradiation at 400 nm, green fluorescence is restored.
Photoactivatable Green Fluorescent Proteins
One of the first optical highlighters resulted from a photo-induced conversion (photoconversion) within the wild-type (wt) green fluorescent protein (GFP;(Yokoe and Meyer, 1996)). The wt GFP chromophore population exists as neutral phenols when Y66 within the chromophore is protonated and as anionic phenolates when Y66 is deprotonated, and these different populations produce a major absorbance peak at ∼397 nm and a minor absorbance peak at 475 nm, respectively. After irradiation at ∼400 nm, the absorbance at 475 nm increases, and this leads to an increase in the fluorescence upon excitation at 475 nm. This phenomenon was utilized in developing a GFP mutant with decreased absorbance at 488 nm (compared to wt GFP) that could also undergo photoconversion when irradiated at ∼400 nm (Patterson and Lippincott-Schwartz, 2002). The first of these mutants was T203I, which has a mostly neutral phenol chromophore population giving a major absorbance peak at ∼400 nm and little absorbance at the common 488-nm laser line used for excitation (Heim et al., 1994; Ehrig et al., 1995). Substitution of the threonine at position 203 on the protein led to the T203H mutant, finally named PA-GFP (Fig.1A), which gave the highest fluorescence emission contrast after photoactivation compared to nonphotoactivated protein (Patterson and Lippincott-Schwartz, 2002).
Figure 1.

Fluorescence excitation and emission spectra of selected photoactivatable fluorescent proteins show the diversity that is now available from this class of fluorescent proteins. (A) The excitation spectra for PA-GFP are shown prior to (closed circles) and after (open circles) photoactivation. The emission spectrum after photoactivation is shown in open squares. (B) The excitation spectra for PS-CFP2 are shown prior to (closed circles) and after (open circles) photoactivation. The emission spectra for PS-CFP2 are shown prior (closed squares) to photoactivation and after (open squares) photoactivation. (C) The excitation spectra (open circles) and emission spectrum (open squares) are shown for photoactivated KFP1. (D) The excitation spectrum for Dronpa are shown prior to (closed circles) photoswitching “ON”. The emission spectrum before (closed squares) after (open squares) photoswitching “ON” is shown in open squares. (E) The excitation spectra for KikGR are shown prior to (closed circles) and after (open circles) photoconversion. The emission spectra for KikGR are shown prior to (closed squares) and after (open squares) photoconversion.
Contrary to the previous examples, photoswitchable-cyan fluorescent protein (PS-CFP) displays a shift of both absorbance and emission from cyan to green fluorescent protein ((Chudakov et al., 2004); Fig. 1B). It exhibits initial excitation at 402 nm and emission at 468 nm (Chudakov et al., 2004) which shift to 490 nm and 511 nm, respectively, and give ∼1500-fold increase in the green-to-cyan fluorescence ratio upon photoactivation at ∼400 nm. It is designated as photoactivatable because the proposed mechanism is similar to that of wtGFP and PA-GFP.
Photoactivatable Red Fluorescent Proteins
The monomeric red fluorescent protein, mRFP1(Campbell et al., 2002), was converted into a series of photoactivatable fluorescent proteins, PAmRFP1-1, PAmRFP1-2, and PAmRFP1-3(Verkhusha and Sorkin, 2005). The brightest, PAmRFP1-1, has little fluorescence before activation and gives ∼70-fold increase in red fluorescence upon ∼400-nm irradiation (Verkhusha and Sorkin, 2005). The DsRed variant, mCherry(Shaner et al., 2004), was developed into PAmCherry1(Subach et al., 2009) which is turned on with 405 nm light with up to 4000-fold contrast for pure proteins. The conventional fluorescent protein, TagRFP(Merzlyak et al., 2007), was also developed in PATagRFP(Subach et al., 2010), which is about 3 times brighter than PAmCherry1.
Less well known is asFP595 (emission λmax = 595 nm), a protein isolated from the sea anemone, Anemonia sulcata. Its red fluorescence can be enhanced by exposure to green light and quenched by exposure to blue light (Lukyanov et al., 2000). Kindling fluorescent protein (KFP1) is an improved variant of asFP595 (Chudakov et al., 2003). Activation with 532-nm laser light increases its red fluorescence ∼30-fold, and it can revert to the inactive state or be stabilized in the photoactivated state, depending on the intensity of the photoactivation light. (Fig. 1C)
Photoswitchable Green Fluorescent Proteins
Dronpa is a green optical highlighter derived from Pectimiidae (Ando et al., 2004). Its spectra (Fig. 1D) resemble those of PA-GFP, except that it has a much higher extinction coefficient and the photoactivation is reversible. Initially, Dronpa displays green fluorescence with excitation at 503 nm and emission at 518 nm. Excitation at 490 nm (0.4 W/cm2) results in loss of the absorbance at 503 nm and an increase at ∼390 nm. However, after irradiation at 400 nm (0.14 W/cm2), 503-nm absorbance is increased and the green fluorescence is restored.
Dronpa has been further developed into Dronpa2(Ando et al., 2007), Dronpa3(Ando et al., 2007), rsFastlime(Stiel et al., 2007), Padron(Andresen et al., 2008), and bsDronpa(Andresen et al., 2008). The Dronpa-2 and -3 are more easily switched to the off state than Dronpa, whereas rsFastlime switches both on and off more rapidly. The bsDronpa has a blue-shifted and broadened excitation spectrum and Padron displays photoswitching wavelength dependence opposite that of the other derivatives, 405 nm turns it of and 488 nm turns it on.
Photoswitchable Red Fluorescent Proteins
Several red fluorescent proteins having photoswitchable properties are now available. Two versions of mCherry, rsCherry and rsCherryRev (Stiel et al., 2008), can be reversibly switched using yellow and blue light. For rsCherry, yellow light (561nm) switches from the off-to-on state and the blue light switches from the on-to-off state, whereas the rsCherryRev displays the opposite wavelength dependence for switching. The rsCherry behavior is similar to that of Padron in that the more red-shifted irradiation turns it on while the more blue-shifted wavelength turns it off, although under some conditions, the on-off cycling behavior can reverse to resemble that of rsCherryRev(Subach et al., 2009). Nevertheless, these molecules provide red photoswitchable markers were shown to function well in both diffraction-limited and molecular localization imaging. (Stiel et al., 2008)
Photoconvertible Green-to-Red Fluorescent Proteins
Many of the naturally occurring and engineered photoactivatable fluorescent proteins exhibit a spectral shift from a green fluorescent protein into a red fluorescent protein (see Fig. 1D). The first of these to be discovered is Kaede, from a stony coral, Trachyphyllia geoffroyi (Ando et al., 2002). Kaede absorbs maximally at 508 nm and emits at 518 nm, is photoactivated by irradiation at ∼400 nm, and then exhibits absorbance at 572 nm and emission at 582 nm afterward. Since both the excitation and emission peaks are shifted, ratio imaging results in a >2000 fold increase in the red-to-green ratio. KiKGR is another fluorescent protein from coral that was engineered to undergo green-to-red photoactivation (Tsutsui et al., 2005). KikGR is an obligate tetramer, but has been developed into a monomeric protein, mKikGR,(Habuchi et al., 2008) for use in protein localization and tracking experiments (Fig. 1E). EosFP, from another stony coral, Lobophyllia hemprichii, also exhibits a green-to-red fluorescence photoconversion after exposure to ultraviolet or near ultraviolet light (Wiedenmann et al., 2004). EosFP has a preactivated excitation maximum at 506 nm with emission at 516 nm, which shift to 571 nm and 581 nm, respectively, after photoactivation. EosFP comes in several varieties, dimer, tandem dimer, and monomer. However, the original monomer folded inefficiently at 37°C, but a new version, mEos2(McKinney et al., 2009), corrects this problem and displays increased brightness over the original. Another derivative of EosFP is IrisFP(Adam et al., 2008). It is also photoconverted from green-to-red using 405 nm light but interestingly can also be grouped with the photoswitchable fluorescent proteins since it can be photoswitched on and off in both green and red states. Dendra from Dendronephthya sp., which gives up to a 4500-fold increase in the red-to-green ratio after photoactivation (Gurskaya et al., 2006). Uniquely, Dendra can be activated with potentially less phototoxic wavelengths (∼488 nm) in addition to the ∼400-nm light required by the other green-to-red optical highlighters.
Requirements for Hightlighting Fluorescent Proteins
These general procedures can be applied to most optical highlighters, but the varied characteristics of each protein must be considered when addressing many of the points listed below.
Locating Positive Cells and Structures
Molecules that have little fluorescence prior to photoactivation produce the first obstacle, which is locating a cell and a specific region of a cell to activate. This is straightforward for the green-to red and cyan-to-green optical highlighters because the unactivated green or cyan emission wavelength can be used for targeting. Initially, the Dronpa protein has a 503-nm excitation peak and 518-nm fluorescence, but after irradiation with light in the 470- to 510-nm wavelength range, it is rendered nonfluorescent and develops absorbance at 390 nm. The PA-GFP emits green fluorescence when excited with low levels of ∼400-nm light, and this is often sufficient to locate positive cells and structures. Alternatively, cotransfection with a second red or cyan fluorescent protein can usually be used to locate transfected cells.
Turning “ON” the Fluorescent Protein
Photoactivation generally requires a separate excitation source (or at least a different filter set) than that used for imaging. For most of the molecules listed in Table 1, ∼400-nm irradiation is required. Notable exceptions are the KFP1, which uses green (∼532 nm) light, Dendra, which can be activated with ∼400-nm light and ∼490-nm light, Padron, which photoswitches on with 488 nm light, IrisFP, which photoswitches in both green and red states, and the two mCherry derivatives, rsCherry and rsCherryRev, which photoswitch with blue and yellow light.
Imaging the Highlighted Fluorescent Protein
Imaging of these molecules is similar to imaging any other molecule, with a few differences that can be advantageous or disadvantageous, depending on the experiment. Unlike conventional fluorescent proteins in which the starting level of fluorescence is usually the highest level that will be observed, the level of fluorescence after photoactivation must be estimated to avoid detector saturation at a given imaging excitation power and detector gain. A simple procedure for estimation of post-photoactivation fluorescence involves taking a pre-photoactivation image, measuring the mean fluorescence within the region to be photoactivated, background subtracting that mean and multiplying that number by the x-fold increase in fluorescence emission reported for the protein being used (See contrast column in Table 1). This will give a rough estimation of the pixel values after photoactivation, but a little trial and error with various detector gain settings (contrast or voltage settings) will help the experimenter gain an appreciation of the levels of fluorescence to be expected with each experiment.
Optimization of the Highlighting Step
Regardless of the excitation source, some effort must be expended to obtain optimal photoactivation. Photoactivation is dependent on the duration of excitation power per unit area. In addition to photobleaching during the imaging after the photoactivation, photobleaching can also be encountered during the photoactivation event. Therefore, to maximize the contrast between pre- and post-photoactivation, power and duration of the photoactivation must be optimized such that the highest level of photoactivation is obtained with the least amount of photobleaching. The power and duration dependence are most easily demonstrated and optimized with cells expressing the optical highlighter. A straightforward approach to optimization is to image a cell or field of cells between brief exposures to the activation light. This procedure is analogous to a fluorescence loss in photobleaching (FLIP) experiment, with the major difference being that fluorescence is increased instead of lost. As long as similar parameters (objective, zoom factor, photoactivation wavelength) are used, this optimization routine should suffice. However, fluctuations in laser power and alignment or changes in the parameters listed above will require additional optimizations. For a more detailed procedure, see below.
Optimization Procedures
Set the instrument light path configurations for imaging both the highlighted and inactive molecule, if desired. This will require proper mirrors and emission filters to detect the green and/or red fluorescence depending on the protein in use. It is important to remember that a dichroic mirror capable of reflecting the photoactivation light source to the sample is also required. For most of the optical highlighters, this will be light of ∼400 nm for one-photon excitation, with the notable exceptions of KFP1 and Dendra, discussed earlier. If two-photon excitation is to be used, be aware that the dichroic mirror should reflect the near-infrared wavelengths being used.
Set the imaging parameters (magnification or zoom factor, pixel dwell time or scan time, imaging excitation power, detector gain) required for the experiment. Some of these parameters may change for different experiments, but if the light path (mirrors, alignment etc.), magnification, and pixel dwell time remain similar, optimized photoactivation conditions should be similar for each experiment.
Locate a cell or cells expressing the optical highlighter. As discussed earlier, this will depend on the protein being used.
Set the photoactivation light power level to be used for the experiment. This parameter will have to be optimized depending on the requirements of the experiment and the light sensitivity of the cell type. Photoactivation light levels can affect both cell viability and the function of the protein of interest, but this depends on the protein as well as the cell type. It is the investigator's responsibility to test both possibilities. Regardless of the light level required, the procedure outlined here will be the same. To begin, use the maximum power available. This will lead to conditions for the most rapid photoactivation.
Set the photoactivation time. Depending on the instrument used, this may depend on the pixel dwell time or scan time used for imaging, or it may be set separately. Set this to a period of photoactivation as short as possible.
Set up an experiment with the following steps: (a) Image the optical highlighter in the photoactivated spectrum before photoactivation, perform one photoactivation using the desired photoactivation power level, and image the optical highlighter in the photoactivated spectrum again. (b) Repeat the photoactivation and imaging steps in sequence 10 to 100 times. Over the course of the experiment, the fluorescence should increase to a maximum and perhaps begin to decrease gradually. If the fluorescence is quantified within the photoactivated cells, the maximum level represents the optimal contrast that is obtainable over the inactivated optical highlighters. The number of photoactivation cycles required to reach the maximum fluorescent signal determines the optimal photoactivation time required for this photoactivation power level, magnification, and light path. An example of such an optimization is shown in Figure 2 using two-photon excitation for the photoactivation. A note of caution here is that if the photoactivation power level is too high for use with the optical highlighter of choice, it may be photoactivated and photobleached during the same iteration. Thus, it may appear that no photoactivation is observed during the optimization. Performing the same protocol with lower photoactivation power levels will test for this possibility and help the experimenter find optimal photoactivation conditions.
Figure 2.
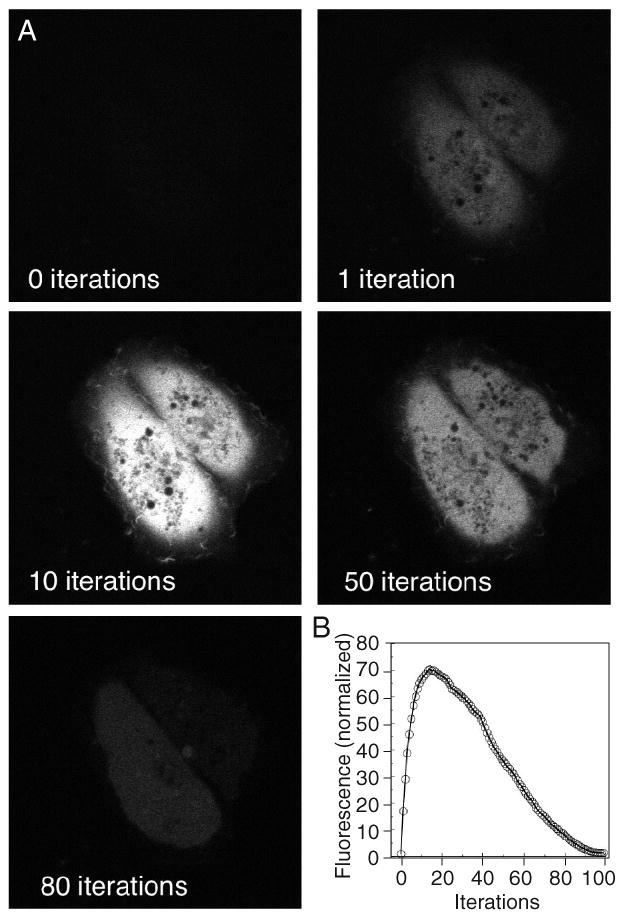
Photoactivation of PA-GFP. (A) COS-7 cells expressing PA-GFP were repeatedly imaged using 488-nm excitation. Between images, the entire field was briefly irradiated by two-photon excitation using 800-nm laser light. The total number of two-photon irradiations is shown in the lower left corner of the image. (B) The 488-nm excited fluorescence within the photoactivated region was normalized to the initial value and displayed as a function of the accumulated irradiations.
General Photoactivation Experiment
An example of a photoactivation experiment using the following procedure is shown in Figure 3. This protocol is written for PA-GFP using a laser scanning confocal microscope, but it should work for most of the optical highlighters in Table 1 and on other types of microscopes with appropriate alterations. Most commercial confocal systems have a software module devoted to performing photobleaching or photoactivation experiments. These can often be used for photoactivation experiments as well, as long as the laser wavelength used for the photoactivation event is close to ∼400 nm rather than the normal imaging laser line.
Figure 3.
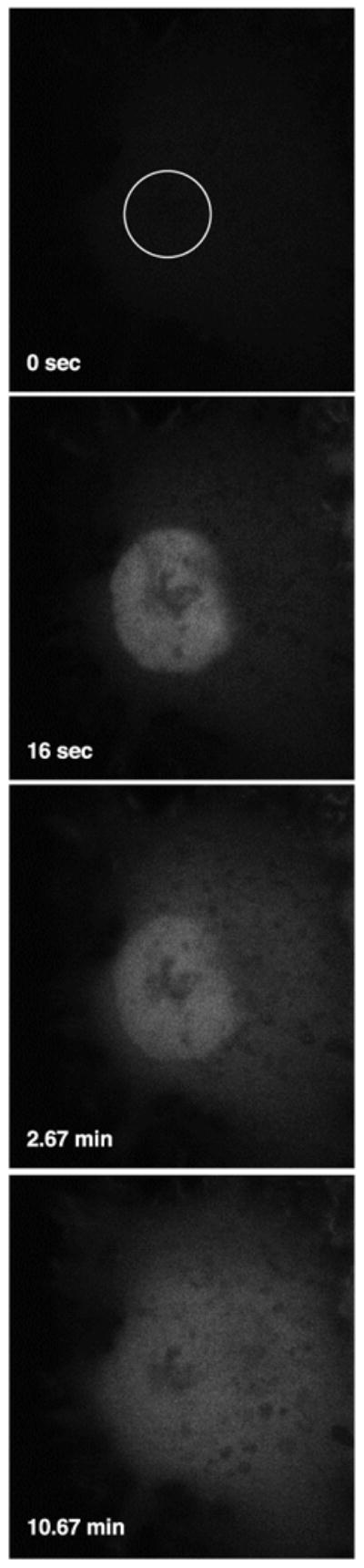
Selective photoactivation of PA-GFP. A COS-7 cell expressing PA-GFP was imaged using low levels of 488-nm light before and after photoactivation of the nuclear region (indicated by the white circle) with ∼1 sec of 413-nm light. The images were acquired at 15.75-sec intervals.
Locate an expressing cell and the region of interest. The photobleaching or perhaps photoactivation software module generally has a feature for selecting the region to be photobleached or photoactivated. Select the region using the appropriate function.
Adjust the 488-nm laser power and detector gain for the expected fluorescence increase. This may require some of the trial and error indicated earlier.
Using the time series software module, program a time-lapse image experiment that will encompass the entirety of the process under study. This should collect images at the desired intervals, which will depend on the time scale of the process of interest.
Begin the time-lapse experiment by initially imaging the cell for one to ten frames with 488 nm to establish the baseline fluorescence.
Switch the irradiation wavelength to ∼400 nm. For one photon, the increasingly common 405-nm diode lasers work well. A mercury lamp with a diaphragm can be used, but it is less efficient and has more limited temporal resolution. For two-photon excitation, published reports indicate 800 to 820 nm works well (Post et al., 2005; Schneider et al., 2005).
Irradiate the region of interest using the activation power levels and activation times determined in the optimization above.
Switch excitation back to ∼488 nm and image the cell at the desired time intervals until the end of the experiment.
Uses of Optical Highlighter Fluorescent Proteins
Some examples making use of optical highlighters are introduced here to convey the diverse applications of optical highlighters. With the exception of photoactivated localization microscopy (PALM; see below), where generally only a few molecules are activated at a time, the procedures discussed earlier should assist or at least serve as an appropriate starting point for optimization of many optical highlighter experiments.
Protein Dynamics
One of the most common uses of optical highlighters is complementary to the photobleaching technique, fluorescence photobleaching recovery (FPR) or fluorescence recovery after photobleaching (FRAP). However, a distinct difference between FRAP and photoactivation is that in photoactivation, rather than monitoring the recovery of fluorescence within a region, a decrease in fluorescence can monitored when molecules are activated in one region and move to another. When monitoring rapid movement, this potentially provides two helpful characteristics compared to photobleaching. First, photoactivation can often rapidly and efficiently produce high contrast between highlighted and nonhighlighted molecules. Second, the highest signal-to-background ratio is found in the earliest points of the photoactivation experiment. Thus, the movement of proteins by diffusion or transport within a cell or organelle, movement of organelles within a cell, or even movement of a cell within an organism, can be readily monitored.
One disadvantage in photoactivation is that often the signal generated in one region dissipates as it moves throughout surrounding regions. To compensate for this signal dilution, it is possible to repeatedly photoactivate the same region, which is a variation similar to the photobleaching technique, fluorescence loss in photobleaching (FLIP). It relies on repeatedly photoactivating within the same region while monitoring the fluorescence increases in another region (Kim et al., 2006).
Fluorescence Pulse Labeling
New protein synthesis is often a concern when monitoring dynamics of fluorescent protein–tagged molecules of interest. This can often lead to artifacts in experiments unless synthesis is inhibited. However, photoactivation overcomes this problem because it affects only the protein that is already synthesized and properly folded at the time of photoactivation. Proteins synthesized before photoactivation fluoresce at the photoactivated wavelengths, while those that are synthesized after photoactivation do not. This characteristic of optical highlighters has been used to monitor the fate of cells during embryogenesis with KFP1 (Chudakov et al., 2003), the movements of chromatin loci in Drosophila embryos (Post et al., 2005), and the formation of new peroxisomes in cell culture (Kim et al., 2006).
This fluorescence pulse labeling ability also introduces an approach to monitoring protein turnover. A brief pulse with the activation wavelength of the tagged proteins of interest labels a population of molecules, and protein degradation is monitored by imaging the loss of fluorescence. It must be realized that the population of fluorescent molecules labeled under these conditions includes only those optical highlighters that are fully synthesized and properly folded, and the optical highlighter must be degraded along with the protein of interest for proper read-out. Biochemical pulse-labeling thus should be carried out in parallel experiments to verify results. The temporal resolution of fluorescence pulse-labeling is essentially limited by the instrument parameters (usually milliseconds-seconds are required for activation), has subcellular spatial resolution (dependent on the optics used for imaging), and allows study of protein turnover in a single living cell.
Photoquenching Fluorescence Resonance Energy Transfer (PQ-FRET)
Fluorescent proteins have contributed to the study of protein-protein interactions within living cells by Förster resonance energy transfer (FRET; (Day et al., 2001)). Since FRET requires that the distance between the donor and acceptor fluorophores be <10 nm, the energy transfer can be interpreted as an interaction of the tagged proteins of interest. An interesting approach to the study of protein-protein interactions in cells has been termed photoquenching FRET (PQ-FRET) and relies on the use of photoactivated PA-GFP as an acceptor to quench the fluorescence of a donor fluorophore, cyan fluorescent protein (CFP; (Demarco et al., 2006)). In the inactive state, the absorbance spectrum of PA-GFP has little overlap with the emission spectrum of CFP, but after photoactivation of PA-GFP it exhibits spectral overlap that makes FRET possible. Using PQ-FRET, the authors were able to monitor both the movement of the proteins and the interactions of the heterochromatin protein (HP1α) and the transcription factor CCAAT/enhancer binding protein alpha (C/EBPα) within distinct domains of the nuclei of living cells (Demarco et al., 2006). Thus, PQ-FRET offers the capability of monitoring the dynamic interactions of molecules at steady state within different regions of the cell.
Photoactivated Localization Microscopy (PALM)
Unlike the applications mentioned above, the high-resolution imaging technique, photoactivated localization microscopy (PALM), does not benefit from optimizing optical highlighter photoactivation and actually requires only a few molecules to be activated with each activation cycle. Fluorescent proteins tagged to a protein of interest offer spatial information that is precise to within a few nanometers. Yet, the limit of resolution for conventional optical techniques is ∼100× more than the size of the fluorescent proteins. PALM provides near molecular resolution of proteins (Betzig et al., 2006). PALM involves imaging of molecules individually and then localizing them to high precision by determining their centers of fluorescence emission. This is achieved by performing a statistical fit of their measured photon distributions with ideal point spread functions. When the background noise is negligible compared to the molecular signal, the error in the fitted position is where s is the standard deviation of a Gaussian distribution approximating the point spread function of the objective, and N is the total number of detected photons (Cheezum et al., 2001; Thompson et al., 2002).
Determination of protein localization using PALM is usually limited to molecules that are separated by the distance required of conventional optics (∼250 nm). However, within most fluorescent specimen, hundreds or thousands of molecules may be present within one point spread function. PALM approaches this problem using both spectral and temporal means to isolate individual molecules. It relies on the serial photoactivation (using low levels of photoactivation light) of a small population of optical highlighters and their subsequent imaging until photodestruction. The photoactivation of small populations ensures that the density of imaged molecules remains much less than one molecule per normal resolution limit, and their subsequent photodestruction allows a new subset of molecules to be imaged. By fitting the fluorescent signal from each molecule to a two-dimensional Gaussian distribution, the coordinates for the location of the molecule and its uncertainty are determined. Each molecule is then rendered in a new image as a two-dimensional Gaussian distribution of standard deviation based on its uncertainty centered at the determined coordinates.
Reversible Saturable Optical Fluorescence Transitions (RESOLFT)
Reversible saturable optical fluorescence transitions (RESOLFT; (Hofmann et al., 2005)), relies on the switching behavior of the reversible optical highlighters such as asFP495 ((Lukyanov et al., 2000)) or Dronpa (Ando et al., 2004) to increase obtainable resolutions to the 50- to 100-nm range. This technique relies on the photoactivation of the reversible optical highlighter within the normal optical diffraction–limited spot in conjunction with photoswitching off of the optical highlighter in the same diffraction limited spot with a zero node inactivation. This produces a region of inactivation that leaves a region of photoactivated optical highlighter smaller than the diffraction limited spot.
Application of Optical Highlighter Fluorescent Proteins in Cytometry
The plethora of optical highlighters, specifically their broad wavelength range, is a boon for use in cytometry where just as with conventional fluorescent proteins(Morozova et al., 2010), cell populations expressing several different varieties can be easily separated. The potential power of such an approach is displayed an elegant use of an optical highlighter is demonstrated in a recent paper by Victora et. al. (Victora et al., 2010) where two photon microscopy was used to photoactivate PA-GFP in B cells within specific regions of the germinal centers located in the lymph nodes of transgenic mice. Cells from these lymph nodes were purified by cell sorting for further characterization. A key feature for this experiment was that the cells originating from the different regions of the germinal centers could be distinguished based on their photoactivation state. It is anticipated that this technique will be further utilized in coming years for molecular analysis of cells in deep tissue in immunology, neurobiology, and embryonic development studies.
Future Directions of Optical Highlighter Fluorescent Proteins
A long-term goal in optical imaging is to monitor the behavior of individual molecules of interest within the living cells of an organism, and the developments using optical highlighters such the ones discussed above have certainly advanced this goal (see (Hofmann et al., 2005; Betzig et al., 2006)). If the trend in fluorescent protein development continues, molecules with more defined absorbance cross sections, narrower emission spectra, higher fluorescence quantum efficiencies, more photostability, and less blinking and/or flickering are likely to be introduced in the near future. These will undoubtedly serve to advance the techniques mentioned above and lead to new and clever optical approaches in studies of cell biology.
Literature Cited
- Adam V, Lelimousin M, Boehme S, Desfonds G, Nienhaus K, Field MJ, Wiedenmann J, McSweeney S, Nienhaus GU, Bourgeois D. Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proc Natl Acad Sci U S A. 2008;105:18343–8. doi: 10.1073/pnas.0805949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando R, Flors C, Mizuno H, Hofkens J, Miyawaki A. Highlighted generation of fluorescence signals using simultaneous two-color irradiation on Dronpa mutants. Biophys J. 2007;92:L97–9. doi: 10.1529/biophysj.107.105882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:12651–6. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando R, Mizuno H, Miyawaki A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–3. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- Andresen M, Stiel AC, Folling J, Wenzel D, Schonle A, Egner A, Eggeling C, Hell SW, Jakobs S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat Biotechnol. 2008;26:1035–40. doi: 10.1038/nbt.1493. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–5. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–82. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheezum MK, Walker WF, Guilford WH. Quantitative comparison of algorithms for tracking single fluorescent particles. Biophys J. 2001;81:2378–88. doi: 10.1016/S0006-3495(01)75884-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Belousov VV, Zaraisky AG, Novoselov VV, Staroverov DB, Zorov DB, Lukyanov S, Lukyanov KA. Kindling fluorescent proteins for precise in vivo photolabeling. Nat Biotechnol. 2003;21:191–4. doi: 10.1038/nbt778. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Photoswitchable cyan fluorescent protein for protein tracking. Nat Biotechnol. 2004;22:1435–9. doi: 10.1038/nbt1025. [DOI] [PubMed] [Google Scholar]
- Day RN, Davidson MW. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 2009;38:2887–921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day RN, Periasamy A, Schaufele F. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 2001;25:4–18. doi: 10.1006/meth.2001.1211. [DOI] [PubMed] [Google Scholar]
- Demarco IA, Periasamy A, Booker CF, Day RN. Monitoring dynamic protein interactions with photoquenching FRET. Nat Methods. 2006;3:519–24. doi: 10.1038/nmeth889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrig T, O'Kane DJ, Prendergast FG. Green-fluorescent protein mutants with altered fluorescence excitation spectra. FEBS Lett. 1995;367:163–6. doi: 10.1016/0014-5793(95)00557-p. [DOI] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Habuchi S, Tsutsui H, Kochaniak AB, Miyawaki A, van Oijen AM. mKikGR, a monomeric photoswitchable fluorescent protein. PLoS One. 2008;3:e3944. doi: 10.1371/journal.pone.0003944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 1994;91:12501–4. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci U S A. 2005;102:17565–9. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PK, Mullen RT, Schumann U, Lippincott-Schwartz J. The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J Cell Biol. 2006;173:521–32. doi: 10.1083/jcb.200601036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Patterson GH. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–65. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Innovation: Photoactivatable fluorescent proteins. Nat Rev Mol Cell Biol. 2005;6:885–91. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]
- Lukyanov KA, Fradkov AF, Gurskaya NG, Matz MV, Labas YA, Savitsky AP, Markelov ML, Zaraisky AG, Zhao X, Fang Y, Tan W, Lukyanov SA. Natural animal coloration can Be determined by a nonfluorescent green fluorescent protein homolog. J Biol Chem. 2000;275:25879–82. doi: 10.1074/jbc.C000338200. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–3. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TW, Chudakov DM. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods. 2007;4:555–7. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- Morozova KS, Piatkevich KD, Gould TJ, Zhang J, Bewersdorf J, Verkhusha VV. Far-red fluorescent protein excitable with red lasers for flow cytometry and superresolution STED nanoscopy. Biophys J. 2010;99:L13–5. doi: 10.1016/j.bpj.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–7. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Post JN, Lidke KA, Rieger B, Arndt-Jovin DJ. One- and two-photon photoactivation of a paGFP-fusion protein in live Drosophila embryos. FEBS Lett. 2005;579:325–30. doi: 10.1016/j.febslet.2004.11.092. [DOI] [PubMed] [Google Scholar]
- Schneider M, Barozzi S, Testa I, Faretta M, Diaspro A. Two-photon activation and excitation properties of PA-GFP in the 720-920-nm region. Biophys J. 2005;89:1346–52. doi: 10.1529/biophysj.104.054502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–72. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Stiel AC, Andresen M, Bock H, Hilbert M, Schilde J, Schonle A, Eggeling C, Egner A, Hell SW, Jakobs S. Generation of monomeric reversibly switchable red fluorescent proteins for far-field fluorescence nanoscopy. Biophys J. 2008;95:2989–97. doi: 10.1529/biophysj.108.130146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiel AC, Trowitzsch S, Weber G, Andresen M, Eggeling C, Hell SW, Jakobs S, Wahl MC. 1.8 A bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochem J. 2007;402:35–42. doi: 10.1042/BJ20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott-Schwartz J, Verkhusha VV. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods. 2009;6:153–9. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Renz M, Lippincott-Schwartz J, Verkhusha VV. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132:6481–91. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–83. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Karasawa S, Shimizu H, Nukina N, Miyawaki A. Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep. 2005;6:233–8. doi: 10.1038/sj.embor.7400361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhusha VV, Sorkin A. Conversion of the monomeric red fluorescent protein into a photoactivatable probe. Chem Biol. 2005;12:279–85. doi: 10.1016/j.chembiol.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler KD, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci U S A. 2004;101:15905–10. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoe H, Meyer T. Spatial dynamics of GFP-tagged proteins investigated by local fluorescence enhancement. Nat Biotechnol. 1996;14:1252–6. doi: 10.1038/nbt1096-1252. [DOI] [PubMed] [Google Scholar]