

Abstract
Increases in oxidative stress in the heart play an important role in mediating hypertrophy, apoptosis, fibrosis, mitochondrial dysfunction and the consequent development of heart failure. Although it has been widely believed that electron leakage from the mitochondrial electron transport chain is the primary source of oxidative stress in the failing heart, increasing lines of evidence suggest that enzymes which produce reactive oxygen species (ROS) may also contribute to it. NADPH oxidases are transmembrane enzymes dedicated to producing superoxide (O2-) by transferring an electron from NAD(P)H to molecular oxygen. Nox4 is a major NADPH oxidase isoform expressed in the heart. Nox4 is localized primarily at mitochondria in cardiac myocytes, and upregulation of Nox4 hypertrophic stimuli enhances O2- production, apoptosis, and mitochondrial dysfunction, thereby playing an important role in mediating cardiac dysfunction. Since Nox4 could be a key molecule mediating oxidative stress and pathological hypertrophy, it may serve as an important target of heart failure treatment. In this review, the importance of NADPH oxidases as sources of increased oxidative stress in the failing heart and the role of Nox4 in mediating growth and death of cardiac myocytes are discussed.
Keywords: NADPH oxidase, cardiac failure, O2-, reactive oxygen species, apoptosis, cardiac myocytes, hypertrophy, mitochondrial dysfunction
Reactive oxygen species (ROS) are defined as molecules derived from oxygen with characteristic instability and chemical reactivity, and include both free radicals, such as superoxide (O2-) and hydroxyl radical (OH-), and non-radicals, such as hydrogen peroxide (H2O2) and singlet oxygen (1O2). Excessive production of ROS causes damage in tissue/cellular constituents, including DNA, proteins, and lipids, leading to cellular/organ dysfunction and consequent cell death [1,2]. However, regulated generation of ROS at low levels mediates physiological functions, such as cell survival, growth, differentiation, and metabolism. The NADPH oxidase (Nox) family enzymes are a major cellular source of O2-, and of H2O2, into which O2- is rapidly converted [3,4]. ROS generated by Noxs may be involved in various physiological and pathological processes in the heart, such as hypertrophy, apoptosis, heart failure and hypoxic adaptation [5]. The purpose of this review is to highlight the function of Nox4, an isoform of the Nox family abundantly expressed in the heart, and to discuss its potential as a target for drug therapy in heart failure patients.
NADPH oxidases
The first identified example of an enzyme dedicated to producing O2- was an NADPH oxidase expressed in mammalian phagocytes [3,6]. During the engulfment of invading microbes, the phagocyte NADPH oxidase becomes activated to reduce molecular oxygen to O2−, a precursor of microbicidal ROS, in conjunction with oxidation of NADPH. The catalytic core of the phagocyte NADPH oxidase is gp91phox, a membrane-integrated glycoprotein with a molecular mass of 91 kD. gp91phox contains two hemes in the N-terminal transmembrane region, and NADPH- and FAD-binding domains in the C-terminal cytoplasmic region, forming a complete electron-transferring apparatus through which an electron provided by NADPH is transferred to molecular oxygen on the other side of membrane. Searches in genome databases led to the identification of novel homologs of gp91phox known as Nox (NADPH oxidase) or Duox (dual oxidase) [3,6]. The human genome contains seven genes encoding gp91phox homologs: Nox1-Nox5, where gp91phox is renamed Nox2, and the distantly related oxidases Duox1 and Duox2. Nox1-Nox4 share conserved structural features, including two hemes and FAD- and NADPH-binding domains, and form a complex with p22phox, another membrane-integrated protein. Although Nox1-Nox3 require additional cytosolic factors for their ROS producing activity, Nox4 does not [7,8] (Fig. 1). For example, activation of Nox2 requires stimulus-evoked membrane translocation of cytosolic factors, including p47phox, p67phox, p40phox, and Rac, a small GTPase, and formation of an active oxidase complex at the membrane. Although the activity of Nox4 may also be modulated by interaction with unique proteins, such as Poldip2 [9], the contributions of such mechanisms to the overall activity of Nox4 remain to be elucidated. Due to the difference in the activation mechanism, it is believed that O2- production by Nox2 is inducible, whereas that by Nox4 is constitutive. Since O2- production by Nox2 during phagocytosis is quick and robust, it is called a “respiratory burst”. On the other hand, changes in O2- production by Nox4 take place primarily at the level of mRNA (transcription) [10,11]. The heart and cardiac myocytes therein primarily express Nox2 [12-14] and Nox4 [15,16]. In order to elucidate the function of endogenous Nox4 in an isoform specific manner, we have recently generated cardiac-specific Nox4 -/- mice (c-Nox4 -/- mice). SOD-inhibitable O2- production in whole heart homogenates was markedly less in c-Nox4 -/- mice than in wild-type controls, indicating that Nox4 in cardiac myocytes is a major source of basal O2- production in the mouse heart [17].
Figure 1.
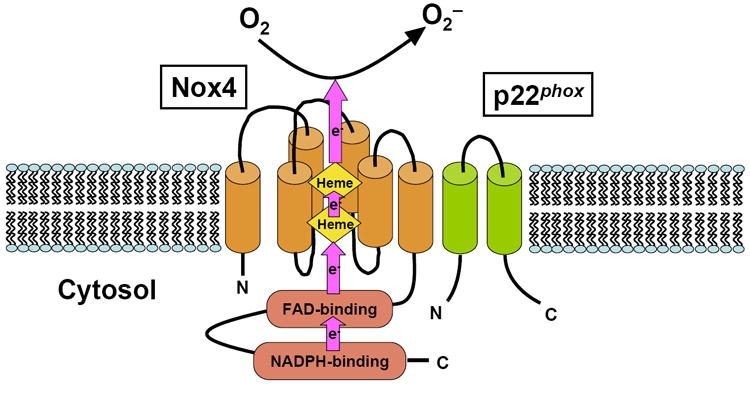
The molecular structure of Nox4. Nox4 forms a heterodimer with p22phox. Nox family members, including Nox4, have a complete electron-transferring apparatus within their own molecules. Arrows indicate flow of electrons. Cylinders represent six transmembrane α-helices.
Cardiac hypertrophy
Cardiac hypertrophy is an independent risk factor for the development of heart failure and a major cause of mortality [18]. ROS play an essential role in mediating hypertrophy in response to α-adrenergic agonists [14], angiotensin II (Ang II) [19], endothelin-1 [20], and TNF-α [19] in cultured cardiac myocytes. Increasing lines of evidence suggest that activation of pro-hypertrophic transcription factors, including MEF2, NFAT and GATA4, and the nuclear exit of class II histone deacetylases (HDACs), such as HDAC5, play an essential role in mediating pathological hypertrophy [21,22]. We have shown recently that nuclear exit of class II HDACs is mediated not only through phosphorylation by HDAC kinases, such as PKD, Ca2+/CaM kinase and GRK5, but also by oxidation of evolutionarily conserved cysteine residues [23]. Stimulation of cultured cardiac myocytes with phenylephrine induces formation of a disulfide bond between Cysteines 667 and 669 in HDAC4, which in turn induces nuclear exit of HDAC4 through a Crm-1-dependent nuclear export mechanism. At present, however, the origins of the ROS responsible for oxidation of HDAC4 remain to be shown. Considering the fact that phenylephrine induces cysteine oxidation of HDAC4 rapidly (within 5 min), the involvement of enzymes allowing rapid production of ROS, such as Noxs, is plausible.
NADPH oxidases are involved in α-adrenergic agonists-induced cardiac hypertrophy [14]. NADPH oxidase activity is enhanced in cardiac hypertrophy induced by pressure overload, which is accompanied by upregulation of Nox2 in both cardiomyocytes and endothelial cells [24]. However, cardiac hypertrophy in response to aortic banding is not attenuated or enhanced in systemic Nox2 -/- mice compared to wild type (WT) controls [16,25]. Increases in total NADPH oxidase activity under pressure overload are also similar between systemic Nox2 -/- and WT mice [16], suggesting the involvement of Nox4, the other major Nox isoform in the heart. In our preliminary studies, increases in oxidative stress in response to pressure overload were significantly reduced in c-Nox4 -/- mice compared to WT mice. c-Nox4 -/- mice exhibited significantly less hypertrophy and better cardiac function than WT mice in response to pressure overload. These results suggest that ROS produced by Nox4 may contribute to the formation of hypertrophy and exacerbation of cardiac dysfunction in response to pressure overload. It should be noted that mice with cardiac-specific overexpression of Nox4 induce cardiac dysfunction without obvious cardiac hypertrophy at the organ level, raising the possibility that the contribution of Nox4 to cardiac hypertrophy may be secondary to cardiac dysfunction. In fact, overexpression of Nox4 induces apoptotic cell death but not hypertrophy in cultured cardiac myocytes, suggesting that the primary effect of Nox4 is cell death rather than cell growth in cardiac myocytes. Interestingly, cardiac hypertrophy in response to Ang II infusion is markedly inhibited in Nox2 -/- mice [12]. Cardiac hypertrophy after chronic myocardial infarction is significantly attenuated in p47phox -/- mice [26]. Since p47phox is an essential cytosolic cofactor for Nox2, this result supports the idea that Nox2 plays a role in mediating cardiac remodeling after myocardial infarction. These results suggest that Nox2 and Nox4 have distinct roles in mediating hypertrophy. A possible explanation for these distinct roles could be that Nox2 preferentially mediates cardiac hypertrophy in which autocrine/paracrine factors and G protein signaling are involved. As we discuss below, Nox2 and Nox4 have distinct subcellular localizations. Identifying the specific signaling molecule whose activity is regulated by each Nox isoform may allow us to elucidate the molecular mechanisms of cardiac hypertrophy and to develop specific modalities to treat pathological hypertrophy.
Apoptosis
Loss of terminally differentiated cardiac myocytes contributes to the development of heart failure [27]. Oxidative stress induces apoptosis either through damaging DNA, lipids, and proteins, or modulating proapoptotic signaling pathways such as ASK-1, JNK, ERK1/2, and p38 MAPK [28]. Induction of apoptosis in cultured cardiac myocytes by Ang II was abolished in the presence of apocynin, indicating that NADPH oxidase is involved. However, since apocynin may not be entirely specific against NADPH oxidases, the involvement of other apoptotic mechanisms cannot be excluded [29]. Ang II-induced apoptosis is mediated partly through NADPH oxidase-dependent peroxynitrite formation and consequent DNA damage [30]. Although O2- production from Nox2 is acutely increased in response to Ang II stimulation, protein expression of Nox4 is also increased chronically. Thus, it remains to be elucidated which Nox isoform is involved in Ang II-induced apoptosis. Apoptosis was increased in transgenic mice with cardiac-specific overexpression of Nox4 compared to non-transgenic controls. Increased expression of Nox4 potently induces apoptosis in cultured cardiac myocytes, suggesting that the proapoptotic effect of Nox4 is cell autonomous [31]. These results suggest that chronic upregulation of Nox4 by hypertrophic stimuli and aging increases apoptosis, which may contribute to gradual decreases in LV function. Nox4-induced apoptosis in cardiac myocytes was accompanied by cytochrome c release and prevented in the presence of Bcl-xL, suggesting that the mitochondrial apoptotic pathway is activated [31]. As we discuss below, we have shown recently that Nox4 is predominantly localized in mitochondria. Local production of ROS, including H2O2, OH- and peroxynitrate, efficiently induces mitochondrial damage, thereby leading to MPTP opening and cell death. Identifying which mitochondrial target of Nox4 critically mediates apoptosis is of great interest.
Fibrosis
Interstitial and perivascular fibrosis are characteristic features of pathological cardiac hypertrophy [32]. Oxidative stress is associated with increases in fibrosis in many organs, including lungs [33], liver [34], and kidneys [35,36]. Increased Nox activity has been implicated in vascular fibrosis as well [37,38]. In the heart, interstitial fibrosis induced by Ang II is significantly attenuated in systemic Nox2 -/- mice [12,16]. Nox2 -/- mice also exhibit significantly less fibrosis in response to pressure overload than WT mice, despite the fact that they develop a similar degree of hypertrophy. Nox2 critically regulates profibrotic mechanisms, including activation of NF-κB and upregulation of connective tissue growth factor (CTGF) and matrix metalloproteinase-2 (MMP-2) [39]. Since these observations were made with systemic Nox2 -/- mice, it remains unknown whether fibrosis is regulated by Nox2 in cardiac myocytes or non-myocytes. Nox4 plays a critical role in mediating cardiac fibroblast proliferation and transformation into myofibroblasts [40]. We have recently shown that cardiac-specific overexpression of Nox4 induces more fibrosis in an age-dependent manner, whereas that of dominant negative Nox4 leads to significantly less fibrosis, indicating that Nox4 is also involved in interstitial fibrosis. Since the activity of Nox4 was modulated in a myocyte specific manner in these experiments, the direct consequence of Nox4 expression in fibroblasts remains to be elucidated. Since Nox4 promotes apoptosis in cardiac myocytes, it would be interesting to evaluate whether Nox4 promotes cell growth/proliferation in fibroblasts. Nox4 induces cell proliferative responses in various cell types, including endothelial cells [41], VSMCs [42], and mesangial cells [43]. Thus, it is possible that upregulation of Nox4 induces proliferation rather than apoptosis in cardiac fibroblasts.
Where are ROS generated in the failing heart?
Experimental and clinical studies have suggested that ROS in the heart are increased during heart failure [44-47]. The extent of oxidative stress in the heart is affected by the balance between the production and the clearance of ROS. Although some reports described that antioxidants are downregulated in failing hearts [48,49], others have shown that O2- dismutase (SOD), catalase, and glutathione peroxidase (GSHPx) are not necessarily decreased [50]. ROS are deleterious byproducts of aerobic metabolism, and the mitochondrial electron transport chain is considered to be the primary source of this type of ROS production [51]. Among the various cell types in the human body, cardiac myocytes have the highest volume density of mitochondria in order to meet the high energy demand [52]. Under physiological conditions, the small amount of ROS formed through mitochondrial respiration is efficiently removed by endogenous antioxidants, including MnSOD [52]. However, in the failing heart, suppression/damage of the mitochondrial electron transport at Complex I and Complex III stimulates leakage of electrons and accumulation of ROS [53]. In addition, some enzyme molecules, such as xanthine oxidases [54,55] and uncoupled endothelial NO synthase [56,57], actively produce O2- during heart failure. We have shown that expression of Nox4 in the heart is upregulated by hypertrophic stimuli, including pressure overload [31]. In a mouse model of chronic myocardial infarction, increases in ROS are suppressed in p47phox -/- mice, suggesting that the p47phox-dependent Nox, most likely Nox2, is responsible for the increased oxidative stress during cardiac remodeling [26]. Since the activity of NADPH oxidases is increased in human heart failure [58], we speculate that either Nox2 or Nox4 contributes to the increased oxidative stress in the failing heart.
O2- produced by Noxs rapidly dismutates to H2O2, which in turn easily spreads through either plasma or intracellular membranes. Although Nox2 is localized primarily at the plasma membrane, Nox4 is expressed on intracellular membranes [7,31]. Activation of Noxs could lead to local elevation of O2- and diffuse increases in H2O2 levels in cells. Prolonged oxidative stress in the failing myocardium results in damage to mitochondrial DNA and proteins, which further stimulates ROS generation (so called “ROS-induced ROS release” [53]) and cellular injury, leading to the functional decline in the heart. Thus, mitochondria are both the source and the target of oxidative stress in the failing heart. Mice with a cardiac/skeletal muscle specific deficiency in SOD develop progressive heart failure, secondary to excess O2- production, and defects in mitochondrial respiration [59], suggesting the importance of mitochondria as a source of ROS and/or a facilitator of the ROS-induced ROS release. We have shown that Nox4 is predominantly expressed in mitochondria in cardiac myocytes [31]. When expression of Nox4 in mitochondria is upregulated by aging, hypertrophic stimuli and heart failure, increased production of O2- in mitochondria leads to iron release from mitochondrial proteins containing iron-sulfur clusters and consequent mitochondrial dysfunction. We have shown that oxidation of the MPTP complex during pressure overload is attenuated by overexpression of thioredoxin 1 in the mouse heart [60], suggesting that mitochondrial proteins are highly sensitive to the redox status in cells. Furthermore, we have shown recently that cysteine residues in NADH dehydrogenase flavoprotein I, a component of Complex I, and in ANT1, a key component of the MPTP complex, together with aconitase-2, an established redox sensitive protein, are highly oxidized and their function is inhibited in transgenic mice with cardiac specific overexpression of Nox4 [31], suggesting that they may be directly modulated by O2- generated by Nox4.
It should be noted that activation of Noxs induces activation of other ROS producing enzymes, such as xanthine oxidase and uncoupled endothelial NO synthase, and oxidation/inactivation of anti-oxidants, such as thioredoxin1 [26,56,61]. Thus, activation of Noxs amplifies oxidative stress through multiple mechanisms. Suppressing a key element in the amplification mechanism may allow one to efficiently extinguish the increase in oxidative stress. We believe that Nox4 may be such a key molecule driving oxidative stress in the failing heart.
Conclusions
Increasing lines of evidence suggest that NADPH oxidases in the heart, including Nox4, are major producers of O2- in cardiac myocytes, and have a crucial role in the development of cardiac failure (Fig. 2). Nox4 may be involved in aging, hypertrophy, apoptosis, fibrosis and regulation of mitochondrial function in the heart and cardiac myocytes and non-myocytes therein. However, many questions remain unanswered regarding the isoform specific function of Nox4 in the heart. These include but are not limited to: “What is the molecular mechanism by which expression of Nox4 is upregulated in response to hypertrophic stimuli?”, “Is any cytosolic factor involved in the regulation of Nox4?”, “What are the downstream targets whose function is modulated by O2- produced by Nox4?”, “What is the physiological function of basal O2- produced by Nox4?”, and “What is the role of Nox4 in cardiac fibroblasts?” Although Nox2 and Nox4 appear to have both common and distinct functions in the heart, their functions should be compared side by side in order to better understand the isoform specific functions of Noxs. As development of an inhibitor for HDAC kinases effectively suppressing pathological hypertrophy is actively pursued, small molecules inhibiting oxidation or stimulating reduction of class II HDACs may also be considered for prevention/treatment of heart failure. Considering the fact that Nox4 promotes cardiac myocyte apoptosis, mitochondrial dysfunction and cardiac dysfunction, Nox4 is a promising target for treatment of heart failure. Although the specificity of currently available small molecule inhibitors for Noxs appears to be too low for clinical use [62-64], future development of specific inhibitors for Nox4 may allow for treatment of heart failure patients by effectively extinguishing cellular sources of ROS and halting the vicious cycle of oxidative stress during heart failure.
Figure 2.
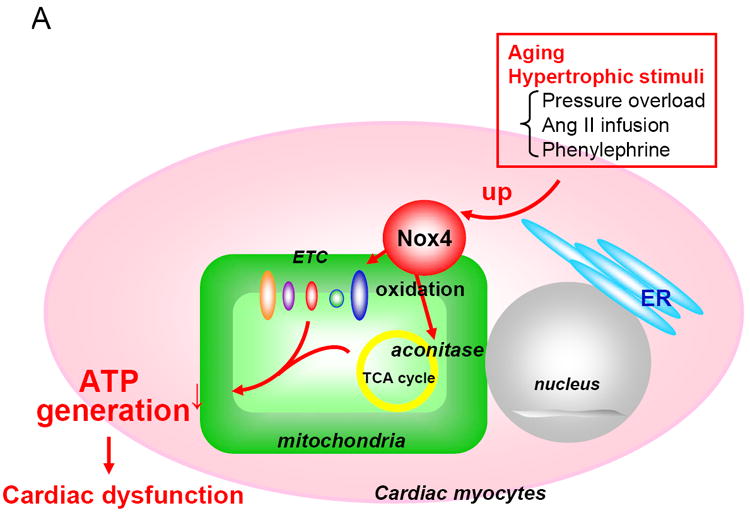
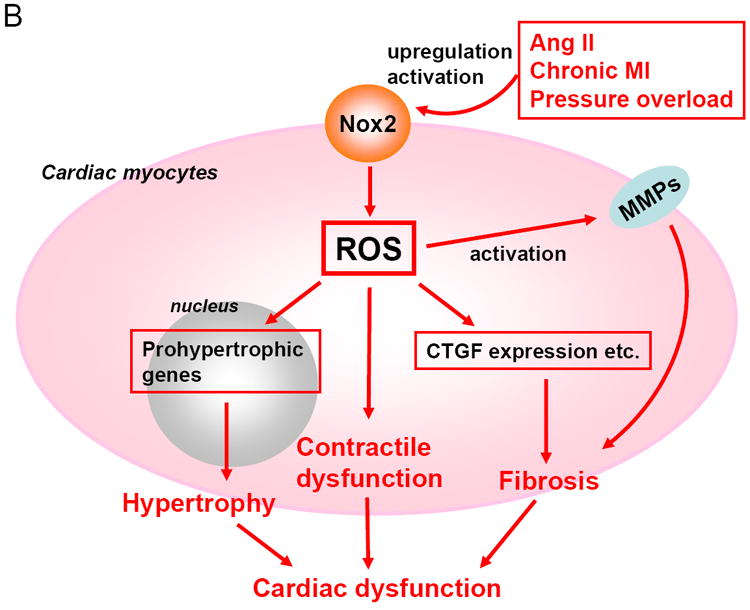
Proposed functions of Nox4 in the heart. Nox4 is localized primarily at mitochondria in cardiac myocytes. Nox4 is upregulated by aging and hypertrophic stimuli. Upregulation of Nox4 increases production of ROS in mitochondria, thereby inducing oxidation of mitochondrial proteins, such as aconitase, and mitochondrial dysfunction, which may play an important role in mediating cardiac dysfunction.
Table.
The involvement of Nox isoforms in cardiac pathophysiology
| Hypertrophic stimuli (models) | Description | References |
|---|---|---|
| Pressure overload (Aortic banding) | Nox2 is upregulated in both cardiac myocytes and endothelial cells. | Xiao et al. (11) |
| Cardiac hypertrophy is not affected in systemic Nox2 -/- mice. | Byrne et al. (13), Maytin et al. (22) | |
| Nox2 -/- mice exhibit significantly less fibrosis. | Byrne et al. (13) | |
| Nox4 is upregulated in cardiac myocytes. | Ago et al. (28) | |
| Oxidative stress, hypertrophy, fibrosis and LV dysfunction are attenuated in cardiac specific Nox4 -/- mice. | Kuroda et al. (Submitted) | |
|
| ||
| Ang II infusion | Cardiac hypertrophy is markedly inhibited in Nox2 -/- mice. | Bendall et al. (10) |
| Interstitial fibrosis is significantly attenuated in systemic Nox2 -/- mice. | Bendall et al. (10), Byrne et al. (13) | |
| Nox4 is upregulated in cardiac myocytes. | Ago et al. (28) | |
|
| ||
| Chronic MI | Cardiac hypertrophy is significantly attenuated in p47phox -/- mice. | Doerries et al. (23) |
| Oxidative stress is suppressed in p47phox mice. | ||
|
| ||
| Aging | Cardiac specific overexpression of Nox4 exacerbates LV function, oxidative stress, apoptosis, and fibrosis in transgenic mice. | Ago et al. (28) |
Acknowledgments
The authors thank Daniela Zablocki for critical reading.
Sources of Funding This work was supported in part by U.S. Public Health Service Grants HL 59139, HL67724, HL69020, HL91469, and AG27211.
Footnotes
Disclosures None
References
- 1.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115(3):500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15(2):247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 3.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Akki A, Zhang M, Murdoch C, Brewer A, Shah AM. NADPH oxidase signaling and cardiac myocyte function. J Mol Cell Cardiol. 2009;47(1):15–22. doi: 10.1016/j.yjmcc.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338(1):677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- 7.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47(9):1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275(13):3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 9.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, et al. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105(3):249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res. 2005;65(2):495–504. doi: 10.1016/j.cardiores.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406(1):105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105(3):293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 13.Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. 2006;26(3):180–191. doi: 10.1152/physiolgenomics.00029.2005. [DOI] [PubMed] [Google Scholar]
- 14.Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol. 2002;282(4):C926–934. doi: 10.1152/ajpcell.00254.2001. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, et al. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell. 2006;17(9):3978–3988. doi: 10.1091/mbc.E05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93(9):802–805. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 17.Kuroda J, Ago T, Sadoshima J. Abstract 2275: Nox4 is a Major Source of Superoxide Production in the Mouse Heart. Circulation. 2009;120(18 Supplement):S614. [Google Scholar]
- 18.Artham SM, Lavie CJ, Milani RV, Patel DA, Verma A, Ventura HO. Clinical impact of left ventricular hypertrophy and implications for regression. Prog Cardiovasc Dis. 2009;52(2):153–167. doi: 10.1016/j.pcad.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98(8):794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 20.Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105(4):509–515. doi: 10.1161/hc0402.102863. [DOI] [PubMed] [Google Scholar]
- 21.Oka S, Ago T, Kitazono T, Zablocki D, Sadoshima J. The role of redox modulation of class II histone deacetylases in mediating pathological cardiac hypertrophy. J Mol Med. 2009;87(8):785–791. doi: 10.1007/s00109-009-0471-2. [DOI] [PubMed] [Google Scholar]
- 22.Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure. Expert Opin Ther Targets. 2009;13(7):767–784. doi: 10.1517/14728220902939161. [DOI] [PubMed] [Google Scholar]
- 23.Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133(6):978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 24.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40(4):477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 25.Maytin M, Siwik DA, Ito M, Xiao L, Sawyer DB, Liao R, et al. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation. 2004;109(9):1168–1171. doi: 10.1161/01.CIR.0000117229.60628.2F. [DOI] [PubMed] [Google Scholar]
- 26.Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, et al. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res. 2007;100(6):894–903. doi: 10.1161/01.RES.0000261657.76299.ff. [DOI] [PubMed] [Google Scholar]
- 27.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111(10):1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal. 2005;7(3-4):472–481. doi: 10.1089/ars.2005.7.472. [DOI] [PubMed] [Google Scholar]
- 29.Qin F, Patel R, Yan C, Liu W. NADPH oxidase is involved in angiotensin II-induced apoptosis in H9C2 cardiac muscle cells: effects of apocynin. Free Radic Biol Med. 2006;40(2):236–246. doi: 10.1016/j.freeradbiomed.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Grishko V, Pastukh V, Solodushko V, Gillespie M, Azuma J, Schaffer S. Apoptotic cascade initiated by angiotensin II in neonatal cardiomyocytes: role of DNA damage. Am J Physiol Heart Circ Physiol. 2003;285(6):H2364–2372. doi: 10.1152/ajpheart.00408.2003. [DOI] [PubMed] [Google Scholar]
- 31.Ago T, Kuroda J, Zhai P, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Submitted. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 33.Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172(4):417–422. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Aspects Med. 2000;21(3):49–98. doi: 10.1016/s0098-2997(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 35.Ha H, Lee HB. Reactive oxygen species and matrix remodeling in diabetic kidney. J Am Soc Nephrol. 2003;14(8 Suppl 3):S246–249. doi: 10.1097/01.asn.0000077411.98742.54. [DOI] [PubMed] [Google Scholar]
- 36.Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am J Hypertens. 2004;17(7):597–603. [PubMed] [Google Scholar]
- 37.Rey FE, Pagano PJ. The reactive adventitia: fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol. 2002;22(12):1962–1971. doi: 10.1161/01.atv.0000043452.30772.18. [DOI] [PubMed] [Google Scholar]
- 38.An SJ, Boyd R, Zhu M, Chapman A, Pimentel DR, Wang HD. NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts. Cardiovasc Res. 2007;75(4):702–709. doi: 10.1016/j.cardiores.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006;20(9):1546–1548. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 40.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97(9):900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 41.Peshavariya H, Dusting GJ, Jiang F, Halmos LR, Sobey CG, Drummond GR, et al. NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedebergs Arch Pharmacol. 2009;380(2):193–204. doi: 10.1007/s00210-009-0413-0. [DOI] [PubMed] [Google Scholar]
- 42.Menshikov M, Plekhanova O, Cai H, Chalupsky K, Parfyonova Y, Bashtrikov P, et al. Urokinase plasminogen activator stimulates vascular smooth muscle cell proliferation via redox-dependent pathways. Arterioscler Thromb Vasc Biol. 2006;26(4):801–807. doi: 10.1161/01.ATV.0000207277.27432.15. [DOI] [PubMed] [Google Scholar]
- 43.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Nox4 mediates angiotensin II-induced activation of Akt/protein kinase B in mesangial cells. Am J Physiol Renal Physiol. 2003;285(2):F219–229. doi: 10.1152/ajprenal.00414.2002. [DOI] [PubMed] [Google Scholar]
- 44.Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65(5):245–248. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol. 1996;148(1):291–300. [PMC free article] [PubMed] [Google Scholar]
- 46.Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86(2):152–157. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- 47.Hill MF, Singal PK. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96(7):2414–2420. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- 48.Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A, Bristow MR, et al. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005;11(6):473–480. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 49.Baumer AT, Flesch M, Wang X, Shen Q, Feuerstein GZ, Bohm M. Antioxidative enzymes in human hearts with idiopathic dilated cardiomyopathy. J Mol Cell Cardiol. 2000;32(1):121–130. doi: 10.1006/jmcc.1999.1061. [DOI] [PubMed] [Google Scholar]
- 50.Tsutsui H, Ide T, Hayashidani S, Suematsu N, Utsumi H, Nakamura R, et al. Greater susceptibility of failing cardiac myocytes to oxygen free radical-mediated injury. Cardiovasc Res. 2001;49(1):103–109. doi: 10.1016/s0008-6363(00)00197-8. [DOI] [PubMed] [Google Scholar]
- 51.Tsutsui H. Mitochondrial oxidative stress and heart failure. Intern Med. 2006;45(13):809–813. doi: 10.2169/internalmedicine.45.1765. [DOI] [PubMed] [Google Scholar]
- 52.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81(3):449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 53.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85(4):357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 54.George J, Struthers AD. The role of urate and xanthine oxidase inhibitors in cardiovascular disease. Cardiovasc Ther. 2008;26(1):59–64. doi: 10.1111/j.1527-3466.2007.00029.x. [DOI] [PubMed] [Google Scholar]
- 55.Bergamini C, Cicoira M, Rossi A, Vassanelli C. Oxidative stress and hyperuricaemia: pathophysiology, clinical relevance, and therapeutic implications in chronic heart failure. Eur J Heart Fail. 2009;11(5):444–452. doi: 10.1093/eurjhf/hfp042. [DOI] [PubMed] [Google Scholar]
- 56.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Otani H. The role of nitric oxide in myocardial repair and remodeling. Antioxid Redox Signal. 2009;11(8):1913–1928. doi: 10.1089/ars.2009.2453. [DOI] [PubMed] [Google Scholar]
- 58.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41(12):2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 59.Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem. 2006;281(44):33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- 60.Fu C, Wu C, Liu T, Ago T, Zhai P, Sadoshima J, et al. Elucidation of thioredoxin target protein networks in mouse. Mol Cell Proteomics. 2009;8(7):1674–1687. doi: 10.1074/mcp.M800580-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McNally JS, Saxena A, Cai H, Dikalov S, Harrison DG. Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterioscler Thromb Vasc Biol. 2005;25(8):1623–1628. doi: 10.1161/01.ATV.0000170827.16296.6e. [DOI] [PubMed] [Google Scholar]
- 62.Williams HC, Griendling KK. NADPH oxidase inhibitors: new antihypertensive agents? J Cardiovasc Pharmacol. 2007;50(1):9–16. doi: 10.1097/FJC.0b013e318063e820. [DOI] [PubMed] [Google Scholar]
- 63.Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol Ther. 2008;120(3):254–291. doi: 10.1016/j.pharmthera.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 64.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11(10):2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]