

Abstract
Rationale
Overexpression of muscle atrophy F-box (MAFbx/atrogin-1), an E3 ubiquitin ligase, induces proteasomal degradation in cardiomyocytes. The role of endogenous MAFbx in regulating cardiac hypertrophy and failure remains unclear.
Objective
We investigated the role of MAFbx in regulating cardiac hypertrophy and function in response to pressure overload. Transverse aortic constriction (TAC) was applied to MAFbx KO and wild type (WT) mice.
Methods and Results
Expression of MAFbx in WT mice was significantly increased by TAC. TAC-induced increases in cardiac hypertrophy were significantly smaller in MAFbx KO than in WT mice. There was significantly less lung congestion and interstitial fibrosis in MAFbx KO than in WT mice. MAFbx KO also inhibited β-adrenergic cardiac hypertrophy. DNA microarray analysis revealed that activation of genes associated with the transcription factor binding site for the NF-κB family were inhibited in MAFbx KO mice compared with WT mice after TAC. Although the levels of IκB-α were significantly decreased after TAC in WT mice, they were increased in MAFbx KO mice. MAFbx regulates ubiquitination and proteasomal degradation of IκB-α in cardiomyocytes. In primary cultured rat cardiomyocytes, phenylephrine-induced activation of NF-κB and hypertrophy were significantly suppressed by MAFbx knock-down, but were partially rescued by overexpression of NF-κB p65.
Conclusions
MAFbx plays an essential role in mediating cardiac hypertrophy in response to pressure overload. Downregulation of MAFbx inhibits cardiac hypertrophy in part through stabilization of IκB-α and inactivation of NF-κB. Taken together, inhibition of MAFbx attenuates pathological hypertrophy, thereby protecting the heart from progression into heart failure.
Keywords: E3 ligase, ubiquitin proteasome system, cardiac hypertrophy, NF-κB
Under conditions of increased hemodynamic load, including pressure overload (PO) caused by high blood pressure and volume overload after myocardial infarction, cardiomyocytes undergo hypertrophy in order to adapt to increased workload and reduce wall stress 1. However, the prolonged existence of cardiac hypertrophy is an independent risk factor for cardiac morbidity and mortality 2. Cardiac hypertrophy is mediated by the coordinated action of cellular signaling mechanisms, hypertrophy gene expression programs, sarcomere assembly, and alteration in cellular energy metabolism 3, 4. One key element of cardiac remodeling is altered protein turnover, namely changes in both protein synthesis and degradation 5, 6. While a large body of evidence has shown that increases in protein synthesis play an important role in mediating cardiac hypertrophy, the role of protein degradation is less well understood.
The ubiquitin-proteasome system (UPS) is a major protein degradation system critically important for the maintenance of cellular homeostasis in eukaryotic cells 5–7. Proteins degraded by the UPS are covalently linked to four or more ubiquitin molecules. Poly-ubiquitin conjugation involves three components, an ubiquitin-activation enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin ligase (E3). Each E3 ligase recognizes specific substrates, thereby conferring specificity to the UPS 6. Muscle atrophy F-box (MAFbx), also known as FBXO32, F-box protein 32, and atrogin-1, is an E3 ubiquitin ligase expressed only in muscle, including skeletal and cardiomyocytes 8, 9. MAFbx was originally identified as a critical mediator of skeletal muscle atrophy 8, 9. Since protein degradation is promoted under conditions of cardiac atrophy and regression of cardiac hypertrophy 10, it was originally postulated that MAFbx may mediate atrophy or regression of hypertrophy in cardiac muscle as well. However, thus far, the role of endogenous MAFbx in mediating regression of cardiac hypertrophy has not been clearly demonstrated. Interestingly, a study conducted with MAFbx KO mice showed that endogenous MAFbx negatively regulates cardiac hypertrophy in response to exercise 11, a model of physiological hypertrophy. In contrast, the role of MAFbx in regulating pathological hypertrophy has been studied only with a gain-of-function animal model in vivo, where overexpressed MAFbx inhibited PO-induced hypertrophy by targeting calcineurin for ubiquitin-mediated proteolysis 12. Thus, the role of endogenous MAFbx in the heart during pathological hypertrophy and failure remains to be studied with a loss-of-function animal model. Cardiac hypertrophy caused by pathological insults is accompanied by increases in protein synthesis and ER stress, which require protein quality control mediated by the UPS 5. However, the role of the UPS in left ventricular hypertrophy and remodeling is controversial 13. Thus, testing whether MAFbx is involved in pathological hypertrophy is important.
We here investigated the role of MAFbx in mediating a pathological form of cardiac hypertrophy. We asked 1) whether endogenous MAFbx is upregulated during hypertrophy induced by PO, 2) whether endogenous MAFbx positively or negatively mediates cardiac hypertrophy induced by PO, and 3) which downstream target mediates the effect of MAFbx upon pathological hypertrophy. Surprisingly, our study using a loss of function mouse model allowed us to identify a novel function of MAFbx, namely to critically mediate, but not inhibit, PO-induced cardiac hypertrophy.
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Mice
MAFbx KO mice have been described 9 (Novartis Institutes for Biomedical Research). All experiments involving animals were approved by the Institutional Animal Care and Use Committee at the New Jersey Medical School.
Adenoviral vectors
Adenovirus harboring rat MAFbx and recombinant adenovirus vector containing the hairpin-forming oligo were constructed, amplified and titered as described 14. The hairpin-forming oligo of sh-MAFbx corresponds to bases 474 to 494 of the rat MAFbx gene.
Statistics
All values are expressed as mean ± SEM except where noted. Statistical analyses were performed by either t-test or ANOVA and the Tukey post-test procedure, with p<0.05 considered significant.
Results
MAFbx plays an important role in mediating PO-induced cardiac hypertrophy
We examined the effect of PO upon expression of MAFbx in the mouse heart. Although protein and mRNA expression of MAFbx is low under basal conditions, it was significantly upregulated one week after transverse aortic constriction (TAC) and further increased at 2 weeks (Fig. 1A). Phosphorylation of FoxO3a, a transcription factor known to upregulate MAFbx, was increased after TAC, which presumably induces nuclear exit of FoxO3a, suggesting that TAC-induced upregulation of MAFbx may be mediated by FoxO3a-independent mechanisms. In order to examine the role of the MAFbx upregulation in mediating PO-induced cardiac hypertrophy, we applied TAC to MAFbx KO and wild type (WT) mice. After 2 weeks, the pressure gradient was not significantly different between MAFbx KO and WT mice (Online Fig. IA, Online Table I). Interestingly, TAC-induced increases in left ventricular weight (LVW)/body weight (BW) and LVW/tibial length (TL) were significantly attenuated in MAFbx KO mice compared to in WT mice (Fig. 1B, Online Fig. IB, Online Table II), as were TAC-induced increases in echocardiographically determined LV wall thickness (Online Fig. IC, Online Table III). TAC-induced increases in LV myocyte cross sectional area were also significantly attenuated in MAFbx KO mice (Fig. 1C). These results suggest that the lack of MAFbx attenuates PO-induced cardiac hypertrophy. We also examined the role of MAFbx in mediating hypertrophy induced by phenylephrine (PE), an α1-adrenergic receptor agonist, in cultured cardiomyocytes. Downregulation of MAFbx by adenovirus harboring shRNA-MAFbx significantly attenuated PE-induced cardiac hypertrophy (Fig. 1D), suggesting that MAFbx plays an essential role in mediating hypertrophy at the myocyte level.
Figure 1. Deletion of MAFbx inhibits PO-induced cardiac hypertrophy.
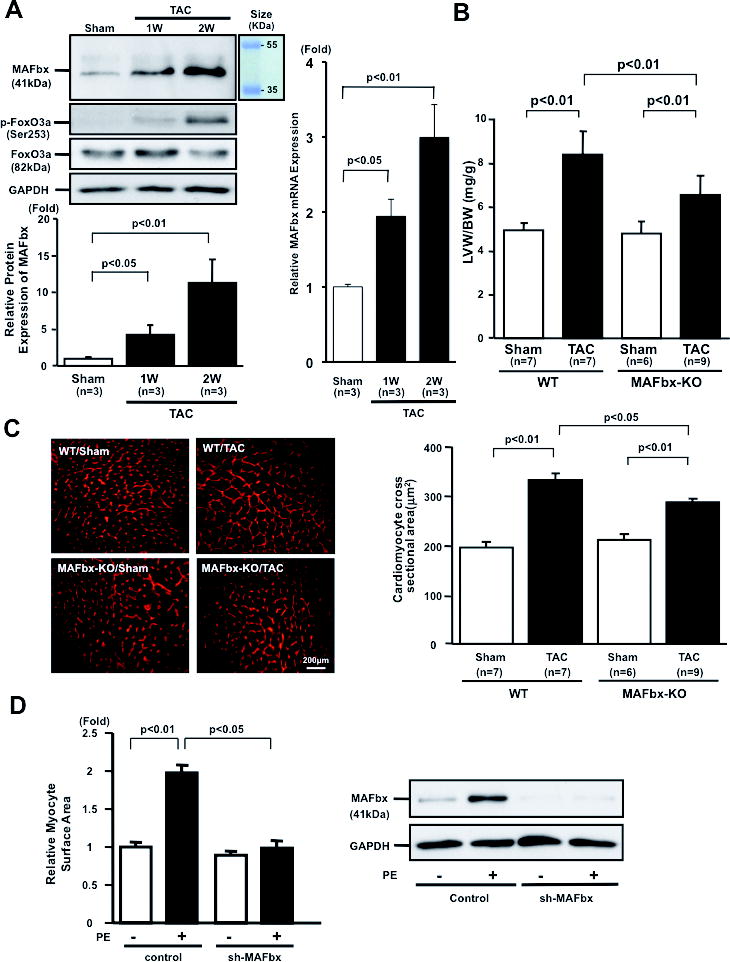
A, Expression of MAFbx, Foxo3a, and phosphorylated Foxo3a in mouse hearts in response to TAC. TAC (1 and 2 weeks) or sham operation was performed on C57/BL6 mice. Protein expression of MAFbx and GAPDH was determined by immunoblotting (left upper panels). The results of the quantitative analysis of MAFbx expression are also shown (left lower panel). mRNA expression of MAFbx gene was determined by quantitative RT-PCR (right panel). B and C, MAFbx KO mice were subjected to TAC or sham operation for 2 weeks. B, Left ventricular weight (LVW)/body weight (BW) after TAC is shown. C, LV cardiomyocyte cross sectional area is shown. Representative pictures of wheat germ agglutinin staining (left panels) and a bar graph showing the results of quantitative analysis (right panel). D, The effect of MAFbx knock-down on phenylephrine (PE)-induced cardiac hypertrophy. Myocytes were transduced with Ad-sh-MAFbx or control adenovirus and stimulated with PE (10 μmol/L) for 48 hours (left panel). Mean myocyte surface area was obtained from 250 myocytes/well. Protein expression of MAFbx and GAPDH was determined by immunoblotting (right panels).
Lack of MAFbx inhibits PO-induced cardiac dysfunction
We examined the effect of MAFbx KO upon cardiac function after TAC for 2 and 4 weeks. The increase in lung weight/BW, an indicator of lung congestion, after TAC was significantly attenuated in MAFbx KO compared to WT mice at 2 weeks (Online Table II) and 4 weeks (Fig. 2A, Online Table IV). Echocardiographic measurements showed that decreases in LV ejection fraction (EF) and fractional shortening (%FS) after TAC were significantly attenuated in MAFbx KO mice at 4 weeks (Fig. 2BCD, Online Table IV). Hemodynamic measurement showed that an increase in LV end diastolic pressure (LVEDP) after TAC was abolished in MAFbx KO mice at 2 weeks (Online Table I) and 4weeks (Fig. 2E). Taken together, these data show that decreases in LV function and the development of heart failure after PO were significantly attenuated in MAFbx KO mice, suggesting that upregulation of MAFbx during PO not only stimulates hypertrophy but also induces LV dysfunction and heart failure.
Figure 2. Deletion of MAFbx inhibits PO-induced cardiac dysfunction.

A, Lung weight/BW after TAC for 4 weeks is shown. B–D, LV systolic function was evaluated by echocardiographic measurement. Representative M-mode tracings are shown in B. C, LV ejection fraction (LVEF). D, fractional shortening (%FS). E, LV end-diastolic pressure (EDP) was obtained by hemodynamic measurement.
Lack of MAFbx inhibits PO-induced cardiac remodeling
TAC-induced hypertrophy was accompanied by upregulation of fetal type genes, including atrial natriuretic factor (ANF) and α-skeletal actin, in WT mice. TAC-induced upregulation of the fetal type genes was significantly attenuated in MAFbx KO mice (Fig. 3A, B). The ratio of mRNA expression of β-MHC/α-MHC was significantly increased after TAC in WT mice. TAC induced upregulation of this ratio was significantly attenuated in MAFbx KO mice (Fig 3C), suggesting that genetic ablation of MAFbx inhibits PO-induced fetal gene expression. Histological analyses showed that TAC-induced increases in interstitial fibrosis were significantly attenuated in MAFbx KO mice (Fig. 3D). The number of TUNEL- and troponin T- double positive nuclei was increased in both WT and MAFbx KO mice after TAC compared to respective sham operated mice. However, the increase in the percentage of the double-positive nuclei was significantly smaller in MAFbx KO than in WT mice (Figure 3E). Thus, the cardiac phenotype commonly observed in pathological hypertrophy was significantly attenuated in MAFbx KO mice compared to WT mice.
Figure 3. Genetic deletion of MAFbx inhibits TAC-induced cardiac hypertrophy.

A, B, and C, mRNA expression of fetal type genes associated with cardiac hypertrophy was determined by quantitative RT-PCR. Atrial natriuretic factor (ANF) (A). α-skeletal actin (αSA) (B). β-MHC/α-MHC (C). D, Picric acid sirius red (PASR) staining of cardiac sections (left). Percentage of PASR positive areas (right). E, Representative images of TUNEL staining of cardiac sections two weeks after TAC counterstained with anti-troponin T antibody (left). Percent TUNEL-positive myocytes (right).
In order to examine whether endogenous MAFbx also plays an important role in mediating cardiac hypertrophy in response to another stimulus, WT and MAFbx KO mice were subjected to continuous infusion of isoproterenol (ISO), an agonist for the β-adrenergic receptor, for 2 weeks. The heart rate and mean blood pressure after ISO infusion were not significantly different between WT and MAFbx KO mice (Online Fig. IIA, B). The ISO-induced cardiac hypertrophy and lung congestion observed in WT mouse hearts were significantly attenuated in MAFbx KO hearts (Online Fig. IIC–E). ISO-induced increases in ANF expression (Online Fig. II2F), myocyte cross sectional area and fibrosis (Online Fig. IIIA, B) in the heart were attenuated in MAFbx KO hearts. These results suggest that MAFbx also plays an important role in mediating β-adrenergic cardiac hypertrophy and cardiac dysfunction in vivo.
Deletion of MAFbx normalizes changes in the gene expression profile induced by PO
In order to further characterize the effect of MAFbx KO upon phenotypic changes in the heart in response to TAC in an unbiased manner, a DNA microarray analysis was conducted, using total RNA prepared from WT and MAFbx KO mouse hearts subjected to TAC. In order to examine the effect of MAFbx KO upon TAC-induced changes in gene expression, we plotted the relationship between changes induced by TAC in the WT heart and changes induced by MAFbx KO in hearts subjected to TAC for all genes. The graph showed that TAC-induced changes in gene expression in WT for each gene (X-axis in Fig. 4A) inversely correlated with differences in gene expression after TAC between MAFbx KO and WT (Y-axis in Fig. 4A), indicating attenuation of gene regulation in MAFbx KO in response to TAC. Transcription factor binding site (TFBS) analysis was conducted using the microarray data. The results of the TFBS analysis suggested that genes with binding sites for the NF-κB family of transcription factors in the promoter region were significantly downregulated in MAFbx KO mice (Online Table V and Fig. 4B). TAC-induced upregulation of the genes with binding sites for the NF-κB family of transcription factors in the promoter region, including IL-6, Bcl-3, and Tnfrsf1b, was significantly attenuated in MAFbx KO mice compared to WT mice (Online Table VI and Fig. 4C). To further investigate whether the binding of NF-κB to the target genes is regulated by MAFbx, we performed a ChIP assay in heart homogenates obtained from WT or MAFbx-KO mice,using antibody against NF-κB-p65. The ChIP product was subjected to PCR with a pair of primers designed to amplify the fragment of the IL-6 gene promoter region containing the NF-κB-binding motif (Figure 4D top 3 panels and Online Fig. IV). A pair of primers designed to amplify the fragment of the IL-6 gene promoter region that does not contain the NF-κB-binding motif were used as a negative control (Figure 4D lower 2 panels). The results showed that NF-κB binds to the IL-6 promoter and PO enhances the DNA binding of NF-κB in WT mice. However, the TAC-induced enhancement of the NF-κB binding to the IL-6 promoter was significantly attenuated in MAFbx KO mice. Collectively, these results suggest that MAFbx facilitates DNA binding of NF-κB and expression of the NF-κB target genes in the heart in response to PO.
Figure 4. Inverse correlation of gene expression profile between MAFbx KO and WT mice after TAC.

A, Gene density plot of the gene expression profiles for comparison. The x-axis is the difference in gene expression between WT sham and WT TAC, and the y-axis is the difference between WT TAC and MAFbx KO TAC. The observed gene density values were normalized to expected values derived from randomized data and plotted in a heat map to represent gene enrichment or depletion. Red indicates gene enrichment and blue gene depletion. The negative correlation is also indicated by Spearman rank correlation (r = −0.39, p < 2.2×10−16). B, Genes associated with the binding site of the NF-κB family were significantly downregulated in MAFbx KO mice after TAC. X-axis indicates log2 (fold change) in MAFbx KO vs. WT after TAC. The black line is for genes associated with the binding site of the NF-κB family (GGGNNTTTCC_V$NFKB_Q6_01), and the gray one is for other genes. The leftward shift of the black line indicates more downregulation compared with the gray one. The P-value was based on the comparison of black and gray lines using the Kolmogorov-Smirnov (KS) test. C, Expression of genes of IL-6, Bcl-3, and Tnfrsf1b was determined by quantitative RT-PCR. D, Left panels: ChIP assay with antibody against NFκB-p65. A parallel ChIP assay was performed with rabbit IgG as a ChIP assay control. DNA was amplified and quantitated by PCR with specific primers flanking the mouse IL-6 gene promoter containing the NF-κB-binding motif (Online Fig. IV) and a pair of control primers that does not contain the NF-κB-binding motif. PCR using input DNA as template serves as an internal control. Right panel: Quantitative analysis of the data shown in left panels. Results are expressed as ratios of immunoprecipitated vs. input DNA. *p < 0.05 vs. WT-Sham, #p < 0.05 vs. WT-TAC.
Downregulation of MAFbx inhibits activation of NF-κB by hypertrophic stimuli
In order to examine whether MAFbx KO affects NF-κB-mediated transcription, we examined expression of NF-κB by immunostaining. Although expression of NF-κB is low in sham operated mouse hearts, TAC significantly increased NF-κB nuclear staining in WT hearts. In contrast, TAC-induced increases in NF-κB nuclear staining were significantly attenuated in MAFbx KO hearts (Fig. 5A and Online Figure V). Consistently, although protein expression of IκB-α, an inhibitor of NF-κB, was significantly decreased after TAC in WT hearts, it was significantly increased in sham operated MAFbx KO hearts and was further enhanced after TAC in the MAFbx KO hearts (Fig. 5B). Phosphorylation of IκB-α was significantly increased after TAC in WT hearts, consistent with the decrease in IκB-α in response to TAC. In MAFbx KO mice, the level of phosphorylated IκB-α was significantly increased in sham-operated hearts and it was further increased after TAC, indicating that IκB-α is stabilized even in the presence of phosphorylation. MAFbx KO did not significantly affect the level of total IκB kinase-α (IKK-α) or phosphorylated IKK/total IKK after Sham and TAC (Fig. 5B) mRNA expression of IκB-α was increased significantly after TAC in both WT and MAFbx KO hearts but there was no significant difference between WT and MAFbx KO hearts (Fig. 5C). PE-induced upregulation of NF-κB in the nucleus was also attenuated in the presence of shRNA-MAFbx in cultured cardiomyocytes (Fig. 5D). In addition, PE-induced increases in the transcriptional activity of NF-κB, as evaluated by reporter gene assays, were attenuated in the presence of shRNA-MAFbx (Fig. 5E). These results suggest that endogenous MAFbx plays an essential role in mediating upregulation of NF-κB-mediated transcription by hypertrophic stimuli in cardiomyocytes, and that these effects of MAFbx are cell autonomous in cardiomyocytes.
Figure 5. Deletion of MAFbx inhibits NF-κB activationin vivo andin vitro.

A-C, MAFbx-KO and WT mice were subjected to TAC or sham operation for 2 weeks. A, Myocardial sections were stained with anti-NF-κB-p65 antibody (green), anti-troponin (T antibody (red) and DAPI (blue). Arrows indicate nuclear localization of NF-κB. B, The expression levels of IκB-α, phosphorylated IκB-α, IκB kinase-α (IKK-α) and phosphorylated IKK were determined by immunoblotting (upper panels). The results of the quantitative analysis of IκB-α, phosphorylated IκB-α, phosphorylated IKK (lower panels). C, Expression of IκB-α gene was determined by quantitative RT-PCR. D, The nuclear translocation of NF-κB was evaluated with immunostaining. Myocytes were transduced with either Ad-LacZ or Ad-shRNA-MAFbx and stimulated with PE for 48 hours. E, The effect of MAFbx knock-down on PE-induced NF-κB activation. Myocytes were transfected with NF-κB/luciferase reporter together with either control plasmid or pDC311-sh-MAFbx. Myocytes were stimulated with PE for 48 hours.
Since IκB-α is accumulated in the absence of endogenous MAFbx, we hypothesized that proteasome degradation of IκB-α is regulated by MAFbx. Co-immunoprecipitation assays indicated that myc-MAFbx and IκBα physically associate with one another in cardiomyocytes in vitro (Fig. 6A). To test whether MAFbx directly ubiquitinates I-κB, in vitro ubiquitination assays were performed with recombinant proteins. GST-MAFbx and cdc34, an E2 ubiquitin-conjugating enzyme, were incubated with biotin-ubiquitin, a ubiquitin activating enzyme (E1), and GST-IκB-α, and the reaction mixture was subjected to immunoblotting with anti-ubiquitin and anti-IκB-α antibodies. Ubiquitination of I-κB, as assessed by the detection of high-molecular-weight polyubiquitin chains of ubiquitin and monoubiquitinated IκB-α, was detected in a reaction, containing E1, cdc34, MAFbx and ubiquitin (Fig. 6B). Deletion of any single component in this reaction abolished ubiquitination of IκB-α. The extent of IκB-α ubiquitination was evaluated using the GST-Tandem Ubiquitin Binding Entity assay 15. Although the total amount of ubiquitin-conjugated proteins was similar among the samples analyzed, the level of ubiquitinated IκB-α after TAC was significantly greater in the WT mouse heart than in the MAFbx KO heart (Fig. 6C). Overexpression of MAFbx decreased expression of IκB-α in cardiomyocytes, an effect that was reversed in the presence of MG132, a proteasome inhibitor (Fig. 6D). These results suggest that MAFbx induces destabilization of IκB-α through ubiquitination and proteasomal degradation.In order to test whether accumulation of IκB-α and downregulation of NF-κB play an important role in mediating the suppression of cardiac hypertrophy in MAFbx knock-down myocytes, a rescue experiment was conducted. Overexpression of p65, one of the NF-κB subunits, in the presence of MAFbx knock-down, partially restored expression of p65 and attenuated the inhibition of PE-induced cardiomyocyte hypertrophy by MAFbx downregulation (Fig. 6E). These results suggest that the lack of MAFbx attenuates PE-induced cardiac hypertrophy in part through downregulation of the NF-κB pathway. Interestingly, overexpression of MAFbx also inhibited PE-induced cardiomyocyte hypertrophy. Overexpression of MAFbx markedly enhanced PE-induced accumulation of p65, suggesting that exogenous MAFbx appears to inhibit cardiac hypertrophy through IκB-NF-κB-independent mechanisms.
Figure 6. MAFbx induces proteasomal degradation of IkB, thereby inducing cardiac hypertrophy.

A, Lysates from cardiomyocytes transduced with Ad-myc-MAFbx or Ad-LacZ were subjected to immunoprecipitation with anti-myc or control IgG. The immunoprecipitates were immunoblotted with anti-IκBα antibody. The left panel shows an immunoblot of the input with anti-IκB-α antibody. B, In vitro ubiquitination reactions were performed with indicated materials. IκB-α is ubiquitinated by MAFbx as demonstrated with immunoblot analyses with anti-ubiquitin (upper panel) and anti-IκB-α antibodies (lower panel). C, Heart homogenates (500 μg) were incubated with 20 μg of GST-TUBE2 to obtain ubiquitinated proteins. The samples were subjected to immunoblot with anti-IκB-α antibody. The lower panel shows an immunoblot of the input with anti-ubiquitin antibody. D, Myocytes were transduced with Ad-MAFbx or Ad-LacZ and pretreated with or without MG132. Cell lysates were subjected to immunoblot using anti-IκB-α antibody. E, Myocytes were transduced with Ad-sh-MAFbx, Ad-p65 or Ad-MAFbx alone or in combination and stimulated with PE for 48 hours. Myocyte surface area was obtained from 250 myocytes/well. Right panel shows expression of MAFbx, NF-κB-p65 and GAPDH. The results are from 4–5 experiments.
Discussion
Our results suggest that endogenous MAFbx/atrogin-1 plays a key role in mediating the development of pathological hypertrophy. Expression of MAFbx is increased in response to PO, and hypertrophy induced by either PO or β-adrenergic stimulation is significantly inhibited in MAFbx KO mice. Furthermore, downregulation of MAFbx inhibits LV dysfunction in response to PO and β-adrenergic stimulation. These findings are in contrast with the role of MAFbx in skeletal muscle, where MAFbx mediates denervation-induced atrophy 9, as well as with the inhibitory role of MAFbx in exercise-induced cardiac hypertrophy in the heart 11. We propose that MAFbx mediates pathological hypertrophy in part through proteasomal degradation of IκB-α and stabilization of NF-κB.
MAFbx is strongly upregulated by PO in the heart at both the mRNA and protein levels. MAFbx was originally identified as a gene upregulated when skeletal muscle undergoes denervation-induced atrophy 11. However, mRNA expression of fetal type genes and genes encoding the UPS are generally upregulated in the heart during both hypertrophy and atrophy 16. In cardiomyocytes, both FoxO3a 17 and TNF-α 18 upregulate expression of MAFbx, whereas Akt 19 and exercise 19 reduce it. FoxO3a induces atrophy of the heart 17. Since FoxO3a is phosphorylated and presumably excluded from the nucleus by PO, it is unlikely that FoxO3a mediates upregulation of MAFbx in our TAC model. The level of TNF-α is increased by PO and in the failing heart, and TNF-α could stimulate hypertrophy 20. Thus, it will be interesting to elucidate the role of TNF-α in mediating upregulation of MAFbx in response to PO.
Our results suggest that upregulation of MAFbx during PO positively mediates hypertrophy and cardiac dysfunction. To our knowledge, the role of MAFbx in mediating pathological hypertrophy has not been investigated with a loss-of-function mouse model. The fact that the lack of MAFbx reverses the changes in the gene expression profile that accompany PO-induced hypertrophy, as evaluated by unbiased DNA microarray analyses, confirms the reversal of the hypertrophy phenotype in the MAFbx KO mice, supporting our hypothesis. Since selective knock-down of MAFbx also inhibited PE-induced cardiac hypertrophy in cardiomyocytes in vitro, the role of MAFbx in mediating cardiac hypertrophy must be cell autonomous.
It should be noted that Li et al reported that overexpression of MAFbx in the heart inhibits TAC-induced cardiac hypertrophy. Overexpressed MAFbx directly binds to calcineurin and promotes its degradation 12. Since calcineurin plays an important role in mediating cardiac hypertrophy, the authors proposed that overexpression of MAFbx may inhibit cardiac hypertrophy through suppression of calcineurin 21. These results are in marked contrast with our finding that MAFbx mediates cardiac hypertrophy in response to PO. Furthermore, upregulation of calcineurin in response to PO was significantly inhibited in MAFbx KO mice in our hands (Online Fig. VI). This difference may be due to the fact that Li et al used a gain-of-function model 12, while we used a loss-of-function model of MAFbx. In fact, we found that overexpression of MAFbx partially inhibited PE-induced hypertrophy in cultured cardiomyocytes despite marked accumulation of p65-NF-κB. This raises a possibility that exogenously overexpressedMAFbx regulates additional targets, thereby inhibiting cardiac hypertrophy. Although expression of MAFbx is low at baseline, it is markedly increased by PO. The loss-of-function model would be more useful for evaluating the role of endogenous MAFbx upregulation in regulating hypertrophy during PO. Li et al also demonstrated that MAFbx negatively regulates physiological hypertrophy using MAFbx KO mice 11. Previous studies showed that several molecules, such as Akt 22 and ASK 23, not only promote physiological hypertrophy but also suppress pathological hypertrophy. Thus, it is possible that the role of MAFbx in suppression of physiological hypertrophy and mediation of PO-induced hypertrophy may involve a common mechanism. At present, however, this hypothesis remains to be tested.
In this report, we showed that MAFbx regulates pathological hypertrophy in part through activation of the NF-κB pathway. Transcription factor binding site analyses showed that genes with DNA-binding sites for the NF-κB family were significantly downregulated compared to genes without NF-κB binding sites. Subsequent analyses showed that ubiquitination of IκB-α, a negative regulator of NF-κB, in the PO heart was significantly attenuated in MAFbx KO mice. Furthermore, MAFbx and IκB-α physically interact with one another and MAFbx induces proteasomal degradation of IκB-α. MAFbx directly uniquitinates IkB in vitro. Although further investigation is needed to prove that IκB-α is a direct and physiological substrate of MAFbx in the heart in vivo, these results suggest that endogenous MAFbx regulates UPS-induced degradation of IκB-α in cardiomyocytes.
Pharmacologic blockade of NF-κB activation with pyrrolidine dithiocarbamate (PDTC) inhibits cardiac hypertrophy and cardiac remodeling 24. Additionally, cardiac-specific NF-κB inhibition by expression of a stabilized IκB-α mutant attenuates angiotensin II-induced hypertrophy 25. Furthermore, the targeted disruption of the p50 subunit of NF-κB has been shown to attenuate myocardial hypertrophy 26. These findings suggest that NF-κB plays an important role in the development of cardiac hypertrophy, as well as cardiac dysfunction. It should be noted that the heart weight and cardiomyocyte size in MAFbx KO mice are not significantly different from those in WT mice at baseline, despite significant accumulation of IκB-α. We speculate that other mechanisms override the MAFbx - NF-κB pathway to prevent baseline hypertrophy. NF-κB is one of the most important signaling pathways linked to the loss of skeletal muscle mass during aging, disuse, space travel, AIDS, sepsis, cancer, and chronic heart failure 27. This dichotomic function of NF-κB in cardiac and skeletal muscle may partially explain why MAFbx KO attenuates pathological hypertrophy in cardiac muscles but attenuates atrophy in skeletal muscle.
Although the complete suppression of PE-induced cardiac hypertrophy in the presence of MAFbx knock-down was reversed when NF-κB was overexpressed, the reversal was partial. We speculate that MAFbx may regulate additional molecules to mediate cardiac hypertrophy. Recent evidence suggests that MAFbx regulates expression of MPK-1, a phosphatase, thereby inducing persistent activation of JNKs and apoptosis of cardiomyocytes 28. We have shown previously, however, that activation of the MEKK1-JNK pathway during PO negatively regulates cardiac hypertrophy and cardiac dysfunction 29. Thus, the role of the MPK1-JNK pathway in mediating the effect of MAFbx during PO remains to be elucidated.
Our findings have significant clinical implications. First, endogenous MAFbx not only mediates hypertrophy, but also induces cardiac dysfunction during PO. Second, expression of MAFbx is increased in skeletal muscle in chronic heart failure 30. MAFbx is implicated in the development of muscle atrophy during cardiac cachexia 30. We therefore speculate that intervention to attenuate the function of MAFbx during cardiac hypertrophy and heart failure may not only ameliorate cardiac remodeling and dysfunction, but also prevent skeletal muscle atrophy, a significant complication in heart failure patients.
In conclusion, endogenous MAFbx plays an essential role in mediating pathological cardiac hypertrophy in response to PO, in part through nuclear accumulation of NF-κB (Online Fig. VII). We propose that MAFbx and its downstream signaling molecules, including NF-κB, could be important targets for future treatment of heart failure patients.
Supplementary Material
Acknowledgments
None
Sources of Funding
This work was supported in part by U.S. Public Health Service Grants HL59139, HL67724, HL69020, HL91469, HL102738, AG27211 (to JS), and HL98802 (to BT), an American Heart Association grant 0340123N (to JS), a Grant-in-Aid for Young Scientists B (22790697 to SU) from the Ministry of Education, Culture, Sports, Science and Technology, the Japan Heart Foundation/Novartis Grant for Research Award in molecular cellular cardiology, 2010 (to SU), and the Foundation of Leducq Transatlantic Network of Excellence (to JS).
Non-standard Abbreviations and Acronyms
- Ad
adenovirus
- ANF
atrial natriuretic factor
- αSA
α-skeletal actin
- BW
body weight
- EF
ejection fraction
- FS
fractional shortening
- GST-TUBE
glutathione S transferase- tandem ubiquitin binding entity
- ISO
isoproterenol
- LV
left ventricular
- MAFbx
muscle atrophy F-box
- PASR
picric acid sirius red
- PDTC
pyrrolidine dithiocarbamate
- PE
phenylephrine
- PO
pressure overload
- TAC
transverse aortic constriction
- TFBS
transcription factor binding sites
- TL
tibial length
- UPS
ubiquitin-proteasome system
- WT
wild type
Footnotes
Disclosures
None
References
- 1.Morisco C, Sadoshima J, Trimarco B, Arora R, Vatner DE, Vatner SF. Is treating cardiac hypertrophy salutary or detrimental: the two faces of Janus. Am J Physiol (Heart Circ Physiol) 2003;284:H1043–H1047. doi: 10.1152/ajpheart.00990.2002. [DOI] [PubMed] [Google Scholar]
- 2.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 3.Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 6.Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–478. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomes AV, Zong C, Ping P. Protein degradation by the 26S proteasome system in the normal and stressed myocardium. Antioxid Redox Signal. 2006;8:1677–1691. doi: 10.1089/ars.2006.8.1677. [DOI] [PubMed] [Google Scholar]
- 8.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 10.Stansfield WE, Tang RH, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294:H645–650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- 11.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patterson C, Ike C, Willis PWt, Stouffer GA, Willis MS. The bitter end: the ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. doi: 10.1161/CIRCULATIONAHA.106.649863. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto H, Takagi H, Tamamoti-Adachi T, Kitajima S, Sadoshima J. Phosphorylation of GSK-3α is essential for myocyte proliferation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–20905. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009;10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma S, Ying J, Razeghi P, Stepkowski S, Taegtmeyer H. Atrophic remodeling of the transplanted rat heart. Cardiology. 2006;105:128–136. doi: 10.1159/000090550. [DOI] [PubMed] [Google Scholar]
- 17.Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, Lecker S, Taegtmeyer H, Goldberg AL, Walsh K. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814–20823. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams V, Linke A, Wisloff U, Doring C, Erbs S, Krankel N, Witt CC, Labeit S, Muller-Werdan U, Schuler G, Hambrecht R. Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: Effect on myocardial contractility. Cardiovasc Res. 2007;73:120–129. doi: 10.1016/j.cardiores.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 19.Adams V, Linke A, Gielen S, Erbs S, Hambrecht R, Schuler G. Modulation of Murf-1 and MAFbx expression in the myocardium by physical exercise training. Eur J Cardiovasc Prev Rehabil. 2008;15:293–299. doi: 10.1097/HJR.0b013e3282f3ec43. [DOI] [PubMed] [Google Scholar]
- 20.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119:1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 23.Taniike M, Yamaguchi O, Tsujimoto I, Hikoso S, Takeda T, Nakai A, Omiya S, Mizote I, Nakano Y, Higuchi Y, Matsumura Y, Nishida K, Ichijo H, Hori M, Otsu K. Apoptosis signal-regulating kinase 1/p38 signaling pathway negatively regulates physiological hypertrophy. Circulation. 2008;117:545–552. doi: 10.1161/CIRCULATIONAHA.107.710434. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Ha T, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. NF-kappaB activation is required for the development of cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol. 2004;287:H1712–1720. doi: 10.1152/ajpheart.00124.2004. [DOI] [PubMed] [Google Scholar]
- 25.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation. 2005;111:2319–2325. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 26.Kawano S, Kubota T, Monden Y, Kawamura N, Tsutsui H, Takeshita A, Sunagawa K. Blockade of NF-kappaB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res. 2005;67:689–698. doi: 10.1016/j.cardiores.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 27.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- 28.Xie P, Guo S, Fan Y, Zhang H, Gu D, Li H. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J Biol Chem. 2009;284:5488–5496. doi: 10.1074/jbc.M806487200. [DOI] [PubMed] [Google Scholar]
- 29.Sadoshima J, Montagne O, Wang QM, Yang GP, Warden J, Liu J, Takagi G, Karoor V, Hong C, Johnson GL, Vatner DE, Vatner SF. The MEKK1-JNK pathway plays a protective role in pressure overload, but does not mediate cardiac hypertrophy. J Clin Invest. 2002;110:271–279. doi: 10.1172/JCI14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li P, Waters RE, Redfern SI, Zhang M, Mao L, Annex BH, Yan Z. Oxidative phenotype protects myofibers from pathological insults induced by chronic heart failure in mice. Am J Pathol. 2007;170:599–608. doi: 10.2353/ajpath.2007.060505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.